

Abstract
Whereas the role of mammalian thioredoxin (Trx) as an intracellular protein cofactor is widely appreciated, its function in the extracellular environment is not well understood. Only few extracellular targets of Trx-mediated thiol-disulfide exchange are known. For example, Trx activates extracellular transglutaminase 2 (TG2) via reduction of an intramolecular disulfide bond. Because hyperactive TG2 is thought to play a role in various diseases, understanding the biological role of extracellular Trx may provide critical insight into the pathogenesis of these disorders. Starting from a clinical-stage asymmetric disulfide lead, we have identified analogs with >100-fold specificity for Trx. Structure-activity relationship and computational docking model analyses have provided insights into the features important for enhancing potency and specificity. The most active compound identified had an IC50 below 0.1µM in cell culture, and may be appropriate for in vivo use to interrogate the role of extracellular Trx in health and disease.
INTRODUCTION
Mammalian thioredoxin-1 (Trx) is an archetypal protein cofactor. In partnership with thioredoxin reductase (TrxR), it plays a crucial role in many intracellular redox reactions and regulatory processes.1 Additionally, mammalian cells are also known to secrete Trx, although the mechanism and biological implications of this phenomenon are poorly understood.1–5 For example, the TRPC ion channel6 and the HIV-1 envelope protein gp1207 are activated by extracellular Trx. Selective small molecule inhibitors could therefore be useful tools for decoding the biology of extracellular Trx.
Our own interest in extracellular thioredoxin was motivated by the related observations that, not only does this cofactor activate transglutaminase 2 (TG2) via disulfide bond reduction (Figure 1A), but also that interferon-γ (IFN-γ) induces Trx secretion from monocytic cells in amounts that are sufficient to activate TG2 in the extracellular matrix.8,9 These observations are especially relevant to celiac disease pathogenesis, because TG2 activation is required for post-translational modification of antigenic gluten peptides in the small intestine,10–12 and IFN-γ is the principal inflammatory cytokine secreted by disease-specific T cells.13 Together, these findings have led to the hypothesis that extracellular Trx is the missing link in a maladaptive, gluten-induced amplificatory loop between inflammatory T cells and TG2 (Figure 2). Because Trx-null mice show embryonic lethality,14 testing of this hypothesis requires the engineering of small molecule inhibitors that inactivate extracellular Trx to the exclusion of its intracellular counterpart.
Figure 1.

(A) Thioredoxin (Trx) mediated activation of transglutaminase 2 (TG2). (B) Reaction of a small molecule asymmetric disulfide with Trx.19
Figure 2.

Proposed pathogenic role of Trx in celiac disease. In celiac disease patients, extracellular transglutaminase 2 (TG2) facilitates a robust T cell (Th1) response to dietary gluten. Proteolytically resistant, Pro- and Gln-rich peptides derived from gluten accumulate in the gut lumen. By themselves, these peptides are weak antigens; their affinity for HLA-DQ2 or DQ8 is markedly enhanced by TG2-catalyzed deamidation of selected Gln residues. However, extracellular TG2 in the small intestinal mucosa is maintained in an inactive state via an intramolecular disulfide bond between vicinal Cys residues. TG2 is activated by Th1 cell-derived IFN-γ, which promotes Trx release. In turn, secreted Trx reduces the disulfide bond in TG2 with high specificity. Thus, Trx is proposed as the missing link that mutually amplifies Th1 activity and TG2 activation in celiac disease, leading to an inflammatory response.
All Trx proteins have a highly conserved WCGPC catalytic motif. The Cys residues of this motif are responsible for reducing disulfide bonds in oxidized proteins. Inside the cell, the overall catalytic cycle is completed when the resulting intramolecular disulfide bond of Trx is reduced by TrxR with the concomitant consumption of one NADPH molecule.1 Thus, intracellular Trx acts as a protein cofactor for disulfide bond reduction. In contrast, because the extracellular environment lacks TrxR or NADPH, secreted Trx is presumably a stoichiometric reagent outside the cell. This singular difference could be exploited in the design of small molecule inhibitors that oxidize extracellular Trx via selective disulfide bond exchange.
One such agent is 1 (PX-12, 1-methyl-1-propyl-2-imidazolyl disulfide),15,16 which has undergone clinical evaluation in cancer patients.17,18 1 oxidizes the active site cysteines of Trx into a disulfide bond (Figure 1B).19 Using a combination of novel assay development and structure-activity relationship analysis, we have identified analogs with markedly improved potency, selectivity, and activity in cell culture systems. Additionally, a docking model was developed to rationalize the observed improvements against human Trx.
RESULTS
Design, synthesis, and biochemical analysis of human Trx inhibitors
The general synthesis of Trx inhibitors is outlined in Scheme 1. Our starting point for developing a pharmacologically useful inhibitor of extracellular Trx was 1 (Table 1), a small molecule Trx inhibitor that has entered human clinical trials in cancer patients.17,18 1 oxidatively inactivates extracellular Trx without appreciably altering the levels of reduced intracellular Trx, because the latter pool of Trx has access to TrxR whereas the former does not. Unfortunately, the compound has an extremely short half-life in humans,18 which precludes derivation of a reliable correlation between drug dose and pharmacological response. We have confirmed this short half-life in our own preliminary studies in rats (data not shown). Additionally, the release of sec-butyl thiol from the Trx reaction (Figure 1B) induces a noxious response, thereby limiting the maximum tolerable dose of 1.
Scheme 1a.
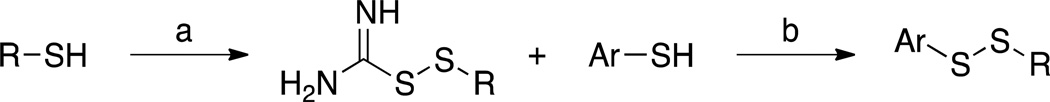
aReagents and conditions: (a) thiourea, HCl, H2O2, H2O/EtOH, 0 °C, 11–91%; (b) NaHCO3, MeOH, 58–93%.
Table 1.
Structures and Trx inhibitory activities of various asymmetric disulfides.
| Inhibitor Structure | Cmpd | R = | Trx kinh/Ki (µM−1min−1) |
Trx/DTT | T84 IC50 (µM) |
|---|---|---|---|---|---|
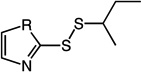 |
1 | N | 0.076 | 7 | 2.11 ± 1.05 |
| 2 | S | 0.025 | 30 | - | |
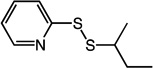 |
3 | - | 0.240 | 44 | - |
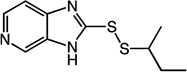 |
4 | - | 0.085 | 71 | - |
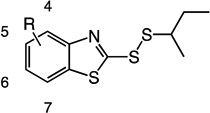 |
5 | 6-H | 0.078 | 65 | - |
| 6 | 6-F | 0.098 | 98 | - | |
| 7 | 6-Cl | 0.055 | 120 | - | |
| 8 | 6-I | 0.002 | 48 | - | |
| 9 | 4-Br | 0.041 | 96 | - | |
| 10 | 5-Br | 0.008 | 28 | - | |
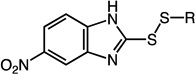
| 11 | 6-NO2 | 0.240 | 77 | - | |
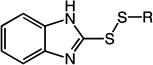 |
12 | CH2CH3 | 1.100 | 65 | 0.34 ± 0.18 |
| 13 | C(CH3)3 | 0.001 | 8 | 7.31 ± 2.06 | |
| 14 | CH(CH3)CH2CH3 | 0.130 | 29 | - | |
| 15 | CH(CH3)2 | 0.110 | 26 | - | |
| 16 | cyclopentyl | 0.110 | 50 | - | |
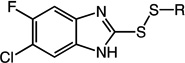
 |
17 | N | 0.160 | 72 | - |
| 18 | S | 0.031 | 52 | 0.51 ± 0.18 | |
| 19 | O | 0.270 | 63 | - | |
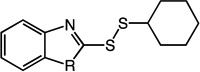
 |
20 | CH(CH3)CH2CH3 | 0.240 | 140 | - |
| 21 | cyclohexyl | 0.140 | 200 | 0.17 ± 0.04 | |
 |
22 | CH(CH3)CH2CH3 | 0.280 | 140 | - |
| 23 | cyclohexyl | 0.340 | 170 | 0.09 ± 0.01 | |
 |
24 | - | 0.270 | 110 | - |
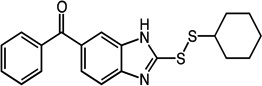 |
25 | - | 0.076 | 150 | 0.24 ± 0.06 |
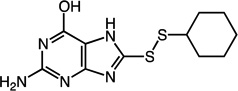 |
26 | - | 0.072 | 120 | 0.29 ± 0.07 |
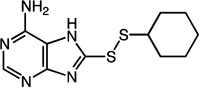 |
27 | - | 0.089 | 74 | - |
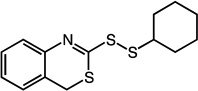 |
28 | - | 0.160 | 65 | - |
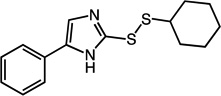 |
29 | - | 0.087 | 73 | 0.45 ± 0.15 |
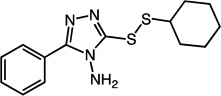 |
30 | - | 0.180 | 42 | 0.54 ± 0.28 |
To quantify the specificity of a small molecule disulfide for human Trx, we established a kinetic assay to measure the bimolecular rate constant (kinh/Ki) of this reaction. Reduced dithiothreitol (DTT) was used as a representative control capable of undergoing intramolecular dithiol → disulfide oxidation. Representative data for 1 and DTT is shown in Figure S1. From these results, it can be estimated that 1 has 7-fold specificity for human Trx over DTT (Table 1).
An initial set of 1 analogs was synthesized, in which both halves of the asymmetric disulfide were systematically varied (Table 1). Comparison of imidazolyl 1 versus thiazolyl 2 compounds suggested that the latter functional group was moderately preferred. Similarly, comparison of the imidazolyl 1 and benzimidazolyl 14 compounds, or alternatively thiazolyl 2 and benzothiazolyl 5 compounds suggested that polycyclic aromatic functional groups could be readily accommodated without penalty. We therefore synthesized and tested a series of substituted benzothiazole derivatives 5 – 11. In general, electron withdrawing substituents showed a modest increase in activity, whereas electron donating groups were slightly deleterious. The 6-Cl analog 7 had the highest specificity, with a Trx/DTT reactivity ratio of 120.
Previous studies by Kirkpatrick et al. together with our LC-MS data suggested that the thiol-disulfide exchange reaction between 1 and Trx involves an initial attack that results in the release of 2-mercaptoimidazole.19 To assess the influence of alternative alkyl substituents on molecular recognition by human Trx, we synthesized analogues of compound 14 bearing alternative primary, secondary, and tertiary thiol substituents (12 – 17, Table 1). The primary thiol 12 was most reactive against Trx while the tertiary thiol 13 was least reactive. The reaction rates of secondary thiols did not vary significantly as a function of size or conformation. Cyclic alkanes showed moderately improved specificity, a feature that was verified through the synthesis and evaluation of two other heterocycles 18 – 19. Additionally, cycloalkanethiols would also have reduced odor-related toxicity due to higher boiling points. For these reasons, the cyclohexanethiol moiety was selected as the preferred alkyl substituent for future analogs.
To seek further improvement in specificity, an additional series of compounds 20 – 30 (Table 1) was synthesized, and promising new analogs were identified. Of greatest interest are compounds 21 and 23, both of which show more than 20-fold enhanced specificity than 1. Moreover, these efforts also yielded a series of compounds (e.g., 21, 23, 25, 26, 29, and 30) with systematic differences in potency and specificity. It was therefore anticipated that, not only would this compound set provide insight regarding the most important physicochemical determinants of biological activity, but it could also facilitate development of a computational model for Trx-inhibitor interactions.
Biological evaluation of Trx inhibitors
Our strategy for designing a biological assay for Trx inhibitors that would be informative in the context of celiac disease is summarized in Figure 3. Our previous studies had shown that monolayers of mature T84 enterocytic cells naturally express TG2 in their extracellular matrix, but that the enzyme is maintained in a predominantly inactive state.20 We therefore sought to induce extracellular TG2 activity in this cultured cell system by adding recombinant human Trx (Figure 3A). TG2 activity was quantified by the concomitant addition of 5-biotinamido-pentylamine (5BP), an amine substrate of TG2 that undergoes covalent attachment to TG2 substrates in the extracellular matrix (Figure 3B). As seen in Figure 3C, extracellular TG2 was rapidly activated within an hour, and reached peak activity 3 h following Trx addition. Dose-response analysis revealed an EC50 of 25 nM for Trx (Figure 3D), further reinforcing the exquisite specificity of this protein-protein interaction. The selectivity of the 5BP assay was verified by addition of ERW1041E, a selective TG2 inhibitor,21 which fully suppressed attachment of this amine to extracellular matrix protein (Figure 3D). Fluorescence microscopy was undertaken to verify the logic of this assay. As can be seen in Figure 4, TG2 is abundant in the extracellular matrix of T84 cells. (Compare the distribution of E-cadherin and TG2.) Whereas the amount of TG2 protein (green) remains unchanged as a result of exposure to Trx, its activity (blue) is induced in a dose-dependent manner.
Figure 3.

An assay for Trx-mediated TG2 activation. (A) T84 enterocytic cells are grown in flat bottom, 48-well plates for 7 days, and then treated with 0–10 µM Trx for a variable duration along with 200 µM 5BP for 3 h. Trx activates inactive extracellular TG2 (Figure 1). (B) Once activated, TG2 cross-links a biotinylated amine, 5BP, to selected Gln residues in proteins such as fibronectin in the extracellular matrix. Using streptavidin conjugated to horseradish peroxidase, the amount of 5BP incorporated is quantified as a measure of TG2 activity. (C) Trx-mediated TG2 activity over 0–24 h where the last 3 h were in the presence of 5BP. Maximal TG2 activity occurred within 2–4 h of Trx exposure. The maturity of the T84 monolayer did not significantly influence the result (Figure S2). (D) At the 3 h time-point, the EC50 of Trx-mediated TG2 activation was 25±3 nM. Addition of 25 µM TG2 inhibitor, ERW1041E, completely blocked Trx mediated 5BP incorporation. Experiments were performed in triplicate. Measurements are normalized to TG2 activity in the absence of Trx, and are reported as average +/− standard error.
Figure 4.

Fluorescence microscopy of T84 monolayers treated with Trx. T84 enterocytic cells were treated with 0–10 µM Trx in the presence of 200 µM 5BP for 3 h with or without 25 µM of the selective TG2 inhibitor, ERW1041E. Cultures were stained with antibodies against E-cadherin (red) and TG2 (green) proteins, as well as with a streptavidin conjugate (blue). Images were obtained using a Zeiss LSM 510 Meta confocal microscope with an oil immersion compatible 40× objective capable of digital imaging correlation. White bars in each panel correspond to 50 µm. Each experiment was performed in triplicate. All images were processed in the same manner.
Using the above assay, the biological activity of selected Trx inhibitors was quantified based on their ability to block 5BP incorporation. Their IC50 values are summarized in Table 1 (see Figure S3 for titration data). Of particular interest is compound 23, which has the lowest IC50 value of 0.09 ± 0.01 µM amongst all inhibitors tested. This inhibitor is 24-fold more selective for Trx than 1, a value that agrees well with biochemical data (Table 1).
Our results from the T84 enterocyte assay system also reveal that the potency of an inhibitor in cell culture correlates both with the absolute magnitude of its bimolecular rate constant (kinh/Ki) as well as its specificity for Trx over DTT. For example, compound 23 is more potent than the less reactive but comparably specific compounds 21 or 25. Similarly, compounds 1, 29, 26, and 25 are comparably reactive toward DTT; they show progressive improvement in potency in a manner that correlates well with Trx specificity. To verify that DTT is indeed a representative non-specific thiol, the reactivity of selected inhibitors with DTT, cysteine, and glutathione was compared (Table S1). The specificity of this class of asymmetric disulfides for Trx is evident from the data summarized in Table S1.
Computational and mutagenic analysis of inhibitor-Trx interactions
Thioredoxin reduces disulfide bonds in proteins via a bimolecular thiol-disulfide exchange mechanism. The thiol-disulfide exchange reaction is a fundamental process in biology, and has therefore been extensively investigated via experiment and theory.22–25 It is generally accepted that a thiolate anion is the attacking species, and that the reaction proceeds via an SN2 transition state involving back-attack of the disulfide bond by the thiolate (Figure 5A). The S-S bond length in the reactant and product is 2.1 Å, while the S-S bond length in the transition state is estimated to be slightly elongated to 2.6–2.9 Å.25 Water molecules stabilize the transition state by forming hydrogen bonds with partially charged sulfur atoms.
Figure 5.

A model for the binding of asymmetric disulfide 14 to human Trx. (A) Thiol-disulfide SN2 exchange mechanism. Attack by the thiolate leads to a linear S⋯S⋯S transition state. (B) Top view of the inhibitor docking onto the active site of Trx. The benzene ring of the benzimidazolyl group interacts with a tryptophan indole near the Trx active site.
To identify a putative binding site on Trx for compound 14, we used the SwissDock server to derive a docking model. (See Experimental section for details.) Given that each asymmetric disulfide consists of two rotamers that likely have differential affinity and reactivity for Trx, both rotamers were docked using the SwissDock server. Docking solutions were assessed based on three criteria. First, the distance between the Cys32 thiol in Trx and the sec-butyl thiol in compound 14 should be less than 4.5 Å, which would allow thiol-disulfide exchange to occur. Second, the angle of S−S⋯S(Cys32) atoms should be greater than 135°, which allows back-attack of the incoming thiolate. Third, Trx interacts predominantly with the benzimidazolyl group and not significantly with the alkyl group, as suggested by our SAR data.
By screening hundreds of docking solutions, the most favored one was identified and is shown in Figure 5B. Docking the othe r rotamer of 14 in the Trx active site was unfavorable. In this model, the distance between Cys32 thiol and sec-butyl thiol is 3.7 Å, only 0.8 Å longer than transition state. An SN2 back-attack by Cys32 is also supported by a 160° S−S⋯S angle. This model also accounts for our SAR data, namely, the distance between benzimidazolyl moiety and the C̃H group of Cys73 is only ~3.4 Å—close enough for thiol-π interaction26,27—and is thus consistent with recognition of the aromatic rings of our best inhibitors by Trx. An alternate docking model also satisfies the screening criteria (figure not shown), in which the aromatic ring is oriented towards the Trp31 residue and sec-butyl group points in the opposite direction. This model is less favored with a 145° S−S⋯S angle. Furthermore, the former model is consistent with our observation that compounds 5, 12, and 14, would react more readily with Trx than compounds 2, 13, and 1, respectively, due to interactions with residues in the vicinity of the Trx active site.
To test the proposed docking model, a mutant W31A was engineered in human Trx. This mutation was predicted to reduce steric hindrance for the sec-butyl group of 14. The insulin reduction assay revealed that the mutant had comparable activity (78%) to wild-type Trx, a value consistent with an analogous mutation in E. coli Trx.28 However, its reactivity toward compound 14 showed an eleven-fold increase (kinh/Ki = 1.42 µM−1 min−1 as compared to 0.13 µM−1 min−1). Notwithstanding the results of the in silico model, it is possible that both rotamers are responsible for activity in vitro, such that the calculated kinh/Ki would be a composite value.
DISCUSSION AND CONCLUSIONS
Although the role of thioredoxin (Trx) in intracellular redox homeostasis has been extensively investigated, the function of this protein cofactor in the extracellular environment is poorly understood.29 Recent observations have suggested that activation of extracellular transglutaminase 2 (TG2) is a major consequence of Trx secretion.9 TG2 is a ubiquitous but tightly regulated multifunctional enzyme.30 Its aberrant activity has been implicated in a number of pathological conditions,31 the most well studied example being celiac disease.32 Thus, a small molecule approach to interrogate the role of extracellular Trx in celiac disease has the potential to provide fundamentally new insights into inflammatory biology. In turn, small molecule Trx inhibitors could also become drug leads due to their ability to indirectly block TG2 activity.
Two major challenges can be anticipated in the development of a small molecule inhibition strategy for extracellular Trx biology. First, an inhibitor must inactivate secreted Trx while leaving intracellular Trx unperturbed. Second, unlike enzymes that have well defined pockets, Trx has a relatively shallow active site pocket, making it more difficult to engineer highly specific small molecule inhibitors. In this study we addressed both challenges, starting from a previously reported asymmetric disulfide inhibitor, 1. SAR analysis of 1 suggested that half of the molecule should ideally be an extended aromatic group with electron withdrawing substituents. These insights led to substantial improvements over 1, both with respect to activity and possibly also mitigation of malodorous dose-limiting toxicity.
A novel biological assay that mimicked celiac disease pathogenesis was developed to test candidate inhibitors. A model was also developed to rationalize SAR data utilizing computational simulation and site-directed mutagenesis; this model suggested an important protein – small molecule interaction at Trp31. Together, these accomplishments have yielded a new class of chemical tools to study an emerging facet of the biology of an important human protein.
Although the role of extracellular Trx in human biology and pathophysiology are poorly understood, it appears to play a role in allosterically regulating several extracellular proteins including the TRPC ion channel, gp120 from HIV-1 and transglutaminase 2 (TG2). Using the last of these targets to devise a cell-based assay, we have engineered a class of asymmetric disulfides to identify potent and selective inhibitors of extracellular human Trx. Our chemical tools could be used to understand the role of Trx in celiac disease, HIV, and diseases of the synovial tissue.
EXPERIMENTAL METHODS
Materials
All chemicals used in the syntheses described below were from Sigma-Aldrich or Chemi-Block. DTT was from Invitrogen; SDS-polyacrylamide gradient gels (5–20%) were from Bio-Rad; Ni-NTA resin was from Qiagen; the HiTrap-Q anion exchange column was from GE Healthcare; and the 7K MWCO spin columns were from Pierce. T84 cells were from American Type Culture Collection (ATCC). Tissue culture plates were from Corning Life Sciences. The TG2 inhibitor ERW1041E ((S)-quinolin-3-ylmethyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate) was synthesized, as previously described.21 Tetramethylbenzidine (TMB) ready mix substrate was from Sigma Aldrich. Vectashield Mounting Media was purchased from Vector Laboratories. Cell culture medium, fetal bovine serum, antibiotics, trypsin-EDTA, sterile PBS, goat anti-rabbit (H-L) and goat anti-mouse (H-L) secondary antibodies, Streptavidin Alexa Fluor 647, and Streptavidin-HRP were from Invitrogen. Primary antibodies mouse anti-TG2 (TG100) and rabbit anti-E-cadherin were from Thermo Scientific and Cell Signaling Technology, respectively.
Preparation of Recombinant Human Thioredoxin
Trx was expressed and purified in E. coli Rosetta 2/pCK11 as previously described.9 Briefly, expression of Trx was induced by 0.2 mM IPTG at 18 °C for 12 h. The recombinant protein was then purified by Ni-NTA and anion exchange chromatography at 4 °C with the presence of 1 mM DTT at all times. Purified Trx was then flash frozen and stored at −80 °C. Trx mutants W31A and C73A were generated from pCK11 by site-directed mutagenesis, introduced to E. coli Rosetta 2, and purified by the same procedure as the wild-type protein.
Synthesis of Disulfide Inhibitors
The alkyl asymmetrical disulfides were synthesized by reacting substituted thioisothioureas as described by Kirkpatrick et al. with minor modification (Scheme 1).33 LC-MS and 1H NMR methods were employed to determine the purity of the synthesized compounds. All new compounds tested had purity at or above 95%.
Cyclohexyl carbamo(dithioperoxo)imidate hydrochloride (A)
Cyclohexanethiol (7.9519 g, 65 mmol) and thiourea (3.806 g, 50 mmol) were dissolved in 22.5 mL H2O and 65 mL ethanol. The flask was immersed in an ice-water bath, and concentrated hydrochloric acid (5.5 mL) was carefully added. Hydrogen peroxide (30%, 6.5 mL ice-cold) was added drop-wise over 30 min with vigorous stirring, and the solution was continually stirred for another 3 h. The solvent was removed in vacuo and the residue was dissolved in 10 mL ethanol, diluted with ether, and cooled overnight. Filtration followed by recrystallization from ethanol-water yielded cyclohexyl carbamo(dithioperoxo)imidate hydrochloride (10.3373 g, 91%) as white crystals. 1H NMR (400 MHz, D2O): δ 0.96 – 1.28 (m, 5H); 1.41 – 1.44 (m, 1H); 1.58 – 1.61 (m, 2H); 1.80 – 1.83 (m, 2H); 2.83 – 2.90 (m, 2H).
Sec-butyl carbamo(dithioperoxo)imidate hydrochloride (B)
Compound B (121 mg, 0.60 mmol, 12%) was prepared by the method used for A to yield a white solid. 1H NMR (400 MHz, D2O): δ 0.77 – 0.81 (t, 3H, J = 7.2); 1.13 – 1.14 (d, 2H, J = 6.8); 1.35 – 1.55 (m, 2H); 2.86 – 2.93 (m, 1H).
Ethyl carbamo(dithioperoxo)imidate hydrochloride (C)
Compound C (330 mg, 1.9 mmol, 38%) was prepared by the method used for A to yield a white solid. 1H NMR (400 MHz, D2O): δ 1.12 – 1.16 (t, 3H, J = 7.6); 2.70 – 2.75 (d, 2H, J = 7.2).
Tert-butyl carbamo(dithioperoxo)imidate hydrochloride (D)
Compound D (111 mg, 0.55 mmol, 11%) was prepared by the method used for A to yield a white solid. 1H NMR (400 MHz, D2O): δ 1.20 (s, 9H).
Cyclopentyl carbamo(dithioperoxo)imidate hydrochloride (E)
Compound E (160 mg, 0.76 mmol, 15%) was prepared by the method used for A to yield a white solid. 1H NMR (400 MHz, D2O): δ 1.40 – 1.59 (m, 4H); 1.81 – 1.90 (m, 2H); 3.29 – 3.36 (m, 1H).
Isopropyl carbamo(dithioperoxo)imidate hydrochloride (F)
Compound F (383 mg, 2.1 mmol, 41%) was prepared by the method used for A to yield a white solid. 1H NMR (400 MHz, D2O): δ 1.14 – 1.16 (d, 6H, J = 6.8); 3.06 – 3.16 (sep, 1H, J = 6.8).
2-(sec-butyldisulfanyl)-1H-imidazole (1)
Commercially available 2-mercaptoimidazole (220 mg, 2.2 mmol) and 1-methyl-1-propylthioisothiourea hydrochloride (366 mg, 2.5 mmol) were dissolved in 6 ml methanol. Sodium bicarbonate (286 mg, 3.4 mmol) in 10 mL water was added drop-wise with vigorous stirring at room temperature. The resulting mixture was stirred for an additional 1 h and cooled overnight at 4 °C. The crude product was collected by filtration, recrystallized from an ethanol-water mixture to yield a white powder (316 mg, 1.7 mmol, 76%). 1H NMR (400 MHz, CDCl3): δ 0.908 – 0.945 (t, 3H, J = 7.4); 1.31 (d, 3H, J = 6.8); 1.499 – 1.607 (m, 1H); 1.682 – 1.792 (m, 1H); 2.910 – 2.994 (m, 1H); 7.08 – 7.11 (m, 1H); 9.45 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C7H12N2S2, 188.0452, found, 188.0451 (M + H)+.
2-(sec-butyldisulfanyl)thiazole (2)
Compound 2 (172 mg, 0.84 mmol, 84%) was prepared by the method used for 1 to yield a yellow liquid. 1H NMR (400 MHz, DMSO): δ 0.911 – 0.948 (t, 3H, J = 7.4); 1.31 (m, 3H); 1.512 – 1.718 (m, 2H); 3.072 – 3.156 (m, 1H); 7.740 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C7H11NS3, 205.0054, found, 205.0053 (M + H)+.
2-(sec-butyldisulfanyl)pyridine (3)
Compound 3 (172 mg, 0.86 mmol, 86%) was prepared by the method used for 1 to yield a yellow oil. 1H NMR (300 MHz, DMSO): δ 0.95 (t, 3H); 1.24 (d, 3H); 1.57 (m, 2H); 2.98 (m, 1H); 7.25 (m, 1H); 7.76 (m, 2H); 8.46 (m, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C9H13NS2, 199.0489, found, 199.0491 (M + H)+.
2-(sec-butyldisulfanyl)-3H-imidazo[4,5-c]pyridine (4)
Compound 4 (304 mg, 1.27 mmol, 58%) was prepared by the method used for 1 to yield a gray solid. 1H NMR (300 MHz, CD3OD): δ 0.97 (t, 3H); 1.32 (d, 3H); 1.62 (m, 2H); 3.04 (m, 1H); 7.29 (t, 1H); 7.92 (d, 1H); 8.32 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C10H13N3S2, 239.0551, found, 239.0552 (M + H)+.
2-(sec-butyldisulfanyl)benzo[d]thiazole (5)
Compound 5 (494 mg, 1.94 mmol, 88%) was prepared by the method used for 1 to yield a yellow liquid. 1H NMR (300 MHz, DMSO): δ 1.07 (t, 3H); 1.38 (d, 3H); 1.71 (m, 2H); 3.10 (m, 1H); 7.39 (m, 2H); 7.85 (m, 2H). HRMS (Q-TOF MS ES+) m/z calcd for C11H13NS3, 255.0210, found, 255.02101 (M + H)+.
2-(sec-butyldisulfanyl)-6-fluorobenzo[d]thiazole (6)
Compound 6 (191 mg, 0.7 mmol, 87%) was prepared by the method used for 1 to yield a yellow oil. 1H NMR (400 MHz, DMSO): δ 0.95 – 0.99 (t, 3H, J = 7.6); 1.32 – 1.33 (d, 3H, J = 6.8); 1.52 – 1.75 (m, 2H); 3.14 – 3.23 (m, 1H); 7.32 – 7.37 (td, 1H, J = 2.4, 6.4); 7.83 – 7.87 (dd, 1H, J = 4.8, 4); 7.97 – 8.00 (dd, 1H, J = 2.8, 6).
2-(sec-butyldisulfanyl)-6-chlorobenzo[d]thiazole (7)
Compound 7 (168 mg, 0.58 mmol, 92%) was prepared by the method used for 1 to yield a yellow oil. 1H NMR (400 MHz, CDCl3): δ 0.95 – 0.98 (t, 3H, J = 7.6); 1.31 – 1.33 (d, 3H, J = 6.8); 1.54 – 1.75 (m, 2H); 3.15 – 3.24 (m, 1H); 7.49 – 7.52 (dd, 1H, J = 2.4, 6.4); 7.81 – 7.83 (d, 1H, J = 8.0); 8.21 (d, 1H, J = 2.4).
2-(sec-butyldisulfanyl)-6-iodobenzo[d]thiazole (8)
Compound 8 (153 mg, 0.40 mmol, 80%) was prepared by the method used for 1 to yield a yellow oil. 1H NMR (400 MHz, DMSO): δ 0.95 – 0.98 (t, 3H, J = 7.2); 1.31 – 1.33 (d, 3H, J = 6.8); 1.52 – 1.75 (m, 2H); 3.15 – 3.23 (m, 1H); 7.60 – 7.62 (d, 1H, J = 8.4); 7.75 – 7.78 (dd, 1H, J = 1.6, 6.8); 8.48 (d, 1H, J = 1.6).
4-bromo-2-(sec-butyldisulfanyl)benzo[d]thiazole (9)
Compound 9 (193 mg, 0.58 mmol, 72%) was prepared by the method used for 1 to yield an orange oil. 1H NMR (400 MHz, DMSO): δ 0.96 – 0.99 (t, 3H, J = 7.2); 1.33 – 1.34 (d, 3H, J = 6.8); 1.55 – 1.76 (m, 2H); 3.17 – 3.23 (m, 1H); 7.29 – 7.33 (t, 1H, J = 8); 7.70 – 7.72 (dd, 1H, J = 0.8, 6.8); 8.06 – 8.08 (dd, 1H, J = 0.8, 7.2). HRMS (Q-TOF MS ES+) m/z calcd for C11H12BrNS3, 332.9315, found, 332.9313 (M + H)+.
5-bromo-2-(sec-butyldisulfanyl)benzo[d]thiazole (10)
Compound 10 (156 mg, 0.47 mmol, 59%) was prepared by the method used for 1 to yield an orange oil. 1H NMR (400 MHz, DMSO): δ 0.95 – 0.99 (t, 3H, J = 7.2); 1.32 – 1.33 (d, 3H, J = 6.8); 1.54 – 1.75 (m, 2H); 3.18 – 3.23 (m, 1H); 7.54 – 7.57 (dd, 1H, J = 1.6, 6.8); 8.02 – 8.05 (m, 2H). HRMS (Q-TOF MS ES+) m/z calcd for C11H12BrNS3, 332.9315, found, 332.9317 (M + H)+.
2-(sec-butyldisulfanyl)-6-nitrobenzo[d]thiazole (11)
Compound 11 (132 mg, 0.44 mmol, 88%) was prepared by the method used for 1 to yield a yellow solid. 1H NMR (400 MHz, DMSO): δ 0.97 – 1.00 (t, 3H, J = 7.6); 1.34 – 1.36 (d, 3H, J = 6.8); 1.57 – 1.77 (m, 2H); 3.22 – 3.30 (m, 1H); 7.99 – 8.01 (d, 1H, J = 8.8); 8.31 (dd, 1H, J = 2.8, 6.4); 9.12 (d, 1H, J = 2.4). HRMS (Q-TOF MS ES+) m/z calcd for C11H12N2O2S3, 300.0061, found, 300.0062 (M + H)+.
2-(ethyldisulfanyl)-1H-benzo[d]imidazole (12)
Compound 12 (326 mg, 1.55 mmol, 70%) was prepared by the method used for 1 to yield a white solid. 1H NMR (300 MHz, CDCl3): δ 1.37 (t, 3H); 2.88 (m, 2H); 7.42 (m, 1H); 7.68 (m, 1H); 9.78 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C9H10N2S2, 210.0285, found, 210.0286 (M + H)+.
2-(tert-butyldisulfanyl)-1H-benzo[d]imidazole (13)
Compound 13 (299 mg, 1.25 mmol, 60%) was prepared by the method used for 1 to yield a white solid. 1H NMR (300 MHz, CD3OD): δ 1.39 (s, 9H); 7.50 (m, 2H). HRMS (Q-TOF MS ES+) m/z calcd for C11H14N2S2, 238.0598, found, 238.0600 (M + H)+.
2-(sec-butyldisulfanyl)-1H-benzo[d]imidazole (14)
Compound 14 (446 mg, 1.9 mmol, 85%) was prepared by the method used for 1 to yield a white solid. 1H NMR (400 MHz, CDCl3): δ 0.963 – 0.999 (t, 3H, J = 7.2); 1.328 – 1.345 (d, 3H, J = 6.8); 1.525 – 1.634 (m, 1H); 1.720 – 1.826 (m, 1H); 2.937 – 3.021 (m, 1H); 7.216 – 7.254 (m, 2H); 7.394 – 7.439 (m, 1H); 7.647 – 7.692 (m, 1H); 9.57 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C11H14N2S2, 238.0598, found, 238.0600 (M + H)+.
2-(isopropyldisulfanyl)-1H-benzo[d]imidazole (15)
Compound 15 (314 mg, 1.40 mmol, 64%) was prepared by the method used for 1 to yield a white solid. 1H NMR (300 MHz, CD3OD): δ 1.34 (d, 6H); 3.25 (m, 1H); 7.24 (d, 2H); 7.51 (s, 2H). HRMS (Q-TOF MS ES+) m/z calcd for C10H12N2S2, 224.0442, found, 224.0443 (M + H)+.
2-(cyclopentyldisulfanyl)-1H-benzo[d]imidazole (16)
Compound 16 (419 mg, 1.67 mmol, 76%) was prepared by the method used for 1 to yield a white solid. 1H NMR (300 MHz, CDCl3): δ 1.74 (m, 4H); 2.01 (m, 2H); 3.45 (m, 1H); 7.42 (s, 1H); 7.68 (s, 1H); 9.68 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C12H14N2S2, 250.0598, found, 250.0600 (M + H)+.
2-(cyclohexyldisulfanyl)-1H-benzo[d]imidazole (17)
Compound 17 (360 mg, 1.36 mmol, 62%) was prepared by the method used for 1 to yield a white solid. 1H NMR (300 MHz, CD3OD): δ 1.32 (m, 6H); 1.78 (m, 2H); 2.03 (m, 2H); 3.00 (m, 1H); 7.24 (d, 2H); 7.51 (s, 2H). HRMS (Q-TOF MS ES+) m/z calcd for C13H16N2S2, 264.0755, found, 264.0756 (M + H)+.
2-(cyclohexyldisulfanyl)benzo[d]thiazole (18)
Compound 18 (2.047 g, 7.3 mmol, 90%) was prepared by the method used for 1 to yield a yellow oil. 1H NMR (400 MHz, DMSO): δ 1.11 – 1.47 (m, 5H); 1.50 – 1.57 (m, 1H); 1.67 – 1.74 (m, 2H); 1.98 – 2.04 (m, 2H); 3.12 – 3.19 (m, 1H); 7.36 – 7.41 (m, 1H); 7.44 – 7.49 (m, 1H); 7.81 – 7.83 (m, 1H); 8.03 – 8.05 (m, 1H).
2-(cyclohexyldisulfanyl)benzo[d]oxazole (19)
Compound 19 (116 mg, 0.44 mmol, 87%) was prepared by the method used for 1 to yield a red oil. 1H NMR (400 MHz, DMSO): δ 1.12 – 1.43 (m, 5H); 1.50 – 1.57 (m, 1H); 1.67 – 1.73 (m, 2H); 1.96 – 2.02 (m, 2H); 3.13 – 3.20 (m, 1H); 7.34 – 7.41 (m, 2H); 7.67 – 7.75 (m, 2H). HRMS (Q-TOF MS ES+) m/z calcd for C13H16N2S2, 265.0595, found, 265.0597 (M + H)+.
2-(sec-butyldisulfanyl)-6-chloro-5-fluoro-1H-benzo[d]imidazole (20)
Compound 20 (87 mg, 0.30 mmol, 60%) was prepared by the method used for 1 to yield an off-white solid. 1H NMR (400 MHz, CDCl3): δ 0.96 – 1.00 (t, 3H, J = 7.6); 1.32 – 1.34 (d, 3H, J = 6.8); 1.52 – 1.63 (m, 1H); 1.70 – 1.81 (m, 1H); 2.95 – 3.04 (m, 1H); 7.33 – 7.35 (d, 1H, J = 9.2); 7.57 – 7.58 (d, 1H, J = 6.4). HRMS (Q-TOF MS ES+) m/z calcd for C13H14ClFN2S2, 316.0271, found, 316.0271 (M + H)+.
6-chloro-2-(cyclohexyldisulfanyl)-5-fluoro-1H-benzo[d]imidazole (21)
Compound 21 (105 mg, 0.33 mmol, 66%) was prepared by the method used for 1 to yield a beige solid. 1H NMR (400 MHz, DMSO): δ 1.10 – 1.38 (m, 5H); 1.51 – 1.56 (m, 1H); 1.66 – 1.71 (m, 2H); 1.93 – 1.98 (m, 2H); 3.03 – 3.10 (m, 1H); 7.50 – 7.53 (d, 1H, J = 9.6); 7.66 – 7.68 (d, 1H, 6.8). HRMS (Q-TOF MS ES+) m/z calcd for C11H12ClFN2S2, 290.0115, found, 290.0118 (M + H)+.
2-(sec-butyldisulfanyl)-5-nitro-1H-benzo[d]imidazole (22)
Compound 22 (507 mg, 1.79 mmol, 81%) was prepared by the method used for 1 to yield a yellow solid. 1H NMR (400 MHz, CDCl3): δ 0.98 – 1.02 (t, 3H, J = 7.2); 1.35 – 1.36 (d, 3H, J = 6.8); 1.55 – 1.67 (m, 1H); 1.73 – 1.83 (m, 1H); 2.99 – 3.07 (m, 1H); 7.48 – 7.70 (dd, 1H, J = 8.4, 73); 8.18 – 8.21 (dd, 1H, J = 2, 6.8); 8.37 – 8.55 (d, 1H, J = 70); 10.14 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C11H13N3O2S2, 283.0449, found, 283.0453 (M + H)+.
2-(cyclohexyldisulfanyl)-5-nitro-1H-benzo[d]imidazole (23)
Compound 23 (2.310 g, 7.47 mmol, 93%) was prepared by the method used for 1 to yield a brown solid. 1H NMR (400 MHz, DMSO): δ 1.09 – 1.39 (m, 5H); 1.49 – 1.56 (m, 1H); 1.65 – 1.71 (m, 2H); 1.94 – 2.00 (m, 2H); 3.06 – 3.14 (m, 1H); 7.64 – 7.66 (d, 1H, J = 8.8); 8.07 – 8.10 (dd, 1H, J = 2.4, 6.8); 8.35 (d, 1H, J = 2). HRMS (Q-TOF MS ES+) m/z calcd for C13H15N3O2S2, 309.0606, found, 309.0606 (M + H)+.
2-(cyclohexyldisulfanyl)-5-ethoxy-1H-benzo[d]imidazole (24)
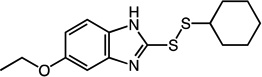
Compound 24 (130 mg, 0.42 mmol, 85%) was prepared by the method used for 1 to yield a black oil. 1H NMR (400 MHz, DMSO): δ 1.10 – 1.38 (m, 8H); 1.48 – 1.57 (m, 1H); 1.63 – 1.73 (m, 2H); 1.93 – 2.00 (m, 2H); 3.01 – 3.08 (m, 1H); 3.98 – 4.03 (m, 2H); 6.76 – 6.79 (dd, 1H, J = 2.4, 6.4); 6.96 (s, 1H); 7.35 – 7.37 (d, 1H, J = 8). HRMS (Q-TOF MS ES+) m/z calcd for C15H20N2OS2, 308.1017, found, 308.1023 (M + H)+.
phenyl(2-thioxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)methanone
Synthesis adapted from Okolotowicz et al.34 (3,4-diaminophenyl)(phenyl)methanone (743 mg, 3.5 mmol) and potassium ethyl xanthate (1074 mg, 6.7 mmol) were dissolved in 1.5 mL H2O and 8 mL ethanol and refluxed for 3 h. Activated carbon was added and the solution was refluxed for 20 m and then filtered. 10 mL H2O was added to the filtrate, followed by 1.5 mL 50% acetic acid. The suspension was cooled overnight at 4 °C and dried in vacuo, yielding the title compound (641 mg, 2.5 mmol, 72%) as an orange solid. 1H NMR (400 MHz, DMSO): δ 7.24 – 7.26 (d, 1H, J = 8.4); 7.45 – 7.46 (d, 1H, J = 1.2); 7.53 – 7.57 (m, 3H); 7.63 – 7.70 (m, 3H); 12.82 (s, 2H).
(2-(cyclohexyldisulfanyl)-1H-benzo[d]imidazol-6-yl)(phenyl)methanone (25)
Compound 25 (88 mg, 0.24 mmol, 72%) was prepared by the method used for 1 to yield an orange solid 1H NMR (400 MHz, DMSO): δ 1.11 – 1.41 (m, 5H); 1.52 – 1.57 (m, 1H); 1.66 – 1.72 (m, 2H); 1.95 – 2.01 (m, 2H); 3.06 – 3.13 (m, 1H); 7.53 – 7.74 (m, 8H); 7.84 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C20H20N2OS2, 368.1017, found, 368.1021 (M + H)+.
2-amino-8-(cyclohexyldisulfanyl)-7H-purin-6-ol (26)
Compound 26 (74 mg, 0.25 mmol, 71%) was prepared by the method used for 1 to yield an off-white solid. 1H NMR (400 MHz, DMSO): δ 1.13 – 1.42 (m, 5H); 1.52 – 1.56 (m, 1H); 1.61 – 1.73 (m, 2H); 1.85 – 1.98 (m, 2H); 2.96 – 3.05 (m, 1H); 6.41 – 6.50 (m, 2H); 10.61 (s, 1H); 12.85 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C11H15N5OS2, 297.0718, found, 297.0720 (M + H)+.
8-(cyclohexyldisulfanyl)-7H-purin-6-amine (27)
Compound 27 (69 mg, 0.24 mmol, 70%) was prepared by the method used for 1 to yield n orange solid. 1H NMR (400 MHz, DMSO): δ 1.11 – 1.42 (m, 5H); 1.50 – 1.57 (m, 1H); 1.63 – 1.73 (m, 2H); 1.93 – 2.02 (m, 2H); 3.00 – 3.09 (m, 1H); 7.20 (s, 2H); 8.07 (s, 1H); 13.25 (s, 1H). HRMS (Q-TOF MS ES+) m/z calcd for C11H15N5S2, 281.0769, found, 281.0772 (M + H)+.
2-(cyclohexyldisulfanyl)-4H-benzo[d][1,3]thiazine (28)
Compound 28 (59 mg, 0.20 mmol, 79%) was prepared by the method used for 1 to yield a brown oil. 1H NMR (400 MHz, DMSO): δ 1.13 – 1.43 (m, 5H); 1.50 – 1.57 (m, 1H); 1.63 – 1.73 (m, 2H); 1.93 – 2.02 (m, 2H); 2.98 – 3.06 (m, 1H); 4.11 (s, 2H); 7.15 – 7.36 (m, 4H).
2-(cyclohexyldisulfanyl)-5-phenyl-1H-imidazole (29)
Compound 29 (115 mg, 0.40 mmol, 79%) was prepared by the method used for 1 to yield an off-white solid. 1H NMR (400 MHz, DMSO): δ 1.14 – 1.39 (m, 5H); 1.52 – 1.57 (m, 1H); 1.65 – 1.72 (m, 2H); 1.94 – 2.02 (m, 2H); 3.00 – 3.07 (m, 1H); 7.18 – 7.22 (t, 1H, J = 7.2); 7.33 – 7.37 (t, 2H, J = 7.6); 7.72 – 7.74 (d, 2H, J = 7.6). HRMS (Q-TOF MS ES+) m/z calcd for C15H18N2S2, 290.0911, found, 290.0914 (M + H)+.
3-(cyclohexyldisulfanyl)-5-phenyl-4H-1,2,4-triazol-4-amine (30)
Compound 30 (119 mg, 0.39 mmol, 78%) was prepared by the method used for 1 to yield a white solid. 1H NMR (400 MHz, DMSO): δ 1.16 – 1.42 (m, 5H); 1.50 – 1.55 (m, 1H); 1.68 – 1.74 (m, 2H); 1.95 – 2.01 (m, 2H); 3.10 – 3.17 (m, 1H); 6.22 (s, 2H); 7.50 – 7.55 (m, 3H); 8.00 – 8.04 (m, 2H). HRMS (Q-TOF MS ES+) m/z calcd for C14H18N4S2, 306.0973, found, 306.0975 (M + H)+.
In Vitro Thioredoxin Activity Assay
Before use, recombinant human Trx was freshly reduced with a ten-fold molar excess of DTT on ice. The excess DTT was removed by passing the solution through a 7K MWCO spin column. Trx concentration was determined by A280nm (ε = 7570 M−1 cm−1) and the protein was used freshly within 2 h. Pre-reduced Trx was reacted with disulfide inhibitors in 0.1 M phosphate-citrate buffer, pH 6.0, at a concentration of 10 µM each at room temperature.
Reduction of the inhibitor disulfide was monitored spectrophotometrically based on an increase in absorbance at 252 nm due to the release of 2-mercaptoimidazole (Δε = 9400 M−1 cm−1) or corresponding substituents. The time dependent absorption curve was recorded until no significant change was observed. From this data, the rate of 2-mercaptoimidazole and oxTrx formation was calculated based on the above Δε value. In a separate measurement, 250 µM DTT was reacted with 10 µM of each inhibitor to calculate bimolecular rate constants for the reaction between the inhibitor and a reference thiol reducing agent. The resulting time-dependent curves were fitted to the following equation to yield kinh/Ki or ki values: [P] = [I]0(1 − e−k[E]t), where [P] is the accumulated concentration of the aromatic reaction product at time t, [I]0 is the initial inhibitor concentration, [E] is the thioredoxin concentration, and k is kinh/Ki for Trx or ki for DTT, glutathione or cysteine.
Insulin Turbidity Assay
The activities of wild-type and the W31A mutant of human Trx were measured by insulin turbidity assay as previously described with slight modification.35 Briefly, 4 µM Trx was added into a mixture of 160 µM bovine insulin (Km = 30 µM for WT Trx) and 1 mM DTT in 0.1 M phosphate-citrate buffer, pH 6.0. Formation of the reduced insulin was monitored by following the appearance of turbidity at 650 nm due to precipitation. The regeneration of Trx by DTT is much faster than direct reduction of insulin by DTT. Previous experiments have shown that ΔA650nm/time is linear over the Trx concentration 0 to 8 µM (data not shown). Therefore, the activity of the W31A mutant was estimated by normalizing its ΔA650nm/time to that of wild-type Trx.
Docking Models
Docking between the structurally characterized reduced human Trx (RSCD 1ERT) and compound 14 was performed on the Swiss Dock server.36 The coordinate file 1ERT was edited to remove all water molecules. Compound 14 was optimized by MM2 in Chem3D. The ligand-binding site was constrained in a 10×10×10 Å3 cube with (x_31, y_12.5, z_4.5) as the center. Flexible side chains were allowed. All rotatable single bonds were allowed to rotate in the ligand. Docking results were screened by Chimera.37
Cell Culture
T84 epithelial cells were grown in Dulbecco’s modified Eagle’s medium/Ham’s F-12 (1:1) supplemented with antibiotics (penicillin/streptomycin) and 5% (v/v) fetal bovine serum. Cells are grown at 37 °C and 5% CO2. Medium was changed every alternate day. When the cells reached greater than 90% confluency, they were passaged using trypsin-EDTA. For the TG2 activity assays, T84 enterocytes were grown in standard flat bottom 48 well plates. For fluorescent microscopy experiments, T84 enterocytes were grown on collagen coated permeable supports (5 µm pore size, 6.5 mm in diameter) as described previously.20 T84 cells were seeded at 3×104 cells/well in all experiments.
Thioredoxin Mediated TG2 Activity Assay
For measurement of TG2 activity, T84 monolayers were grown in standard 48 well flat bottom plates for 1–14 days (Figure S2) as described above. Recombinant human thioredoxin was reduced using a 10 molar excess of DTT, buffer exchanged into T84 cell culture medium, and used fresh as described in the in vitro thioredoxin activity assay procedure above. Thioredoxin was diluted into fresh medium and added to the T84 cells for 1–24 h (Figure 3). For the last 3 h of thioredoxin exposure 200 µM 5BP was spiked into the medium such that each well was exposed to 5BP for a total of 3 h. For thioredoxin treatments less than 3 h, 5BP was spiked into wells prior to thioredxoin exposure. As a control, TG2 activity was inhibited with 25 µM ERW1041E for the duration of 5BP exposure. After 5BP exposure, T84 monolayers were washed with warm PBS three times, and fixed with 4% (w/v) paraformaldehyde in PBS for 15 min at room temperature. Following three washes with PBS, monolayers were blocked overnight at 4°C using 5% (w/v) BSA in PBS supplemented with 0.1% (v/v) Tween-20. Then, monolayers were treated with horseradish peroxidase conjugated Streptavidin, diluted according to manufacturer’s recommendations, in blocking buffer overnight at 4°C. Cells were washed four times with PBS + 0.1% Tween-20. A ready mix solution of tetramethylbenzidine (TMB) substrate was added to the transwell chambers for 5–10 min, after which 100 µL removed and quenched in equal volume of 1 M hydrochloric acid in a 96 well plate format. Absorbance at 450 nm was measured using a 96 well plate reader. The fold-change in 5BP incorporation was normalized to the 0 µM thioredoxin exposure time. Each set of conditions was assayed in at least triplicate wells.
Fluorescent Microscopy
T84 cells were grown to maturity on permeable supports, as described previously.20 For fluorescence staining experiments, recombinant human thioredoxin was reduced using a 10 molar excess of DTT, buffer exchanged into T84 cell culture medium, and used fresh as described in the in vitro thioredoxin activity assay procedure above. Thioredoxin was diluted into fresh medium containing 200 µM 5-biotinamido pentylamine (5BP) and added to both top and bottom compartments of the T84 transwells for 3 h. As a control, TG2 activity was inhibited with 25 µM ERW1041E for the duration of thioredoxin and 5BP exposure. After the 3 h thioredoxin and 5BP incubation, monolayers were washed with warm PBS three times, and fixed with 4% (w/v) paraformaldehyde in PBS for 15min. Cells were then washed three times with PBS, and blocked overnight at 4 °C using 5% (w/v) BSA in PBS supplemented with 0.1% (v/v) Tween-20. Primary antibodies, rabbit anti-E-cadherin IgG MAb (1:200 dilution in blocking buffer) and mouse anti-TG2 IgG MAb (1:200 dilution in blocking buffer), were used to label E-cadherin and TG2, respectively, in an overnight incubation at 4 °C. E-cadherin was used as a marker of cell-cell contacts in the enterocyte monolayer. Cells were washed three times with PBS + 0.1% (v/v) Tween-20, and incubated once again overnight at 4 °C with secondary antibodies, goat anti-rabbit (H-L) IgG Alexa Fluor 555 conjugate (2 µg/mL in blocking buffer) and goat anti-mouse (H-L) IgG Alexa Fluor 488 conjugate (2 µg/mL in blocking buffer). To visualize TG2 activity, streptavidin Alexa Fluor 647 conjugate (2 µg/mL in blocking buffer) was also added. Subsequently, cells were washed four times with PBS + 0.1% (v/v) Tween-20. Permeable supports were cut from their frames with a sterile razor blade and mounted on slides using Vectashield mounting media. Cover-slips were sealed onto the mounting slides with multiple coats of clear nail polish. Sealed slides were stored in the dark at 4 °C, and imaged using a Zeiss LSM 510 Meta Confocal Microscope, Zeiss Immersol 518F oil, and a 40X Plan-Neo/1.3 NA Oil objective lens with Digital Image Correlation (DIC) capability.
Supplementary Material
Acknowledgements
This research was supported by a grant from the NIH (R01 DK063158) to C.K.
Abbreviations Used
- Trx, thioredoxin
thioredoxin-1
- TG2
tissue transglutaminase 2
- TrxR
thioredoxin reductase
- IFN-γ
interferon-γ
- DTT
dithiothreitol
- 5BP
5-biotinamido pentylamine
- SAR
structure activity relationship
Footnotes
ANCILLARY INFORMATION
Supporting Information Available: The supplemental information provided illustrates the specificity of Trx for TG2 compared to DTT, the growth time of T84 cells and Trx ability to activate extracellular TG2, biological activity curves for Trx inhibitors, and comparison of Trx inhibitor reactivity for DTT, glutathione, and cysteine. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions: Authors Thomas R. DiRaimondo, Nicholas M. Plugis, and Xi Jin contributed equally.
REFERENCES
- 1.Holmgren A, Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010;396:120–124. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 2.Gromer S, Urig S, Becker K. The thioredoxin system–from science to clinic. Med. Res. Rev. 2004;24:40–89. doi: 10.1002/med.10051. [DOI] [PubMed] [Google Scholar]
- 3.Lillig CH, Holmgren A. Thioredoxin and related molecules–From biology to health and disease. Antioxid. Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 4.Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J. Biol. Chem. 1992;267:24161–24164. [PubMed] [Google Scholar]
- 5.Nakamura H. Thioredoxin as a key molecule in redox signaling. Antioxid. Redox Signal. 2004;6:15–17. doi: 10.1089/152308604771978309. [DOI] [PubMed] [Google Scholar]
- 6.Xu S-Z, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang L-H, Emery P, Sivaprasadarao A, Beech DJ. TRPC channel activation by extracellular thioredoxin. Nature. 2008;451:69–72. doi: 10.1038/nature06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azimi I, Matthias LJ, Center RJ, Wong JWH, Hogg PJ. Disulfide bond that constrains the HIV-1 gp120 V3 domain is cleaved by thioredoxin. J. Biol. Chem. 2010;285:40072–40080. doi: 10.1074/jbc.M110.185371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Oh J, Choi J, Jang J, Kang M. Identification of human thioredoxin as a novel IFN-gamma induced factor: Mechanism of induction and its role in cytokine production. BMC Immunol. 2008;9:1–15. doi: 10.1186/1471-2172-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin X, Stamnaes J, Klöck C, Diraimondo TR, Sollid LM, Khosla C. Activation of extracellular transglutaminase 2 by thioredoxin. J. Biol. Chem. 2011;286:37866–37873. doi: 10.1074/jbc.M111.287490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molberg O, Mcadam SN, Körner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Norén O, Roepstorff P, Lundin KE, Sjöström H, Sollid LM. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 11.van de Wal Y, Kooy Y, van Veelen P, Peña S, Mearin L, Papadopoulos G, Koning F. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J. Immunol. 1998;161:1585–1588. [PubMed] [Google Scholar]
- 12.Siegel M, Strnad P, Watts RE, Choi K, Jabri B, Omary MB, Khosla C. Extracellular transglutaminase 2 Is catalytically inactive, but Is transiently activated upon tissue injury. PLoS ONE. 2008;3 doi: 10.1371/journal.pone.0001861. e1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut. 1995;37:766–776. doi: 10.1136/gut.37.6.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol. 1996;178:7–7. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 15.Kirkpatrick DL, Ehrmantraut G, Stettner S, Kunkel M, Powis G. Redox active disulfides: the thioredoxin system as a drug target. Oncol. Res. 1997;9:351–356. [PubMed] [Google Scholar]
- 16.Kunkel MW, Kirkpatrick DL, Johnson JI, Powis G. Cell line-directed screening assay for inhibitors of thioredoxin reductase signaling as potential anti-cancer drugs. Anticancer Drugs. 1997;12:659–670. [PubMed] [Google Scholar]
- 17.Ramanathan RK, Kirkpatrick DL, Belani CP, Friedland D, Green SB, Chow H-HS, Cordova CA, Stratton SP, Sharlow ER, Baker A, Dragovich T. A Phase I pharmacokinetic and pharmacodynamic study of PX-12, a novel inhibitor of thioredoxin-1, in patients with advanced solid tumors. Clin. Cancer Res. 2007;13:2109–2114. doi: 10.1158/1078-0432.CCR-06-2250. [DOI] [PubMed] [Google Scholar]
- 18.Ramanathan RK, Abbruzzese J, Dragovich T, Kirkpatrick L, Guillen JM, Baker AF, Pestano LA, Green S, Hoff DD. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer Chemother. Pharmacol. 2011;67:503–509. doi: 10.1007/s00280-010-1343-8. [DOI] [PubMed] [Google Scholar]
- 19.Kirkpatrick DL, Kuperus M, Dowdeswell M, Potier N, Donald LJ, Kunkel M, Berggren M, Angulo M, Powis G. Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides. Biochem. Pharmacol. 1998;55:987–994. doi: 10.1016/s0006-2952(97)00597-2. [DOI] [PubMed] [Google Scholar]
- 20.Diraimondo TR, Klöck C, Khosla C. Interferon-γ activates transglutaminase 2 via a phosphatidylinositol-3-kinase-dependent pathway: implications for celiac sprue therapy. J. Pharmacol. Exp. Ther. 2012;341:104–114. doi: 10.1124/jpet.111.187385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watts RE, Siegel M, Khosla C. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J. Med. Chem. 2006;49:7493–7501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson J, Bayer R, Hupe D. Structure-reactivity correlations for the thiol-disulfide interchange reaction. J. Am. Chem. Soc. 1977;99:7922–7926. [Google Scholar]
- 23.Szajewski R, Whitesides G. Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J. Am. Chem. Soc. 1980;102:2011–2026. [Google Scholar]
- 24.Houk J, Whitesides G. Structure-reactivity relations for thiol-disulfide interchange. J. Am. Chem. Soc. 1987;109:6825–6836. [Google Scholar]
- 25.Fernandes PA, Ramos MJ. Theoretical insights into the mechanism for thiol/disulfide exchange. Chemistry. 2004;10:257–266. doi: 10.1002/chem.200305343. [DOI] [PubMed] [Google Scholar]
- 26.Tauer TP, Derrick ME, Sherrill CD. Estimates of the ab initio limit for sulfur-π interactions: the H2S-benzene dimer. J. Phys. Chem. A. 2005;109:191–196. doi: 10.1021/jp046778e. [DOI] [PubMed] [Google Scholar]
- 27.Ringer AL, Senenko A, Sherrill CD. Models of S/π interactions in protein structures: comparison of the H2S benzene complex with PDB data. Protein Sci. 2007;16:2216–2223. doi: 10.1110/ps.073002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krause G, Holmgren A. Substitution of the conserved tryptophan 31 in Escherichia coli thioredoxin by site-directed mutagenesis and structure-function analysis. J. Biol. Chem. 1991;266:4056–4066. [PubMed] [Google Scholar]
- 29.Burke-Gaffney A, Callister MEJ, Nakamura H. Thioredoxin: friend or foe in human disease? Trends Pharmacol. Sci. 2005;26:398–404. doi: 10.1016/j.tips.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Klöck C, Khosla C. Regulation of the activities of the mammalian transglutaminase family of enzymes. Protein Sci. 2012;21:1781–1791. doi: 10.1002/pro.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gundemir S, Colak G, Tucholski J, Johnson GVW. Transglutaminase 2: a molecular Swiss army knife. Biochim. Biophys. Acta. 2012;1823:406–419. doi: 10.1016/j.bbamcr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sollid LM, Jabri B. Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr. Opin. Immunol. 2011;23:732–738. doi: 10.1016/j.coi.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirkpatrick D, Jimale M, King K, Chen T. Synthesis and evaluation of imidazolyl disulfides for selective cytotoxicity to hypoxic EMT6 tumor cells in vitro. Eur. J. Med. Chem. 1992;27:33–37. [Google Scholar]
- 34.Okolotowicz KJ, Shi R, Zheng X, MacDonald M, Reed JC, Cashman JR. Selective benzimidazole inhibitors of the antigen receptor-mediated NF-κB activation pathway. Bioorg. Med. Chem. 2010;18:1918–1924. doi: 10.1016/j.bmc.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 35.Arnér ES, Holmgren A. Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. 2001;24:7.4.1–7.4.14. doi: 10.1002/0471140856.tx0704s05. [DOI] [PubMed] [Google Scholar]
- 36.Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.