

Abstract
Rationale
Oxidation of cysteine residues in class II histone deacetylases (HDACs), including HDAC4, causes nuclear exit, thereby inducing cardiac hypertrophy. The cellular source of reactive oxygen species (ROS) responsible for oxidation of HDAC4 remains unknown.
Objective
We investigated whether Nox4, a major NADPH oxidase, mediates cysteine oxidation of HDAC4.
Methods and Results
Phenylephrine (PE, 100 μM), an α1 adrenergic agonist, induced upregulation of Nox4 (1.5-fold, p<0.05) within 5 min, accompanied by increases in O2− (3.5-fold, p<0.01) from the nuclear membrane and nuclear exit of HDAC4 in cardiomyocytes (CM). Knockdown of Nox4, but not Nox2, attenuated O2− production in the nucleus and prevented PE-induced oxidation and nuclear exit of HDAC4. After continuous infusion of PE (20 mg/kg/day) for 14 days, wild-type (WT) and cardiac-specific Nox4 knockout (c-Nox4 KO) mice exhibited similar aortic pressures. Left ventricular (LV) weight/tibial length (5.7 ±0.2 vs. 6.4 ±0.2 mg/mm, p<0.05) and CM cross-sectional area (223 ±13 vs. 258 ±12 μm2, p<0.05) were significantly smaller in c-Nox4 KO than in WT mice. Nuclear O2− production in the heart was significantly lower in c-Nox4 KO than in WT mice (4116 ±314 vs. 7057 ±1710 RLU, p<0.05), and cysteine oxidation of HDAC4 was decreased. HDAC4 oxidation and cardiac hypertrophy were also attenuated in c-Nox4 KO mice 2 weeks after transverse aortic constriction.
Conclusions
Nox4 plays an essential role in mediating cysteine oxidation and nuclear exit of HDAC4, thereby mediating cardiac hypertrophy in response to PE and pressure overload.
Keywords: reactive oxygen species, oxidative stress, free radicals, hypertrophy, remodeling
Introduction
Cardiac hypertrophy is an adaptive mechanism triggered in the heart in response to increased mechanical stress. However, the prolonged presence of hypertrophy can cause cardiac dysfunction, and cardiac hypertrophy is therefore regarded as one of the major risk factors for heart failure.1 Experimental and clinical studies have demonstrated that the cellular status of reduction and oxidation (redox) is intimately involved in the pathogenesis of cardiac hypertrophy and failure.2 Class II histone deacetylases (HDACs), including HDAC4, directly interact with key transcription factors that mediate hypertrophy, including nuclear factor of activated T cells (NFAT) and myocyte enhancer factor 2 (MEF2), thereby inhibiting their activity through histone deacetylation.3 Hypertrophic stimuli activate HDAC kinases, including protein kinase D (PKD), calcium/calmodulin-dependent kinase II (CaMKII), and G protein-coupled receptor kinase 5 (GRK5), which phosphorylate conserved serine residues and induce cytoplasmic translocation of class II HDACs. This removes the negative constraint of class II HDACs on the hypertrophic transcription factors and causes hypertrophy. Importantly, in addition to the phosphorylation-dependent mechanisms, oxidation of conserved cysteine residues in class II HDACs also induces their cytoplasmic translocation,4 and the latter mechanism appears more important than the former in some forms of cardiac hypertrophy, such as β-adrenergic hypertrophy.5 In addition, thioredoxin1 (Trx1), an antioxidant, prevents cardiac hypertrophy by inhibiting HDAC4 cysteine oxidation.4 Despite its importance, however, the molecular mechanism through which HDAC4 is oxidized in response to hypertrophic stimuli is not well understood.
Among the potential sources of reactive oxygen species (ROS) within the heart, NAD(P)H oxidases (Noxes) are the major sources of superoxide (O2−) production and are involved in both physiological and pathological processes in cells. The fact that Noxes actively produce O2− distinguishes them from the other major source of O2−, mitochondria, where O2− is produced through a leakage of electrons, a passive mechanism. Thus far, seven members of the Nox family of proteins (Nox1 to 5 and Duox1 and 2) have been identified.6 Nox2 and Nox4 are the major isoforms in the heart.7 Despite their structural similarity, these proteins have distinct characteristics. For example, although Nox2 needs cytosolic factors for its activation,8 Nox4 may not.9 Nox2 and Nox4 also have distinct subcellular localizations, although these appear to be cell type-dependent. In general, Nox2 is localized primarily at the plasma membrane whereas Nox4 is found at intracellular membranes, including the nuclear membrane.10, 11
Previous studies have suggested the involvement of Noxes in cardiac hypertrophy. For example, Nox2 plays an important role in mediating angiotensin II-induced hypertrophy, but not pressure overload (PO)-induced hypertrophy.12 Nox4 is known to be upregulated in response to several hypertrophic stimuli, such as angiotensin II (AngII), α-adrenergic agonists and PO, and is a major source of ROS in hypertrophic hearts.10, 11 Cardiac-specific, but not systemic, downregulation of Nox4 attenuated pathological hypertrophy after PO through inhibition of mitochondrial dysfunction and apoptosis.11 Importantly, though, the molecular mechanisms through which each Nox isoform mediates hypertrophy are not well understood.
Given the fact that the cysteine residues of HDAC4 are oxidized by hypertrophic stimuli in a regulated manner, it is reasonable to hypothesize that enzymes purposefully producing ROS, such as Noxes, might be involved in the cysteine oxidation of class II HDACs. Since Nox4 is localized at intracellular membranes and is intimately involved in increases in oxidative stress during cardiac hypertrophy, we speculated that Nox4 is responsible for cysteine oxidation of HDAC4. Accordingly, the purpose of this study was to examine the roles of Nox2 and Nox4 in mediating cysteine oxidation of HDAC4 and cardiac hypertrophy, using both in vitro and in vivo models of cardiac hypertrophy.
Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org
Mice
Nox4−/− mice were generated using lox-P and homologous recombination strategies. Cardiac-specific deletion of Nox4 (c-Nox4 KO) was achieved using αMHC-Cre.11 Mice with cardiac-specific overexpression of Nox4 (Tg-Nox4) were generated as described previously.10 All experiments were conducted in 2–3-month-old male mice. All protocols concerning the use of animals were approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey.
Continuous infusion of phenylephrine
Continuous infusion of phenylephrine (20 mg/kg/day) or vehicle control was conducted with osmotic mini-pumps (model 2002, Alza Corp., Palo Alto, CA, USA) as described previously.13 Control mice received pumps filled with 0.9% sodium chloride (saline).
Detection of thiolate cysteines of HDAC4 and HDAC5
Myocytes transduced with HDAC4 adenoviruses were treated with phenylephrine (PE) in the presence of knockdown or overexpression of Nox4 and lysed with lysis buffer containing 200 μM biotinylated–iodoacetamide (IAM). Heart homogenates from mice treated with or without PE or transverse aortic constriction (TAC) were also used. Biotinylated proteins were pulled down with streptavidin beads (PIERCE). Biotinylated HDAC4 and HDAC5, representing the reduced form, were detected by immunoblotting.
Statistical Analysis
Data are expressed as mean ± SEM. Comparison of means between groups was performed by one-way ANOVA, followed by t-tests. Bonferroni’s correction was carried out for multiple comparisons of means. P<0.05 was considered to be statistically significant.
Results
Nox4 is Involved in PE-Induced Hypertrophy in Cultured Cardiomyocytes
We examined the cell-autonomous effect of Nox4 in mediating cardiac hypertrophy using phenylephrine (PE)-induced cardiac hypertrophy in cultured cardiomyocytes, a well-established in vitro model of cardiac hypertrophy. PE increased Nox4 protein expression in a dose-dependent manner (Figure 1A, B). As shown previously, PE also induced cardiomyocyte hypertrophy in a dose-dependent manner (Figure 1C). To evaluate the direct effect of Nox4 upon PE-induced cardiac hypertrophy, we transduced adenoviruses harboring either wild-type Nox4 or short hairpin RNA for Nox4 into cardiomyocytes. As we reported previously, overexpression of Nox4 (1.5-fold, p<0.05, data not shown) was not sufficient to induce cardiac hypertrophy, as evaluated by cell size, protein/DNA content, and expression of ANF, a fetal-type gene. However, overexpression of Nox4 significantly enhanced PE-induced hypertrophy in cardiomyocytes (Figure 1D, E, and F). Conversely, although downregulation of Nox4 (0.2-fold, p<0.05, data not shown) did not affect cardiac hypertrophy at baseline, it significantly attenuated PE-induced cardiomyocyte hypertrophy (Figure 1D, E, and F). These results indicate that endogenous Nox4 is necessary for PE-induced cardiac hypertrophy and that upregulation of Nox4 enhances PE-induced cardiac hypertrophy.
Figure 1. Nox4 is required for PE-induced cardiac hypertrophy in vitro.

A and B. Expression levels of Nox4 and GAPDH in cultured neonatal rat cardiomyocytes 48 hours after PE treatment at multiple doses (n=3). C. Mean surface area of cardiomyocytes 48 hours after PE treatment at multiple doses was obtained from 200 myocytes per sample (n=6). D, E, and F. Cultured cardiac myocytes were transduced with Ad-LacZ, Ad-Nox4, or Ad-shNox4. D. Cell surface area of myocytes treated with the indicated adenoviruses in the presence or absence of PE for 48 hours was examined (n=6). E. Protein and DNA content of myocytes treated with the indicated adenoviruses in the presence or absence of PE for 48 hours was examined (n=8). F. Expression of mRNA for atrial natriuretic factor (ANF) in myocytes treated with the indicated adenoviruses in the presence or absence of PE for 48 hours was measured by quantitative RT-PCR (n=8). *P<0.05, **P<0.01. N.S.: not significant.
Overexpression of Nox4 Exacerbates PE-Induced Cardiac Hypertrophy in Mice
We examined the effect of Nox4 upon PE-induced cardiac hypertrophy in vivo. Cardiac hypertrophy was induced in mice by continuous infusion with a subpressor dose of PE (20 mg/kg/day). PE increased Nox4 expression in the left ventricle (LV) in both nontransgenic (NTg) and Tg-Nox4 mice. However, the Nox4 expression level was significantly higher in Tg-Nox4 mice than in NTg mice after PE stimulation (Figure 2A and B). There was no difference in blood pressure (BP) among the four groups (Figure 2C). Although Tg-Nox4 mice showed the same myocyte cross-sectional area (MCSA) as NTg mice at baseline, they exhibited a significantly greater MCSA than NTg mice after PE treatment (Figure 2D and E). PE-induced increases in ANF expression were also enhanced significantly in Tg-Nox4 mice (Figure 2F). These results suggest that upregulation of Nox4 exacerbated PE-induced cardiac hypertrophy in vivo. Despite the enhancement of cardiac hypertrophy at a cardiomyocyte level, PE-induced increases in LV weight (LVW)/tibial length (TL) were not significantly enhanced in Tg-Nox4 mice (Figure 2G). Tg-Nox4 mice also did not show the apparant difference in LV morphology after PE infusion (Online Figure IA). These may be due in part to the enhancement of apoptosis, since there were significantly more TUNEL-positive cardiomyocytes in Tg-Nox4 than in NTg mice in the presence of PE (Online Figure IB), which is reminiscent of the phenotype observed in old Tg-Nox4 mice.10 There was no difference in collagen volume fraction in the LV among the four groups (Online Figure IC).
Figure 2. Nox4 enhances PE-induced cardiac hypertrophy in vivo.

A and B. Expression levels of Nox4 and GAPDH in NTg and Tg-Nox4 mouse hearts subjected to either PE or saline infusion for 2 weeks. The results of the quantitative analysis of Nox4 expression are shown (n=5). C. The effect of PE and overexpression of Nox4 on blood pressure was measured by tail cuff (n=6). D and E. LV myocyte cross-sectional area, as evaluated using wheat germ agglutinin (WGA) staining (n=4). Bar=20 μm. F. Expression of ANF mRNA in NTg and Tg-Nox4 mouse hearts in the presence or absence of PE infusion was evaluated by quantitative RT-PCR (n=6). G. LVW/TL after PE stimulation was determined (n=6). *P<0.05, **P<0.01. N.S.: not significant.
Cardiac-Specific Nox4 Knockout Attenuates PE-Induced Cardiac Hypertrophy in Mice
In order to investigate the role of endogenous Nox4 in mediating PE-induced cardiac hypertrophy in vivo, cardiac-specific Nox4 knockout (c-Nox4 KO) mice were used. c-Nox4 KO mice exhibited a significantly lower level of Nox4 expression in the heart in both the saline- and PE-infusion groups (Figure 3A and B). There was no difference in BP among the four groups (Figure 3C). The PE-induced increase in MCSA was significantly attenuated in c-Nox4 KO mice (Figure 3D and E). The level of ANF expression and LVW/TL after PE treatment were also significantly lower in c-Nox4 KO mice than in wild-type (WT) mice (Figure 3F and G). The cross section of the LV also showed that PE-induced cardiac hypertrophy was attenuated in c-Nox4 mice (Online Figure IIA). There was no significant difference in apoptosis or interstitial fibrosis in the LV among the four groups (Online Figure IIB and C). Echocardiographic measurements showed that, although fractional shortening did not vary significantly among the four groups, PE-induced increases in the wall thickness were significantly attenuated in c-Nox4 KO mice (Online Figure IIIA-C). Hemodynamic measurements showed that c-Nox4 KO tended to have a lower LV end-diastolic pressure and minimum dP/dt, indexes of cardiac diastolic function, than WT after PE treatment (Online Table I, p=0.085 and p=0.055 vs. WT, respectively). These results indicate that endogenous Nox4 plays an important role in mediating PE-induced cardiac hypertrophy and diastolic dysfunction in the mouse heart.
Figure 3. Endogenous Nox4 plays an essential role in mediating PE-induced cardiac hypertrophy.

A and B. Expression levels of Nox4 and GADPH in wild-type (WT) and c-Nox4 KO mouse hearts subjected to either PE or saline infusion for 2 weeks. The results of the quantitative analysis of Nox4 expression are shown (n=3). C. The effect of PE and knockdown of Nox4 on blood pressure was measured by tail cuff (n=6). D and E. LV myocyte cross-sectional area, as evaluated using WGA staining (n=4). Bar=20 μm. F. ANF mRNA normalized by GAPDH mRNA in WT and c-Nox4 KO mouse hearts in the presence or absence of PE infusion was evaluated by quantitative RT-PCR (n=6). G. LVW/TL after PE stimulation was determined (n=6). *P<0.05, **P<0.01.
Knockdown of Nox4, but not Nox2, Attenuates PE-Stimulated Nuclear ROS Production
We examined the localization of Nox4 in cardiomyocytes. Using immunostaining, Nox4 was found to be localized in the perinuclear region and on the surface of the nucleus in cardiomyocytes (Figure 4A). Immunoblot analysis showed that Nox4 is found not only at mitochondria and microsomes but also in the nuclear fraction (Figure 4B). On the other hand, Nox2 was expressed mostly in the microsomal fractions (Figure 4B). The level of Nox4 increased rapidly, within 5 minutes, after PE stimulation in cardiomyocytes (Figure 4C), accompanied by increases in O2−, as evaluated by the lucigenin assay, and H2O2 production, as evaluated with Hyper-nuc fluorescence, in the nucleus (Figure 4D and E). Overexpression of Nox4 further increased ROS production, whereas downregulation of Nox4 attenuated it (Figure 4D and E). In contrast, downregulation of Nox2 did not affect nuclear O2− or H2O2 production after PE stimulation (Figure 4D, Online Figure IV).
Figure 4. Endogenous Nox4 mediates PE-stimulated nuclear ROS production.

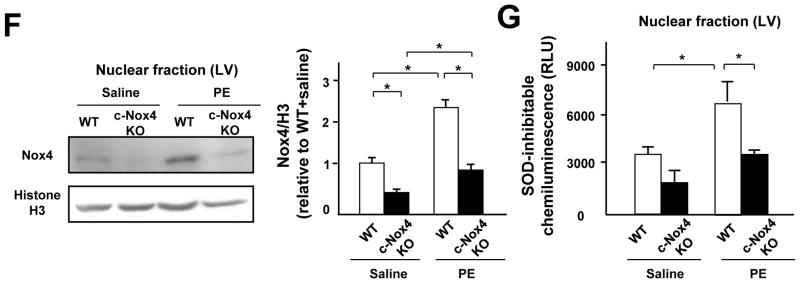
A. Cultured myocytes were co-stained with rabbit anti-Troponin T antibody and mouse anti-NOX4 antibody, followed by anti-rabbit AlexaFluor-568 conjugated secondary antibody and anti-mouse AlexaFluor-488 conjugated secondary antibody (upper panel), or with anti-Troponin T and secondary antibodies alone (lower panel). Nuclei were stained with DAPI. Merged images are also shown. Bar=10 μm. B. Cytosolic, mitochondrial, microsomal, and nuclear fractions were prepared from cultured myocytes. Immunoblot analyses were conducted with anti-Nox4 antibody and anti-Nox2 antibody. The purity of the mitochondrial fraction was confirmed by the absence of histone H3 or GAPDH staining. The purity of the nuclear fraction was confirmed by the absence of COX IV and GAPDH. C. Expression level of Nox4 in the nucleus in cardiomyocytes after PE stimulation. Protein expression of Nox4 and GAPDH was determined by immunoblotting. The results of the quantitative analysis of Nox4 expression are also shown (n=4). D. Nuclear fractions were prepared from myocytes transduced with the indicated adenoviruses. NADPH-dependent O2− release was measured by the lucigenin method. The SOD-inhibitable component of O2− release from the nuclear fraction was determined (n=6). E. Myocytes were transduced with Ad-Hyper-nuc and Ad-LacZ or Ad-shNox4 in the presence or absence of PE for 5 minutes. Hyper-nuc is an indicator of hydrogen peroxide localized in the nucleus. Green staining indicates H2O2 production (n=8). Nuclei were stained with DAPI. Bar=20 μm. F and G. Nuclear fractions were prepared from WT or c-Nox4 KO mouse hearts. F. Expression levels of Nox4 and histone H3 in the nuclear fraction from WT or c-Nox4 KO mouse hearts. G. NADPH-dependent and SOD-inhibitable O2− release was measured by the lucigenin method (n=4). *P<0.05, **P<0.01. N.S.: not significant.
Nox4 in the nuclear fraction was upregulated after PE treatment in control mouse hearts (Figure 4F and Online Figure VA). Tg-Nox4 mice showed a further increase in nuclear Nox4 expression after PE treatment, whereas PE-treated c-Nox4 KO mice exhibited a Nox4 expression level similar to that of control WT mice (Figure 4F and Online Figure VA). We also evaluated nuclear O2− production in the heart. Consistent with the level of Nox4 in the nucleus, overexpression of Nox4 increased PE-induced O2− production in the nuclear fraction (Online Figure VB), whereas downregulation of Nox4 attenuated it (Figure 4G). Taken altogether, these data suggest that Nox4, not Nox2, is the main Nox isoform mediating nuclear ROS production in cardiomyocytes.
Downregulation of Nox4 Inhibits Nuclear Exit of HDAC4 after PE Treatment in a Redox-Sensitive Manner
To elucidate the mechanism by which downregulation of Nox4 inhibits cardiac hypertrophy, we examined the nuclear exit of HDAC4. Immunostaining showed that PE induces nuclear exit of HDAC4 within 5 minutes in cardiomyocytes (Figure 5A). Importantly, downregulation of Nox4 prevented the nuclear exit of HDAC4 after PE treatment (Figure 5A). Consistent with this result, immunoblotting showed that the nuclear exit of HDAC4 was prevented by downregulation of Nox4 in cardiomyocytes and was enhanced in Nox4-overexpressing myocytes (Figure 5B and C). In contrast, downregulation of Nox2 did not affect the nuclear exit of HDAC4 (Figure 5D). These results indicate that endogenous Nox4 plays an important role in the nuclear exit of HDAC4 in cardiomyocytes.
Figure 5. Endogenous Nox4 mediates nuclear export of HDAC4 after PE treatment.

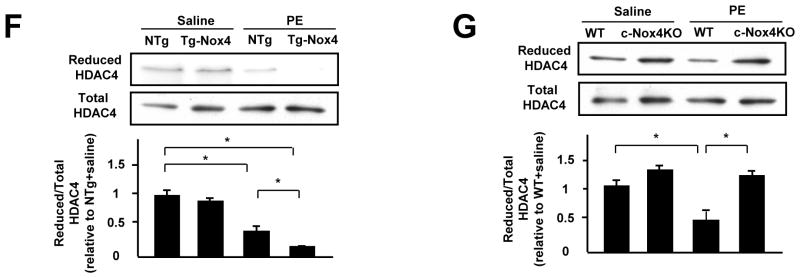
A. Myocytes transduced with the indicated adenoviruses were stained with anti-HDAC4 antibody (green), anti-Troponin T antibody (red), and DAPI (Blue). Bar=10 mm. The result shown is representative of 8 experiments. The results of the quantitative analysis of the nuclear/total HDAC4 ratio are shown (n=8). B. Protein levels of HDAC4 in the nucleus, cytosol, and whole cell fractions from myocytes transduced with the indicated adenoviruses 5 minutes after PE stimulation were determined by immunoblot. C. The results of the quantitative analysis of the nuclear/total HDAC4 ratio at baseline and cytosol/total HDAC4 ratio after PE stimulation are shown (n=4). D. Myocytes transduced with the indicated adenoviruses were stained with anti-HDAC4 antibody (green), anti-Troponin T antibody (red), and DAPI (Blue). Bar=10 μm. The result shown is representative of 8 experiments. E. Myocytes transduced with the indicated adenoviruses were treated with PE for the indicated time periods. The extent of cysteine reduction in HDAC4 was evaluated with iodoacetamide. The reduced HDAC4 protein level was normalized by the total HDAC4 protein level (n=4). F and G. The extent of cysteine reduction in HDAC4 in the indicated mouse hearts was detected. *P<0.05. **P<0.01.
The nuclear exit of HDAC4 is induced by oxidation (disulfide bond formation) of Cysteines 667 and 669. We therefore evaluated the extent of cysteine oxidation in HDAC4. When myocytes were treated with PE, the levels of free thiols in HDAC4 were significantly decreased within 5 minutes (Figure 5E). The level of reduced cysteines in the whole-cell fraction was preserved in Ad-sh-Nox4-transduced myocytes, but not in Ad-sh-Nox2-transduced ones (Figure 5E, Online Figure VIA). The level of reduced cysteines in the nuclear fraction was also preserved in Ad-sh-Nox4-transduced myocytes (Online Figure VIB). These results suggest that Nox4 is the main oxidizer of HDAC4 during PE stimulation.
Another important mechanism involved in the nuclear exit of HDAC4 is phosphorylation of three critical serines (Ser-246, Ser-467, and Ser-632) by HDAC kinases, including CaMKII. To modulate the nuclear exit of HDAC4 through the phosphorylation-dependent mechanism, we used adenoviruses harboring HDAC4 mutants in which these three serines were substituted with alanines (3SA) or aspartic acids (3SD). As reported previously,14 the 3SD mutant was localized in the cytosol even without PE stimulation (Online Figure VIIA). Although the 3SA mutant was localized in the nucleus under unstimulated conditions, it was exported from the nucleus to the cytosol after PE stimulation. Downregulation of Nox4 inhibited the PE-induced nuclear exit of the 3SA mutant, suggesting that the nuclear exit of HDAC4 by PE takes place via a Nox4-dependent mechanism (Online Figure VIIA). PE induced phosphorylation of the three serine residues in HDAC4 after 120 minutes, at a later time point than oxidation of HDAC4 (Online Figure VIIB).4 Importantly, downregulation of Nox4 did not affect PE-induced phosphorylation of HDAC4 (Online Figure VIIB) or PE-induced phosphorylation of the HDAC kinase CaMKII (Online Figure VIIC). These findings indicate that Nox4 induces the nuclear exit of HDAC4 independently of the phosphorylation-mediated mechanisms. We confirmed that a lower dose of PE (10μM) also induces oxidation and the nuclear exit of HDAC4 (Online Figure IXA), suggesting that oxidation and the nuclear exit of HDAC4 are physiological responses to PE stimulation in cardiomyocytes. Cysteine residues subjected to oxidation are conserved in the class II HDAC family, including HDAC5. PE treatment also induced cysteine oxidation in HDAC5 in cardiomyocytes (Online Figure IXB).
In order to elucidate the role of Nox4 localized in mitochondria and the nucleus in mediating oxidation of HDAC4, we generated adenovirus harboring either Peroxiredoxin-3 (Prx3), a mitochondrial isoform of the Prx family, or nuclear-localized catalase (Nuc-catalase) (Online Figure VIIIA and B). Treatment with N-acetyl-cysteine (NAC) or overexpression of Nuc-catalase, but not overexpression of Prx3, prevented the nuclear exit of HDAC4 after PE stimulation in cardiomyocytes (Online Figure VIIIC). These data indicate that a global increase in ROS or ROS derived from the nucleus rather than mitochondria are critical for oxidation of HDAC4.
Downregulation of Nox4 Prevents PE-Induced Upregulation of NFAT Target Genes
Nuclear exit of HDAC4 activates NFAT. We examined NFAT transcriptional activity using luciferase assays. PE increased the activity of the NFAT reporter in cardiomyocytes, an effect that was significantly increased by overexpression of Nox4 and decreased by downregulation of Nox4 (Figure 6A). To further investigate the effect of Nox4 upon hypertrophy-associated gene expression in an unbiased manner, we conducted a DNA microarray analysis using total RNA prepared from WT and c-Nox4 KO mouse hearts after PE treatment. The pathway analysis showed that PE upregulated extracellular matrix-related and actin- and sarcolemma-related genes (Figure 6B). The PE-induced changes in these pathways were all normalized in c-Nox4 KO mice, indicating the involvement of Nox4 in these processes. PE treatment increased Nox4 mRNA levels in WT but not in c-Nox4 KO mouse hearts (Figure 6C). PE-induced increases in Nppa (natriuretic peptide precursor A) and Nppb (natriuretic peptide precursor B), markers of cardiac hypertrophy, were attenuated in c-Nox4 KO mouse hearts (Figure 6C), also indicating the validity of the assays. Although the mRNA levels of Nfat and Mef2 were not significantly different between groups (Figure 6C), PE treatment upregulated NFAT downstream genes, including Myh7, Rcan1, and Wisp1, which were normalized in c-Nox4 KO mouse hearts (Figure 6D). Collectively, these data show that downregulation of Nox4 prevents upregulation of the known NFAT targets in the mouse heart.
Figure 6. Nox4 regulates NFAT target genes in response to PE.

A. Myocytes transduced with the indicated adenovirus were treated with or without PE for 48 hours. NFAT luciferase activity was measured (n=8). B–D. Total RNA was subjected to microarray analyses. B. The pathway analysis showed that PE upregulated extracellular matrix-related, actin-related and sarcolemma-related genes. C. Trend of expression levels of Nox4, Nppa, Nppb, Nfatc4, and Mef2c in the indicated mouse hearts. D. Trend of expression levels of downstream targets of NFAT.
The NF-κBInduced Nox4 Upregulation
To elucidate the mechanism mediating the rapid upregulation of Nox4, we evaluated the level of Nox4 mRNA. PE treatment increased the mRNA level of Nox4 within 5 minutes (Figure 7A). The activated NF-κB complex binds DNA at NF-κB-binding motifs such as GGGRNNYYCC or HGGARNYYCC 3 (H: A, C, or T; R: A or G purine; Y: C or T pyrimidine) and induces gene expression. We found an NF-κB binding site upstream of the transcriptional start point of rat Nox4 gene (from −1628 to −1618: GGGGGTTTCC, Online Figure XA). In addition, PE increased the activity of an NF-κB luciferase reporter (Figure 7B) in cardiomyocytes. PE rapidly decreased expression of IκB and increased the nuclear expression of NF-κB (Figure 7C) in cardiomyocytes. In addition, PE increased phosphorylated NF-κB, which is an active form of NF-κB, in mouse hearts (Online Figure XB). To further investigate whether NF-κB binds to the Nox4 gene, we performed a chromatin immunoprecipitation (ChIP) assay in lysates obtained from myocytes treated with or without PE using an antibody against NF-κB-p65. The ChIP product was subjected to a polymerase chain reaction to amplify a fragment of the upstream region of the Nox4 gene containing the NF-κB-binding motif (Online Figure XA). The results showed that NF-κB binds to the upstream region of Nox4 and that PE enhances the DNA binding of NF-κB (Figure 7D). These data raise the possibility that PE upregulates Nox4 through activation of NF-κB. We next evaluated the role of the IκB-NF-κB pathway in regulating Nox4 expression. Transduction of cardiomyocytes with an adenovirus harboring constitutively active IκB (IκB-SA) inhibited PE-induced phosphorylation of NF-κB, which was accompanied by a decrease in PE-induced Nox4 expression (Figure 7E). These results suggest that the IκB-NF-κB pathway plays an essential role in mediating PE-induced upregulation of Nox4.
Figure 7. NF-κB mediates PE-induced Nox4 upregulation.

A. Myocytes were treated with PE for the indicated time periods. mRNA expression of Nox4 was determined by quantitative RT-PCR (n=5). B. Myocytes were treated with PE for 48 hours. NF-κB-luciferase activity was measured (n=8). C. Lysates were prepared from cardiomyocytes treated with PE for the indicated time periods. Protein expression of IκB and GAPDH was determined by immunoblotting. Nuclear fractions were prepared from myocytes treated with PE for the indicated time periods. Protein expression of NF-κB and histone H3 in the nuclear fraction was determined by immunoblotting (n=4). D. Chromatin immunoprecipitation (ChIP) assays with antibody against NF-κB (p65). A parallel ChIP assay was performed with rabbit IgG as an assay control. DNA was amplified and quantified by PCR with specific primers flanking the region of the rat Nox4 gene promoter containing the NF-κB-binding motif and with a pair of control primers that do not amplify the region containing the NF-κB-binding motif. PCR using input DNA as a template served as an internal control. The result shown is representative of 3 experiments. E. Myocytes transduced with the indicated adenoviruses were treated with PE for 48 hours. A representative analysis of the expression levels of phosphorylated NF-κB, NF-κB, IκB, Nox4 and GAPDH in the cultured neonatal rat cardiomyocytes and quantitative analyses of phosphorylated NF-κB/NF-κB and Nox4/GAPDH are shown (n=4). *P<0.05.
We also investigated whether PE rapidly induces nuclear exit of HDAC4 in a Nox4-dependent manner in the mouse heart. The expression of Nox4 was upregulated within 5 min of intravenous injection of PE, which was accompanied by nuclear exit and oxidation of HDAC4. c-Nox4KO mice did not show oxidation and nuclear exit of HDAC4, suggesting that Nox4 is required for the nuclear exit and oxidation of HDAC4 in response to acute injection of PE in vivo. The expression level of IκB was downregulated and phosphorylation of NF-κB was upregulated within 2.5min of PE injection, consistent with the notion that the upregulation of Nox4 is mediated by an NF-κB-dependent mechanism (Online Figure XI).
Downregulation of Nox4 Prevents Pressure Overload-Induced Cardiac Hypertrophy by Inhibiting Oxidation of HDAC4
We investigated whether Nox4 is involved in the oxidation of HDAC4 in response to pressure overload (PO). Analyses were conducted 2 weeks after transverse aortic constriction (TAC), when systolic function was still maintained, in order to avoid the secondary effect of cardiac dysfunction upon cardiac hypertrophy. Nuclear expression of Nox4 was significantly increased in response to TAC in the LVs of control mice, while the upregulation was decreased in those of c-Nox4 KO mice (Figure 8A and B). Although PO significantly increased nuclear O2− production in the LVs of control mice, this was attenuated in c-Nox4 KO mice (Figure 8C). Iodoacetamide labeling experiments showed that the level of the reduced form of HDAC4 was significantly decreased during PO in whole-cell and nuclear fractions. Importantly, downregulation of Nox4 in c-Nox4 KO mice prevented the cysteine oxidation of HDAC4 in response to PO (Figure 8D and Online Figure XII) and decreased nuclear exit of HDAC4 (Figure 8E and 8F). We also evaluated S632 phosphorylation of HDAC4, another mechanism mediating its nuclear exit. After pressure overload, downregulation of Nox4 did not affect phosphorylation of HDAC4 (Figure 8G). PO-induced increases in MCSA were significantly attenuated in c-Nox4 KO mice (Online Figure XIIIA and B). PO-induced increases in LVW/TL and wall thickness were also significantly attenuated in c-Nox4 KO mice (Online Figure XIIIC and D). Collagen volume fraction and apoptosis in the LV were increased after PO, but these increases were attenuated in c-Nox4 KO mice (Online Figure XIVA and B). Hemodynamic measurements showed that LVEDP was significantly elevated in WT after TAC, but the increase was attenuated in c-Nox4 KO mice (Online Table II). These results suggest that Nox4 in cardiomyocytes plays an important role in mediating PO-induced cardiac hypertrophy and diastolic dysfunction in the mouse heart.
Figure 8. Nox4 plays an essential role in mediating oxidative stress in the nucleus and pressure overload-induced cardiac hypertrophy.

A and B. Expression levels of Nox4 and histone H3 in nuclear fractions from WT and c-Nox4 KO mouse hearts subjected to either sham or TAC operation. Protein expression of Nox4 and histone H3 was determined by immunoblotting (n=3). C. Nuclear fractions were prepared from the indicated mouse hearts. NADPH-dependent and SOD-inhibitable O2− release was measured by the lucigenin method (n=4). D. Homogenates were prepared from the indicated mouse hearts. The extent of cysteine reduction in HDAC4 was detected with biotinylated-iodoacetamide (n=4). The reduced HDAC4 protein level was normalized by the total HDAC4 protein level. E. Protein levels of HDAC4 in the nucleus and the cytosol in the indicated mouse hearts subjected to either sham or TAC operation were determined by immunoblot (n=3). F. Fourteen days after TAC, specimens of mouse hearts were stained with HDAC4 antibody with the use of a horseradish-peroxidase system. Arrows indicate HDAC4 staining in the nucleus. Bar=10 μm. The result shown is representative of 4 experiments. G. The expression level of HDAC4 and phosphorylation of HDAC4 in the indicated mouse hearts subjected to either sham or TAC operation were examined by immunoblot. *P<0.05.
Discussion
HDAC4 is a member of the class II family of HDACs, which are expressed only in terminally differentiated cells, including cardiomyocytes.3 Oxidation of HDAC4 at Cys-667 and Cys-669 and of conserved cysteine residues in other members of the class II HDAC family leads to nuclear exit of the class II HDACs and induction of cardiac hypertrophy. Thus far, thioredoxin1 is known to prevent cardiac hypertrophy by inhibiting the oxidation of HDAC4 in myocytes.4 However, the important question of how HDAC4 is oxidized in the presence of hypertrophic stimuli remains unresolved. Our study shows that Nox4 plays an important role in mediating oxidation of HDAC4 in response to phenylephrine and pressure overload. Although the involvement of ROS in cardiac hypertrophy is well established, the specific sources of ROS and their molecular targets have yet to be discovered. Our results have established a link between Nox4, a source of ROS, and HDAC4, a cellular target of ROS, in the development of cardiac hypertrophy (Online Figure XV).
Some ROS, including H2O2, are membrane-permeable and diffusible in cells. Thus, even if they originate from other (non-nuclear) subcellular sources, they could, in theory, contribute to oxidative damage in the nucleus. Nox2 and Nox4 equally induce oxidative stress in the heart in response to ischemia/reperfusion when oxidative stress is evaluated at the whole-cell level (manuscript in preparation). Here, however, we found that knockdown of Nox4, but not Nox2, attenuates oxidation and prevents nuclear export of HDAC4 after PE treatment. These findings suggest that the compartmentalization of Nox4 may play an essential role in regulating the redox state of HDAC4 and hypertrophic signaling.
The isoform-specific actions of Noxes depend upon their subcellular localizations. We have shown previously that Nox4 is localized not only at mitochondria but also at the nucleus in myocytes, whereas Nox2 is expressed predominantly in the microsomal fraction. The possibility that Nox4 exhibits nuclear localization is supported by the fact that Nox4 has nuclear localization signals in its 90–96 and 451–457 residues.15 Using confocal microscopy, we showed here that a Nox4-specific signal can be observed on the nuclear membrane. Importantly, expression of Nox4 in each fraction, including the nuclear fraction, is upregulated by hypertrophic stimuli. These findings suggest that PE-induced ROS production in the nuclear fraction may be derived from Nox4 localized on the nuclear membrane and rapidly upregulated by hypertrophic stimuli. Although it is difficult to exclude the possibility that Nox4 localized on other intracellular membranes also contributes to the PE-induced increases in oxidative stress in the nucleus, the ineffectiveness of Nox2 knockdown in inhibiting the nuclear exit of HDAC4 suggests that localization of Nox4 at intracellular membranes is critical. Furthermore, the fact that nuc-catalase more effectively suppressed PE-induced nuclear exit of HDAC than Prx3 suggests that production of ROS in the nucleus, most likely from Nox4, is important.
The activity of Nox2 is regulated by posttranslational modification of cytosolic factors, such as p47phox, p67phox, and Rac1.16 On the other hand, Nox4 does not need cytosolic factors for its activation. Recent evidence suggests that a Nox4-interacting protein, polymerase (DNA-directed) delta-interacting protein 2 (Poldip2), modifies the activity/function of Nox4 in smooth muscle cells.17 Nevertheless, although Nox4-interacting proteins, including Poldip2, may affect the function of Nox4, the ROS-producing activity of Nox4 is regulated primarily by its expression level.18 Thus, the involvement of Nox4 in the PE-induced rapid oxidation of HDAC4 was unexpected, since it would require rapid upregulation of Nox4. However, Ang II upregulates Nox4 protein levels within 2.5 minutes in mesangial cells.19 Similarly, we found that PE rapidly (within 5 minutes) upregulates Nox4 protein levels in cardiomyocytes. The promoter region of Nox4 contains an NF-κB-binding motif in humans.20 PE decreases IκB protein levels and increases NF-κB expression in the nucleus within 1 minute. Furthermore, ChIP assays showed that PE stimulation rapidly induces binding of NF-κB to the Nox4 promoter in rat cardiac myocytes. Although rapid upregulation of Nox4 can still occur through an increased stability of protein/mRNA, our results suggest that expression of Nox4 can be rapidly regulated by PE stimulation through NF-κB, a rapid-acting primary transcription factor which does not require new protein synthesis for its activation.21 Since ROS also promote NF-κB activation,22 NF-κB-Nox4-ROS may form a positive feedback loop, thereby rapidly amplifying the upregulation of Nox4.
We have shown previously that adenovirus-mediated overexpression of Nox4 at 30 MOI or 1.3–2.0-fold overexpression of Nox4 in the Tg-Nox4 heart was not sufficient to induce cardiac hypertrophy.10 Interestingly, however, Nox4 significantly enhanced PE-induced cardiac hypertrophy. One possibility is that the level of Nox4 expression in Tg-Nox4 is not sufficient for oxidizing HDAC4. In fact, both the level of Nox4 expression and the amount of ROS production were lower in Tg-Nox4 hearts than in NTg hearts treated with PE (Fig. 2AB and Fig. 4D). The availability of the antioxidant system may affect the extent of oxidation in HDAC4. We have shown previously that Tg-Nox4 mice exhibit upregulation of catalase, thereby effectively preventing oxidative stress in the heart at 3–4 months of age.10 Another possibility is that upregulation of Nox4 is necessary, but not sufficient, to induce cardiac hypertrophy. The presence of signaling mechanisms originating from both PE and Nox4 may cooperatively stimulate cardiac hypertrophy. Further investigations are required to elucidate the mechanism by which the function of Nox4 is modulated through posttranslational modification during cardiac hypertrophy.
Zhang et al. reported that Nox4 mediates protection against PO by enhancing angiogenesis in mice.23 This result appears to contradict our findings that Nox4 mediates cardiac hypertrophy and cell death. However, one of the most critical differences between the two studies is the fact that Zhang et al. used systemic Nox4 KO mice, whereas we used cardiac-specific Nox4 KO mice. In fact, we found that the suppression of cardiac hypertrophy/dysfunction after TAC observed in c-Nox4 KO mice was no longer observed in systemic Nox4 KO mice, and systemic Nox4 KO mice showed more cardiac fibrosis and diastolic dysfunction than WT mice (Online Figure XVI). We speculate that Nox4 may affect a different group of proteins in cardiac non-myocytes. In fact, the class II HDACs are not expressed in cardiac non-myocytes.
Phosphorylation of HDAC4 by CaMKII, an HDAC kinase, is an important mechanism for mediating cardiac hypertrophy. Recent evidence suggests that CaMKII is activated not only through Ca2+-calmodulin-dependent mechanisms, but also by methionine oxidation in a Ca2+-calmodulin-independent manner,24 and that Nox2 plays an important role in mediating Ang II-induced activation of CaMKII.24 However, downregulation of Nox2 did not affect PE-induced nuclear O2− production or the nuclear export of HDAC4 in cardiomyocytes. The serine phosphorylation of CaMKII after PE stimulation was not changed by downregulation of Nox4, nor was phosphorylation of HDAC4 affected by downregulation of Nox4. These findings suggest that Nox4 plays an important role in mediating oxidation of HDAC4, independently of CaMKII.
We have shown previously that modulation of HDAC4 by oxidation and reduction overrides its modulation by phosphorylation. Namely, an oxidation-resistant mutant of HDAC4 stays in the nucleus even if the critical serine residues known to stimulate 14–3–3 binding and nuclear export are phosphorylated. We therefore speculate that suppression of the cysteine oxidation in class II HDACs may suppress the progression of cardiac hypertrophy even in the presence of hypertrophic stimuli known to activate HDAC kinases. Consistently, downregulation of Nox4 in c-Nox4 KO mice suppressed both PE- and TAC-induced cardiac hypertrophy without affecting phosphorylation of HDAC4.
In conclusion, Nox4 plays an essential role in mediating increases in ROS in the nucleus in response to hypertrophic stimuli and induces cysteine oxidation and nuclear export of HDAC4, a class II HDAC. Thus, our results suggest that therapies designed to interfere with nuclear oxidative stress could be beneficial for preventing and treating pathological cardiac hypertrophy and that Nox4, in particular, may be a promising target for medical treatment.
Supplementary Material
Acknowledgments
We thank Daniela Zablocki and Christopher D. Brady for critical reading of the manuscript.
Funding Statement
This work was supported in part by U.S. Public Health Service Grants HL67724, HL91469, HL102738, AG23039, and HL112330, and the Foundation of Leducq Transatlantic Network of Excellence.
List of abbreviations
- Ad
adenovirus
- CaMKII
calcium/calmodulin-dependent kinase II
- ChIP
chromatin immunoprecipitation
- CVF
collagen volume fraction
- GRK5
G protein-coupled receptor kinase 5
- HDAC
histone deacetylase
- LV
left ventricle (or left ventricular)
- LVW/BW
left ventricular weight/body weight ratio
- MCSA
myocyte cross-sectional area
- MEF2
myocyte enhancer factor 2
- NFAT
nuclear factor of activated T cells
- NLS
nuclear localization signal
- Nox
NADPH oxidase
- Nppa
natriuretic peptide precursor A
- Nppb
natriuretic peptide precursor B
- NTg
nontransgenic
- PE
phenylephrine
- PKD
protein kinase D
- ROS
reactive oxygen species
- sh-RNA
short hairpin RNA
- TAC
transverse aortic constriction
- Tg
transgenic
Footnotes
Competing Interest Statement
None
References
- 1.Okin PM, Devereux RB, Nieminen MS, Jern S, Oikarinen L, Viitasalo M, Toivonen L, Kjeldsen SE, Dahlof B. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: The losartan intervention for endpoint reduction in hypertension (life) study. Circulation. 2006;113:67–73. doi: 10.1161/CIRCULATIONAHA.105.569491. [DOI] [PubMed] [Google Scholar]
- 2.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 3.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. Cam kinase ii selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J. A redox-dependent pathway for regulating class ii hdacs and cardiac hypertrophy. Cell. 2008;133:978–993. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 5.Haworth RS, Stathopoulou K, Candasamy AJ, Avkiran M. Neurohormonal regulation of cardiac histone deacetylase 5 nuclear localization by phosphorylation-dependent and phosphorylation-independent mechanisms. Circ Res. 2012;110:1585–1595. doi: 10.1161/CIRCRESAHA.111.263665. [DOI] [PubMed] [Google Scholar]
- 6.Sumimoto H. Structure, regulation and evolution of nox-family nadph oxidases that produce reactive oxygen species. FEBS J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 7.Maejima Y, Kuroda J, Matsushima S, Ago T, Sadoshima J. Regulation of myocardial growth and death by nadph oxidase. J Mol Cell Cardiol. 2011;50:408–416. doi: 10.1016/j.yjmcc.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, Nunoi H, Sumimoto H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing nadph oxidases. J Biol Chem. 2003;278:25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- 9.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of nox1 and nox4 in basal and angiotensin ii-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. Nadph oxidase 4 (nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, Shah AM. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol. 2006;47:817–826. doi: 10.1016/j.jacc.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 13.Zhai P, Galeotti J, Liu J, Holle E, Yu X, Wagner T, Sadoshima J. An angiotensin ii type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin ii-mediated cardiac hypertrophy. Circ Res. 2006;99:528–536. doi: 10.1161/01.RES.0000240147.49390.61. [DOI] [PubMed] [Google Scholar]
- 14.Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- 15.Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, Chappell MC. The angiotensin ii-at1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun. 2009;384:149–154. doi: 10.1016/j.bbrc.2009.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedard K, Krause KH. The nox family of ros-generating nadph oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 17.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of nox4 reveals unique characteristics compared to other nadph oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 19.Block K, Eid A, Griendling KK, Lee DY, Wittrant Y, Gorin Y. Nox4 nad(p)h oxidase mediates src-dependent tyrosine phosphorylation of pdk-1 in response to angiotensin ii: Role in mesangial cell hypertrophy and fibronectin expression. J Biol Chem. 2008;283:24061–24076. doi: 10.1074/jbc.M803964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu X, Murphy TC, Nanes MS, Hart CM. Ppar{gamma} regulates hypoxia-induced nox4 expression in human pulmonary artery smooth muscle cells through nf-{kappa}b. Am J Physiol Lung Cell Mol Physiol. 2010;299:L559–566. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Usui S, Maejima Y, Pain J, Hong C, Cho J, Park JY, Zablocki D, Tian B, Glass DJ, Sadoshima J. Endogenous muscle atrophy f-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappab. Circ Res. 2011;109:161–171. doi: 10.1161/CIRCRESAHA.110.238717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Murphy TM, Chitano P, Hoidal JR. Nadph oxidase promotes nf-kappab activation and proliferation in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2002;282:L782–795. doi: 10.1152/ajplung.00206.2001. [DOI] [PubMed] [Google Scholar]
- 23.Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. Nadph oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of camkii by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.