

Abstract
Many xenobiotics containing a furan ring are toxic and/or carcinogenic. The harmful effects of these compounds require furan ring oxidation. This reaction generates an electrophilic intermediate. Depending on the furan ring substituents, the intermediate is either an epoxide or a cis-enedione with more ring substitution favoring epoxide formation. Either intermediate reacts with cellular nucleophiles such as protein or DNA to trigger toxicities. The reactivity of the metabolite determines which cellular nucleophiles are targeted. The toxicity of a particular furan is also influenced by the presence of competing metabolic pathways or efficient detoxification routes. GSH plays an important role in modulating the harmful effects of this class of compound by reacting with the reactive metabolite. However, this may not represent a detoxification step in all cases.
Keywords: furan, metabolism, reactive metabolites, epoxide, enedione
1 Introduction
Furan-containing compounds are abundant in food, synthetic and herbal medicines, industrial processes and the environment.1–5 Many but not all of these compounds are toxic, causing concerns when human exposure is likely (Table 1). Small changes in structure affect which organ is targeted as well as what toxicity is observed. The harmful effects result from cytochrome P450 (P450) catalyzed furan ring oxidation.1,6 This oxidation leads to the formation of a reactive metabolite which alkylates proteins and, in some cases, DNA. Adduct formation is thought to be an important trigger to the toxic effects with the reactive intermediate’s chemical nature playing an influential role in which cellular targets are hit. Because furan is a structure alert for medicinal chemists and risk assessors,7,8 understanding how structure influences the formation, lifetime and targets of the reactive intermediate is critical for the design of safe drugs and the determination of which furan-containing compounds may represent a human health risk.
Table 1.
Representative Furan-containing Compounds: Their source, toxicity and the nature of the reactive intermediate
| Compound | Source | Toxicity | Reactive Intermediate |
|---|---|---|---|
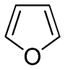 Furan |
Present in processed foods, smog, wood smoke, tobacco smoke and car exhaust20–24 | Rat and mouse liver toxicant and carcinogen when dosed orally25,41–43 | enedialdehyde14,57,58 |
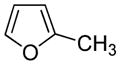 2-Methylfuran |
Present in foods, coffee and tobacco smoke.21,22,66 | Rat lung and liver toxicant when dosed i.p.67 | enedione10 |
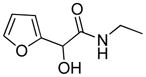 2-(N-ethylcarbamoyl-hydroxymethyl)-furan |
Synthetic | Rat lung toxicant when dosed orally18 | Unknown furanyl epoxide unlikely18 |
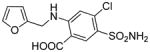 Furosemide |
Pharmaceutical | Not toxic to humans at therapeutic dose Associated with hypersensitivity and jaundice in a few cases.72,73 Not toxic to rats or hamsters75,76 Mouse liver toxicant at high i.p. doses74,75 |
Furanyl epoxide16 and enedione75 |
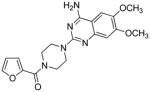 Prazosin |
Pharmaceutical | Not toxic | enedione19 |
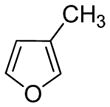 3-Methylfuran |
Present in indoor and outdoor air pollution,4,5 food,88 and tobacco smoke.89,90 Mold metabolite.5 |
Lung toxicant when inhaled or given by i.p.91,93–97 | enedione10 |
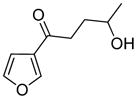 4-Ipomeanol |
Produced by moldy sweet potatoes.98 | Lung toxicant in lab animals98,100,101 Liver toxicant in humans102–104 |
unclear |
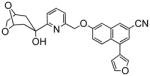 L-739,010 |
Synthetic | No overt toxic properties.117 Binds to proteins.116,118 |
enedione116,118 |
 Teucrin A |
Germander extracts120 | Germander extracts are human and mouse liver toxicant.120–128 Mouse liver toxicant129 |
Furanyl epoxide128,131 and/or enedione15 |
 Menthofuran |
Pulegone, pennyroyal oil136–139 | Mouse liver and lung toxicant133,140 Rat liver toxicant.141 Pennyroyal oil is human toxicant134,135 Mechanism based inactivator of P450146,147 |
Furanyl epoxide13 and/or enedione141,141,143,144 |
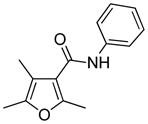 Methfuroxam |
Fungicide | furanyl epoxide12 | |
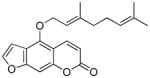 Bergamottin |
Grapefruit juice152–157 | Mechanism based inactivator of P450154–157,159–161 | unclear |
 L-754,394 |
Synthetic | Mechanism based inactivator of P450.162 | unclear |
The structure of the reactive intermediate resulting from furan ring oxidation is somewhat ambiguous. Two structures have been proposed: an epoxide or a cis-enedione (Scheme 1). Furan oxidation by P450 enzymes is thought to proceed through one of two general mechanisms.9,10 The first involves the direct formation of an epoxide. The other involves the addition of the high valent iron(IV)-oxospecies to the π-system of the furan ring to produce a tetrahedral intermediate or cationic σ complex that can rearrange to yield either an epoxide (1) or a zwitterionic intermediate (2, Scheme 1). Either intermediate 1 or 2 can rearrange to form a cis-enedione (3, Scheme 1).10
Scheme 1.
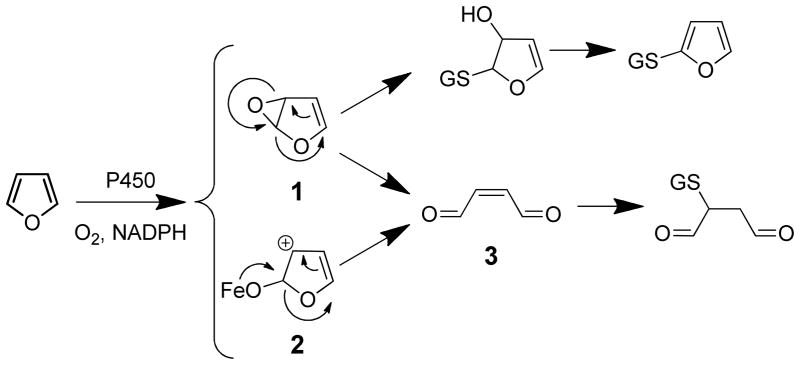
Oxidation of furan by P450 enzymes to either an epoxide 1 or a cis-enedione 3 metabolite and their reaction with GSH.9,10
Evidence for the involvement of furanyl epoxide intermediates has been sought in several indirect ways since they are difficult to isolate for chemical characterization. The potential for their formation can be determined through the reaction of the parent compound with oxidizing agents that generate epoxides, such as dimethyldioxirane.11 Transient formation of furanyl epoxides have been reported for a few furan-containing compounds, including methfuroxam (2,4,5-trimethyl-N-phenyl-3-furancarboxamide)12 and menthofuran [(R)-3,6-dimethyl-4,5,6,7-tetrahydrobenzofuran] (Table 1).13 Both compounds have multiple substitutions on the furan ring. Therefore, the epoxide generated is sterically hindered, possibly stabilizing the reactive intermediate. No epoxide intermediates have been detected for a number for furans, including furan11,14 and teucrin A.15 For these compounds, the products of furanyl oxidation are cis-enediones. 11,14,15 The inability to detect the epoxide intermediates may be due to their extreme instability and ease at which they rearrange to a cis-enedione.
Additional support for the participation of an epoxide in the toxicity of furanyl compounds comes from epoxide hydrolase inhibition studies performed in vitro with 1,2-epoxy-3,3,3-trichloropropane (TCPO). The results of these experiments have been compound dependent. For example, TCPO increased the protein binding of furosemide 2-fold16 whereas it had no effect on the protein binding of 4-ipomeanol17 or 2-(N-ethylcarbamoylhydroxymethyl)-furan.18 The inability of TCPO to inhibit protein binding of an epoxide can be interpreted several ways. First, it may mean that there is no epoxide intermediate. However, a negative result does not rule out the involvement of an epoxide in the formation of protein or DNA damage. It is possible that it is not a substrate for epoxide hydrolase or that it is too reactive to be detoxified by epoxide hydrolase.
Trapping experiments indicate that cis-enedione intermediates are reactive metabolites of many furans. Inclusion of semicarbazide in microsomal incubations of a number of furan-containing compounds results in the trapping of the reactive metabolite as either a bis-semicarbazone or a pyridazine derivative.14,19 Glutathione (GSH) trapping reactions also are consistent with the involvement of a cis-enedione intermediate with preferential formation of a 1,4-addition product as opposed to that resulting from GSH addition to the carbon adjacent to the furanyl oxygen (Scheme 1). All these observations do not preclude initial epoxide formation since it may rapidly rearrange to an enedione prior to reaction with nucleophiles.
This article reviews the involvement of reactive metabolites in the metabolism of a variety of compounds containing furan rings. It is focused on furan-containing compounds where there have been multiple studies exploring the characteristics of the reactive intermediate and its role in the compound’s toxicity. In general, the nature of the reactive intermediate is controlled by the substitution on the furanyl ring, with more substitution favoring an epoxide intermediate. In addition, not all furan-containing molecules are toxic despite extensive oxidation of the furan ring. In these cases, the potentially toxic metabolite is rapidly neutralized by further metabolism or it is so reactive that it does not alkylate many targets beyond the enzyme catalyzing the oxidation reaction. Therefore, the structural features of the parent compound are important determinants as to whether the presence of the furanyl ring represents true toxic potential.
2 Furan
Furan is the parent compound for this class of toxic compounds. It is an important industrial chemical.20 A product of incomplete combustion, it is also present in the environment as a component of smog, wood smoke, tobacco smoke and car exhaust and has been detected in food and beverages.20–24 Furan is a potent liver toxicant and carcinogen in rats and mice.25 It also induces cholangiocarcinomas in rats.25 Because of its carcinogenic potency in animal models and the ubiquitous presence of furan in the environment, furan is listed as a possible human carcinogen (class 2B) by the National Toxicology Program26 and the International Agency for Research on Cancer. 20
The mechanism of tumor induction by furan is unknown. The prevailing view is that furan is a nongenotoxic carcinogen. Furan is not mutagenic in mutagenesis assays.25,27–29 In addition, it does not elicit a DNA repair response30 and induces a toxic response followed by compensatory cell proliferation,30 similar to what has been observed with other nongenotoxic liver carcinogens.31,32 However, a genotoxic mechanism cannot be excluded. There is evidence that furan induces liver tumors in mice via a genotoxic mechanism since furan induced unique mutations in activated oncogenes.33 In addition, low levels of DNA adducts have been detected in liver DNA from [14C]furan-treated rat.34 Furthermore, furan induced liver DNA damage as detected by the comet assay.29,34,35 Finally, a metabolite of furan is mutagenic in a number of model systems28,36 and reacts with DNA.37–40
Cytotoxicity is likely to contribute to the hepatocarcinogenic properties of furan. It causes toxicity in livers of rodents at doses as low as 0.12 mg/kg.25,41–43 A recent report indicates that furan targets cytosolic and mitochondrial proteins involved in energy production, redox regulation and protein folding, providing a possible mechanistic link between protein adduct formation and cytotoxicity resulting from the disruption of normal homeostasis.44 Furan is a strong inducer of cell proliferation in both mouse and rat liver as a consequence of its toxic activity.30,41,42,45,46 Genes involved in stress response, DNA damage and cell proliferation are up-regulated in rat liver following furan exposure (4 daily doses of 2, 4, 8, 12 or 16 mg/kg or 1, 3, 7, or 14 doses of 4 or 40 mg/kg or 3 months of 30 mg/kg).35,47,48 Genes related to cell cycle regulation and DNA repair were down regulated.35 Alterations in cell cycle and apoptotic genes were observed in the livers of rats treated daily with 0.1 mg/kg for four weeks.49 Toxicity and proliferation precedes the development of cholangiofibrosis and subsequent cholangiocarcinomas in rats.46,48,50 These studies support a mechanism of carcinogenesis in which chronic toxicity with secondary cell proliferation triggers cancer in furan-exposed rodents.
It is well established that furan’s toxic effects require metabolism. Furan is transformed into a protein-binding intermediate via a P450-dependent process both in vivo and in vitro.51,52 Furan depletes GSH and reduces cell viability at μM concentrations in freshly isolated hepatocytes.53 Furan also depletes ATP in isolated hepatocytes and uncouples oxidative phosphorylation both in vitro and in vivo.54 These effects are much reduced in the presence of P450 2E1 inhibitors such as 1-phenylimidazole and enhanced by acetone pretreatment (induction of P450 2E1), paralleling the effects of inhibitors and inducers of furan metabolism.55
The initial product of P450-catalyzed oxidation of furan is cis-2-butene-1,4-dial (BDA, Scheme 2).14,56,57 The primary P450 enzyme responsible for the conversion of furan to BDA is P450 2E1.57,58 While it is conceivable that there is an epoxide formed at the P450’s active site, there is no evidence that this intermediate has a significant lifetime. The epoxide is not a detectable product of furan oxidation by dimethyldioxirane.11,14 Semicarbazide trapping experiments in microsomes indicate that BDA is a reactive metabolite of furan with the major product detected being cis-2-butene-l,4-dia1 bis-semicarbazone (4, Scheme 2).14 GSH-trapping experiments generated both mono- and bis-GSH reaction products (5 and 6, respectively, Scheme 2).57 The regiochemistry of the bis-GSH reaction products formed from reaction of GSH with the P450-generated metabolite was similar to that observed with synthetic BDA.57 This observation suggests that the primary product of furan oxidation is BDA and not furanyl epoxide; the latter metabolite is expected to generate primarily the 2-substituted reaction product whereas the 3-substituted reaction product dominates in the reaction of GSH with BDA.56 S-(2-Furanyl)-glutathione was not detected as a metabolite of furan.
Scheme 2.

Trapping of the reactive metabolite formed in the P450-catalyzed oxidation of furan in microsomes.14,57
Characterization of the in vivo rat metabolites of furan indicates that BDA plays a central role in its overall metabolism (Scheme 3). Oxidation of furan to BDA is likely the first step in the overall conversion of furan to its major metabolite, carbon dioxide; this metabolite represent 26% of a 8 mg/kg dose in rats.51 All of the characterized urinary metabolites are derived from the reaction of BDA with cellular nucleophiles. 59–63 The mono-GSH-BDA reaction product 5 and its downstream metabolites 7 and 8 result from the reaction of BDA with GSH (Scheme 3).59,60 R-2-Acetylamino-6-(2,5-dihydro-2-oxo-1H-pyrrol-1-yl)-1-hexanoic acid (9) is derived from the reaction of BDA with lysine to form pyrrolinone adducts.60,61 A variety of urinary metabolites are derived from the cysteine-BDA-lysine cross-S-[1-(5-amino-5-carboxypentyl)-1H-pyrrol-3-yl]-L-cysteine (10).60–62 Biotransformation of the cysteine portion of 10 generated metabolite 11.60,62 Metabolites 12–19 result from cysteine N-acetylation as well as metabolism of the lysine part of the cross-link (Scheme 3).60–62 N-Acetylcysteine-BDA-spermidine cross-links 20 and 21 have also been detected in the urine from furan-treated rats (Scheme 3).63 Similarly, the identified biliary metabolites are derived from GSH-BDA reaction products such as 5 and downstream metabolites of 6 (GSH-BDA-glutamate, CysGly-BDA-GSH, CysGly-BDAglutamate, and Cys-BDA-GSH).64 Metabolite 14 was also detected in bile.64
Scheme 3.

The cysteine in the cysteine-BDA-lysine cross-links observed in vivo has several possible sources: free or protein bound cysteine or GSH. Studies in rat hepatocytes indicate that a major source of the cysteine-BDA-lysine cross-link metabolites is a GSH-BDA-lysine cross-link, S-[1-(5-amino-5-carboxypentyl)-1H-pyrrol-3-yl]-glutathione (22, Scheme 4).61 This observation indicates that once BDA is formed, it reacts with GSH to form 2-(S-glutathionyl)butanedial (23), which then reacts with lysine to form GSH-BDA-lysine cross-links (22, Scheme 4). Both α- and ε-amino groups of lysine react with GSH-BDA to form cross-links and both products (22a and 22b) are observed in hepatocytes.61 Interestingly, the ratio of 22a to 22b drops over time, indicating a further enrichment of the ε-amino cross-link (22b).61 This shift is consistent with an increased contribution of degraded GSH-BDA-protein lysine cross-links since GSH-BDA-lysine protein cross-links will only occur with the ε-amino group of lysine as the α-amino group is involved in a peptide bond. Subsequent metabolism of the GSH and lysine portions of this cross-link results in the observed urinary metabolites, 11–19 (Scheme 3). Consistent with the source of these metabolites being GSH-BDA-protein cross-links, immunoblot analysis with anti-GSH antibodies demonstrated that GSH is covalently cross-linked to liver proteins in a furan- and metabolism-dependent manner.61 It is not surprising to consider that a major portion of the urinary and biliary metabolites are degraded protein adducts since 13% of an 8 mg/kg dose was covalently bound to liver proteins 24 h after treatment.51
Scheme 4.

Characterization of other hepatocyte metabolites indicated that 23 is an important reactive intermediate in furan metabolism. In addition to lysine, 23 reacted with other cellular amines including glutamine, ornithine, putrescine, and spermidine generating GSH-BDA-amine cross-links 24–26 (Scheme 4).61,63 Based on these observations, it is clear that reaction of BDA with GSH does not completely deactivate BDA as previously suggested.56 Earlier studies indicated that GSH significantly blocked P450 catalyzed protein binding of [14C]furan in microsomes.52 It was hypothesized that GSH is a good trap for the reactive metabolite because it has both a thiol and an amino group and, therefore, is able to neutralize all the reactive characteristics of BDA.56 However, the abundance of GSH-BDA-amine cross-links indicates that the intramolecular reaction to form the mono-GSH reaction product 5 is not fast enough to protect against alkylation of other cellular nucleophiles. In fact, our data indicate that 23 might have a sufficiently long half-life that it may migrate across membranes and alkylate amine groups distant from the site of its formation.61 The alkylation of cellular nucleophiles by 23 likely contributes to the overall toxic properties of furan.
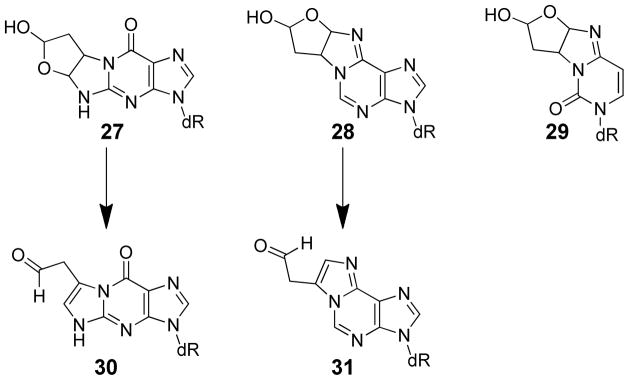
BDA itself is toxic and mutagenic in bacteria and mammalian cell lines.28,36 It reacts readily with the exocyclic and endocyclic nitrogens of dGuo, dAdo and dCyd to form relatively stable diastereomeric oxadiazabicyclo(3.3.0)octaimine adducts (27, 28, and 29; Scheme 5).37,38 The presence of the masked aldehyde in all of the BDA-derived adducts indicate that these adducts retain electrophilic reactivity. The dGuo and dAdo adducts rapidly rearrange under physiological conditions to generate substituted etheno adducts with a reactive aldehydic group (30 and 31, Scheme 5).39 The dCyd and dAdo adducts were detected in bacteria treated with mutagenic concentrations of BDA.40 However, they were not detected in liver DNA isolated from furan-treated rats.34,65 Studies with [14C]furan in rats indicate that the levels of furan-derived DNA damage is low;34,51 a dose of 0.1 mg/kg or 2 mg/kg [14C]furan (20 mCi/mmol) generated 1.7 ± 0.7 adducts/108 nucleotides and 3.3 ± 2.1 adducts/107 nucleotides, respectively, in liver DNA as determined by accelerator mass spectrometry.34 This radioactivity did not co-elute with standards for 27, 28, or 29.34 The chemical structure of these DNA adducts was not determined. The levels of protein adduct formation far exceed the levels of DNA damage,51 indicating that furan’s reactive metabolites preferentially react with protein. BDA may be so reactive that only low quantities of this metabolite reach nuclear DNA, explaining the low levels of BDA-derived DNA damage in furan-treated rats. In addition, reaction of BDA with GSH to form 23 may also reduce the availability of BDA for reaction with DNA.
Scheme 5.
Based on these observations, we can start to make some predictions about what reactive metabolites are important to the overall toxic and mutagenic effects of furan and develop molecular mechanisms for how furan exerts its toxic effects. BDA has been considered the ultimate reactive metabolite of furan. This chemical is very reactive; it readily alkylates protein, polyamine and DNA nucleophiles in vitro.38–40,56 However, a number of the cellular reaction products are derived from the reaction of GSH-BDA (23) with various amines such as lysine, protein amino groups and polyamines. Further studies are required to determine the relative contribution of the reactive metabolites BDA and 23 to the overall toxic and carcinogenic properties of furan.
3 2-Substituted Furans
3.1 2-Methylfuran
2-Methylfuran has been detected in many foods, coffee and cigarette smoke.21,22,66 It induces liver necrosis and pulmonary bronchiolar lesions in rats following a single i.p. injection (≥ 100 mg/kg).67 The toxic effects of long-term exposure to 2-methylfuran have not been investigated. The toxicity of 2-methylfuran requires metabolism. Pretreatment of rats with the P450 inhibitor N-octylimidazole blocked its toxicity and the P450 inducer phenobarbital enhanced its toxicity.67 Pretreatment of rats with 3-methylcholanthrene had no effect.67
Metabolism led to the covalent binding of 2-methylfuran to protein and DNA in vivo.67 The level of [14C]2-methylfuran binding was highest in the liver; significantly lower levels of binding were also observed in kidney, lung and blood.67 Pretreatment of rats with phenobarbital increased the covalent binding of [14C]2-methylfuran to DNA and proteins in all tissues whereas the P450 inhibitor, N-octylimidazole, reduced covalent binding. Similar observations were reported when [14C]2-methylfuran was incubated with either rat liver or lung microsomes.68 These results suggest the involvement of a reactive metabolite in the in vivo toxicity of 2-methylfuran.
The initial product of P450 catalyzed oxidation of 2-methylfuran is 4-oxo-2-pentenal (32, Scheme 6). This enedione metabolite is a potent inhibitor of 2-methylfuran metabolism.68 It was trapped as the bis-semicarbazone derivative 33 in microsomal incubations of 2-methylfuran (Scheme 6).10 The inclusion of semicarbazide in the microsomal incubations inhibited 80% of the 2-methylfuran-derived protein binding indicating that 4-oxo-2-pentenal is a likely candidate for the reactive metabolite formed in vivo.10,68 Interestingly, GSH and cysteine blocked 80% and 95% of the protein binding whereas N-acetylcysteine (NAC) or lysine were only able to block 40–50% of the binding. This observation indicates that both a thiol and an amino group may be required for efficient blocking of protein binding by the proposed reactive metabolite, 4-oxo-2-pentenal. However, the GSH conjugate of 4-oxo-2-pentenal was not stable enough for chemical characterization.10
Scheme 6.
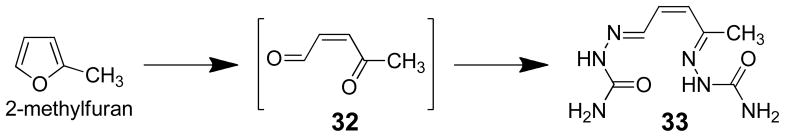
Trapping of the reactive metabolite formed in the P450-catalyzed oxidation of 2-methylfuran in microsomes.10
2-Methylfuran may be activated as a result of GSH conjugation. Pretreatment of rats with L-2-oxothiazolidine-4-carboxylate (OTZ), an inducer of liver GSH, enhanced the liver toxicity of 2-methylfuran as well as increased the amount of radiolabeled 2-methylfuran covalently bound to liver proteins.69 OTZ decreased the amount of liver DNA binding of this compound.69 These observations are consistent with protein, not DNA, adduction as a trigger for toxicity. They also indicate that 4-oxo-2-pentenal is likely responsible for DNA adduct formation. In vitro, it generates 1,N2-substituted adducts with dGuo (34 and 35, Scheme 7).70,71 It is possible that a GSH-aldehyde reaction product is responsible for protein adduct formation, similar to what has been proposed for furan.61 Characterization of the urinary and/or hepatocyte metabolites would provide some insight into the involvement of a GSH-aldehyde reaction product in this process. Attempts have been made to characterize the urinary metabolites of 2-methylfuran but these efforts were complicated by difficulties in separation as well as by the large number of metabolites detected.69 One of the urinary metabolites was thought to be a cysteinyl derivative.69 More in-depth characterization of the metabolites of 2-methylfuran are warranted.
Scheme 7.
3.2 Furosemide
Furosemide (4-chloro-2-(furan-2-ylmethylamino)-5-sulfamoylbenzoic acid, Scheme 8) is a 2-substituted furan that is a diuretic agent used to treat hypertension and edema. At therapeutic doses, furosemide is not toxic in humans. However, it has been associated with hypersensitivity and jaundice.72,73 High intraperitoneal doses of furosemide (≥ 200 mg/kg) generated necrosis in the midzonal and centrilobular areas of the liver in mice.74,75 Furosemide-treated rats or hamsters showed little evidence of hepatotoxicity.75,76 In mice, necrosis was preceded by the formation of covalently modified proteins.74 The toxicity does not involve dysregulation of mitochondrial function77 or depletion of GSH.74,77 Furosemide is toxic to both mouse and rat hepatocytes, with the toxicity accompanied by a slow depletion of GSH78,79 and protein thiols.79 The difference between the in vitro and in vivo observations is likely explained by the rapid elimination of the compound in vivo, reducing its conversion to the reactive metabolite.79
Scheme 8.

P450-mediated oxidation of furosemide initiates toxicity in mice. Pretreatment with the P450 inhibitors piperonyl butoxide, cobalt chloride, α-napthylisothiocyanate or 1-aminobenzotriazole, reduced the hepatotoxicity as well as the covalent binding of the drug to liver proteins.74,76 Pretreatment of mice with phenobarbital shifted the necrosis to the midzonal region and increased the incidence but not severity of necrosis.76 Consistent with the induction of furosemide metabolism, phenobarbital pretreatment increased the rate of furosemide clearance and slightly enhanced the extent of furosemide covalently bound to liver proteins.76
Studies with microsomal preparations confirmed that P450 enzymes are involved in the generation of a protein-reactive furosemide metabolite.16,76 Rat liver microsomal preparations were more active than mouse liver microsomes in converting furosemide to a protein-binding intermediate; both species were much more active than human liver microsomes.75,80 This binding required oxygen and NADPH and was inhibited by a NADPH-cytochrome c reductase antibody or a carbon monoxide atmosphere. Microsomes from mice pretreated with piperonyl butoxide or cobalt chloride were less effective at converting furosemide to a protein-binding metabolite.16,76 Microsomal protein binding was enhanced by pretreatment with phenobarbital but not 3-methylcholanthrene.16
Strong support for an epoxide intermediate in furosemide metabolism was the observation that the epoxide hydrolase inhibitor, TCPO, significantly enhanced the microsomal protein binding of furosemide.16 Consistent with this hypothesis, the furan ring is required for the formation of a protein-reactive metabolite; the reduced analog, [35S]tetrahydrofurosemide, did not bind to microsomal proteins.16 Furthermore, dual labeling studies established that the furan portion of furosemide but not the sulphamoylanthranilic acid segment of the molecule is covalently attached to protein both in vitro and in vivo.16,74,76
Thiols are good trapping agents for the reactive metabolite; GSH, cysteine or cysteamine reduced microsomal binding by >90%.75,76,80 The toxicity of this drug in hepatocytes was blocked by NAC.78 Consistent with the trapping of a reactive intermediate, several GSH conjugates are formed when GSH is included in microsomal mixtures.80 The chemical structure of these metabolites was not determined. Inclusion of NAC and N-acetyl-lysine (NAL) in microsomal incubations led to the formation of a double NAC/NAL furosemide adduct whose mass spectral characteristics were supportive of a pyrrole ring structure with the amino group of NAL providing the pyrrole nitrogen (36, Scheme 8).75 This latter observation suggests the formation of a γ-ketoenal intermediate (37) following oxidation of the furan ring.
Attempts to synthesize the epoxide or γ-ketoenal metabolite by oxidizing furosemide with dimethyldioxirane led to the detection of 9-chloro-2,6-dioxo-9-sulfamoyl-1,2,4a,6-tetrahydro-1H-benzo[d]pyrido[2,1-b][1,3]oxazine (38) which converted to 4-chloro-2-(3′-hydroxypyridinium-1′-yl)-5-sulfamoylbenzoic acid (39, Scheme 8).81 There was no evidence of an epoxide or γ-ketoenal intermediate in this process. This compound resulted from initial intramolecular condensation of the amino group with the aldehyde of the presumed γ-ketoenal intermediate. This pyridinium ion was detected in rat liver microsomal incubations of furosemide in the presence of the required cofactors.81
Characterization of the in vivo and hepatocyte metabolites of furosemide in rats and mice indicated that there are three major routes of metabolism.75 They include acyl glucuronidation to form 40, N-dealkylation to generate 2-amino-4-chloro-5-sulfamoylbenzoic acid (41), and furan ring oxidation to produce a GSH conjugate 42 and a γ-ketocarboxylic acid metabolite, 4-chloro-2-(4-carboxy-2-oxobutylamino)-5-sulfamoylbenzoic acid (43, Scheme 9). Metabolites 42 and 43 were formed through P450 mediated pathways since their levels were reduced by the P450 inhibitor, 1-aminobenzotriazole.75 P450 enzymes 2C11, 2E1, 3A1 and 3A2 are involved in the overall metabolism of furosemide in rats.82 Wistar rats metabolize furosemide more rapidly than CD-1 mice but have significantly less covalent binding of the compound to hepatic liver proteins.75 In these studies, the products of N-dealkylation (41) and furan ring oxidation (43) were the major biliary metabolites in rats representing 21 and 22% of the excreted dose, respectively.75 The acyl glucuronide 40 accounted for 13% of the excreted dose. In mice, furosemide represented roughly 90% of the excreted dose. The remaining 10% was attributed to roughly equal amounts of metabolites 40, 41 and 43.75 The GSH conjugate was not detected in mice.
Scheme 9.

Biliary and hepatocyte metabolites of furosemide.75
These same metabolites were observed in hepatocytes from CD1 mice and Wistar rats.75 Rat hepatocytes were more active than mouse hepatocytes in converting furosemide to metabolites, with the major pathway being acyl glucuronidation (~60% of the recovered radioactivity). The extent of glucuronidation in mouse hepatocytes was roughly one half that observed in rats. Similar to the in vivo results, the GSH conjugate 42 was not observed in mouse hepatocytes. The levels of P450-dependent metabolism were similar in both species.
Collectively, these observations indicate that epoxide and γ-ketoenal intermediates are important in the bioactivation of furosemide to a toxic metabolite. The structures of the γ-ketocarboxylic acid metabolite may require the formation of an γ-ketoenal intermediate. Either intermediate will lead to the formation of the NAC/NAL adduct and the pyridinium metabolite observed in microsomal preparations.
Species differences in toxicity between rat and mouse may be explained by the relative turnover of parent drug to stable metabolites: 66% turnover in rats as compared to 10% turnover in mice.75 This is predicted to increase the exposure of the mouse liver to furosemide. The lack of hepatotoxicity in humans results in part from a reduced ability to generate the protein-reactive intermediate.75,80 The cause of the decreased protein binding is not known; it could be either low furan oxidase activity or increased detoxification of the reactive intermediate. Furthermore, glucuronidation is the dominant pathway of metabolism in humans,83–85 reducing the amount of drug available for oxidation. In addition, the efficacious dose in humans (1–2 mg/kg) is well below the doses required for toxicity in rodents (>200 mg/kg). Finally, the reactive metabolite of furosemide may be inactivated by intramolecular trapping; however, it is not known if this metabolite is generated in vivo.
3.3 Prazosin
Prazosin (2-[4-(2-furoyl)piperazin-1-yl]-6,7-dimethoxyquinazolin-4-amine) is an antihypertensive agent that is also used as a treatment for post-traumatic stress disorder.86 This drug has been in use since 1976 with no evidence of safety issues despite the presence of the 2-substituted furan ring. Furan ring oxidation is a major in vivo pathway of metabolism for this drug,86 which indicates that furanyl ring oxidation does not necessarily lead to toxicity. The most abundant metabolite detected in urine and feces of prazosin-treated rats is the ring-opened carboxylic acid derivative 44 (Scheme 10).86 GSH conjugation to the furanyl group is a minor pathway.
Scheme 10.

Metabolites derived from furanyl ring oxidation of prazosin.19,86
Studies in liver microsomes confirmed that furan ring opening is a major metabolic pathway of prazosin.19 Both the acid 44 and alcohol 45 products of the ring-opening reaction were observed (Scheme 10). Levels of these two metabolites were significantly reduced when semicarbazide was included in the incubation mixture.19 The intermediate was trapped as a pyrazidine product 46 which is consistent with the involvement of a γ-ketoenal intermediate 47 (Scheme 10). GSH trapping led to the formation of GSH reaction products with molecular ions at m/z 709.19 This mass is consistent with the addition of GSH to a γ-keto-α,β-unsaturated alcohol 48. These data indicate that the γ-ketoenal intermediate is rapidly reduced to an alcohol in microsomes. A recent report indicates that P450s are good catalysts for the reduction of α,β-unsaturated aldehydes.87 Therefore, the swift reduction/oxidation of the reactive metabolite is likely responsible for the absence of toxicity observed with prazosin despite the extensive metabolism of the furanyl ring.
4 3-Substituted Furans
4.1 3-Methylfuran
3-Methylfuran is a component of indoor and outdoor air pollution,4,5 as well as a volatile mold metabolite.5 It has been detected in food88 as well as in the exhaled breath from smokers and passive smokers.89,90 When inhaled, 3-methylfuran causes acute pulmonary toxicity in mice (≥ 14 μmol/L for 1 h),91–94 rats (148 μmol/L for 1 h) and hamsters (322 μmol/L for 1 h).91 It also induces acute olfactory necrosis in rats92 and mice93 and kidney toxicity in mice.93 When administered intraperitoneally, it is a lung but not liver or kidney toxicant.94,95 Exposure to 3-methylfuran by inhalation (344 μmol/L for 2 h once a week for 10 weeks) did not led to any adverse effects 10 months or 2 years later.96
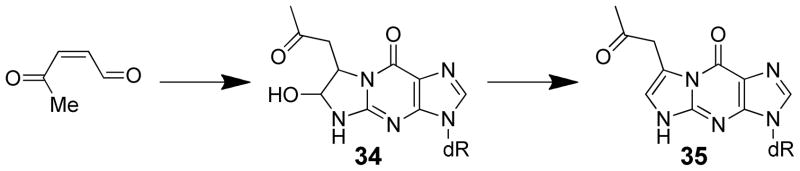
The toxicity of 3-methylfuran requires metabolism by P450 enzymes. The general P450 inhibitor, piperonyl butoxide, blocked both pulmonary macromolecular binding and necrosis induced by the inhalation of 3-methylfuran.97 Binding of 3-methylfuran to lung microsomal proteins required NADPH and O2 and was inhibited by carbon monoxide or piperonyl butoxide.10,97 Inclusion of semicarbazide in hepatic and pulmonary microsomal incubations trapped the proposed reactive metabolite, 2-methyl-cis-2-butene-1,4-dial (49), as a bis-semicarbazone derivative 50 (Scheme 11) and protected against the protein binding of 3-methylfuran in vitro,10 supporting the hypothesis that 2-methyl-cis-2-butene-1,4-dial is the reactive metabolite responsible for the toxic effects of this compound.
Scheme 11.
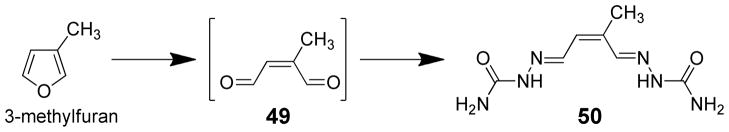
Trapping the reactive metabolite of 3-methylfuran as a bis-semicarbazone.10
4.2 4-Ipomeanol
4-Ipomeanol [1-(3′-furyl)-4-hydroxy-1-pentanone, Scheme 12] and its structural analogs, ipomeanine [1-(3′-furyl)-1,4-pentanedione], 1-ipomeanol [1-(3′-furyl)-1-hydroxy-4-pentanone] and 1,4-ipomeadiol [1-(3′-furyl)-1,4-pentanediol] are cytotoxicants produced by moldy sweet potatoes.98 Ipomeanine and 1,4-ipomeadiol are also metabolites of 4-ipomeanol.99 They are lung specific toxicants in laboratory animals.98,100,101 4-Ipomeanol is a liver toxicant in humans.102–104
Scheme 12.

Structures of the GSH reaction products formed in GSH-fortified microsomal incubations of 4-ipomeanol.99,108,114
The toxicity of these compounds requires P450 oxidation of the furan ring.100,105 Tissue selective P450 expression likely contributes to the species differences in target organ toxicity caused by 4-ipomeanol. P450 4B1 catalyzes the oxidation of 4-ipomeanol in rodent lungs.106–108 The human homolog of this enzyme is functionally defective,109 explaining why 4-ipomeanol is not a pulmonary toxicant in humans. Human liver P450s 1A2, 2C19, 2D6 and 3A4 are all able to catalyze the oxidation of 4-ipomeanol to a reactive metabolite;106,108 the activities of these enzymes are low in human lung.110
Consistent with the proposal that metabolism is important for the toxicity of 4-ipomeanol, a shift in target organ toxicity to the liver was observed in rats when they were pretreated with 3-methylcholanthrene.100 3-Methylcholanthrene induces liver but not lung microsomal metabolism of 4-ipomeanol.105 A similar shift in organ specificity is not observed with phenobarbital pretreatment despite increased liver microsomal metabolism of 4-ipomeanol.105 This seemingly contradictory response can be explained by the likely simultaneous induction of detoxification enzymes; phenobarbital, but not 3-methylcholanthrene, induced a large increase in urinary level of ipomeanol-4-glucuronide in rats.111
Oxidation of 4-ipomeanol led to the formation of protein and DNA adducts.17,106,112 The structures of these adducts have not been characterized. Consistent with the hypothesis that metabolism drives the toxicity of this chemical, levels of protein binding were higher in lung versus liver microsomes from rats.112 Phenobarbital and 3-methylcholanthrene pretreatment increased microsomal covalent binding of 4-ipomeanol in rat liver microsomes but not lung microsomes, paralleling the effect of these inducers on its microsomal metabolism.112 4-Ipomeanol is a mechanism-based inhibitor of P450 3A4 (Table 2) but not the other P450s known to metabolize this furan.108,113 The stoichiometry of binding between the radioactive substrate and enzyme was ~1.5:1. GSH provided only minor protection (10%) against inactivation indicating that the metabolite that inhibits the enzyme is very reactive.
Table 2.
Kinetic parameters for the inhibition of P450s by furan-containing compounds
| Chemical | Enzyme | Ki (μM) | kinact (min−1) | Partition ratioa |
|---|---|---|---|---|
| 4-Ipomeanol | Recombinant P450 3A4113 | 20 | 0.15 | 257 |
| Menthofuran | Recombinant P450 2A6146 | 0.84 | 0.25 | 3.5 ± 0.6 |
| Recombinant P450 2A6147 | 2.2 | 1 | 30 | |
| P450 2A6 activity in HLM146 | 2.5 | 0.22 | ||
| Bergamottin | Recombinant P450 3A4154,159 | 7.7 | 0.3 | ~27 |
| Recombinant P450 2B6159 | 5.2 | 0.087 | ~2 | |
| Recombinant ΔP450 2B6 K262R160 | 8.2 | 0.23 | ||
| Recombinant P450 3A5159 | 20 | 0.045 | ~20 | |
| L-754,394 | P450 3A4 activity in HLM163 | 7.5 | 1.62 | 1.4 |
| P450 2D6 activity in HLM 163 | 32 | 0.18 | 40 | |
| recombinant P450 3A4165 | 7.5 | 4 |
nmoles product formed per nmoles enzyme inactivated
The chemical structure of the reactive intermediate is not clearly established. Addition of the epoxide hydrolase inhibitor TCPO had little effect on covalent binding,17 suggesting that an epoxide intermediate may not participate in this reaction. GSH protects against protein binding and toxicity in vitro and in vivo.17,100,112 Correspondingly, GSH conjugates have been detected in GSH-enriched microsomal incubations of 4-ipomeanol.99,108,112,114,115 Inclusion of cytosol does not influence the levels of GSH-conjugates indicating that the reactive metabolite is either very reactive or it is not a good substrate for cytosolic GSTs.112
Chemical characterization of the GSH-trapped metabolites demonstrates a complex picture. Buckpitt and Boyd reported the observation of two GSH reaction products in rat liver and lung microsomes incubated in the presence of 4-ipomeanol, NADPH and GSH.112 These metabolites lacked UV absorbance, indicating that the furan chromophore of the parent compound had been disrupted but the structures of these products were not determined. Alvarez-Diez and Zheng detected one microsomal GSH-4-ipomeanol reaction product whose MS data were consistent with the addition of one molecule of GSH to an epoxide or enedione intermediate (m/z 492).114 Baer et al. did not observe the mono-GSH reaction product. However, they reported two bis-GSH-reaction products whose MS data were consistent with the formation of 4-ipomeanol mediated cross link between the thiol of one GSH and the amino group of another (51 or 52, Scheme 12).108 Chen et al. demonstrated that the reactive metabolite of 4-ipomeanol can react with either one or two molecules of GSH to form multiple mono- or bis-GSH reaction products 51–56 (Scheme 12).99 The structure of these products can be rationalized by either initial 1,2- or 1,4-addition to the proposed enedial intermediate 57. These addition products then react with the amino group of the same or different GSH molecule to form pyrrole cross-links. Based on the balance of product formation, 1,2-addition is the major pathway. Similar findings were reported for the trapping of the reactive intermediate with NAC and NAL where the major product was a 2-(S-N-acetylcysteinyl)-4-(4-hydroxypentanoyl)-pyrrole adduct of NAL (58, Scheme 13).108 These products could also be formed by thiol attachment of an epoxide intermediate. Presumably this reaction would occur primarily on the carbon adjacent to the furanyl oxygen ring. Model reactions with the proposed enedial intermediate have been stymied by the difficult synthesis of this possible reactive intermediate.99
Scheme 13.

Trapping of reactive intermediate formed in N-acetylcysteine (NAC)/N-acetyllysine (NAL) fortified microsomal incubations of 4-ipomeanol.108
4.3 L-739,010
L-739,010, [1S,5R]-3-cyano-1-(3-furyl)-6-(6-[3-(3α-hydroxy-6,8-dioxabicyclo[3.2.1]-octanyl)]pyridin-2-yl-methoxyl)naphthalene, is a direct inhibitor of 5-lipoxygenase. While the plasma half-life of this drug is about 4h, the plasma half-life of radioactivity after i.v. treatment with the [14C]labeled drug was 2.7 and 4 days in rats and rhesus monkeys, respectively.116 The radioactivity was covalently associated with plasma proteins. While L-739,010 caused a dose-dependent elevation in alanine transaminase levels in dogs, suggesting that it induced hepatotoxicity,116 histopathological analysis showed no indication of liver toxicity.117 It initiated lipidosis in the gall bladder.117
Binding of L-739,010 to proteins requires metabolism. Incubation of L-739,010 with liver microsomes results in the NADPH-dependent loss of parent compound without significant formation of detectable metabolites.116,118 There were species differences in the rate of protein binding (monkey > rat > human > dog).116,118 Most of the unaccounted-for-material was covalently bound to microsomal proteins.116 P450 3A enzymes are implicated as the critical enzyme for this process since protein adduct formation was induced by phenobarbital and dexamethasone and was inhibited by the P450 inhibitors triacetyloleandomycin and diethyldithiocarbamate.116,119 Inclusion of GSH and methoxyamine reduced the covalent binding by ~50% and ~80%, respectively.116
Furanyl ring oxidation to a reactive enedial (59) is an important step for the covalent binding of L-739,010 to proteins (Scheme 14). Inclusion of methoxyamine led to the formation of five methyloxime adducts.116,118 The mono- and bis-methyloxime adducts of the furan ring-opened metabolite of L-739,010 were observed (60 and 61, respectively). The presence of the mono-methyloxime adduct suggests that one of the aldehydes is more reactive than the other. The major product was the Z isomer of the 1-O-methyloxime-2-butene-4-alcohol derivative of L-739,010 (62). This product results from the reduction of the enedial metabolite of L-739,010 to an alcohol. The fifth methyloxime adduct was to the 6,8-dioxabicyclo[3.2.1]octanyl portion of the molecule.116,118 Furan oxidation was the dominant pathway in rat liver microsomes.116 The absence of any detectable metabolites in the absence of trapping agents indicates that the enedial intermediate 46 or possible epoxide precursor is very reactive with protein nucleophiles.
Scheme 14.
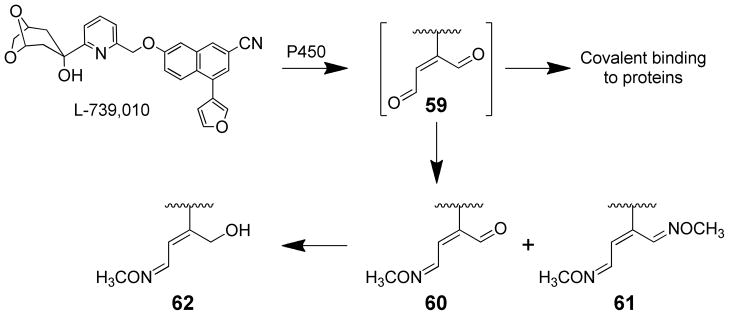
Products formed during the microsomal oxidation of L-739,010 in the presence of semicarbazide and required co-factors.116,118
4.4 Teucrin A
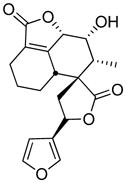
The 3-substituted furan, teucrin A, is the major germander neoclerodane diterpenoid present in germander extracts.120 Germander has been used as an herbal antiseptic and anticholeretic medicine as well as a bitter flavoring of wine and spirits.120 In the early 1990s, there were numerous reports of cytolytic hepatitis resulting from germander capsule or tea consumption.120–127 A number of the cases showed signs of an immunoallergic type of hepatitis since a rechallenge caused early recurrence of the toxicity. Germander-derived human liver toxicity was replicated in the mouse and shown to be caused by furan-containing neoclerodane diterpenoids.128 A major member of this group, teucrin A, produced the same midzonal hepatic necrosis in mice as extracts enriched in these compounds.129
Metabolism of the furan ring was required for teucrin A-induced hepatotoxicity. Toxicity was enhanced by P450 3A inducers and blocked by P450 3A inhibitors.128,129 It was accompanied by the depletion of intracellular GSH and alkylation of protein thiols.129,130 GSH depletion enhanced the toxic effects of both teucrin A and teucrin A containing extracts.128–130 These data indicate a GSH-reactive metabolite is likely responsible for the liver toxicity. Consistent with the formation of a reactive metabolite, teucrin A becomes covalently associated with hepatocellular proteins in a dose-dependent manner.130 The tetrahydrofuran analog of teucrin A was not a hepatotoxicant in mice, providing strong evidence that oxidation of the furan ring was critical for toxicity.129
Several pieces of data are consistent with a furanyl epoxide of teucrin A as the reactive metabolite (63, Scheme 15). First, teucrin A’s hepatotoxicity was reduced in animals pretreated with either butylated hydroxyanisole or clofibrate, two compounds that induce epoxide hydrolase.128 Additional support was obtained in vitro. P450 3A4 converted teucrin A into a single uncharacterized metabolite which was converted into two uncharacterized products when epoxide hydrolase was included in the incubation mixtures.131 In these reactions, epoxide hydrolase was inactivated in a time dependent manner. These observations are consistent with P450 3A4-catalyzed formation of a teucrin A furanyl epoxide intermediate, which is hydrolyzed by epoxide hydrolase. Since serum antibodies from patients who chronically consumed germander tea recognized human epoxide hydrolase both modified and unmodified with teucrin A, it has been postulated that the P450 3A4- and teucrin A-dependent modification of epoxide hydrolase triggers autoantibody formation.131 This mechanism could explain the immunoallergic nature of the toxicity in some patients.
Scheme 15.

Reactive intermediates in teucrin A metabolism and the reaction products between teucrin A enedial and N-acetylcysteine (NAC) and/or N-acetyl-lysine (NAL).15
Attempts to generate the furanoepoxide of teucrin A were unsuccessful.15 The product of dimethyldioxirane oxidation of teucrin A is the cis-2-ene-dialdehyde derivative of teucrin A 64, not the epoxide (Scheme 15). This observation does not rule out the possible intermediacy of an epoxide in the P450 catalyzed oxidation of teucrin A because it is expected to rapidly rearrange to the enedial. Reaction of the synthetic enedial of teucrin A with model protein nucleophiles, NAC and/or NAL, led to adducts (Scheme 15).15 Reaction with NAL led to N-alkyl-3-pyrrolin-2-one reaction products with the more sterically hindered product, N-alkyl-3-alkyl-3-pyrrolin-2-one (65) as the major product. Reaction of the enedial with NAC led to the formation of unstable products as well as a stable N-acetyl-S-2-furanyl-L-cysteine adduct (66). When the enedial was incubated with NAC and NAL, a number of products were observed with the major product being a 1,2,4-subsubstituted pyrrole (67). Small amounts of tetrasubstituted pyrroles were also observed. In all cases, epimerization occurred at the C12 of teucrin A, providing support for the involvement of enedial and hydroxyenal tautomers in the reactions of this compound with nucleophiles.
Protein adduction by teucrin A appears to be an important trigger of its toxicity. Specific protein targets were identified using antibodies generated against teucrin A enedial-treated peptides.132 The major teucrin A targets in rat livers were mitochondrial and endoplasmic reticulum associated proteins in addition to enzymes involved in cell maintenance and cellular metabolism. A number of the endoplasmic reticulum proteins were xenobiotic metabolizing enzymes such as P450 enzymes (P450s 2A1, 2A2, 2C7, 2D1 and 2D3), UDP-glucuronosyl transferase 1-1 and epoxide hydrolase. GSH S-transferase was a cytosolic target of teucrin A. The widespread alkylation of mitochondrial proteins may suggest that teucrin A causes its toxicity by triggering mitochondrial dysfunction. The toxicological significance of these reactions remains to be determined.
5 2,3-Substituted Furans
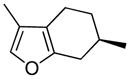
5.1 Menthofuran
Pennyroyal oil is used as a flavoring agent or fragrance component as well as an herbal medicine for abortion and menses induction.133 High doses of pennyroyal oil used for the latter purpose cause hepatoxicity and death in humans.134,135 Menthofuran is a minor constituent of pennyroyal oil as well as a primary metabolite of R-(+)-pulegone in rat, the major terpene constituent of pennyroyal oil.136–139 Menthofuran is a liver and lung toxicant in mice133,140 and a liver toxicant in rats.141 It induces midzonal hepatoxicity in Swiss-Webster mice140 and centrilobular necrosis in BALB/c mice.133
Like other furans, menthofuran-induced toxicity requires P450-mediated oxidation. The toxicity was increased by pretreating animals with phenobarbital140,142 and blocked by pretreatment with piperonyl butoxide,140 indicating the importance of P450 enzymes in the metabolic activation of menthofuran. 3-Methylcholanthrene did not influence menthofuran toxicity in rats.142 The metabolism of [14C]menthofuran converts it into a protein-binding metabolite in vivo.140 Interestingly, while phenobarbital pretreatment caused a doubling of the liver toxicity, it slightly reduced the levels of menthofuran covalently bound to liver proteins.140 The reasons for this discrepancy are not understood. Liver GSH levels are only modestly reduced following menthofuran exposure.133 Depletion of GSH with diethylmaleate or buthionine sulfoximine had no effect on menthofuran toxicity.133,141
Incubation of [14C]menthofuran with rat, mouse or human liver microsomes led to the formation of protein adducts.140 This binding required NADPH and was blocked by the inclusion of semicarbazide. This binding was reduced by the pretreatment of mice with piperonyl butoxide and enhanced by the pretreatment of mice with phenobarbital.140 All these data support the involvement of P450 enzymes in the formation of a reactive metabolite.
A metabolite with the mass expected for the γ-ketoenal metabolite, 2-Z-(2′-oxo-4′-methylcyclohexylidene)propanal (68) was detected by GC-MS as a metabolite of menthofuran in rat liver microsomes (Scheme 16).143,144 This metabolite was converted to 5,6,7,8-tetrahydro-4,7-dimethyl-7H-cinnoline (69) upon inclusion of semicarbazide in rat, mouse or human liver microsomes,140,143,144 indicating that 68 or some precursor to 68 is the protein-binding metabolite of menthofuran. 2-Hydroxymenthofuran (70) was also detected as a product of P450 catalyzed oxidation of menthofuran.145 This metabolite rearranges to diastereomeric mintlactones 71, additional products of microsomal menthofuran oxidation.140,145 2-Hydroxymenthofuran did not react with semicarbazide to form 69.
Scheme 16.

Mechanisms of metabolite formation from the proposed epoxide intermediate of menthofuran.140,143–145
Oxygen-18 and 2H-labeling studies provide support for initial epoxide formation in the P450 catalyzed formation of 2-hydroxymenthofuran.145 The epoxide intermediate can either directly rearrange to form 70 or γ-ketoenal 68 and/or react with water to form the 2,3-diol which then can dehydrate to form either 70 or rearrange to the γ-ketoenal 68 (Scheme 16). Support for this mechanism was obtained in chemical model studies.13,145 NMR studies of the chemical oxidation of menthofuran by dimethyldioxirane at low temperatures indicated that the epoxide 72 is the initial oxidation product and it undergoes a subsequent sigmatropic rearrangement to the γ-ketoenal 68.13 When the dimethyldioxirane reaction mixture was quenched with water, 2-hydroxymenthofuran and the mintlactones were formed.145 When semicarbazide was present in the quenching solution, 5,6,7,8-tetrahydro-4,7-dimethyl-7H-cinnoline (69) was observed.145 Based on this mechanism, either the epoxide 72 or the γ-ketoenal 68 could be responsible for the protein binding and toxic effects of menthofuran. Further research is required to address this question.
P450s 1A2, 2A6, 2C19 and 2E1 are catalysts for the oxidation of menthofuran in humans.145 P450 2E1 was the most efficient catalyst of this reaction. Menthofuran is a mechanism based inactivator of P450 2A6, but not P450 1A2, 2E1 or 2A13.146,147 The kinetic parameters of this inactivation are displayed in Table 2. The inactivation of P450 2A6 was not blocked by GSH or NAC. However, GSH, methoxylamine and semicarbazide but not NAL protected against adduction to NADPH-P450 reductase.146 Covalent binding to P450 2A6 was not influenced by these trapping agents,146 indicating that the reactive metabolite responsible for the inactivation does not escape the enzyme’s active site.
Chemical characterization of the in vivo metabolites of menthofuran indicates that the oxidation of the furan ring is an important pathway in vivo.143,144,148,149 In vivo metabolites of [2-14C]menthofuran in rats indicates that the furan ring is a target for oxidation and the reactive intermediate in this process, furanoepoxide or γ-ketoenal, reacts with water, GSH or taurine.148,149 Subsequent metabolism of these products generates a complex mixture of metabolites.148
Menthofuran is a metabolite of pulegone and a number of studies indicate that the metabolism of pulegone to menthofuran followed by the subsequent oxidation of menthofuran’s furanyl ring is responsible for the toxic effects of pennyroyal.137,141,143–145,150 Incubation of pulegone with mouse liver microsomes in the presence of semicarbazide led to the formation of 5,6,7,8-tetrahydro-4,7-dimethyl-7H-cinnoline (69) which is consistent with the P450 catalyzed conversion of pulegone to menthofuran which subsequently is oxidized to an γ-ketoenal intermediate (Scheme 16).151 The presence of semicarbazide reduced the protein binding of [14C]pulegone,151 indicating that 68 or 72 are involved in the covalent binding of pulegone to proteins. The importance of this pathway to the toxic effects observed in humans consuming high doses of pennyroyal is supported by the detection of menthofuran-modified proteins in liver samples from an individual who died from the consumption of pennyroyal oil.135 However, pennyroyal and R-(+)-pulegone resulted in extensive depletion of GSH.133 Moreover, GSH depletion enhanced the toxicity of these two treatments.133 These results are different from what has been reported for menthofuran which does not deplete GSH nor is its toxicity enhanced by GSH depletion.133,141 There are likely other pathways contributing to pennyroyal toxicity in addition to the toxicity triggered by menthofuran.
5.2 Furanocoumarins
5.2.1 Bergamottin
Bergamottin is the parent compound for a variety of furanocoumarins present in grapefruit juice.152–157 Consumption of grapefruit juice resulted in increased bioavailability of a variety of drugs, which results in altered drug activity as well as toxicity.158 This interaction is thought to occur via irreversible inhibition of intestinal P450s, resulting in increased absorption of the drug.158 All of the furanocoumarins in grapefruit juice are mechanism-based inhibitors of P450 3A4 as well as other P450s.154–157,159–161 The furan ring is required for inactivation.157
The inactivation of P450s by bergamottin is time- and concentration-dependent and requires NADPH.154,159,160 The kinetic parameters for the inactivation are displayed in Table 2. GSH does not block the irreversible inhibition suggesting that the reactive metabolite does not escape the active site prior to inactivation.154 The stoichiometry of bergamottin binding to the apoprotein is ~0.5:1.159 The P450 reduced carbon monoxide spectrum was decreased and the native heme destroyed for all inactivated P450s.154,159 The reactive intermediate reacts with the apoprotein; no products of heme alkylation were observed.154,159
Mass spectral analysis of the inactivated enzyme indicated the addition of bergamottin plus 2–3 oxygen atoms.159 Metabolism studies performed with P450 2B6 and P450 3A5 in the presence of GSH indicated that oxidation of the furan double bond leads to the formation of a GSH conjugate (73) in which GSH has added to an epoxide intermediate (74, Scheme 17).161 The corresponding dihydroxymetabolite resulting from the reaction of an epoxide intermediate with water was not detected. The main site of oxidation is the geranyloxy side chain which is readily hydroxylated by P450s. Based on these observations, it was proposed that bergamottin is first oxidized on the geranyloxy side chain and then undergoes oxidation of the furan ring to generate the reactive intermediate which results in enzyme activation.159 Lin et al. proposed either a furanoepoxide or γ-ketoenal intermediates as possible structures for the reactive metabolite.159
Scheme 17.
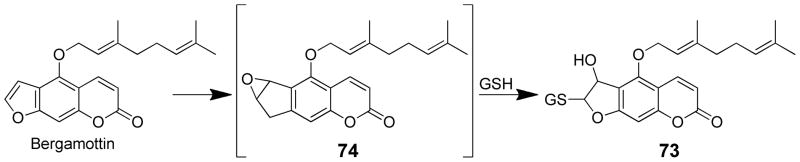
Metabolism of bergamottin to a reactive metabolite.161
5.2.2 L-754,394
L-754,394 (N-[2(R)-hydroxy-1(S)-indanyl]-5-[2(S)-[((1,1-dimethylethyl)amino)-carbonyl]-4-[(furo[2,3-b]pyridin-5-yl)methyl]piperazin-1-yl]-4(S)-hydroxy-2(R)-(phenyl-methyl) pentanamide) is an experimental anti-AIDS drug that is a potent and selective inhibitor of HIV-1 protease. Preclinical pharmacokinetic studies indicated that it was a mechanism based inhibitor of P450.162
Studies in rat, dog, monkey and human liver microsomal preparations demonstrated that L-754,394 caused an NADPH-dependent and time-dependent reduction in the CO-binding spectrum of P450.162,163 P450 2C11 and P450 3A1/2 are the targets in rat liver microsomes with the activity of P450 2C11 decreased by 60–70% in liver microsomes from rats given a single iv dose of 10 mg/kg.162 L-754,394 is a mechanism based inhibitor of human P450 3A4163–165 as well as P450 2D6 (Table 2).163 The low partition ratio (≤ 4) indicates that L-754,394 is a potent inactivator of P450 3A4. The binding stoichiometry was 1.08 ± 0.5 moles drug covalently bound/moles P450 inactivated in studies performed with recombinant P450 3A4.165 While GSH, NAC and methoxyamine did not protect against L-754,394-mediated P450 inactivation, these trapping agents modestly reduced (15–25%) the NADPH-dependent binding of [14C]L-754,394 to microsomal proteins.164 The epoxide hydrolase inhibitor, TPCO, did not affect microsomal protein binding when present during the microsomal incubations.164 These data indicate that the metabolic intermediate in the inactivation process is very reactive. Studies performed with recombinant P450 3A4 indicate that very little of the reactive metabolite escapes the enzyme’s active site.165
The furan ring is required for the inactivation; the dihydrofuran derivative and the analog that lacks the furan ring were not irreversible inhibitors of P450.164 Chemical characterization of the microsomal metabolites of L-754,394 provides evidence for a furanopyridine epoxide intermediate 75 (Scheme 18). However, further experimentation is required to eliminate the involvement of a γ-ketoenal intermediate 76.165 Mass spectral characterization of the trapped GSH/NAC reaction products indicates that they are GSH/NAC adducts of a mono-oxygenated L-754,394 metabolite with the proposed structure 77 as shown in Scheme 18.164 In rat and monkey liver microsomes, a metabolite was detected whose MS was consistent with a dihydrodiol 78 which was converted into an O-methyloxime 79 when methoxyamine was included in the incubation mixtures (Scheme 18).164
Scheme 18.

Products of furanyl ring oxidation of L-754,394 in the presence and absence of trapping agents.164
Attempts to characterize the L-754,394-P450 adduct were complicated by its instability to protein purification conditions. The covalent modification was unstable to protein purification and hydrolysis conditions.165 However, Lighting et al demonstrated that the radioactivity was specifically associated with the apoprotein and that the radioactivity released from the protein during CNBr generation of peptides had a retention time similar to the dihydrodiol metabolite 78.165 The peptide fraction associated with L-754,394-derived radioactivity was tentatively identified as an active site peptide. Subsequent LC/MS studies performed with the intact adducted P450 3A4 indicated that the inactivation of P450 was accompanied by the addition of 685 ± 3 Da. This mass increase indicates that the mass of the apoprotein has increased by the weight of two oxygen atoms and L-754,394, not by the addition of one oxygen atom and L-754,394 as expected for enzyme inactivation by a furanopyridine epoxide or γ-ketoenal intermediate.166 These data suggest that L-754,394 undergoes a double oxidation or an oxidation plus addition of water in the inactivation reaction. Therefore, simple reaction of a protein nucleophile with an oxidized intermediate of L-754-394 is ruled out as a possible mechanism of enzyme inactivation. Further experiments are necessary to elucidate the mechanism of L-754,394-mediated irreversible inhibition of P450 3A4.
6 Conclusions and Future Directions
Several common themes are highlighted in this literature review. One is that the toxicity of furan ring containing compounds requires oxidation of the furan ring. The target organ toxicity exhibited by this group of compounds is often driven by tissue-specific expression of the P450 enzymes responsible for the oxidation of the furan ring. There are at least three factors that impact the overall toxicity of a particular furan-containing compound. First, the involvement of other metabolic pathways can influence its toxicity. Furans that are more actively metabolized through other pathways are less toxic than those compounds whose primary metabolic pathway involves furan ring oxidation. For example, furosemide is relatively non-toxic since its major route of metabolism is glucuronidation, not furan ring oxidation. Second, rapid detoxification of the reactive intermediate results in a less toxic compound. This explains why prasozin is a safe drug despite the fact that furan ring oxidation is a major pathway of metabolism; rapid reduction of the α,β-unsaturated system of the intermediate results in an nontoxic metabolite. Third, the reactivity of the furan ring oxidized metabolite will influence the cellular targets and, consequently, the overall toxicity of the compound. The most reactive metabolites result in enzyme inhibition such as bergamottin and L-754,394. The less reactive migrate beyond the site of formation and alkylate targets in other subcellular locations.
Structure also determines the relative stability of the intermediate in furan ring oxidation. Less substituted furans generate less stable epoxides and are more likely to rearrange to a reactive enedione intermediate. The strongest evidence for the intermediacy of an epoxide was obtained with more substituted furans such as menthofuran, bergamottin and L-754,394. In addition, the more rigid the furan ring, the longer the lifetime of the epoxide intermediate.
GSH plays an important role in modulating the toxicity. It readily reacts with the reactive intermediates to form GSH conjugates, in most cases, in the absence of glutathione S-transferases. For most furans, this reaction protects cells against the harmful effects of the furan; consistently, GSH depletion enhanced the toxicity of these compounds. However, GSH may enhance the toxicity for the less substituted furans. 2-Methylfuran was more toxic when GSH levels were elevated. The mechanism behind this enhancement has not been investigated. Furan, one of the most toxic in this chemical class, is converted to many GSH-BDA-amine cross-links. Whether GSH plays a role in the toxicity of furan is a question still needs investigation.
Future studies will define how these reactive intermediates cause their toxicity. It is clear that the molecular structure of the reactive intermediate influences the location and types of cellular nucleophiles targeted. Alkyl substitutions on the enedione reactive intermediate alter the rate at which it reacts with cellular nucleophiles and has some influence on its toxicity.167 For example, the reactive intermediate(s) in furan bioactivation primarily target proteins whereas the addition of a methyl group results in significant formation of DNA as well as protein adducts.34,51,67,69 The cellular targets will determine the toxicological outcome; DNA adduct formation is critical for cancer initiation whereas protein alkylation is important for toxicity. A catalog of how furan structure alters the cellular targets will provide insights into the toxic mechanisms of this class of chemicals and will provide valuable information for the design of safe drugs as well as aid in human risk assessment when exposed to furan-containing compounds.
Acknowledgments
Funding Sources
This research was partially funded by ES-10577 from the National Institutes of Health.
I thank Bob Carlson for his assistance with the preparation of this manuscript. I also thank Drs. Stephen Hecht and Sharon Murphy as well as members of the Murphy and Peterson research groups for their helpful comments.
Abbreviations
- BDA
cis-2-butene-1,4-dial
- NAC
N-acetylcysteine
- NAL
N-acetyl-lysine
- OTZ
L-2-oxothiazolidine-4-carboxylate
- TCPO
1,2-epoxy-3,3,3-trichloropropane
Reference List
- 1.Burka LT, Boyd MR. Bioactivation of Foreign Compounds. Academic Press; NY: 1985. Furans; pp. 243–257. [Google Scholar]
- 2.Williams GM, Mattia A, Renwick A. Furan-substituted aliphatic hydrocarbons, alchohols, aldehydes, ketones, carboxylic acids and related esters, sulfides, disulfides and ethers (addendums) WHO Food Additives Series. 2009;60:481–532. [Google Scholar]
- 3.Zhou S, Koh HL, Gao Y, Gong ZY, Lee EJ. Herbal bioactivation: the good, the bad and the ugly. Life Sci. 2004;74:935–968. doi: 10.1016/j.lfs.2003.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saunders RA, Griffith JR, Saalfield FE. Identification of some organic smog components based on rain water analysis. Biomed Mass Spectrom. 1974;1:192–194. doi: 10.1002/bms.1200010309. [DOI] [PubMed] [Google Scholar]
- 5.Sunesson AL, Vaes WHJ, Nilsson CA, Blomquist G, Andersson B, Carlson R. Identification of volatile metabolites from five fungal species cultivated on two media. Appl Environ Microbiol. 1995;61:2911–2918. doi: 10.1128/aem.61.8.2911-2918.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem Res Toxicol. 2002;15:269–299. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 7.Kalgutkar AS, Gardner I, Obach RS, Shaffer CL, Callegari E, Henne KR, Mutlib AE, Dalvie DK, Lee JS, Nakai Y, O’Donnell JP, Boer J, Harriman SP. A comprehensive listing of bioactivation pathways of organic functional groups. Curr Drug Metab. 2005;6:161–225. doi: 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- 8.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 9.Guengerich FP. Cytochrome P450 oxidations in the generation of reactive electrophiles: epoxidation and related reactions. Arch Biochem Biophys. 2003;409:59–71. doi: 10.1016/s0003-9861(02)00415-0. [DOI] [PubMed] [Google Scholar]
- 10.Ravindranath V, Burka LT, Boyd MR. Reactive metabolites from the bioactivation of toxic methylfurans. Science. 1984;224:884–886. doi: 10.1126/science.6719117. [DOI] [PubMed] [Google Scholar]
- 11.Adger BM, Barrett C, Brennan J, McKervey MA, Murray RW. Oxidation of furans with dimethyldioxirane. J Chem Soc, Chem Comm. 1991:1553–1554. [Google Scholar]
- 12.Ruzo LO, Casida JE, Holden I. Direct NMR detection of an epoxyfuran intermediate in peracid oxidation of the fungicide methfuroxam. J Chem Soc, Chem Comm. 1985:1642–1643. [Google Scholar]
- 13.Oishi S, Nelson SD. Evidence for the formation of heterocyclic arene oxides and a γ-keto enal by reaction of menthofuran with dimethyldioxirane. JOC. 1992;57:2744–2747. [Google Scholar]
- 14.Chen LJ, Hecht SS, Peterson LA. Identification of cis-2-butene-1,4-dial as a microsomal metabolite of furan. Chem Res Toxicol. 1995;8:903–906. doi: 10.1021/tx00049a001. [DOI] [PubMed] [Google Scholar]
- 15.Druckova A, Marnett LJ. Characterization of the amino acid adducts of the enedial derivative of teucrin A. Chem Res Toxicol. 2006;19:1330–1340. doi: 10.1021/tx060143k. [DOI] [PubMed] [Google Scholar]
- 16.Wirth PJ, Bettis PJ, Nelson WL. Microsomal metabolism of furosemide evidence for the nature of the reactive intermediate involved in covalent binding. Mol Pharmacol. 1975;12:759–768. [PubMed] [Google Scholar]
- 17.Boyd MR, Burka LT, Wilson BJ, Sasame HA. In vitro studies on the metabolic activation of the pulmonary toxin, 4-ipomeanol, by rat lung and liver microsomes. JPET. 1978;207:677–686. [PubMed] [Google Scholar]
- 18.Guengerich FP. Studies on the activation of a model furan compound - toxicity and covalent binding of 2-(N-ethyl-carbamoylhydroxymethyl)furan. Biochem Pharmacol. 1977;26:1909–1915. doi: 10.1016/0006-2952(77)90165-4. [DOI] [PubMed] [Google Scholar]
- 19.Erve JC, Vashishtha SC, DeMaio W, Talaat RE. Metabolism of prazosin in rat, dog, and human liver microsomes and cryopreserved rat and human hepatocytes and characterization of metabolites by liquid chromatography/tandem mass spectrometry. Drug Metab Dispos. 2007;35:908–916. doi: 10.1124/dmd.106.013219. [DOI] [PubMed] [Google Scholar]
- 20.International Agency for Research on Cancer. Dry Cleaning, Some Chlorinated Solvents and Other Industrial Chemicals. Vol. 393. IARC; Lyon, France: 1995. Furan. [Google Scholar]
- 21.Capurro PU. Effects of exposure to solvents caused by air pollution with special reference to CCl4 and its distribution in air. Clin Toxicol. 1973;6:109–124. doi: 10.3109/15563657308991049. [DOI] [PubMed] [Google Scholar]
- 22.Maga J. Furans in foods. Crit Rev Food Sci Nutr. 1979;11:355–366. doi: 10.1080/10408397909527268. [DOI] [PubMed] [Google Scholar]
- 23.Vranova J, Ciesarova Z. Furan in food - a review. Czech J Food Sci. 2009;27:1–10. [Google Scholar]
- 24.Food and Drug Administration. Exploratory data on furan in food: individual food products. 2009 http://www.fda.gov/Food/FoodSafety/FoodContaminantsAdulteration/ChemicalContaminants/Furan/ucm078439.htm.
- 25.National Toxicology Program. Toxicology and carcinogenesis studies of furan in F344/N rats and B6C3F1 mice vol NTP Technical Report No 402. US Department of Health and Human Services, Public Health Service, National Institutes of Health; Research Triangle Park, NC: 1993. [Google Scholar]
- 26.National Toxicology Program. 12th Report on Carcinogens. US Department of Health and Human Services; Washington DC: 2011. [Google Scholar]
- 27.Mortelmans K, Haworth S, Lawlor T, Speck W, Tainer B, Zeiger E. Salmonella mutagenicity tests. II. Results from the testing of 270 chemicals. Environ Mutagen. 1986;7(suppl):1–119. [PubMed] [Google Scholar]
- 28.Kellert M, Brink A, Richter I, Schlatter J, Lutz WK. Tests for genotoxicity and mutagenicity of furan and its metabolite cis-2-butene-1,4-dial in L5178Y tk+/− mouse lymphoma cells. Mutat Res. 2008;657:127–132. doi: 10.1016/j.mrgentox.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 29.McDaniel LP, Ding W, Dobrovolsky VN, Shaddock JG, Jr, Mittelstaedt RA, Doerge DR, Heflich RH. Genotoxicity of furan in Big Blue rats. Mutat Res. 2012;742:72–78. doi: 10.1016/j.mrgentox.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Wilson DM, Goldsworthy TL, Popp JA, Butterworth BE. Evaluation of genotoxicity, pathological lesions, and cell proliferation in livers of rats and mice treated with furan. Environ Mol Mutagen. 1992;19:209–222. doi: 10.1002/em.2850190305. [DOI] [PubMed] [Google Scholar]
- 31.Melnick RL, Huff J. Liver carcinogenesis is not a predicted outcome of chemically induced hepatocyte proliferation. Toxicol Indust Health. 1993;9:415–438. doi: 10.1177/074823379300900303. [DOI] [PubMed] [Google Scholar]
- 32.Melnick RL, Kohn MC, Portier CJ. Implications for risk assessment of suggested nongenotoxic mechanisms of chemical carcinogenesis. Environ Health Perspect. 1996;104(Supplement 1):123–134. doi: 10.1289/ehp.96104s1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reynolds SH, Stowers SJ, Patterson RM, Maronpot RR, Aaronson SA, Anderson MW. Activated oncogenes in B6C3F1 mouse liver tumors: Implications for risk assessment. Science. 1987;237:1309–1316. doi: 10.1126/science.3629242. [DOI] [PubMed] [Google Scholar]
- 34.Neuwirth C, Mosesso P, Pepe G, Fiore M, Malfatti M, Turteltaub K, Dekant W, Mally A. Furan carcinogenicity: DNA binding and genotoxicity of furan in rats in vivo. Mol Nutr Food Res. 2012 doi: 10.1002/mnfr.201200226. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding W, Petibone DM, Latendresse JR, Pearce MG, Muskhelishvili L, White GA, Chang CW, Mittelstaedt RA, Shaddock JG, McDaniel LP, Doerge DR, Morris SM, Bishop ME, Manjanatha MG, Aidoo A, Heflich RH. In vivo genotoxicity of furan in F344 rats at cancer bioassay doses. Toxicol Appl Pharmacol. 2012;261:164–171. doi: 10.1016/j.taap.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 36.Peterson LA, Naruko KC, Predecki D. A reactive metabolite of furan, cis-2-butene-1,4-dial, is mutagenic in the Ames assay. Chem Res Toxicol. 2000;13:531–534. doi: 10.1021/tx000065f. [DOI] [PubMed] [Google Scholar]
- 37.Gingipalli L, Dedon PC. Reaction of cis- and trans-2-butene-1,4-dial with 2′-deoxycytidine to form stable oxadiazabicyclooctaimine adducts. JACS. 2001;123:2664–2665. doi: 10.1021/ja0056421. [DOI] [PubMed] [Google Scholar]
- 38.Byrns MC, Predecki DP, Peterson LA. Characterization of nucleoside adducts of cis-2-butene-1,4-dial, a reactive metabolite of furan. Chem Res Toxicol. 2002;15:373–379. doi: 10.1021/tx0101402. [DOI] [PubMed] [Google Scholar]
- 39.Byrns MC, Vu CC, Peterson LA. The formation of substituted 1,N6-etheno-2′-deoxyadenosine and 1,N2-etheno-2′-deoxyguanosine adducts by cis-2-butene-1,4-dial, a reactive metabolite of furan. Chem Res Toxicol. 2004;17:1607–1613. doi: 10.1021/tx049866z. [DOI] [PubMed] [Google Scholar]
- 40.Byrns MC, Vu CC, Neidigh JW, Abad JL, Jones RA, Peterson LA. Detection of DNA adducts derived from the reactive metabolite of furan, cis-2-butene-1,4-dial. Chem Res Toxicol. 2006;19:414–420. doi: 10.1021/tx050302k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moser GJ, Foley J, Burnett M, Goldsworthy TL, Maronpot R. Furan-induced dose-response relationships for liver cytotoxicity, cell proliferation, and tumorigenicity (furan-induced liver tumorigenicity) Exp Toxicol Pathol. 2009;61:101–111. doi: 10.1016/j.etp.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Gill S, Bondy G, Lefebvre DE, Becalski A, Kavanagh M, Hou Y, Turcotte AM, Barker M, Weld M, Vavasour E, Cooke GM. Subchronic oral toxicity study of furan in Fischer-344 rats. Toxicol Pathol. 2010;38:619–630. doi: 10.1177/0192623310368978. [DOI] [PubMed] [Google Scholar]
- 43.Gill S, Kavanagh M, Barker M, Weld M, Vavasour E, Hou Y, Cooke GM. Subchronic oral toxicity study of furan in B6C3F1 Mice. Toxicol Pathol. 2011;39:787–794. doi: 10.1177/0192623311412980. [DOI] [PubMed] [Google Scholar]
- 44.Moro S, Chipman JK, Antczak P, Turan N, Dekant W, Falciani F, Mally A. Identification and pathway mapping of furan target proteins reveal mitochondrial energy production and redox regulation as critical targets of furan toxicity. Toxicol Sci. 2012;126:336–352. doi: 10.1093/toxsci/kfs005. [DOI] [PubMed] [Google Scholar]
- 45.Fransson-Steen R, Goldsworthy TL, Kedderis GL, Maronpot RR. Furan-induced liver cell proliferation and apoptosis in female B6C3F1 mice. Toxicology. 1997;118:195–204. doi: 10.1016/s0300-483x(97)03618-4. [DOI] [PubMed] [Google Scholar]
- 46.Sirica AE. Biliary proliferation and adaptation in furan-induced rat liver injury and carcinogenesis. Toxicol Pathol. 1996;24:90–99. doi: 10.1177/019262339602400113. [DOI] [PubMed] [Google Scholar]
- 47.Hamadeh HK, Jayadev S, Gaillard ET, Huang Q, Stoll R, Blanchard K, Chou J, Tucker CJ, Collins J, Maronpot R, Bushel P, Afshari CA. Integration of clinical and gene expression endpoints to explore furan-mediated hepatotoxicity. Mutat Res. 2004;549:169–183. doi: 10.1016/j.mrfmmm.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 48.Hickling KC, Hitchcock JM, Oreffo V, Mally A, Hammond TG, Evans JG, Chipman JK. Evidence of oxidative stress and associated DNA damage, increased proliferative drive, and altered gene expression in rat liver produced by the cholangiocarcinogenic agent furan. Toxicol Pathol. 2010;38:230–243. doi: 10.1177/0192623309357946. [DOI] [PubMed] [Google Scholar]
- 49.Chen T, Mally A, Ozden S, Chipman JK. Low doses of the carcinogen furan alter cell cycle and apoptosis gene expression in rat liver independent of DNA methylation. Environ Health Perspect. 2010;118:1597–1602. doi: 10.1289/ehp.1002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hickling KC, Hitchcock JM, Chipman JK, Hammond TG, Evans JG. Induction and progression of cholangiofibrosis in rat liver injured by oral administration of furan. Toxicol Pathol. 2010;38:213–229. doi: 10.1177/0192623309357945. [DOI] [PubMed] [Google Scholar]
- 51.Burka LT, Washburn KD, Irwin RD. Disposition of [14C]furan in the male F344 rat. J Toxicol Environ Health. 1991;34:245–257. doi: 10.1080/15287399109531564. [DOI] [PubMed] [Google Scholar]
- 52.Parmar D, Burka LT. Studies on the interaction of furan with hepatic cytochrome P-450. J Biochem Toxicol. 1993;8:1–9. doi: 10.1002/jbt.2570080103. [DOI] [PubMed] [Google Scholar]
- 53.Carfagna MA, Held SD, Kedderis GL. Furan-induced cytolethality in isolated rat hepatocytes: Correspondence with in vivo dosimetry. Toxicol Appl Pharmacol. 1993;123:265–273. doi: 10.1006/taap.1993.1245. [DOI] [PubMed] [Google Scholar]
- 54.Mugford CA, Carfagna MA, Kedderis GL. Furan-mediated uncoupling of hepatic oxidative phosphorylation in Fischer-344 rats: an early event in cell death. Toxicol Appl Pharmacol. 1997;144:1–11. doi: 10.1006/taap.1997.8121. [DOI] [PubMed] [Google Scholar]
- 55.Kedderis GL, Carfagna MA, Held SD, Batra R, Murphy JE, Gargas ML. Kinetic analysis of furan biotransformation by F-344 rats in vivo and in vitro. Toxicol Appl Pharmacol. 1993;123:274–282. doi: 10.1006/taap.1993.1246. [DOI] [PubMed] [Google Scholar]
- 56.Chen LJ, Hecht SS, Peterson LA. Characterization of amino acid and glutathione adducts of cis-2-butene-1,4-dial, a reactive metabolite of furan. Chem Res Toxicol. 1997;10:866–874. doi: 10.1021/tx9700174. [DOI] [PubMed] [Google Scholar]
- 57.Peterson LA, Cummings ME, Vu CC, Matter BA. Glutathione trapping to measure microsomal oxidation of furan to cis-2-butene-1,4-dial. Drug Metab Dispos. 2005;33:1453–1458. doi: 10.1124/dmd.105.004432. [DOI] [PubMed] [Google Scholar]
- 58.Gates LA, Lu D, Peterson LA. Trapping of cis-2-butene-1,4-dial to measure furan metabolism in human liver microsomes by cytochrome P450 enzymes. Drug Metab Dispos. 2012;40:596–601. doi: 10.1124/dmd.111.043679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peterson LA, Cummings ME, Chan JY, Vu CC, Matter BA. Identification of a cis-2-butene-1,4-dial-derived glutathione conjugate in the urine of furan-treated rats. Chem Res Toxicol. 2006;19:1138–1141. doi: 10.1021/tx060111x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kellert M, Wagner S, Lutz U, Lutz WK. Biomarkers of furan exposure by metabolic profiling of rat urine with liquid chromatography-tandem mass spectrometry and principal component analysis. Chem Res Toxicol. 2008;21:761–768. doi: 10.1021/tx7004212. [DOI] [PubMed] [Google Scholar]
- 61.Lu D, Sullivan MM, Phillips MB, Peterson LA. Degraded protein adducts of cis-2-butene-1,4-dial are urinary and hepatocyte metabolites of furan. Chem Res Toxicol. 2009;22:997–1007. doi: 10.1021/tx800377v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu D, Peterson LA. Identification of furan metabolites derived from cysteine-cis-2-butene-1,4-dial-lysine cross-links. Chem Res Toxicol. 2010;23:142–151. doi: 10.1021/tx9003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peterson LA, Phillips MB, Lu D, Sullivan MM. Polyamines are traps for reactive intermediates in furan metabolism. Chem Res Toxicol. 2011;24:1924–1936. doi: 10.1021/tx200273z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamberger C, Kellert M, Schauer UM, Dekant W, Mally A. Hepatobiliary toxicity of furan: identification of furan metabolites in bile of male F344/N rats. Drug Metab Dispos. 2010;38:1698–1706. doi: 10.1124/dmd.109.031781. [DOI] [PubMed] [Google Scholar]
- 65.Byrns MC. PhD Thesis. University of Minnesota; 2005. Determination of the role of DNA alkylation by cis-2-butene-1,4-dial in furan-induced carcinogenesis. [Google Scholar]
- 66.Newsome JR, Norman V, Keith CH. Vapor phase analysis of tobacco smoke. Tobacco International. 1965;9:102–110. [Google Scholar]
- 67.Ravindranath V, McMenamin MG, Dees JH, Boyd MR. 2-Methylfuran toxicity in rats - Role of metabolic activation in vivo. Toxicol Appl Pharmacol. 1986;85:78–91. doi: 10.1016/0041-008x(86)90389-3. [DOI] [PubMed] [Google Scholar]
- 68.Ravindranath V, Boyd MR. Metabolic activation of 2-methylfuran by rat microsomal systems. Toxicol Appl Pharmacol. 1985;78:370–376. doi: 10.1016/0041-008x(85)90242-x. [DOI] [PubMed] [Google Scholar]
- 69.Ravindranath V, Boyd MR. Effect of modulations of glutathione synthesis on the hepatotoxicity of 2-methylfuran. Biochem Pharmacol. 1991;41:1311–1318. doi: 10.1016/0006-2952(91)90102-b. [DOI] [PubMed] [Google Scholar]
- 70.Hecht SS, Young-Sciame R, Chung FL. Reaction of α-acetoxy-N-nitrosopiperidine with deoxyguanosine: Oxygen-dependent formation of 4-oxo-2-pentenal and a 1,N2-ethenodeoxyguanosine adduct. Chem Res Toxicol. 1992;5:706–712. doi: 10.1021/tx00029a018. [DOI] [PubMed] [Google Scholar]
- 71.Liu Z, Young-Sciame R, Hecht SS. Liquid chromatography-electrospray ionization mass spectrometric detection of an ethenodeoxyguanosine adduct and its hemiaminal precursors in DNA reacted with α-acetoxy-N-nitrosopiperdine and cis-4-oxo-2-pentenal. Chem Res Toxicol. 1996;9:774–780. doi: 10.1021/tx950206r. [DOI] [PubMed] [Google Scholar]
- 72.Noce R, Paredes BE, Pichler WJ, Krahenbuhl S. Acute generalized exanthematic pustulosis (AGEP) in a patient treated with furosemide. Am J Med Sci. 2000;320:331–333. doi: 10.1097/00000441-200011000-00006. [DOI] [PubMed] [Google Scholar]
- 73.Dargie HJ, Dollery CT. Adverse reactions to diuretic drugs. In: Dukes MNG, editor. Meyler’s Side Effects of Drugs. Exerpta Medica; Amsterdam, The Netherlands: 1975. p. 483. [Google Scholar]
- 74.Mitchell JR, Potter WZ, Hinson JA, Jollow DJ. Hepatic necrosis caused by furosemide. Nature. 1974;251:508–511. doi: 10.1038/251508a0. [DOI] [PubMed] [Google Scholar]
- 75.Williams DP, Antoine DJ, Butler PJ, Jones R, Randle L, Payne A, Howard M, Gardner I, Blagg J, Park BK. The metabolism and toxicity of furosemide in the Wistar rat and CD-1 mouse: a chemical and biochemical definition of the toxicophore. J Pharmacol Exp Ther. 2007;322:1208–1220. doi: 10.1124/jpet.107.125302. [DOI] [PubMed] [Google Scholar]
- 76.Mitchell JR, Nelson WL, Potter WZ, Sasame HA, Jollow DJ. Metabolic activation of furosemide to a chemically reactive, hepatotoxic metabolite. J Pharmacol Exp Ther. 1976;199:41–52. [PubMed] [Google Scholar]
- 77.Wong SG, Card JW, Racz WJ. The role of mitochondrial injury in bromobenzene and furosemide induced hepatotoxicity. Toxicol Lett. 2000;116:171–181. doi: 10.1016/s0378-4274(00)00218-6. [DOI] [PubMed] [Google Scholar]
- 78.Massey TE, Walker RM, McElligott TF, Racz WJ. Furosemide toxicity in isolated mouse hepatocyte suspensions. Toxicology. 1987;43:149–160. doi: 10.1016/0300-483x(87)90005-9. [DOI] [PubMed] [Google Scholar]
- 79.Grewal KK, Rafeiro E, Racz WJ. Bromobenzene and furosemide hepatotoxicity: alterations in glutathione, protein thiols, and calcium. Can J Physiol Pharmacol. 1996;74:257–264. [PubMed] [Google Scholar]
- 80.Masubuchi N, Makino C, Murayama N. Prediction of in vivo potential for metabolic activation of drugs into chemically reactive intermediate: correlation of in vitro and in vivo generation of reactive intermediates and in vitro glutathione conjugate formation in rats and humans. Chem Res Toxicol. 2007;20:455–464. doi: 10.1021/tx060234h. [DOI] [PubMed] [Google Scholar]
- 81.Chen LJ, Burka LT. Chemical and enzymatic oxidation of furosemide: formation of pyridinium salts. Chem Res Toxicol. 2007;20:1741–1744. doi: 10.1021/tx700262z. [DOI] [PubMed] [Google Scholar]
- 82.Yang KH, Choi YH, Lee U, Lee JH, Lee MG. Effects of cytochrome P450 inducers and inhibitors on the pharmacokinetics of intravenous furosemide in rats: involvement of CYP2C11, 2E1, 3A1 and 3A2 in furosemide metabolism. J Pharm Pharmacol. 2009;61:47–54. doi: 10.1211/jpp/61.01.0007. [DOI] [PubMed] [Google Scholar]
- 83.Nakahama H, Miwa Y, Yamaji A, Orita Y, Fukuhara Y, Yanase M, Kamada T, Sonoda T, Ishibasi M, Ichikawa Y. The urinary excretion of frusemide and its metabolites by kidney transplant patients. Eur J Clin Pharmacol. 1987;32:313–315. doi: 10.1007/BF00607581. [DOI] [PubMed] [Google Scholar]
- 84.Beermann B, Dalen E, Lindstrom B, Rosen A. On the fate of furosemide in man. Eur J Clin Pharmacol. 1975;9:51–61. doi: 10.1007/BF00613429. [DOI] [PubMed] [Google Scholar]
- 85.Perez J, Sitar DS, Ogilvie RI. Biotransformation of furosemide in patients with acute pulmonary edema. Drug Metab Dispos. 1979;7:383–387. [PubMed] [Google Scholar]
- 86.Erve JC, Vashishtha SC, Ojewoye O, Adedoyin A, Espina R, DeMaio W, Talaat RE. Metabolism of prazosin in rat and characterization of metabolites in plasma, urine, faeces, brain and bile using liquid chromatography/mass spectrometry (LC/MS) Xenobiotica. 2008;38:540–558. doi: 10.1080/00498250802001826. [DOI] [PubMed] [Google Scholar]
- 87.Amunom I, Dieter LJ, Tamasi V, Cai J, Conklin DJ, Srivastava S, Martin MV, Guengerich FP, Prough RA. Cytochromes P450 catalyze the reduction of α,β-unsaturated aldehydes. Chem Res Toxicol. 2011;24:1223–1230. doi: 10.1021/tx200080b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Becalski A, Hayward S, Krakalovich T, Pelletier L, Roscoe V, Vavasour E. Development of an analytical method and survey of foods for furan, 2-methylfuran and 3-methylfuran with estimated exposure. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2010;27:764–775. doi: 10.1080/19440040903473332. [DOI] [PubMed] [Google Scholar]
- 89.Gordon SM, Wallace LA, Brinkman MC, Callahan PJ, Kenny DV. Volatile organic compounds as breath biomarkers for active and passive smoking. Environ Health Perspect. 2002;110:689–698. doi: 10.1289/ehp.02110689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Buszewski B, Ulanowska A, Ligor T, Denderz N, Amann A. Analysis of exhaled breath from smokers, passive smokers and non-smokers by solid-phase microextraction gas chromatography/mass spectrometry. Biomed Chromatogr. 2009;23:551–556. doi: 10.1002/bmc.1141. [DOI] [PubMed] [Google Scholar]
- 91.Haschek WM, Morse CC, Boyd MR, Hakkinen PJ, Witschi HP. Pathology of acute inhalation exposure to 3-methylfuran in the rat and hamster. Exp Mol Pathol. 1983;39:342–354. doi: 10.1016/0014-4800(83)90063-1. [DOI] [PubMed] [Google Scholar]
- 92.Morse CC, Boyd MR, Witschi H. The effect of 3-methylfuran inhalation exposure on the rat nasal cavity. Toxicology. 1984;30:195–204. doi: 10.1016/0300-483x(84)90091-x. [DOI] [PubMed] [Google Scholar]
- 93.Haschek WM, Boyd MR, Hakkinen PJ, Owenby CS, Witschi H. Acute inhalation toxicity of 3-methylfuran in the mouse: pathology, cell kinetics, and respiratory rate effects. Toxicol Appl Pharmacol. 1984;72:124–133. doi: 10.1016/0041-008x(84)90256-4. [DOI] [PubMed] [Google Scholar]
- 94.Gammal LM, Wiley RA, Traiger G, Haschek WM, Baraban S. Toxicity-distribution relationships among 3-alkylfurans in the mouse lung. Toxicology. 1984;30:177–184. doi: 10.1016/0300-483x(84)90128-8. [DOI] [PubMed] [Google Scholar]
- 95.Wiley RA, Traiger GJ, Baraban S, Gammal LM. Toxicity-distribution relationships among 3-alkylfurans in mouse liver and kidney. Toxicol Appl Pharmacol. 1984;74:1–9. doi: 10.1016/0041-008x(84)90263-1. [DOI] [PubMed] [Google Scholar]
- 96.Witschi HP, Tryka AF, Mauderly JL, Haschek WM, Satterfield LC, Bowles ND, Boyd MR. Long-term effects of repeated exposure to 3-methylfuran in hamsters and mice. J Toxicol Environ Health. 1985;16:581–592. doi: 10.1080/15287398509530765. [DOI] [PubMed] [Google Scholar]
- 97.Boyd MR, Statham CN, Franklin RB, Mitchell JR. Pulmonary bronchiolar alkylation and necrosis by 3-methylfuran, a naturally occurring potential atmospheric contaminant. Nature. 1978;272:270–271. doi: 10.1038/272270a0. [DOI] [PubMed] [Google Scholar]
- 98.Boyd MR, Burka LT, Harris TM, Wilson BJ. Lung-toxic furanoterpenoids produced by sweet potatoes (Ipomoea batatas) following microbial infection. Biochim Biophys Acta. 1974;337:184–195. doi: 10.1016/0005-2760(74)90200-8. [DOI] [PubMed] [Google Scholar]
- 99.Chen LJ, DeRose EF, Burka LT. Metabolism of furans in vitro: ipomeanine and 4-ipomeanol. Chem Res Toxicol. 2006;19:1320–1329. doi: 10.1021/tx060128f. [DOI] [PubMed] [Google Scholar]
- 100.Boyd MR, Burka LT. In vivo studies on the relationship between target organ alkylation and the pulmonary toxicity of a chemically reactive metabolite of 4-ipomeanol. J Pharmacol Exp Ther. 1978;207:687–697. [PubMed] [Google Scholar]
- 101.Durham SK, Boyd MR, Castleman WL. Pulmonary endothelial and bronchiolar epithelial lesions induced by 4-ipomeanol in mice. Am J Pathol. 1985;118:66–75. [PMC free article] [PubMed] [Google Scholar]
- 102.Rowinsky EK, Noe DA, Ettinger DS, Christian MC, Lubejko BG, Fishman EK, Sartorius SE, Boyd MR, Donehower RC. Phase I and pharmacological study of the pulmonary cytotoxin 4-ipomeanol on a single dose schedule in lung cancer patients: hepatotoxicity is dose limiting in humans. Cancer Res. 1993;53:1794–1801. [PubMed] [Google Scholar]
- 103.Kasturi VK, Dearing MP, Piscitelli SC, Russell EK, Sladek GG, O’Neil K, Turner GA, Morton TL, Christian MC, Johnson BE, Kelley MJ. Phase I study of a five-day dose schedule of 4-ipomeanol in patients with non-small cell lung cancer. Clin Cancer Res. 1998;4:2095–2102. [PubMed] [Google Scholar]
- 104.Lakhanpal S, Donehower RC, Rowinsky EK. Phase II study of 4-ipomeanol, a naturally occurring alkylating furan, in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2001;19:69–76. doi: 10.1023/a:1006408803734. [DOI] [PubMed] [Google Scholar]
- 105.Boyd MR, Sasame HA, Franklin RB. Comparison of ratios of covalent binding to total metabolism of the pulmonary toxin, 4-ipomeanol, in vitro in pulmonary and hepatic microsomes, and the effects of pretreatments with phenobarbital or 3-methylcholanthrene. Biochem Biophys Res Commun. 1980;93:1167–1172. doi: 10.1016/0006-291x(80)90612-9. [DOI] [PubMed] [Google Scholar]
- 106.Czerwinski M, McLemore TL, Philpot RM, Nhamburo PT, Korzekwa K, Gelboin HV, Gonzalez FJ. Metabolic activation of 4-ipomeanol by complementary DNA-expressed human cytochromes P-450: evidence for species-specific metabolism. Cancer Res. 1991;51:4636–4638. [PubMed] [Google Scholar]
- 107.Verschoyle RD, Philpot RM, Wolf CR, Dinsdale D. CYP4B1 activates 4-ipomeanol in rat lung. Toxicol Appl Pharmacol. 1993;123:193–198. doi: 10.1006/taap.1993.1237. [DOI] [PubMed] [Google Scholar]
- 108.Baer BR, Rettie AE, Henne KR. Bioactivation of 4-ipomeanol by CYP4B1: Adduct characterization and evidence for an enedial intermediate. Chem Res Toxicol. 2005;18:855–864. doi: 10.1021/tx0496993. [DOI] [PubMed] [Google Scholar]
- 109.Zheng YM, Fisher MB, Yokotani N, Fujii-Kuriyama Y, Rettie AE. Identification of a meander region proline residue critical for heme binding to cytochrome P450: implications for the catalytic function of human CYP4B1. Biochemistry. 1998;37:12847–12851. doi: 10.1021/bi981280m. [DOI] [PubMed] [Google Scholar]
- 110.Hukkanen J, Pelkonen O, Hakkola J, Raunio H. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol. 2002;32:391–411. doi: 10.1080/20024091064273. [DOI] [PubMed] [Google Scholar]
- 111.Statham CN, Boyd MR. Effects of phenobarbitol and 3-methylcholanthrene on the in vivo distribution, metabolism and covalent binding of 4-ipomeanol in the rat; implications for target organ toxicity. Biochem Pharmacol. 1982;31:3973–3977. doi: 10.1016/0006-2952(82)90643-8. [DOI] [PubMed] [Google Scholar]
- 112.Buckpitt AR, Boyd MR. The in vitro formation of glutathione conjugates with the microsomally activated pulmonary bronchiolar alkylating agent and cytotoxin, 4-ipomeanol. JPET. 1980;215:97–103. [PubMed] [Google Scholar]
- 113.Alvarez-Diez TM, Zheng J. Mechanism-based inactivation of cytochrome P450 3A4 by 4-ipomeanol. Chem Res Toxicol. 2004;17:150–157. doi: 10.1021/tx034143l. [DOI] [PubMed] [Google Scholar]
- 114.Alvarez-Diez TM, Zheng J. Detection of glutathione conjugates derived from 4-ipomeanol metabolism in bile of rats by liquid chromatography-tandem mass spectrometry. Drug Metab Dispos. 2004;32:1345–1350. doi: 10.1124/dmd.104.000406. [DOI] [PubMed] [Google Scholar]
- 115.Wolf CR, Statham CN, McMenamin MG, Bend JR, Boyd MR, Philpot RM. The relationship between the catalytic activities of rabbit pulmonary cytochrome P-450 isozymes and the lung-specific toxicity of the furan derivative, 4-ipomeanol. Mol Pharmacol. 1982;22:738–744. [PubMed] [Google Scholar]
- 116.Zhang KE, Naue JA, Arison B, Vyas KP. Microsomal metabolism of the 5-lipoxygenase inhibitor L-739,010: Evidence for furan bioactivation. Chem Res Toxicol. 1996;9:547–554. doi: 10.1021/tx950183g. [DOI] [PubMed] [Google Scholar]
- 117.Molon-Noblot S, Gillet JP, Durand-Cavagna G, Huber AC, Patrick DH, Duprat P. Lipidosis induced in the dog gallbladder by a direct 5-lipoxygenase inhibitor. Toxicol Pathol. 1996;24:231–237. doi: 10.1177/019262339602400211. [DOI] [PubMed] [Google Scholar]
- 118.Chauret N, Nicoll-Griffith D, Friesen R, Li C, Trimble L, Dube D, Fortin R, Girard Y, Yergey J. Microsomal metabolism of the 5-lipoxygenase inhibitors L-746,530 and L-739,010 to reactive intermediates that covalently bind to protein: the role of the 6,8-dioxabicyclo[3.2.1]octanyl moiety. Drug Metab Dispos. 1995;23:1325–1334. [PubMed] [Google Scholar]
- 119.Eagling VA, Tjia JF, Back DJ. Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes. Br J Clin Pharmacol. 1998;45:107–114. doi: 10.1046/j.1365-2125.1998.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Larrey D, Vial T, Pauwels A, Castot A, Biour M, David M, Michel H. Hepatitis after germander (Teucrium chamaedrys) administration: another instance of herbal medicine hepatotoxicity. Ann Intern Med. 1992;117:129–132. doi: 10.7326/0003-4819-117-2-129. [DOI] [PubMed] [Google Scholar]
- 121.Pauwels A, Thierman-Duffaud D, Azanowsky JM, Loiseau D, Biour M, Levy VG. Acute hepatitis caused by wild germander. Hepatotoxicity of herbal remedies Two cases. Gastroenterol Clin Biol. 1992;16:92–95. [PubMed] [Google Scholar]
- 122.Mostefa-Kara N, Pauwels A, Pines E, Biour M, Levy VG. Fatal hepatitis after herbal tea. Lancet. 1992;340:674. [PubMed] [Google Scholar]
- 123.Mattei A, Bizollon T, Charles JD, Debat P, Fontanges T, Chevallier M, Trepo C. Liver damage induced by the ingestion of a product of phytotherapy containing wild germander. Four cases Gastroenterol Clin Biol. 1992;16:798–800. [PubMed] [Google Scholar]
- 124.Legoux JL, Maitre F, Labarriere D, Gargot D, Festin D, Causse X. Cytolytic hepatitis and wild Germander: a new case with reintroduction. Gastroenterol Clin Biol. 1992;16:813–815. [PubMed] [Google Scholar]
- 125.Castot A, Larrey D. Hepatitis observed during a treatment with a drug or tea containing Wild Germander. Evaluation of 26 cases reported to the Regional Centers of Pharmacovigilance. Gastroenterol Clin Biol. 1992;16:916–922. [PubMed] [Google Scholar]
- 126.Diaz D, Ferroudji S, Heran B, Barneon G, Larrey D, Michel H. Fulminant hepatitis caused by wild germander. Gastroenterol Clin Biol. 1992;16:1006–1007. [PubMed] [Google Scholar]
- 127.Ben Yahia M, Mavier P, Metreau JM, Zafrani ES, Fabre M, Gatineau-Saillant G, Dhumeaux D, Mallat A. Chronic active hepatitis and cirrhosis induced by wild germander. 3 cases. Gastroenterol Clin Biol. 1993;17:959–962. [PubMed] [Google Scholar]
- 128.Loeper J, Descatoire V, Letteron P, Moulis C, Degott C, Dansette P, Fau D, Pessayre D. Hepatotoxicity of germander in mice. Gastroenterology. 1994;106:464–472. doi: 10.1016/0016-5085(94)90606-8. [DOI] [PubMed] [Google Scholar]
- 129.Kouzi SA, McMurtry RJ, Nelson SD. Hepatotoxicity of germander (Teucrium chamaedrys L.) and one of its constituent neoclerodane diterpenes teucrin A in the mouse. Chem Res Toxicol. 1994;7:850–856. doi: 10.1021/tx00042a020. [DOI] [PubMed] [Google Scholar]
- 130.Lekehal M, Pessayre D, Lereau JM, Moulis C, Fouraste I, Fau D. Hepatotoxicity of the herbal medicine germander: metabolic activation of its furano diterpenoids by cytochrome P450 3A depletes cytoskeleton-associated protein thiols and forms plasma membrane blebs in rat hepatocytes. Hepatology. 1996;24:212–218. doi: 10.1002/hep.510240134. [DOI] [PubMed] [Google Scholar]
- 131.De Berardinis V, Moulis C, Maurice M, Beaune P, Pessayre D, Pompon D, Loeper J. Human microsomal epoxide hydrolase is the target of germander-induced autoantibodies on the surface of human hepatocytes. Mol Pharmacol. 2000;58:542–551. doi: 10.1124/mol.58.3.542. [DOI] [PubMed] [Google Scholar]
- 132.Druckova A, Mernaugh RL, Ham AJ, Marnett LJ. Identification of the protein targets of the reactive metabolite of teucrin A in vivo in the rat. Chem Res Toxicol. 2007;20:1393–1408. doi: 10.1021/tx7001405. [DOI] [PubMed] [Google Scholar]
- 133.Gordon WP, Forte AJ, McMurtry RJ, Gal J, Nelson SD. Hepatotoxicity and pulmonary toxicity of pennyroyal oil and its constituent terpenes in the mouse. Toxicol Appl Pharmacol. 1982;65:413–424. doi: 10.1016/0041-008x(82)90387-8. [DOI] [PubMed] [Google Scholar]
- 134.Sullivan JB, Jr, Rumack BH, Thomas H, Jr, Peterson RG, Bryson P. Pennyroyal oil poisoning and hepatotoxicity. JAMA. 1979;242:2873–2874. [PubMed] [Google Scholar]
- 135.Anderson IB, Mullen WH, Meeker JE, Khojasteh B, Oishi S, Nelson SD, Blanc PD. Pennyroyal toxicity: measurement of toxic metabolite levels in two cases and review of the literature. Ann Intern Med. 1996;124:726–734. doi: 10.7326/0003-4819-124-8-199604150-00004. [DOI] [PubMed] [Google Scholar]
- 136.Masada Y. The Analysis of Essential Oils by Gas Chromatography and Mass Spectrometry. Wiley; New York: 1976. [Google Scholar]
- 137.Gordon WP, Huitric AC, Seth CL, McClanahan RH, Nelson SD. The metabolism of the abortifacient terpene, (R)-(+)-pulegone, to a proximate toxin, menthofuran. Drug Metab Dispos. 1987;15:589–594. [PubMed] [Google Scholar]
- 138.Thomassen D, Slattery JT, Nelson SD. Contribution of menthofuran to the hepatotoxicity of pulegone: assessment based on matched area under the curve and on matched time course. J Pharmacol Exp Ther. 1988;244:825–829. [PubMed] [Google Scholar]
- 139.Moorthy B, Madyastha P, Madyastha KM. Metabolism of a monoterpene ketone, R-(+)-pulegone--a hepatotoxin in rat. Xenobiotica. 1989;19:217–224. doi: 10.3109/00498258909034694. [DOI] [PubMed] [Google Scholar]
- 140.Thomassen D, Knebel N, Slattery JT, McClanahan RH, Nelson SD. Reactive intermediates in the oxidation of menthofuran by cytochromes P-450. Chem Res Toxicol. 1992;5:123–130. doi: 10.1021/tx00025a021. [DOI] [PubMed] [Google Scholar]
- 141.Thomassen D, Slattery JT, Nelson SD. Menthofuran-dependent and independent aspects of pulegone hepatotoxicity: roles of glutathione. J Pharmacol Exp Ther. 1990;253:567–572. [PubMed] [Google Scholar]
- 142.Madyastha KM, Raj CP. Effects of menthofuran, a monoterpene furan on rat liver microsomal enzymes, in vivo. Toxicology. 1994;89:119–125. doi: 10.1016/0300-483x(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 143.Madyastha KM, Raj CP. Biotransformations of R-(+)-pulegone and menthofuran in vitro: chemical basis for toxicity. Biochem Biophys Res Commun. 1990;173:1086–1092. doi: 10.1016/s0006-291x(05)80897-6. [DOI] [PubMed] [Google Scholar]
- 144.Madyastha KM, Raj CP. Metabolic fate of menthofuran in rats. Novel oxidative pathways. Drug Metab Dispos. 1992;20:295–301. [PubMed] [Google Scholar]
- 145.Khojasteh-Bakht SC, Chen W, Koenigs LL, Peter RM, Nelson SD. Metabolism of (R)-(+)-pulegone and (R)-(+)-menthofuran by human liver cytochrome P-450s: evidence for formation of a furan epoxide. Drug Metab Dispos. 1999;27:574–580. [PubMed] [Google Scholar]
- 146.Khojasteh-Bakht SC, Koenigs LL, Peter RM, Trager WF, Nelson SD. (R)-(+)-Menthofuran is a potent, mechanism-based inactivator of human liver cytochrome P450 2A6. Drug Metab Dispos. 1998;26:701–704. [PubMed] [Google Scholar]
- 147.Kramlinger VM, von Weymarn LB, Murphy SE. Inhibition and inactivation of cytochrome P450 2A6 and cytochrome P450 2A13 by menthofuran, beta-nicotyrine and menthol. Chem Biol Interact. 2012;197:87–92. doi: 10.1016/j.cbi.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen LJ, Lebetkin EH, Burka LT. Metabolism of (R)-(+)-menthofuran in Fischer-344 rats: identification of sulfonic acid metabolites. Drug Metab Dispos. 2003;31:1208–1213. doi: 10.1124/dmd.31.10.1208. [DOI] [PubMed] [Google Scholar]
- 149.Khojasteh SC, Oishi S, Nelson SD. Metabolism and toxicity of menthofuran in rat liver slices and in rats. Chem Res Toxicol. 2010;23:1824–1832. doi: 10.1021/tx100268g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Madyastha KM, Gaikwad NW. Metabolic fate of S-(−)-pulegone in rat. Xenobiotica. 1998;28:723–734. doi: 10.1080/004982598239146. [DOI] [PubMed] [Google Scholar]
- 151.McClanahan RH, Thomassen D, Slattery JT, Nelson SD. Metabolic activation of (R)-(+)-pulegone to a reactive enonal that covalently binds to mouse liver proteins. Chem Res Toxicol. 1989;2:349–355. doi: 10.1021/tx00011a013. [DOI] [PubMed] [Google Scholar]
- 152.Edwards DJ, Bellevue FH, III, Woster PM. Identification of 6′,7′-dihydroxybergamottin, a cytochrome P450 inhibitor, in grapefruit juice. Drug Metab Dispos. 1996;24:1287–1290. [PubMed] [Google Scholar]
- 153.Schmiedlin-Ren P, Edwards DJ, Fitzsimmons ME, He K, Lown KS, Woster PM, Rahman A, Thummel KE, Fisher JM, Hollenberg PF, Watkins PB. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents. Decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug Metab Dispos. 1997;25:1228–1233. [PubMed] [Google Scholar]
- 154.He K, Iyer KR, Hayes RN, Sinz MW, Woolf TF, Hollenberg PF. Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem Res Toxicol. 1998;11:252–259. doi: 10.1021/tx970192k. [DOI] [PubMed] [Google Scholar]
- 155.Tassaneeyakul W, Guo LQ, Fukuda K, Ohta T, Yamazoe Y. Inhibition selectivity of grapefruit juice components on human cytochromes P450. Arch Biochem Biophys. 2000;378:356–363. doi: 10.1006/abbi.2000.1835. [DOI] [PubMed] [Google Scholar]
- 156.Guo LQ, Fukuda K, Ohta T, Yamazoe Y. Role of furanocoumarin derivatives on grapefruit juice-mediated inhibition of human CYP3A activity. Drug Metab Dispos. 2000;28:766–771. [PubMed] [Google Scholar]
- 157.Wangensteen H, Molden E, Christensen H, Malterud KE. Identification of epoxybergamottin as a CYP3A4 inhibitor in grapefruit peel. Eur J Clin Pharmacol. 2003;58:663–668. doi: 10.1007/s00228-002-0537-3. [DOI] [PubMed] [Google Scholar]
- 158.Seden K, Dickinson L, Khoo S, Back D. Grapefruit-drug interactions. Drugs. 2010;70:2373–2407. doi: 10.2165/11585250-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 159.Lin HL, Kent UM, Hollenberg PF. The grapefruit juice effect is not limited to cytochrome P450 (P450) 3A4: evidence for bergamottin-dependent inactivation, heme destruction, and covalent binding to protein in P450s 2B6 and 3A5. J Pharmacol Exp Ther. 2005;313:154–164. doi: 10.1124/jpet.104.079608. [DOI] [PubMed] [Google Scholar]
- 160.Bumpus NN, Sridar C, Kent UM, Hollenberg PF. The naturally occurring cytochrome P450 (P450) 2B6 K262R mutant of P450 2B6 exhibits alterations in substrate metabolism and inactivation. Drug Metab Dispos. 2005;33:795–802. doi: 10.1124/dmd.105.003749. [DOI] [PubMed] [Google Scholar]
- 161.Kent UM, Lin HL, Noon KR, Harris DL, Hollenberg PF. Metabolism of bergamottin by cytochromes P450 2B6 and 3A5. J Pharmacol Exp Ther. 2006;318:992–1005. doi: 10.1124/jpet.105.099887. [DOI] [PubMed] [Google Scholar]
- 162.Lin JH, Chiba M, Chen IW, Vastag KJ, Nishime JA, Dorsey BD, Michelson SR, McDaniel SL. Time- and dose-dependent pharmacokinetics of L-754,394, an HIV protease inhibitor, in rats, dogs and monkeys. J Pharmacol Exp Ther. 1995;274:264–269. [PubMed] [Google Scholar]
- 163.Chiba M, Nishime JA, Lin JH. Potent and selective inactivation of human liver microsomal cytochrome P-450 isoforms by L-754,394, an investigational human immune deficiency virus protease inhibitor. J Pharmacol Exp Ther. 1995;275:1527–1534. [PubMed] [Google Scholar]
- 164.Sahali-Sahly Y, Balani SK, Lin JH, Baillie TA. In vitro studies on the metabolic activation of the furanopyridine L-754,394, a highly potent and selective mechanism-based inhibitor of cytochrome P450 3A4. Chem Res Toxicol. 1996;9:1007–1012. doi: 10.1021/tx960060b. [DOI] [PubMed] [Google Scholar]
- 165.Lightning LK, Jones JP, Friedberg T, Pritchard MP, Shou M, Rushmore TH, Trager WF. Mechanism-based inactivation of cytochrome P450 3A4 by L-754,394. Biochemistry. 2000;39:4276–4287. doi: 10.1021/bi992412u. [DOI] [PubMed] [Google Scholar]
- 166.Bateman KP, Baker J, Wilke M, Lee J, Leriche T, Seto C, Day S, Chauret N, Ouellet M, Nicoll-Griffith DA. Detection of covalent adducts to cytochrome P450 3A4 using liquid chromatography mass spectrometry. Chem Res Toxicol. 2004;17:1356–1361. doi: 10.1021/tx0498861. [DOI] [PubMed] [Google Scholar]
- 167.Bohme A, Thaens D, Schramm F, Paschke A, Schuurmann G. Thiol reactivity and its impact on the ciliate toxicity of α,β-unsaturated aldehydes, ketones, and esters. Chem Res Toxicol. 2010;23:1905–1912. doi: 10.1021/tx100226n. [DOI] [PubMed] [Google Scholar]