

Abstract
Botulinum neurotoxins (BoNTs) are the most lethal biotoxins known to mankind and are responsible for the neuroparalytic disease botulism. Current treatments for botulinum poisoning are all protein based and thus have a limited window of treatment opportunity. Inhibition of the BoNT light chain protease (LC) has emerged as a therapeutic strategy for the treatment of botulism as it may provide an effective post exposure remedy. Using a combination of crystallographic and modeling studies a series of hydroxamates derived from 1-adamantylacetohydroxamic acid (3a) were prepared. From this group of compounds, an improved potency of about 17-fold was observed for two derivatives. Detailed mechanistic studies on these structures revealed a competitive inhibition model, with a Ki = 27 nM, which makes these compounds some of the most potent small molecule, non-peptidic BoNT/A LC inhibitors reported to date.
Keywords: Botulinum neurotoxin, Protease inhibitor, Adamantane derivatives, Small molecule inhibitor, Zinc-dependent metalloprotease
1. Introduction
Botulinum neurotoxins (BoNTs) produced by the anaerobic spore-forming bacterium Clostridium botulinum are the most lethal human poison.1 Serotype A (BoNT/A) is the most potent of the identified serotypes with an estimated lethality of ~ 1 ng/kg.2 There are seven BoNT serotypes (A-G) and while they differ by up to 70 % at the amino acid level all consist of heavy and light chain subunits. Upon cellular internalization of the holotoxin (via binding of heavy chain to cell surface receptors) the light chain (LC), a 50 kDa Zn(II)-dependent metalloprotease, is released. Toxicity from BoNT poisoning results from the site-specific cleavage of the synaptosomal-associated protein 25 (SNAP-25) by the metalloprotease, preventing acetylcholine-containing vesicles from fusing with the presynaptic neuromuscular junction.3 The consequence of protease cleavage of SNAP-25 is inhibition of acetylcholine release, which leads to flaccid paralysis and eventually to death caused typically by heart or respiratory failure.4
Despite their potentially lethal toxicity, BoNTs have emerged as an extremely valuable therapeutic tool for the treatment of a maladies, including strabismus, migraines, and even facial wrinkles.5 However, the potential use of BoNT in a bioterrorist attack remains imminent and the Center for Disease Control (CDC) classifies this agent as “category A”, placing it among the six highest-priority agents.
Currently, there are no approved pharmacological treatments for BoNT intoxication. Although an effective vaccine is available for immuno-prophylaxis,6 vaccine approaches cannot reverse the effects after the toxin has reached its target inside the cell. A small molecule pharmacological intervention, especially one that would be effective against the etiological agent responsible for BoNT intoxication, the light chain protease, would be highly desirable and could obviate vaccine deficiencies. Most research efforts have been focused on the BoNT/A protease, since this serotype is the most toxic to humans with the longest lasting cellular effect.7 Indeed, a number of small molecule, non-peptidic inhibitors of BoNT/A LC have been reported over past two decades,6,8 however, potency is still lacking (Figure 1). Recently, we communicated a logical attempt to improve the potency of our best BoNT/A LC inhibitor X based upon crystallographic analysis and computational modeling.9 The resulting structure, XI, displayed an almost 2-fold lower inhibition constant than the parent X. The research described herein was directed again using crystallographic and modeling studies, but now to a new scaffold: the adamantane hydroxamate 3a10 (Figure 1). A series of 19 derivatives were prepared with improved potency of about 17-fold for the best two new compounds.
Figure 1.
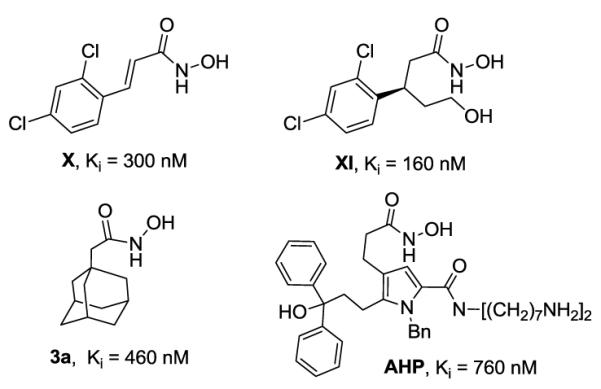
Examples of some of the most active inhibitors of BoNT/A protease: X,11 3a,10 XI,9 AHP.12
2. Results and Discussion
2.1. Crystallography and Modeling Studies
The X-ray crystal structure of the complex between the BoNT/A LC and 1-adamantyl N-hydroxyacetamide (3a) was determined to 2.5Å resolution (PDB ID 4HEV, Figure 2A and S1, Table S1). As observed in other structures of BoNT/A LC complexes with hydroxamate inhibitors,13 the hydroxamate moiety is liganding the Zn2+ ion in a bidentate fashion with the carbonyl and hydroxyl oxygen atoms (2.1 and 2.2Å, respectively). The hydroxamate nitrogen makes a hydrogen bonding interaction with the main-chain carbonyl of Phe163, part of a β-strand that forms one wall of the active site. The adamantyl group, like the phenyl rings of the previously-characterized cinnamyl hydroxamates (e.g. X),13 occupies a largely hydrophobic pocket comprised of the side chains of Ile161, Phe163, Phe194, and Phe369. In structures of BoNT/A LC with no ligands bound, complexed with L-arginine hydroxamate or with arginine-containing peptides,14 Phe369 is rotated out of the active site and Asp370 is rotated in, indicating that 3a induces the same polar-to-hydrophobic change observed for other small, nonpolar ligands.13 While the 250 and 60/70 loops are both largely disordered in the structure, it is clear that there is unoccupied space in the hydrophobic pocket that could be exploited to improve the affinity and potency of the 3a scaffold.
Figure 2.

Stereo view of the adamantyl hydroxamate inhibitor 3a bound within the BoNT/A LC active site (A). Both the 2∣Fo∣-∣Fc∣ and simulated annealing composite omit maps are shown as magenta and green mesh, respectively, contoured at 1.1σ. The active sites after refinement of two substituted adamantane hydroxamates (3i and 3f) are shown in B and C, respectively. The hydroxymethyl and chlorine groups are placed near enough to Arg363 to form favorable non-covalent interactions (2.6 and 3.0Å, respectively).
In order to improve the binding and thus enhance the potency of our lead structure 3a, we decided to perform preliminary modeling studies involving substitution upon the adamantane scaffold (Fig. 2B and C). Probing the active site with 3a, two favorable interactions emerged as a promising platform to build a structure-activity relationship study. Thus, the first interaction identified involved positioning on the core structure of an appropriate polar substituent that could interact with Arg363. The second interaction we viewed to be favorable consisted of a non-polar/aromatic substituent and the hydrophobic pocket comprising three phenylalanines (F163, F194 and F369). Therefore, our proposed studies consisted of a series of derivatives stemming from the parent molecule 3a, which was focused upon exploring interactions with the Arg363 within the enzyme’s active site (Scheme 2), as well as interfacing with the ‘tris-Phe’ hydrophobic pocket (Scheme 3). Having established this rationale, all the substituents were introduced in position 3 relative to the acetohydroxamic moiety and were prepared to contain either varying electronegativity and/or steric requirements. Based on these constraints we anticipated that a halogen atom should favorably interact with the Arg363 residue, while carboxyl (COOH), if ionized to its carboxylate form could generate a stable salt bridge. To further probe this reasoning hydroxyl (3b and 3i), ether (3c) and acetate (3d) were hypothesized to make hydrogen bond donor-acceptor interactions within the Arg363 side pocket. On the other hand, introduction of an aromatic substituent would lead to a π-π stacking with one or more Phe residues inside the hydrophobic pocket.
Scheme 2.
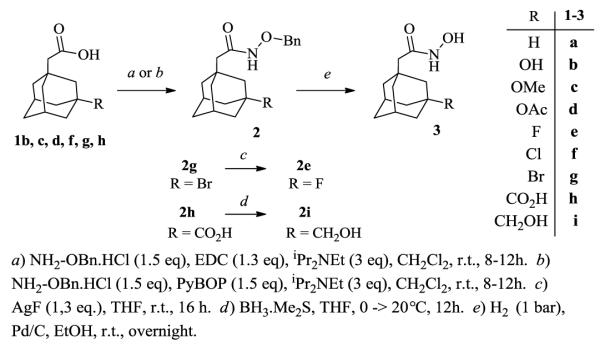
Synthesis of (3-functionalized-1-adamantyl)acetohydroxamic derivatives.
Scheme 3.
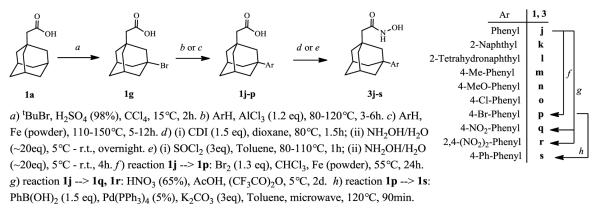
Synthesis of (3-aryl-1-adamantyl)acetic and acetohydroxamic acids.
2.2. Chemistry
The synthesis of (3-halogen-1-adamantyl)acetic acids was initiated from the commercially available and easily accessible 1-adamantylacetic acid (1a). Several methods have been reported for the halogenation of an adamantane,15 and, although chlorination has been somewhat problematic, because of contamination with other chlorinated by-products, there existed a facile and efficient procedure for mono-chlorination of adamantane at the tertiary carbon.16 In accordance with this previous work minor changes included the switch from two components; tBuOH for cation generation and gaseous HCl as source of anion/nucleophile, to simply tBuCl as a singular reagent (Scheme 1, reaction a). Using this same rationale, the tBuBr led to the 3-bromo derivative 1g in excellent yield. The 3-fluoroadamantyl derivative 2e, was made from 2g by simple substitution with AgF (Scheme 2, reaction c). Next, (3-hydroxy-1-adamantyl)acetic acid (1b) was examined for preparation of 3-methoxy-(1c), 3-acetoxy-(1d), and 3-carboxyl-(1h) adamantyl derivatives. Unfortunately, methylation of the 3-hydroxyl group did not proceed either by reaction with MeI nor Me2SO4.
Scheme 1.
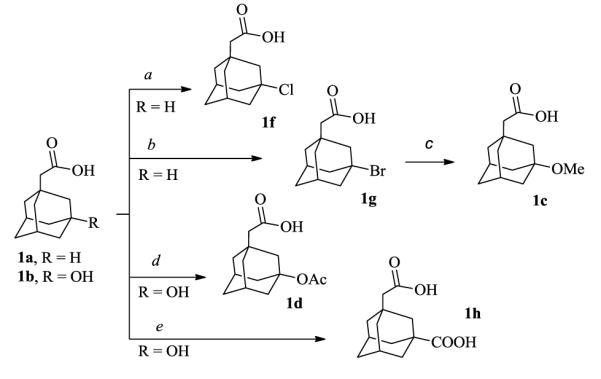
Synthesis of (3-functionalized-1-adamantyl)acetic acids.
a) tBuCl, H2SO4 (98%), CCI4, 10-20°C, 2h. b) tBuBr, H2SO4 (98%), CCI4, 10-20°C, 2h. c) MeONa, AgNO3, MeOH, r.t., 16h. d) AcONa, AcOH, Ac2O, 100°C, 16h. e) HCO2H (98%), H2SO4 (98%), CCI4, 20°C, 2h.
Therefore, acid 1c was prepared by nucleophilic (SN1) reaction of bromide 1g with MeONa in the presence of AgNO3. To complete the series of polar substituents, acetylation could be achieved with Ac2O/AcONa granting 1d, while carboxylation required recourse to the Koch-Haaf reaction,17 yielding 1h. Finally, 3-hydroxymethyl derivative 2i was obtained by reduction of the carboxyl group from intermediate 2h, (Scheme 2, reaction d). Subsequently, all adamantylacetic acids 1b, c, d, f, g, h were converted to the corresponding protected hydroxamate derivatives (2), by coupling with O-benzylhydroxylamine.18 The removal of the benzyl protective group under standard hydrogenation conditions afforded the (3-functionalized-1-adamantyl)acetohydroxamic products (3), (Scheme 2).
In the next iteration of compounds, we utilized the versatile intermediate 1g for introduction of aromatic substituents to the position 3 of 1-adamantyl scaffold (Scheme 3). Thus, the 3-aryl-1-adamantyl acids 1j-p were readily prepared under Friedel-Crafts conditions (AlCl3 catalysis). Interestingly, this adamantylation of aromatic residues could be also catalyzed by use of iron powder, although usually higher reaction temperatures were required.19 Apart from direct alkylation of bromobenzene, the p-bromophenyl acid 1p was also prepared by bromination of 1j. In addition, the bromo-functionality in 1p could be further utilized for diversification of our small library by cross-coupling reactions. Such an option is represented by the biphenyl derivative 1s, (Scheme 3, reaction h). Unfortunately, nitrobenzene did not react with adamantyl bromide; therefore, the nitrophenyl-(1q) along with the dinitrophenyl-(1r) derivatives were prepared by nitration of 1j. Finally, using standard conditions the (3-aryl-1-adamantyl)acetic acids 1j-s were transformed to the corresponding hydroxamates 3j-s via activated acyl species (either acyl imidazole20 or chloride; Scheme 3).
2.3. Kinetic Analysis
Having prepared the adamantane hydroxamates, all compounds were subjected into screening for inhibitory activity of BoNT/A LC (1-425aa). Towards this end we choose the well-established and rapid assay utilizing FRET (fluorescence resonance energy transfer) substrate, SNAPtide™,21 which is a truncated and modified sequence of the native BoNT/A LC substrate, SNAP-25. This 13aa peptide contains a fluorophore/quencher pair separated by the cleavage site; thus producing fluorescence in the presence of an active BoNT/A protease. Preliminary IC50 values from this assay are summarized in Table 1. Unfortunately, introduction of a polar functionality into position 3 of 1-adamantyl core did not improve binding affinity. In fact, the inhibitory potencies of compounds 3b-i were considerably lower than the parent 3a, with exception of chloro-(3f) and bromo-(3g) derivatives where the IC50’s were comparable with the lead molecule (Table 1, entries 2-9). We were especially surprised to see no activity for the 3-carboxyl derivative (3h), which we hypothesized could form an ionic interaction with Arg363 (vide supra). Without further crystallographic data we can only speculate that conformation of residues within the enzyme’s active site have been altered in a way which does not allow formation of these sought after interactions. Such high flexibility of active site residues has been documented previously for BoNT/A protease.13 Gratifyingly, better results were achieved by attaching an aromatic moiety on the adamantyl skeleton (Table 1, entries 10-19). The most profound improvement in activity was seen with p-chloro-(3o), and p-bromophenyl (3p) derivatives with ~ 30-fold lower IC50 values (Table 1, entries 15, 16).
Table 1.
IC50 values of prepared (3-substituted-1-adamantyl)acetohydroxamic derivatives.
| Entry | Compound | Substituent | IC50a (Ki)b/ μM | Entry | Compound | Substituent | IC50a (Ki)b/ μM |
|---|---|---|---|---|---|---|---|
| 1 | 3a | H | 1.0 (0.46) | 10 | 3j | Ph | 0.5 (0.75) |
| 2 | 3b | OH | 23 | 11 | 3k | 2-Naphthyl | 0.3 |
| 3 | 3c | OMe | 5.7 | 12 | 3l | 2-THNc | 1.0 |
| 4 | 3d | OAc | 24 | 13 | 3m | 4-Me-Ph | 0.25 |
| 5 | 3e | F | 3.1 | 14 | 3n | 4-MeO-Ph | 0.25 (0.13) |
| 6 | 3f | Cl | 1.4 | 15 | 3o | 4-Cl-Ph | 0.03 (0.027) |
| 7 | 3g | Br | 1.4 | 16 | 3p | 4-Br-Ph | 0.04 (0.027) |
| 8 | 3h | COOH | > 500 | 17 | 3q | 4-NO2-Ph | 0.5 |
| 9 | 3i | CH2OH | 3.4 | 18 | 3r | 2,4-(NO2)2-Ph | 0.4 |
| 19 | 3s | 4-Ph-Ph | 0.2 |
) determined with 5 μM [SNAPtide] (+ 0.01 % Triton X-100) vs. BoNT/A LC (1-425aa);
) determined with SNAP-66mer (141-206aa) vs. BoNT/A LC (1-425aa);
) 2-THN = 5,6,7,8-Tetrahydronaphthalen-2-yl.
While SNAPtide proved suitable for high-throughput screening and for crude IC50 determinations it is inadequate for finer kinetic analysis such as the evaluation of a kinetic mechanism of inhibition and Ki determinations.10 Therefore, the inhibition profiles of the most potent compounds were examined in more detail using a LC/MS-based assay that employs a substrate consisting of the 66 amino acid long C-terminal domain of SNAP-25 (141-206aa).10 To determine the mechanism of inhibition as well as Ki, concentrations of the inhibitor and the substrate were varied. As expected, obtained data were most consistent with the competitive inhibition model (Supp. Information). The inhibition constants, Ki were determined by a non-linear least squares global fit of a competitive inhibition model to the initial rates of product formation for matrixes of substrate and inhibitor concentrations bracketing KM and Ki, respectively. These results shown that hydroxamates 3o and 3p with Ki = 27 ± 4 nM and 27 ± 2 nM, respectively, are some of the most potent small molecule inhibitors of BoNT/A LC reported to date.
2.4. Biology
As another measure of an LC/A inhibitor profile, a cell-based assay, which monitors intracellular cleavage of SNAP-25 can be used. Accordingly, the cellular efficacy of three of the improved inhibitors found in our study (3n, 3p, and 3q) were investigated using primary rat spinal cord (RSC) cells and stem cell derived neurons, respectively (Supporting Information). In the first assay, BoNT/A was added to the cells in stimulation medium to allow its entry followed by washing and addition of inhibitors. By exposing cells to the toxin prior to inhibitor addition, secondary effects due to cellular changes that would inhibit BoNT/A uptake can be excluded.
In the second assay, the inhibitors were premixed with BoNT/A (~1 pM) and immediately added to stem cell derived neurons. Unfortunately, all three compounds tested showed significant cytotoxicity at concentration ~ 20 μM as judged by a low signal as seen on the Western blot, and did not protect neuronal cells from BoNT/A induced cleavage of SNAP-25 at lower concentrations (Fig. 4, and Supporting Information).
Figure 4.
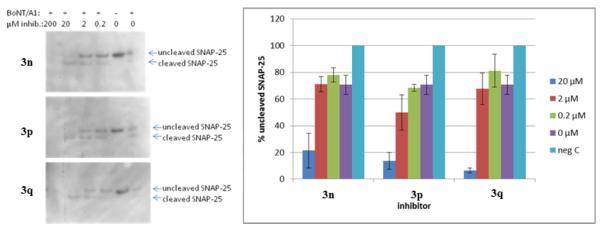
Inhibitors and BoNT/A were premixed and added to stem cell derived neurons.
Having established that these compounds did provide enzymatic inhibition of recombinant protease in vitro, yet, a lack of protection in cells; we surmise is likely due to an inability of the compounds to properly enter the cell cytosol, such hypothesis will be tested in future studies.
3. Conclusion
In summary, a research approach incorporating computational modeling, SAR studies, compound validation, and kinetic characterization was used to successfully improve lead adamantane structure 3a, and afforded (3-substituted-1-adamantyl) acetohydroxamic derivatives. The most potent of these BoNT/A LC inhibitors, 3o and 3p, have Ki values approximately 30 nM, which is 17-fold lower than the parent lead molecule. These findings in fact, represent the most potent small molecule, non-peptidic BoNT/A LC inhibitors reported to date. Unfortunately, when examined in vitro in cell-based assays, these compounds displayed significant cytotoxicity at ~ 20 μM and provided no protection against BoNT/A intoxication at lower concentrations. We propose that the discrepancy between very high activity (low nanomolar) against recombinant LC/A and no apparent activity on the cells (at low micromolar) is due to inefficient cellular uptake and future research will examine this hypothesis in due course.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support of this project by the National Institute of Allergy and Infectious Diseases, National Institute of Health and the Department of Health and Human Services under contract number AI080671.
Footnotes
Supplementary Material Detailed information on the syntheses of all compounds, experimental procedures for enzymatic and cell-based assays, as well as kinetic data analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. Nature Rev. Drug Disc. 2005;4:281–297. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schantz EJ, Johnson EA. Microbiol. Rev. 1992;56:80–99. doi: 10.1128/mr.56.1.80-99.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simpson LL. Annu. Rev. Pharmacol. Toxicol. 2004;44:167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 4.Kongsaengdao S, Samintarapanya K, Rusmeechan S, Wongsa A, Pothirat C, Permpikul C, Pongpakdee S, Puavilai W, Kateruttanakul P, Phengtham U, Panjapornpon K, Janma J, Piyavechviratana K, Sithinamsuwan P, Deesomchok A, Tongyoo S, Vilaichone W, Boonyapisit K, Mayotarn S, Piya-Isragul B, Rattanaphon A, Intalapaporn P, Dusitanond P, Harnsomburana P, Laowittawas W, Chairangsaris P, Suwantamee J, Wongmek W, Ratanarat R, Poompichate A, Panyadilok H, Sutcharitchan N, Chuesuwan A, Oranrigsupau P, Sutthapas C, Tanprawate S, Lorsuwansiri J, Phattana N. Clin. Infect. Dis. 2006;43:1247–1256. doi: 10.1086/508176. [DOI] [PubMed] [Google Scholar]
- 5(a).Hackett R, Kam PC. Med. Chem. 2007;3:333. doi: 10.2174/157340607781024438. [DOI] [PubMed] [Google Scholar]; (b) Truong DD, Jost WH. Parkinsonism Relat. Disord. 2006;12:331. doi: 10.1016/j.parkreldis.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Willis B, Eubanks LM, Dickerson TJ, Janda KD. Angew. Chem. Int. Ed. 2008;47:8360–8379. doi: 10.1002/anie.200705531., and references cited therein.
- 7.Hughes JM, Blumenthal JR, Merson MH, Lombard GL, Dowell VR, Gangarosa E. J. Ann. Intern. Med. 1981;95:442–445. doi: 10.7326/0003-4819-95-4-442. [DOI] [PubMed] [Google Scholar]
- 8.Li B, Peet NP, Butler MM, Burnett JC, Moir DT, Bowlin TL. Molecules. 2011;16:202–220. doi: 10.3390/molecules16010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stowe GN, Šilhár P, Hixon MS, Silvaggi NR, Allen KN, Moe ST, Jacobson AR, Barbieri JT, Janda KD. Organic Lett. 2010;12:756–759. doi: 10.1021/ol902820z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Čapkova K, Hixon MS, McAllister LA, Janda KD. Chem. Commun. (Camb) 2008:3525. doi: 10.1039/b808305c. [DOI] [PubMed] [Google Scholar]
- 11.Boldt GE, Kennedy JP, Janda KD. Org. Lett. 2006;8:1729–1732. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pang YP, Vummenthala A, Mishra RK, Park JG, Wang SH, Davis J, Millard CB, Schmidt JJ. PLoS One. 2009;4:e7730. doi: 10.1371/journal.pone.0007730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD. Chemistry & Biology. 2007;14:533–542. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 14(a).Silvaggi NR, Wilson D, Tzipori S, Allen KN. Biochemistry. 2008;47(21):5736–45. doi: 10.1021/bi8001067. [DOI] [PubMed] [Google Scholar]; (b) Kumaran D, Rawat R, Ludivico ML, Ahmed SA, Swaminathan S. J. Biol. Chem. 2008;283(27):18883–91. doi: 10.1074/jbc.M801240200. [DOI] [PubMed] [Google Scholar]
- 15(a).Stetter H, Mayer J. Chem. Berichte. 1962;95:667–672. [Google Scholar]; (b) Yurchenko RI, Miroshnichenko VV. J. Gen. Chem. USSR (Engl. Transl.) 1992;62:467–468. 379–380. [Google Scholar]; (c) Min KH, Xia Y, Kim EK, Jin Y, Kaur N, Kim ES, Kim DK, Jung HY, Choi Y, Park M-K, Min YK, Lee K, Lee K. Bioorg. Med. Chem. Lett. 2009;19:5376–5379. doi: 10.1016/j.bmcl.2009.07.127. [DOI] [PubMed] [Google Scholar]
- 16.Aigami K, Inamoto Y, Takaishi N, Hattori K. J. Med. Chem. 1975;18:713–721. doi: 10.1021/jm00241a015. [DOI] [PubMed] [Google Scholar]
- 17(a).Stetter H, Schwarz M, Hirschhorn A. Chem. Berichte. 1959;92:1629–1635. [Google Scholar]; (b) Koch H, Haaf W. Angew. Chem. 1960;72:628. [Google Scholar]
- 18.Lienard BMR, Horsfall LE, Galleni M, Frere J-M, Schofield CJ. Bioorg. Med. Chem. Lett. 2007;17:964–968. doi: 10.1016/j.bmcl.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 19.Anderson GL, Kaimari TA-R. 6864264 B1 U.S. Patent. 2005
- 20.Usachova N, Leitis G, Jirgensons A, Kalvinsh I. Synth. Commun. 2010;40:927–935. [Google Scholar]
- 21.Shine NR. 6,504,006 B1 US Patent. 2003
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.