

Abstract
Activation of a cardiac myocyte P2X4 receptor protects in heart failure. 5’-Phosphonate and 5’-phosphate analogues of AMP containing a (N)-methanocarba (bicyclo[3.1.0]hexane) system could protect from heart failure by potentially activating this cardioprotective channel. Phosphoesters and phosphonodiesters were synthesized and administered in vivo via a mini-osmotic pump in a mouse ischemic heart failure model; most significantly increased intact heart contractile function (echocardiography) compared to vehicle-infusion. Several new thio and deuterated phosphate derivatives were protective in a calsequestrin (CSQ)-overexpressing heart failure model. Diethyl (7, MRS4084) and diisopropyl (8, MRS4074) phosphotriesters were highly protective in the ischemic model. Substitution of 2-Cl with iodo reduced protection in the CSQ model. Diisopropyl ester 16 (MRS2978) of (1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonoethylene)-bicyclo[3.1.0]hexane was highly efficacious (CSQ), while lower homologue 1’-phosphonomethylene derivative 14 was inactive. Thus, we identified uncharged carbocyclic nucleotide analogues that represent potential candidates for the treatment of heart failure, suggesting this as a viable and structurally broad approach.
Introduction
Activation of cardiac P2X receptors, which are trimeric ligand-gated ion channels,1 has been explored as a target for the treatment of heart failure.2–4 A P2X receptor on the cardiomyocyte activated by ATP or other potent nucleotide analogues mediates cardioprotection. The P2X4 receptor (P2X4R) is an important subunit of the native cardiac P2X receptor, which mediates an ionic current induced by extracellular ATP, although the precise subunit composition of this receptor channel is unknown.4 Cardiac myocyte-specific overexpression of the P2X4R can mimic the beneficial effects following chronic infusion of P2X agonist analogues, such as (1’S,2’R,3’S,4’R,5’S)-4-(6-amino-2-chloro-9H-purin-9-yl)-1-[phosphoryloxy-methyl]bicyclo[3.1.0]hexane-2,3-diol, (MRS2339, 1, Chart 1). The continuous activation of this ATP-gated, nonselective cation channel under the resting or negative membrane potentials would produce an inward current, while its activation during depolarized portions of the action potential should lead to an outward current. These ionic currents represent a possible ionic mechanism by which the cardiomyocyte P2X channel achieves its protective effect.
Chart 1.

Representative adenine nucleotide derivatives that display cardioprotective activity by activating a P2X ion channel. Both the phosphate derivative 1 and the homologous phosphonate analogues 2 and 3 served as the lead compounds for the present set of ester derivatives.
Other nucleotides, 5’-monophosphates (related to AMP), were also shown to be cardioprotective by the same mechanism. The cardioprotective ability of chronically administered 1 and its phosphonate derivatives 2 (MRS2776) and 3 (MRS2925) was demonstrated using a transgenic mouse overexpressing calsequestrin (CSQ) as a genetic model of heart failure.5 Compound 1 is a methanocarba monophosphate derivative of 2-chloro-AMP that contains a rigid bicyclic ring system (bicyclo[3.1.0]hexane) of the North (N) conformation in place of ribose. For other nucleotide receptors, such as the P2Y1 G protein-coupled receptor for ADP, this constrained bicyclic system consisting of fused cyclopropane and cyclopentane rings maintained a receptor-preferred conformation of the ribose-like moiety of the nucleotide.6 At various P2X receptors, the (N) conformation was also shown to be preferred in receptor activation using rigid methanocarba nucleotide analogues,7 consistent with the high cardioprotective (P2X receptor-dependent) potency of compound 1.
In cardiomyocytes obtained from the CSQ mouse, compound 1 induced a current that is characteristic of the action of the cardiac P2X4R and similar to the response to ATP.4 Compound 1 rescued the hypertrophic and heart failure phenotype in the CSQ-overexpressing mouse.4 It significantly improved standard measures of cardiac function and increased longevity as compared to vehicle-injected mice. The improvement in survival was associated with decreases in the heart weight/body weight ratio and in the cross-sectional area of the cardiac myocytes. Compound 1 was also devoid of any vasodilator action in aorta ring preparations indicating that its salutary effect in heart failure was not due to any vascular unloading.
The active phosphonate derivatives, such 2 and 3, are expected to add an extra measure of stability in vivo. The 5’-monophosphate group of 1, which is associated with P2X receptor recognition, would be subject to cleavage by nucleotidases (although less efficiently than the native riboside6), but this action would be prevented with phosphonate derivatives.5 Nevertheless, the cardioprotective effects of compound 1 chronically administered in vivo were evident in spite of the expected gradual removal of the phosphate moiety.
In this study, we further explored the structure activity relationship (SAR) of 1 and its phosphonate analogues and various protected ester derivatives. We have approached the issues of short half-life and low oral bioavailability typical of charged nucleotide derivatives through the synthesis of simple uncharged phosphoester derivatives. The new derivatives were studied in a phenotypic screen in vivo, i.e. in a model of ischemic heart failure produced by permanent ligation of the left coronary artery in mice. The charged and uncharged nucleotides were administered through chronic infusion via an Alzet mini-osmotic pump, and oral bioavailability was not tested. Most of the derivatives tested alleviated the characteristic heart failure in the ischemic heart failure mouse, suggesting this as a viable and structurally broad approach to use of nucleotides for cardioprotection. Data for some derivatives were also available using the previous CSQ mouse model.
Results
Chemical Synthesis
The reference compounds 1–3 (Chart 1, all containing a 2-chloroadenine nucleobase) were synthesized by the reported methods,5 and their previously reported cardioprotective activity in the CSQ mouse model is listed in Table S1 (Supporting Information).5 Novel, charged 5’-phosphates and phosphonate (compound 4, Table 1 and compounds 5 and 6, Tables 2 and S2) and masked (uncharged) nucleotide phosphoesters (7 – 16, Table 1) in the (N)-methanocarba series were synthesized by the methods shown in Schemes 1–4. Modifications of compound 1 include the introduction of deuterium at the 5’ position (4, Table 1) and substitution of S in place of an O atom (5), and we anticipate that these derivatives might improve stability in vivo.8,9 Compound 6 was prepared as the 2-iodo equivalent of reference 2-chloro 5’-phosphonate 2 (Chart 1). The corresponding masked ester nucleotides (7 – 16) of the reference compounds 1–3 were synthesized using the previously described intermediates 17, 24, 30, and 365 (Schemes 1–4). A series of alkyl, i.e. ethyl, isopropyl and t-butyl, masked 5’-O-phosphate ester derivatives (7 – 9) was synthesized to evaluate their effects on the well-characterized cardioprotective effect of 1.10 In addition, various other modifications on the atoms of 5’-O-phosphate group of reference compound 1, such as dideutero (10), thioate (11, 12) and dithioate (13), have been made in the form of diethyl esters. Finally, we also decided to evaluate the cardioprotective ability of ester analogues 14, 15 and 16 of phosphonate derivatives 2, 6 and 3, respectively.
Table 1.
Novel phosphate and phosphonate analogues: structure and effects on in vivo heart function after subcutaneous infusion for 28 days as determined by echocardiography-derived FS in the LAD ligation model of ischemic heart failure in mice.
| Compound or condition |
Structure | FS in %a | n = | Corresponds to compound: |
|---|---|---|---|---|
| Vehicle control | - | 17.33 ± 1.36 | 10 | - |
| P2X4R transgenic mouse | - | 23.69 ± 1.74* | 9 | - |
| Charged nucleotides | ||||
| 1 | 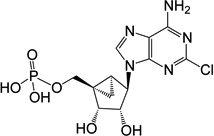 |
22.14 ± 0.68* | 20 | - |
| 4 | 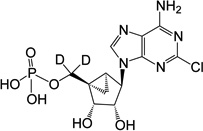 |
23.06 ± 1.14* | 18 | 1 (deuterated) |
| 5 | 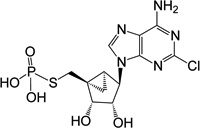 |
ND | 1 (thio) | |
| 6 | 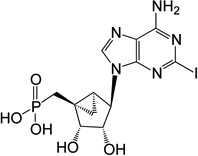 |
ND | 1 (phosphonate, 2-iodo) | |
| Masked nucleotides | Ester derivative of: | |||
| 7 | 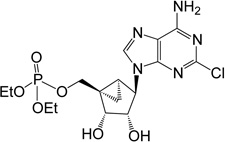 |
24.71 ± 1.42* | 8 | 1 |
| 8 | 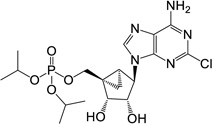 |
25.77 ± 1.10* | 8 | 1 |
| 9 | 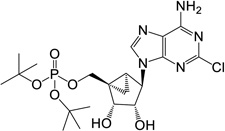 |
24.07 ± 1.64* | 11 | 1 |
| 10 | 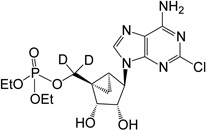 |
21.48 ± 1.38 | 8 | 4 |
| 11 | 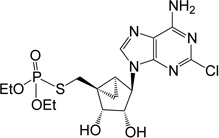 |
24.31 ± 1.45* | 13 | 5 |
| 12 | 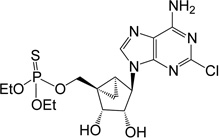 |
ND | 1 (thio) | |
| 13 | 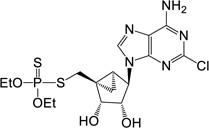 |
24.76 ± 1.32* | 12 | 5 (dithio) |
| 14 | 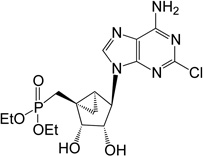 |
ND | 2 | |
| 15 | 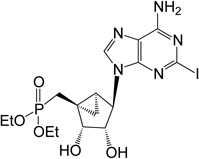 |
23.05 ± 1.45* | 11 | 6 |
| 16 | 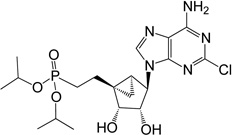 |
ND | 3 | |
at 10 µM, expressed as mean ± SEM.
P<0.05 vs. vehicle for all compounds tested, except compound 10, which displayed P<0.1.
ND – not determined.
Table 2.
Structure and effects on ex vivo heart function of novel phosphate and phosphonate analogues after subcutaneous infusion for 28 days, as determined by isolated working heart preparation in the LAD ligation model of ischemic heart failure in mice. Structures are shown in Table 1.
| Compound | +dP/dt (mmHg/sec)a |
n = |
|---|---|---|
| Vehicle control | 6156 ± 356 | 21 |
| Charged nucleotides | ||
| 1 | 6508 ± 96* | 18 |
| 5 | 6553 ± 435* | 11 |
| 6 | 6581 ± 721* | 8 |
| Masked nucleotides | ||
| 7 | 6711± 726* | 9 |
| 9 | 6681± 528* | 9 |
| 14 | 5946± 337 | 9 |
at 10 µM, expressed as mean ± SEM.
P<0.05 vs. vehicle for all compounds except 14.
ND – not determined.
Scheme 1. Reagents and Conditions.

a) 2M NH3 in i-PrOH, 70 °C, 68%; b) LiBD4, anhydrous THF, reflux, 72%; c) i) Di-t-butylN,N’-diethylphosphoramidite, anhydrous THF, tetrazole; ii) m-chloroperbenzoic acid, 76%; d) diethylchlorophosphate, anhydrous pyridine, rt, 65%; e) Dowex-50 resin, MeOH:H2O (1:1, v/v) 70 °C (4), 55 °C (10, 22), 32% (4), 55% (10), 77% (22); f) LiAlD4, anhydrous THF, 0 °C to rt, 51%, g) diethylchlorophosphate, anhydrous pyridine, rt, 55%.
Scheme 4. Reagents and Conditions.

a) Triphenylphosphine, 6-chloro-2-iodopurine, diisopropyl azodicarboxylate, anhydrous THF, 70%; b) 2M NH3 in i-PrOH, 70 °C, 64%; c) Iodotrimethylsilane, anhydrous CH2Cl2, 49%; d) Dowex-50 resin, MeOH:H2O (1:1, v/v), 55°C, 40% (14), 49% (15).
Synthesis of 2-chloro-5’-dideutero-(N)-methanocarba-adenosine 5’-monophosphate 4 and the corresponding diethyl ester 10 was achieved from previously reported nucleoside 175 and shown in Scheme 1. Initial amination at the C6 position of 2,6-dichloropurine using 2M NH3/i-ProH afforded 6-amino nucleoside 18 in 68% yield. Reduction of the ethyl ester of 18 using LiBD4 in refluxing anhydrous THF provided the 5’-hydroxydideutero nucleoside 19 in 72% yield. The nucleoside 19 was phosphorylated by either with di-t-butyl N,N-diethylphosphoramidite and tetrazole followed by m-chloroperbenzoic acid or with diethylchlorophosphate in pyridine to afford the corresponding di-t-butyl or di-ethyl phosphotriester derivatives 20 and 21, respectively. Both t-butyl groups and acetonide group of 20 were deprotected simultaneously by Dowex-50 resin in a MeOH:H2O mixture with heating at 70 °C to afford the 5’-dideutero monophosphate derivative 4 in 32% yield. On the other hand, diethyl phosphate ester 10 was prepared by chemoselective deprotection of the acetonide group of nucleotide 21 using the same Dowex reaction conditions except heating at 55 °C (Scheme 1). The synthesis of the 5’-dideutero phosphate diethyl ester derivative 10 was also accomplished by an alternative route, in which we used LiAlD4 to reduce the ethyl ester of nucleoside intermediate 18 and incorporate two D atoms (Scheme 1). This route involves initial deprotection of the isopropylidine group of nucleoside 18 to afford 2’,3’-dihydroxy nucleoside 22 in 77% yield. Reduction of 5’-ethyl ester of 22 using LAlD4 in THF afforded the 2’,3’,5’-trihydroxy-5’-dideuteronucleoside 23 in 51% yield. To our surprise, the phosphorylation on this tri-ol nucleoside 23 using diethylchlorophosphate in pyridine yielded the desired 5’-diethylphosphate ester 10 in satisfactory 55% yield (Scheme 1).
Synthesis of various thiophosphate derivatives was achieved from the previously reported common key intermediate 245 and is shown in Scheme 2. 5’-Iodination of the nucleoside 24 using PPh3, I2 and imidazole in THF afforded the 5’-iodo nucleoside 25 in 74% yield. Subsequent deprotection of the acetonide group of 25 using 10% aqueous trifluoroacetic acid afforded the 5’-iodo-2’,3’-dihydroxy nucleoside 26 in 60% yield. Treatment of nucleoside 26 with trisodium thiophosphate in H2O for 3 d afforded the target 5’-S-phosphorothiate 5 in 57% yield. 5’-O-Phosphorothioate diethyl ester 12 was synthesized from nucleoside 24 by initial phosphorylation using O,O′-diethyl chlorothiophosphate in pyridine to get 5’-O-phosphorothioate diethyl ester 27 in very poor yield (13%). Several attempts to improve the yield of this reaction, such as changing the solvent and base, were unsuccessful (data not shown). The target 2’,3’-dihydroxy-5’-O-phosphorothioate diethyl ester 12 was achieved by chemo-selective deprotection of acetonide of compound 27 using acidic Dowex-50 resin at rt. Performing this reaction at 55°C resulted in deprotection of the phosphorothioate moiety to form the corresponding 2’,3’,5’-trihydroxy nucleoside (results not shown). 5’-S-phosphorothioate diester 11 and 5’-S-phosphorodithioate diester 13 were obtained from the common key intermediate 25 using similar reaction conditions. Treating the 5’-iodo nucleoside 25 with O,O-diethyl thiophosphate potassium salt or with O,O′-diethyl dithiophosphate in CH3CN and Et3N as base at rt afforded the phosphorothioate 28 and phosphorodithioate 29, respectively. Deprotection of the acetonide groups of these compounds using Dowex-50 resin resulted in formation of the target 2’,3’-dihydroxy phosphorothioate 11 and phosphorodithioate 13 (Scheme 2), repsectively.
Scheme 2. Reagents and Conditions.

a) Triphenylphosphine, I2, imidazole, anhydrous THF, 74%, b) 10% Aq. trifluoroacetic acid, THF, 65 °C, 60%; c) Trisodium thiophosphate, H2O, 57%; d) O,O′-diethyl chlorothiophosphate, anhydrous pyridine, rt, 13%; e) Dowex-50 resin, MeOH:H2O (1:1, v/v) 55 °C, 12% (12), 48% (11), 59% (13); f) O,O-Diethyl thiophosphate potassium salt, anhydrous CH3CN, Et3N, rt, 52%; g) O,O′-Diethyl dithiophosphate, anhydrous CH3CN, Et3N, rt, 49%;
Synthesis of various phosphate diethyl esters from the previously reported 2,6-dichloropurine nucleoside 305 was carried out as shown in Scheme 3. First, the 5’-hydroxyl of the nucleoside 30 was phosphorylated using diethylchlorophosphate in pyridine at rt to obtain 5’-phosphate diethyl ester 31 in 69% yield. Then, compound 31 was subjected to 2 M NH3 in i-PrOH at 70 °C to give the corresponding 6-amino compound 32 in 65% yield. The target 2’,3’-dihydroxy phosphate diethyl ester 7 was realized upon treating compound 32 with Dowex-50 resin in a mixture of MeOH:H2O (1:1, v/v) at 55°C in 59% yield. Alternately, the corresponding diisopropyl 8 and di-t-butyl 9 esters were synthesized from nucleoside 24. Reaction of nucleoside 24 with diisopropylchlorophosphate in pyridine at rt resulted in the formation of 5’-phosphate diisopropyl ester 33 in 49% yield. Subsequent deprotection of the acetonide of 33 yielded 2’,3’-dihydroxy phosphate diisopropyl ester 8 in 43% yield. To minimize cleaving labile t-butyl groups, we deprotected the acetonide of the previously reported compound 345 using Dowex-50 resin at rt. Even after performing this reaction rt, we observed the formation of substantial amounts of the corresponding monophosphate derivate 1 (Chart 1). As a result we isolated the target 2’,3’-dihydroxy phosphate di-t-butyl ester 8 in very poor yield (9%). The long chain phosphonate diethyl ester 16 was synthesized by deprotection of isopropylidine group of previously reported compound 355 using Dowex-50 resin (Scheme 3).
Scheme 3. Reagents and Conditions.

a) Diethylchlorophosphate, anhydrous pyridine, rt, 69%; b) 2M NH3 in i-PrOH, 70 °C, 65%; c) Dowex-50 resin, MeOH:H2O (1:1, v/v), 55°C (rt for 9), 59% (7), 43% (8), 9% (9), 49% (16); d) Diisopropylchlorophosphate, anhydrous pyridine, rt, 49%.
Synthesis of phosphonate derivatives containing 2-iodo or 2-chloroadenine was performed using the previously reported compound 365 as shown in Scheme 4. Initially, the 6-chloro-2-iodo purine nucleobase was installed at the 1’-position of the (N)-methanocarba sugar 365,6 by a Mitsunobu reaction using PPh3, diisopropyl azodicarboxylate, and 6-chloro-2-iodopurine to generate 6-chloro-2-iodo-purine methanocarba nucleotide 37 in 70% yield. Next, amination at the C6 position of compound 37 using 2M NH3 in i-PrOH afforded the nucleotide 38 in 64% yield. Finally, treating compound 38 either with freshly opened iodotrimethylsilane in CH2Cl2 or with Dowex-50 resin in a mixture of MeOH:H2O resulted in the formation of the target 2’,3’-dihydroxy-5’-phosphonate 6 and corresponding diethyl ester 15, respectively. Similar to the other derivatives, the corresponding 2-chloro derivative of phosphonate diethyl ester 14 was obtained using Dowex-50.
The aqueous solubility of two representative phosphonate esters, diethyl ester 14 and diisopropyl ester 16, was determined to be 60 and 7.25 mM, respectively. Thus, the solubility in this series is substantial and would not be a limiting factor in in vivo application. The stability of 14 and 16 was examined under various conditions (Supporting Information). Compound 16 was stable throughout an incubation of 24 h in aqueous medium at pH 1 and pH 11 (50°C) and in the presence of rat plasma19 or microsomes from mouse liver or rat liver (37°C).20,21 Similarly, compound 14 was stable when incubated in the presence of rat plasma.
Biological Evaluation
Various 5’-phosphate and 5’-phosphonate derivatives were tested for cardioprotection in mice in which heart failure was induced by ligation of the left anterior descending coronary artery (LAD).10
A nucleotide derivative was infused subcutaneously for one month via an Alzet minipump in each mouse, beginning within 24 h of ligation. The in vivo heart function was assessed using echocardiography-derived fractional shortening (FS),5 which is the ratio of the change in the diameter of the left ventricle between the contracted and relaxed states (Table 1). Thus, a higher percentage represents a protection of function. These FS values are not directly comparable to the FS values in the earlier CSQ model (Supporting Information).
The effects of the infused nucleotide derivatives were compared with vehicle-infused control mice and with WT mice overexpressing the P2X4R in the heart. The WT control mice served as the baseline for testing improvement in cardiac function by nucleotide derivatives. The cardiac-specific P2X4R overexpressing transgenic mice are known to be protected in this model of ischemic heart failure and served as a positive control.2 The protective effect was determined as an improvement in left ventricular FS after the coronary ligation. An increase of in vivo echocardiographically-derived FS is an established indication of improved cardiac function in heart failure. Among the compounds tested in the ligation model, only the dideuterated compound 10 did not fully protect as indicated by the absence of a significant increase in FS. There was no difference in infarct size in mice infused with any of the derivatives as compared to vehicle-infused control mice (Fig. 1).
Figure 1. Effects on infarct size following chronic infusion of various agents.

Within 24 hours of LAD ligation, various agents were infused for one month and infarct size determined after measurement of intact heart function as described in Experimental Procedures. As controls, data were also obtained in both vehicle-infused wild type mice and transgenic mice with cardiac specific P2X4 receptor overexpression. There were no differences in the infarct size under any of the drug interventions or in the transgenic mice (one-way ANOVA and post-test comparison, P>0.05).
Further studies were performed using the ex vivo isolated working heart preparation to investigate improvement in cardiac function by selected derivatives. In the same ischemic heart failure model (Table 2), both charged (compounds 5 and 6) and masked (compounds 7 and 9) nucleotides were able to improve function as indicated by a more favorable left ventricular rate of pressure change (+dP/dt). There was no significant change in infarct size for any of these analogues, suggesting that any benefit seen was due to the effect on heart failure progression, consistent with what we previously reported for this class of nucleotides.5 Short chain masked phosphonate 14 did not protect using the ex vivo model to assess protection. This derivative was not evaluated in the LAD model.
Selected analogues were also examined for cardioprotection in the previously examined CSQ mouse model of genetically-induced severe heart failure (Tables S1 and S2, Supporting information). Two-week infusion of the 2-iodo phosphonate derivative 6 (n=5 mice) did not improve FS (Table S2) or prevent LV wall thinning in CSQ-overexpressing mice with heart failure (data not shown). Thus, 2-Cl substitution of the adenine moiety as in phosphonate 2 was essential for activity in this model; substitution with iodo in 6 abolished protection, although the same compound was protective in the ex vivo heart model. A 2-week infusion of thiophosphate 5 (n=5), containing a 5’-thioester, could protect the CSQ mice with a better preservation of LV septal (0.492 ± 0.012 mm) and posterior (0.493 ± 0.016 mm) wall thickness, as compared those obtained in NS-infused (both septal and posterior: 0.450 ± 0.007 mm) CSQ mice (P<0.05, data not shown). Thus, substitution of 5’-oxygen with sulfur was tolerated.
The SAR of the masked (uncharged) nucleotide analogs was explored using the same experimental model. Only some of the ester derivatives of the previously characterized cardioprotective agents 1–3 were shown to act in vivo. These findings implied that there may be an in vivo cleavage step to liberate the charged nucleotide that is known to be active. Among the new derivatives, diisopropyl ester 16 of the longer chain phosphonate (1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonoethylene)-bicyclo[3.1.0]hexane 3 was highly efficacious in the CSQ model. This phosphonate diester resulted in an improved FS as compared to vehicle (Figure S1, Table S2). In mice infused with 16, the LV posterior wall thickness and septal thickness during systole and the LV posterior wall thickness during diastole were greater than those in NS-infused CSQ mice (Figure S2). The activity of diethyl ester 7 was compared to its analogues: isomeric phosphorothioates 11 and 12 and the corresponding dithiophosphate triester 13. The corresponding dideuterated ester 10 was not evaluated in the CSQ mice, but it did not provide statistically significant protection in the LAD model (Table 1).
In order to determine a potential P2X agonist-like activity of charged compounds that were cardioprotective,5 the contractile response to two representative unmasked compounds (5’-monophosphate 1 and 4’-phosphonoethylene derivative 3) was measured in adult WT murine cardiac ventricular myocytes. Both compounds caused a modest increase in contraction shortening (CS), consistent with an agonist-like activity (Fig. 2). Compound 1 was previously shown in murine ventricular myocytes to elicit a P2X-like current.4 Although it is possible that the contractile response to 1 was due to a non-specific effect, an increase of P2X-like current and of CS in ventricular myoctes of the same species (mouse) suggests an agonist effect of this agent. We have not tested these nucleotides in other models of P2X ion channel activity.
Figure 2. Agonist-like effects of compounds 1 and 3 in adult murine cardiac ventricular myocytes.

Ventricular myocytes isolated from 10–12 week old Bl6 wild type mice were studied for contractile response to 1 and 3. CS was measured under control basal condition, drug exposure and washout. Representative CS tracings were shown for both agents (A). Means and standard errors for 1 in 13 myocytes (B) and for 3 in 8 myocytes (C) were shown. Both agents caused a modest increase in C (P<0.05).
Discussion
Initially, we were considering prodrug schemes for in vivo bioavailability of the uncharged phosphate and phosphonate derivatives. Thus, in principle a prodrug could be absorbed intestinally and cleaved to liberate the free drug either in circulation, in the liver, or intracellularly at the site of action. In our case, internalization into the cell is undesirable, because the P2X receptors are ion channels on the surface that are stimulated by extracellular ligands. However, model studies of phosphotriesters and phosphonate diesters have shown that simple esters similar to our uncharged alkyl esters are not readily cleaved in vivo.11–14 Therefore, we examined the stability of representative nucleotide derivatives under biological conditions. Consistently, we were unable to demonstrate any hydrolysis of phosphonate diesters 14 and 16 under various conditions of pH extremes (1 and 11) and the presence of rat plasma or liver microsomes.19–21 Thus, we have no evidence that the protected esters are acting in vivo as prodrugs of the charged derivatives, although we have not examined stability in the presence of cardiac and other tissues. Also, 5’-phosphate 1 was stable to acidic conditions (pH 1.5, 37°C, 24 h) representative of stomach acid (data not shown).
Thus, pronounced in vivo cardioprotective activity of many of the derivatives occurs without evidence of in vivo cleavage. The oral route of administration and the in vivo pharmacokinetics of this series remain to be studied.
The synthetic nucleotide 5’-monophosphate 1 activates a native cardiac P2X receptor, as indicated in electrophysiological experiments with normal cardiac myocytes and those that overexpress CSQ and based on its in vivo ability to improve the heart failure phenotype of these animals. Other agonists of this receptor are 5’-triphosphates,2 and it is not certain if the action of 1 is competitive at the site that recognizes 5’-triphosphates or is allosteric. The rigid carbocyclic ring system contained in this derivative stabilizes nucleotides toward the action of nucleotidases, such as 5’-nucleotidase (CD73), which converts AMP to adenosine.6a
The SAR of the lead molecules was explored, for example with the introduction of a 2-iodoadenine in 6. Also, substitution of monophosphate esters with phosphorothioate groups of various ligands has been found to provide resistance to phosphatase-catalyzed hydrolysis without reducing binding affinity.8 Two sulfur-containing ester derivatives 11 and 13 were protective in the LAD model.
Also, introduction of deuterium in place of hydrogen at strategic locations on labile receptor ligands and other drugs has been shown to increase biological lifetime due to an isotope effect without reducing binding affinity.9 Thus, the 5’-dideutero analogue 4 may prove to be more stable than 1. Curiously, the free phosphate 4 in this series, but not the ester derivative 10, was significantly protective in the LAD model (although 10 was tending toward protection with P<0.1). Further studies will be needed to explore the role of a deuterium isotope effect in the biological activity of these derivatives.
The spacing of the phosphorus relative to the methanocarba ring was important for in vivo activity in the CSQ model. Among phosphonate derivatives, only the longer spacer of 2 carbons in 16 resulted in effective cardioprotection by ester derivatives, even though the precursors or unblocked nucleotides, i.e. 2 and its higher homologue 3, were protective in both cases.5 Thus, both the masked diester 16 and its charged precursor 3 were clearly protective in vivo.
In conclusion, we have chemically modified carbocyclic nucleotide analogues with blocking groups such that the negative charges have been masked. All but one agent evaluated in the LAD model, i.e. the deuterated analogue 4, were able to protect against ischemic heart failure. Using the ex vivo working heart preparation to determine cardiac function in heart failure, only one of the compounds examined, i.e. 14, did not protect. In the CSQ model, in vivo cardioprotective activity was seen with masked nucleotides e.g. 7 and 16, as was observed previously for corresponding unmasked species, e.g. 1 – 3.5 Among the ester derivatives, diethyl (7) and diisopropyl (8) phosphotriesters of were highly protective. Substitution of 2-Cl with iodo reduced protection in the CSQ-overexpressing mouse model. Diisopropyl ester 16 of (1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonoethylene)-bicyclo[3.1.0]-hexane was highly efficacious (CSQ), while lower homologue 1’-phosphonomethylene derivative 14 was inactive. Thus, the uncharged compounds often preserved heart contractile function when compared to their unmasked counterparts in the same models of heart failure, e.g. 8 versus 1 in ischemic heart failure and 16 versus 3 in the CSQ genetic heart failure. Both compounds 1 and 3 appear to have agonist-like effect in adult murine cardiac myocytes, implicating a possible role of cardiac P2X receptors in the protective effect of these two compounds. The lack of difference in infarction size may be multi-factorial, one of which may be an absence of a vascular effect of the compounds. Therefore, these phosphonodiester and phosphotriester derivatives represent potential candidates for the treatment of heart failure and can now be explored in additional models of cardiac failure and cardiomyopathy and in pharmacokinetic/metabolism models.
Experimental Procedures
General methods
All reagents and solvents (regular and anhydrous) were of analytical grade and obtained from commercial suppliers and used without further purification. Compounds 34 and 35 were prepared as described.5,6b Reactions were conducted under an atmosphere of argon whenever anhydrous solvents were used. All reactions were monitored by thin-layer chromatography (TLC) using silica gel coated plates with a fluorescence indicator which were visualized: a) under UV light, b) by dipping in 5% conc. H2SO4 in absolute ethanol (v/v) followed by heating, or c) by dipping in a solution of anisaldehyde:H2SO4 (1:2, v/v) in MeOH followed by heating. Silica gel column chromatography was performed with silica gel (SiO2, 200–400 mesh, 60Å) using moderate air pressure. Evaporation of solvents was carried out under reduced pressure at a temperature below 40 °C. After column chromatography, appropriate fractions were pooled, evaporated and dried at high vacuum for at least 12 h to give the obtained products in high purity. 1H NMR and 31P NMR ascertained sample purity. No corrections in yields were made for solvent of crystallization. 1H NMR and 31P NMR spectra were recorded at 400 MHz and 161.9 MHz, respectively. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane or deuterated solvent as the internal standard (™H: CDCl3 7.26; MeOD-d4 3.31 ppm). Systematic compound names for bicyclic nucleosides are given according to the von Baeyer nomenclature.16 High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine. Observed mass accuracies are those expected on the basis of known performance of the instrument as well as the trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy. Purification of the nucelotide derivatives (4–6) for biological testing was performed by HPLC with a Luna 5µ RP-C18(2) semipreparative column (250 × 10.0 mm; Phenomenex, Torrance, CA) under the following conditions: flow rate of 2 mL/min; 10 mM triethylammonium acetate (TEAA)-CH3CN from 100:0 (v/v) to 70:30 (v/v) in 30 min and isolated in the triethylammonium salt form. Analytical purity of compounds was checked using a Hewlett–Packard 1100 HPLC equipped with Zorbax SB-Aq 5 µm analytical column (50 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 40:60 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 254, 275, and 280 nm. All derivatives tested for biological activity showed >99% purity by HPLC analysis (detection at 254 nm).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[phosphoryloxydideuteromethyl]-2,3-diol-bicyclo[3.1.0]hexane (4)
To a solution containing nucleoside 20 (7.0 mg, 12.8 µmol) in MeOH and H2O (2 mL, 1:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 3 h at 70 °C and the resin removed by filtration. The filtration was then treated with 1 M triethylammonium bicarbonate buffer (1 mL) and lyophilized to dryness. The resulted mixture was purified by purified by semi preparative HPLC (retention time 16.5 min) to get 5'-monophosphate 4 (1.62 mg, 32%) as white solid. ESI-HRMS m/z 392.0493 ([M - H] + (C12H12D2N5O6ClP, calcd 392.0496). 1H NMR (D2O) δ 8.45 (s, 1H), 4.73 (s, 1H), 3.97 (d, 1H, J = 6.5 Hz), 1.72–1.77 (m, 1H), 1.39–1.43 (m, 1H), 0.84–0.90 (m, 1H). 31P NMR (D2O) δ 3.73. Purity >99% by HPLC (retention time: 2.83 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-2,3-(dihydroxy)-1-[monophosporothioate]-bicyclo-[3.1.0]hexane (5)
To the suspension of 5'-iodo nucleoside 26 (3.0 mg, 7.12 µmol) and H2O (0.5 mL), trisodium thiophosphate (10 mg, 55 µmol) was added. After stirring the reaction mixture for 3 d at room temperature under argon atmosphere, reaction mixture was lyophilized and purified by semi preparative HPLC (retention time 19.5 min) to get 5'-monophosphorothioate 5 (1.65 mg, 57%) as a white solid. ESI-HRMS m/z 406.0159 ([M - H]+ (C12H14N5O5SPCl, calcd 406.0165). 1H NMR (D2O) δ 8.39 (s, 1H), 4.62 (s, 1H), 4.63 (s, 1H), 3.97 (d, 1H, J = 6.5 Hz), 3.19–3.26 (m, 1H), 2.79–2.87 (m, 1H), 1.69–1.74 (m, 1H), 1.39–1.43 (m, 1H), 0.84–0.90 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-iodoropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonomethylene)-bicyclo[3.1.0]hexane (6)
Nucleoside 38 (10.0 mg, 0.017 mmol) was coevaporated with anhydrous toluene (3 × 3 mL) and dissolved in anhydrous CH2Cl2 (2 mL). Iodotrimethylsilane (25 µl, 0.17 mmol) was added and after stirring for 17 h, the reaction mixture was cooled to 0 °C followed by the addition of ice-cold H2O (20 mL) and CH2Cl2 (25 mL). The phases were separated and the aqueous phase washed with CH2Cl2 (2 × 35 mL) and diethyl ether (4 × 35 mL). The resulting aqueous phase lyophilized to dryness and purified by HPLC (retention time: 20 min) to afford 6 (4.1 mg, 49%) as a white solid. ESI-HRMS m/z 465.9777 [M - H]−, C12H14IN5O5P−: Calcd. 465.9771); 1H NMR (D2O) δ 8.19 (s, 1H), 4.71 (s, 1H), 4.58 (d, 1H, J = 5.8 Hz), 3.98 (d, 1H, J = 5.8 Hz), 2.17 (t, 1H, 15.5 Hz), 1.90 (t, 1H, 15.5 Hz), 1.68–1.74 (m, 1H), 1.35–1.42 (m, 1H), 0.88–0.94 (m, 1H). 31P NMR (D2O) δ 25.39. Purity >99% by HPLC (retention time: 5.93 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[(diethylphosphate) methyl]-2,3-(dihydroxy)-bicyclo[3.1.0]hexane (7)
To a solution containing 32 (7.0 mg, 14.3 µmol) in MeOH:H2O (2 mL, 1:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 3 h at 55°C and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (0–10% MeOH in CH2Cl2, v/v) to afford nucleoside 7 (3.8 mg, 59%) as a white solid. Rf = 0.5 (8% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 448.1156 ([M + H] + (C16H23N5O6ClP, calcd 448.1153). 1H NMR (MeOD-d4) δ 8.23 (s, 1H), 4.83 (s, 1H), 4.75 (d, 1H, J = 7.2 Hz), 4.79 (d, 1H, J = 7.2 Hz), 4.63–4.69 (m, 1H), 4.13–4.23 (m, 4H), 3.94–4.05 (m, 2H), 1.76–1.82 (m, 1H), 1.66–1.70 (m, 1H), 1.37 (t, 6H,J = 7.2 Hz), 0.91–0.95 (m, 1H). 31P NMR (MeOD-d4) δ −1.25. Purity >99% by HPLC (retention time: 2.52 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[(diisopropylphosphate) methyl]-2,3-(dihydroxy)-bicyclo[3.1.0]hexane (8)
To a solution containing 33 (4.0 mg, 14.3 µmol) in MeOH:H2O (2 mL, 1:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 3 h at 55°C and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (0–10% MeOH in CH2Cl2, v/v) to afford nucleoside 8 (1.63 mg, 43%) as a white solid. Rf = 0.4 (6% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 476.1474 ([M + H] + (C18H28N5O6ClP, calcd 476.1466). 1H NMR (MeOD-d4) δ 8.23 (s, 1H), 4.83 (s, 1H), 4.74 (d, 1H, J = 7.2 Hz), 4.63–4.71 (m, 3H), 3.93–3.99 (m, 2H), 1.75–1.81 (m, 1H), 1.65–1.70 (m,1H), 1.35–1.41 (m, 12H), 0.88–0.96 (m, 1H). 31P NMR (CDCl3) δ −3.03. Purity >99% by HPLC (retention time: 4.69 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-2,3-(dihydroxy)-1-[(t-butylphosphate)methyl]-bicyclo[3.1.0]hexane (9)
To a solution containing 34 (15.0 mg, 36.8 µmol) in MeOH:H2O (3 mL, 2:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 3 h at rt and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (0–6% MeOH in CH2Cl2, v/v) to afford nucleoside 9 (1.61 mg, 9%) as a white solid. Rf = 0.4 (6% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 504.1780 ([M + H] + (C20H32N5O6ClP, calcd 504.1779). 1H NMR (CDCl3) δ 7.89 (s, 1H), 5.83 (s, 2H), 5.47 (s, 1H), 5.03 (d, 1H, J = 6.5 Hz), 4.83 (s, 1H), 4.72 (t, 1H, J = 11.6 Hz), 4.12 (d, 1H, J = 6.5 Hz), 3.92 (t, 1H, J = 11.6 Hz), 3.65 (s, 1H), 1.65–1.70 (m, 1H), 1.55 (s, 18H), 0.90–1.02 (m, 1H). 31P NMR ((MeOD-d4) δ −10.29. Purity >99% by HPLC (retention time: 5.30 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-2,3-(dihydroxyl)-1-[(di-ethylphosphate)dideuteromethyl]bicyclo[3.1.0]hexane (10)
Method A
To a solution containing 21 (8.0 mg, 16.3 µmol) in MeOH:H2O (2 mL, 1:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 2 h at 55°C and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (0–7% MeOH in CH2Cl2, v/v) to afford nucleoside 10 (4.2 mg, 55%) as a white solid.
Method B
Nucleoside 23 (2.5 mg, 7.9 µmol) was coevaporated with anhydrous toluene (3 × 2 mL), dissolved in anhydrous pyridine (0.5 mL) and diethylchlorophosphate (6 µL, 39.8 µmol) was added to this. After stirring at rt for 8 h, the reaction mixture was evaporated to dryness. The resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford nucleoside 10 (1.9 mg, 55%) as a white solid. Rf = 0.3 (6% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 450.1263 ([M + H] + (C16H21D2N5O6Cl·H+, calcd 450.1278). 1H NMR (MeOD-d4) δ 8.14 (s, 1H), 5.35 (d, 1H, J = 6.8 Hz), 4.96 (s, 1H), 4.77 (d, 1H, J = 7.2 Hz), 4.11–4.23 (m, 4H), 1.75–1.80 (m, 1H), 1.53 (s, 3H), 1.33–1.39 (m,6H), 1.27 (s, 3H), 1.21–1.24 (m, 1H), 1.10–1.16 (m, 1H). 31P NMR (D2O) δ 1.22. Purity >99% by HPLC (retention time: 2.92 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[O,O-diethyl S-methyl phosphorothioate]- 2,3-(dihydroxyl)-bicyclo-[3.1.0]hexane (11)
To a solution containing nucleoside 28 (7.0 mg, 13.9 µmol) in MeOH and H2O (2 mL, 1:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 2 h at 55 °C and the resin removed by filtration. The filtrate was then evaporated to dryness and the resulted mixture was purified by purified by silica gel column chromatography (0–7% MeOH in CH2Cl2, v/v) to afford 2’,3’ dihydroxy, 5’-phosphorothioate 11 (3.1 mg, 48%). Rf = 0.4 (7% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 464.0925 ([M+H]+ (C16H24N5O5PSCl·H, calcd 464.0924). 1H NMR (MeOD-d4) δ 8.25 (s, 1H), 4.78 (d, 1H, J = 6.9 Hz), 4.75 (s, 1H), 4.14–4.22 (m, 4H), 4.07 (d, 1H, J = 6.9 Hz), 3.45 (t, 1H, J = 12.9 Hz), 3.24–3.30 (m, 1H), 1.72–1.76 (m, 1H), 1.59–1.63 (m, 1H), 1.30–1.40 (m, 6H), 0.90–0.96 (m, 1H). 31P NMR (MeOD-d4) δ 28.98. Purity >99% by HPLC (retention time: 3.88 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[O,O-diethyl O-methyl phosphorothioate]- 2,3-(O-isopropylidine)-bicyclo-[3.1.0]hexane (12)
To a solution containing nucleoside 27 (3.0 mg, 5.9 µmol) in MeOH and H2O (1 mL, 1:1, v/v) was added Dowex-50 resin (~30 mg). The mixture was stirred for 6 h at rt and the resin removed by filtration. The filtrate was then evaporated to dryness and the resulted mixture was purified by purified by silica gel column chromatography (0–7% MeOH in CH2Cl2, v/v) to afford 2’,3’ dihydroxy, 5’-diethyl phosphorothioate 12 (0.31 mg, 12%). Rf = 0.4 (7% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 464.0926 ([M+H]+ (C16H24N5O5PSCl+ calcd 464.0924). 1H NMR (MeOD-d4) δ 8.31 (s, 1H), 4.84 (s, 1H), 4.69–4.75 (m, 2H), 4.13–4.28 (m, 4H), 3.87–3.93 (m, 2H), 1.76–1.81 (m, 1H), 1.67–1.72 (m, 1H), 1.30–1.40 (m, 6H), 0.90–0.96 (m, 1H). 31P NMR (MeOD-d4) δ 67.9 Purity >99% by HPLC (retention time: 6.41 min).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[O,O-diethyl S-methyl phosphorodithioate]- 2,3-(dihydroxyl)-bicyclo-[3.1.0]hexane (13)
To a solution containing nucleoside 29 (6.0 mg, 11.5 µmol) in MeOH and H2O (2 mL, 1:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 3 h at 45 °C and the resin removed by filtration. The filtrate was then evaporated to dryness and the resulted mixture was purified by purified by silica gel column chromatography (0–5% MeOH in CH2Cl2, v/v) to afford 2’,3’ dihydroxy, 5’-phosphorothioate 13 (3.27 mg, 59%). Rf = 0.3 (5% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 480.0703 ([M+H]+ (C16H24N5O4PS2Cl·H, calcd 480.0696). 1H NMR (MeOD-d4) δ 8.27 (s, 1H), 4.76 (s, 1H), 4.73 (d, 1H, J = 7.2 Hz), 4.09–4.22 (m, 4H), 4.01 (d, 1H, J = 6.9 Hz), 3.56 (t, 1H, J = 13.6 Hz), 3.26 (t, 1H, J = 13.6 Hz), 1.72–1.76 (m, 1H), 1.59–1.63 (m, 1H), 1.30–1.40 (m, 6H), 0.90–0.94 (m, 1H). 31P NMR (MeOD-d4) δ 95.25. Purity >99% by HPLC (retention time: 7.54 min).
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-chloropurin-9-yl)-2’,3’-(hydroxy)-bicyclo[3.1.0]hexane phosphonate (14)
To a solution containing 39 (25 mg, 0.053 mmol) in MeOH (3 mL) and water (3 mL) was added Dowex-50 resin (~100 mg). The mixture was stirred for 3 h at 70°C and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (10% MeOH in EtOAc, v/v) to afford nucleoside 14 (8.3 mg, 40%) as a white solid. Rf = 0.4 (10% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 432.1197 [M + H]+, C16H24ClN5O5P+: Calcd. 432.1204); 1H NMR (MeOD-d4) δ 8.31 (s, 1H), 4.79 (s, 1H), 4.76 (d, 1H, J = 7.1 Hz), 4.10–4.19 (m, 4H), 3.98 (d, J = 7.1 Hz), 2.44–2.54 (m, 1H), 2.27–2.36 (m, 1H), 1.72–1.78 (m, 1H), 1.51–1.57 (m, 1H), 1.28–1.35 (m, 6H), 0.80–0.89 (m, 1H). 31P NMR (MeOD-d4) δ 30.86. Purity >99% by HPLC (retention time: 5.17 min).
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-iodopurin-9-yl)-2’,3’-(dihydroxy)-bicyclo[3.1.0]hexane phosphonate (15)
To a solution containing 38 (6.0 mg, 11.2 µmol) in MeOH:H2O (2 mL, 1:1, v/v) was added Dowex-50 resin (~50 mg). The mixture was stirred for 3 h at 70°C and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (0–8% MeOH in CH2Cl2, v/v) to afford nucleoside 15 (4.5 mg, 49%) as a white solid. Rf = 0.2 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 524.0562 [M + H]+, C16H24IN5O5P+: Calcd. 524.0560); 1H NMR (MeOD-d4) δ 8.25(s, 1H), 4.77–4.81 (m, 2H), 4.10–4.19 (m, 4H), 3.98 (d, 1H, J = 6.8 Hz), 2.35–2.52 (m, 1H), 1.68–1.73 (m, 1H), 1.49–1.52 (m, 1H), 1.29–1.35 (m, 6H), 0.85–0.91 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-chloropurin-9-yl)-1’-[diisopropyl-(E)-ethenylphosphonate]-2,3-(dihydroxy)-bicyclo-[3.1.0]-hexane (16)
To a solution containing 35 (4.0 mg, 7.7 µmol) in MeOH:H2O (1 mL, 1:1, v/v) was added Dowex-50 resin (~25 mg). The mixture was stirred for 3 h at 55 °C and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (0–10% MeOH in CH2Cl2, v/v) to afford nucleoside 16 (1.8 mg, 49%) as a white solid. Rf = 0.3 (10% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 474.1679 [M + H]+, C19H30ClN5O5P·H+: Calcd. 474.1673); 1H NMR (MeOD-d4) δ 8.06 (s, 1H), 4.76 (d, 1H, J = 6.9 Hz), 4.65–4.71 (m, 3H), 4.07 (d, 1H, J = 6.9 Hz), 3.67–3.72 (m, 1H), 3.55–3.59 (m, 1H), 2.10–2.16 (m, 2H), 1.80–2.03 (m, 2H), 1.47–1.52 (m, 1H), 1.28–1.41 (m, 13H), 0.66–0.72 (m, 1H). 31P NMR (MeOD-d4) δ 31.22. Purity >99% by HPLC (retention time: 4.69 min).
Ethyl (1’S,2’R’3’S,4’R,5’S)-4-(6-Amino-2-chloropurin-9-yl]-2,3-O-(isopropylidene)-bicyclo[3.1.0]hexanecarboxylate (18)
Nucleoside 17 (53 mg, 0.13 mmol) was treated with 2M NH3 in i-PrOH (5 mL) and heated to 70 °C. After stirring the reaction for 16 h, reaction mixture evaporated to dryness. The resulting residue purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford nucleoside 18 (35 mg, 68%) as a white solid. Rf = 0.4 (5% MeoH in CH2Cl2, v/v). ESI-HRMS m/z 394.1282 ([M + H] + (C17H21N5O4Cl, calcd 394.1282). 1H NMR (MeOD-d4) δ 8.09 (s, 1H), 5.85 (d, 1H, J = 7.1 Hz), 4.98 (s, 1H), 4.81 (d, 1H, J = 7.1 Hz), 4.20–4.26 (m, 1H), 2.23–2.28 (m, 1H), 1.64–1.67 (m, 1H), 1.52 (s, 3H), 1.34 (t, 3H, J = 7.2 Hz), 1.20–1.29 (m, 4H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[hydroxydeuteromethyl]-2,3-(O-isopropylidine)bicyclo-[3.1.0]hexane (19)
Nucleoside 18 (9.0 mg, 23 µmol) was coevaporated with anhydrous toluene (3 × 10 mL), dissolved in anhydrous THF (10 mL). LiBD4 (3 mg, 115 µmol) was added and after stirring the reaction mixture for 4 h at 70 °C, it is cooled to room temperature and quenched with slow addition of MeOH (3 mL). The resulting reaction mixture was evaporated to dryness, and purified by silica gel column chromatography (0–8% MeOH in CH2Cl2, v/v) to afford nucleoside 19 (6.0 mg, 72%) as a white solid. Rf = 0.4 (10% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 354.1291 ([M + H] + (C15H17D2N5O3Cl, calcd 354.1302). 1H NMR (MeOD-d4) δ 8.26 (s, 1H), 5.37 (d, 1H, J = 6.7 Hz), 4.95 (s, 1H), 4.70 (d, 1H, J = 7.2 Hz), 1.67–1.73 (m, 1H), 1.51 (s, 3H), 1.25 (s, 3H), 1.10–1.15 (m, 1H), 0.94–1.00 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-2,3-(Oisopropylidene)-1-[(di-tert-butylphosphate)dideuteromethyl]-bicyclo[3.1.0]hexane (20)
Nucleoside 19 (6.0 mg, 17 µmol) was coevaporated with anhydrous toluene (3 × 10 mL), dissolved in anhydrous THF (1 mL). Di-t-butyl-N,N’-diethylphosphoramidite (24 µL, 85 µmol) and tetrazole (12 mg, 169 µmol) were added. After stirring at rt for 4 h, the reaction mixture was cooled to −70 °C followed by the addition of m-chloroperbenzoic acid (25 mg, 77%). The reaction mixture was warmed to 0 °C and allowed to stir for 15 min followed by the addition of triethylamine (0.5 mL). The reaction mixture was evaporated to dryness, and the resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford nucleoside 20 (7.0 mg, 76%) as a white solid. Rf = 0.3 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 546.2234 ([M + H] + (C23H34D2N5O6Cl, calcd 546.2217). 1H NMR (MeOD-d4) δ 8.15 (s, 1H), 5.34 (d, 1H, J = 6.8 Hz), 4.98 (s, 1H), 4.77 (d, 1H, J = 7.2 Hz), 1.74–1.77 (m, 1H), 1.48–1.55 (m, 21H), 1.25 (s, 3H), 1.21–1.24 (m, 1H), 1.09–1.13 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-2,3-(Oisopropylidene)-1-[(di-ethylphosphate)dideuteromethyl]-bicyclo[3.1.0]hexane (21)
Nucleoside 19 (20 mg, 54 µmol) was coevaporated with anhydrous toluene (3 × 10 mL), dissolved in anhydrous pyridine (2 mL) and diethylchlorophosphate (24 µL, 161 µmol) was added to this. After stirring at rt for 8 h, the reaction mixture was diluted with EtOAc (25 mL) and washed with aqueous NaHCO3 (2 × 10 mL). The aqueous phase was back extracted with EtOAc (1 × 10 mL) and combined organic phase was evaporated to dryness. The resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford nucleoside 21 (17 mg, 65%) as a white solid. Rf = 0.5 (10% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 489.1537 ([M + H] + (C19H27D2N5O6Cl, calcd 489.1529). 1H NMR (MeOD-d4) δ 8.22 (s, 1H), 4.82 (s, 1H), 4.74 (d, 1H, J = 6.8 Hz), 4.15–4.23 (m, 4H), 3.96 (d, 1H, J = 6.7 Hz), 1.76–1.82 (m, 1H), 1.65–1.70 (m, 3H), 1.37 (t, 6H, J = 7.1 Hz), 0.89–0.95 (m, 1H). 31P NMR (MeOD-d4) δ 1.30.
Ethyl (1’S,2’R’3’S,4’R,5’S)-4-(6-Amino-2-chloropurin-9-yl]-2,3-(dihydroxyl)-bicyclo[3.1.0]hexanecarboxylate (22)
To a solution containing 18 (19 mg, 48.3 µmol) in MeOH:H2O (4 mL, 1:1, v/v) was added Dowex-50 resin (~100 mg). The mixture was stirred for 2 h at 55°C and the resin removed by filtration. Filtrate was evaporated to dryness and resulting crude purified by silica gel column chromatography (0–8% MeOH in CH2Cl2, v/v) to afford nucleoside 22 (13.1 mg, 77%) as a white solid. Rf = 0.6 (10% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 354.0957 ([M + H] + (C14H16N5O4Cl·H+, calcd 354.0969). 1H NMR (MeOD-d4) δ 8.01 (s, 1H), 5.23 (d, 1H, J = 6.7 Hz), 4.77 (s, 1H), 4.25 (q, 2H, J = 7.1 Hz), 4.10 (d, 1H, J = 6.7 Hz), 2.14–2.20 (m, 1H), 1.84–1.86 (m, 1H), 1.60–1.65 (m, 2H), 1.32 (t, 3H, J = 7.1 Hz).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-2,3-(dihydroxyl)-1-[hydroxydeuteromethyl]-bicyclo-[3.1.0]hexane (23)
Nucleoside 22 (6.0 mg, 16.9 µmol) was coevaporated with anhydrous toluene (3 × 10 mL), dissolved in anhydrous THF (3 mL) and cooled to 0 °C. LiAlD4 (3 mg, 115 µmol) was added to ice cold reaction mixture and it is allowed to warm up to room temperature. After stirring the reaction mixture for 6 h and it was quenched with slow addition of MeOH (3 mL). The resulting reaction mixture was evaporated to dryness, and purified by silica gel column chromatography (0–10% MeOH in CH2Cl2, v/v) to afford nucleoside 23 (2.7 mg, 51%) as a white solid. Rf = 0.4 (10% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 314.0982 ([M + H]+ (C12H12D2N5O3Cl, calcd 314.0989). 1H NMR (CDCl3) δ 7.82 (s, 1H), 5.92 (s, 2H) 5.60 (d, 1H, J = 6.4 Hz), 4.78 (s, 1H), 4.68 (d, 1H, J = 7.2 Hz), 3.87 (s, 1H), 1.72–1.76 (m, 1H), 1.70 (s, 2H), 1.56 (s, 3H), 1.27 (s, 3H), 1.13–1.18 (m, 1H), 0.96–1.02 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[iodomethyl]-2,3-(O-isopropylidine) bicyclo-[3.1.0]hexane (25)
Nucleoside 24 (45 mg, 0.128 mmol) was coevaporated with anhydrous toluene (3 × 10 mL) and dissolved in anhydrous THF (3 mL). I2 (66 mg, 0.256 mmol), triphenylphosphine (68 mg, 0.256 mmol), and imidazole (18 mg, 0.256 mmol) were added. After stirring for 17 h, the reaction mixture was diluted with EtOAc (30 mL) and washed with saturated aqueous Na2S2O3 (2 ×15 mL). The phases were separated and aqueous phase was extracted with EtOAc (3 × 25 mL). The combined organic phase was evaporated to dryness, and the resulting residue was purified by silica gel column chromatography (0–80% EtOAc in petroleum ether, v/v) to afford 5’-iodo nucleoside 25 (41 mg, 74%). Rf = 0.4 (5% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 462.0205 ([M + H] + (C15H19N5O2Cl, calcd 462.0194). 1H NMR (CDCl3) δ 8.09 (s, 1H), 6.11 (s, 2H), 5.32 (d, 1H, J = 6.9 Hz), 4.89 (s, 1H), 4.70 (d, 1H, J = 6.9 Hz), 3.63–3.69 (d, 1H, J = 10.5 Hz), 3.53–3.56 (d, 1H, J = 10.5 Hz), 1.65–1.74 (m, 1H), 1.56 (s, 3H), 1.45–1.50 (m, 1H), 1.24–1.32 (s, 4H), 1.10–1.15 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-2,3-(dihydroxy)-1-[iodomethyl]bicyclo-[3.1.0]hexane (26)
5-Iodo nucleoside 25 (106 mg, 0.229 mmol) was dissolved in THF (1 mL) followed by the addition of 10% aqueous trifluoroacetic acid (3.5 mL, v/v). After stirring the reaction mixture at 65 °C for 15 h, reaction mixture was evaporated to dryness and the resulting residue was purified by silica gel column chromatography (0–80% EtOAc in petroleum ether, v/v) to afford 2’,3’-dihydroxy-5’-iodo nucleoside 26 (41 mg, 60%). Rf = 0.3 (10% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 421.9883 ([M + H] + (C12H14N5O2ClI, calcd 421.9881). 1H NMR (MeOD-d4) δ 8.41 (s, 1H), 4.75 (dd, 1H, J = 1.8 Hz), 4.72 (s, 1H), 4.10 (dt, 1H, J = 8 Hz, 2.8 Hz), 3.87–3.91 (d, 1H, J = 10.5 Hz), 3.46–3.52 (d, 1H, J = 10.5 Hz), 1.91–1.95 (m, 1H), 1.671.72 (m, 1H), 1.04–1.09 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[O,O-diethyl O-methyl phosphorothioate]- 2,3-(O-isopropylidine)-bicyclo-[3.1.0]hexane (27)
Nucleoside 24 (18 mg, 51.2 µmol) was dissolved in anhydrous pyridine (3 mL) and O,O′-diethyl chlorothiophosphate (24 µL, 153 µmol) was added to this. After stirring at room temperature for 17 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–5% MeOH in CH2Cl2, v/v) to afford 5’-diethyl phosphorothioate nucleoside 27 (3.2 mg, 13%). ESI-HRMS m/z 504.1234 ([M + H] + (C19H28ClN5O5P, calcd 504.1237). Rf = 0.3 (5% MeoH in CH2Cl2, v/v). 1H NMR (MeOD-d4) δ 8.25 (s, 1H), 5.35 (d, 1H, J = 6.2 Hz), 4.94 (s, 1H), 4.67 (d, 1H, J = 6.2 Hz), 4.06–4.14 (m, 4H), 3.96 (d, 1H, J = 11.5 Hz), 3.63 (d, 1H, J = 11.5 Hz), 1.66–1.71 (m, 1H), 1.51 (s, 3H), 1.30–1.40 (m, 6H), 1.27 (s, 3H), 1.10–1.16 (m, 1H), 0.94–0.99 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[O,O-diethyl S-methyl phosphorothioate]-2,3-(O-isopropylidine) bicyclo-[3.1.0]hexane (28)
5’-iodo nucleoside 25 (11 mg, 0.024 mmol) was dissolved in anhydrous CH3CN (2 mL), O,O-Diethyl thiophosphate potassium salt (24.8 mg, 0.12 mmol) and Et3N (34 µL, 0.24 mmol) were added. After stirring the reaction at room temperature for 22 h, reaction mixture was evaporated to dryness and the resulting crude residue was purified by silica gel column chromatography (0–5% MeOH in CH2Cl2, v/v) to afford 5’-phosphorothioate 28 (7.3 mg, 52%). Rf = 0.4 (5% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 504.1235 ([M + H] + (C19H27N5O5SClP·H, calcd 504.1237). 1H NMR (MeOD-d4) δ 8.14 (s, 1H), 5.32 (d, 1H, J = 7.2 Hz), 4.91 (s, 1H), 4.81 (d, 1H, J = 7.2 Hz), 4.16–4.27 (m, 4H), 3.74 (t, 1H, J = 13.5 Hz), 2.93–3.01 (m, 1H), 1.72–1.76 (m, 1H), 1.53 (s, 3H), 1.30–1.40 (m, 6H), 1.27 (s, 3H), 1.19–1.23 (m, 1H), 1.06–1.11 (m, 1H). 31P NMR (MeOD-d4) δ 28.91.
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-purin-9-yl)-1-[O,O-diethyl S-methyl phosphorodithioate]-2,3-(O-isopropylidine) bicyclo-[3.1.0]hexane (29)
5’-iodo nucleoside 25 (11.0 mg, 0.024 mmol) was dissolved in anhydrous CH3CN (2 mL), O,O′-Diethyl dithiophosphate (19.9 µL, 0.12 mmol) and Et3N (34 µL, 0.24 mmol) were added. After stirring the reaction at room temperature for 22 h, reaction mixture was evaporated to dryness and the resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford 5’-phosphorodithioate 29 (6.8 mg, 49%). Rf = 0.4 (5% MeOH in CH2Cl2, v/v). ESI-HRMS m/z 520.1024 ([M + H] + (C19H27N5O4S2ClP·H, calcd 520.1009). 1H NMR (MeOD-d4) δ 8.15 (s, 1H), 5.30 (d, 1H, J = 7.2 Hz), 4.90 (s, 1H), 4.80 (d, 1H, J = 7.2 Hz), 4.11–4.24 (m, 4H), 3.69 (t, 1H, J = 13.5 Hz), 3.06 (t, 1H, J = 13.5 Hz), 1.69–1.75 (m, 1H), 1.53 (s, 3H), 1.30–1.38 (m, 6H), 1.27 (s, 3H), 1.19–1.23 (m, 1H), 1.06–1.11 (m, 1H). 31P NMR (MeOD-d4) δ 94.27
(1’S,2’R,3’S,4’R,5’S)-4-(2,6-Dichloro-9H-purin-9-yl)-1-[(diethylphosphate) methyl]-2,3-(O-isopropylidene)-bicyclo[3.1.0]hexane (31)
Nucleoside 30 (20 mg, 54 µmol) was coevaporated with anhydrous toluene (3 × 10 mL), dissolved in anhydrous pyridine (2 mL) and diethylchlorophosphate (24 µL, 161 µmol) was added to this. After stirring at rt for 17 h, the reaction mixture was diluted with EtOAc (25 mL) and washed with aqueous NaHCO3 (2 × 10 mL). The aqueous phase was back extracted with EtOAc (1 × 10 mL) and combined organic phase was evaporated to dryness. The resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford nucleoside 31 (19 mg, 69%) as a white solid. Rf = 0.5 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 507.0963 ([M + H] + (C19H26N4O6Cl2P, calcd 507.0967). 1H NMR (CDCl3) δ 8.10 (s, 1H), 5.35 (d, 1H, J = 7.2 Hz), 5.06 (s, 1H), 4.62 (d, 1H, J = 7.2 Hz), 4.43–4.50 (m, 1H), 4.14–4.30 (m, 5H), 1.72–1.77 (m, 1H), 1.57 (s, 3H), 1.35–1.43 (m, 6H), 1.27–1.33 (m, 1H), 1.26 (s, 3H), 1.11–1.16 (m, 1H). 31P NMR (CDCl3) δ −1.30.
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[(diethylphosphate) methyl]-2,3-(O-isopropylidene)-bicyclo[3.1.0]hexane (32)
Nucleoside 31 (17 mg, 33.5 µmol) was dissolved in 2M NH3/i-PrOH (5 mL). After stirring at 70 °C for 17 h, the reaction mixture was evaporated to dryness and resulting crude residue was purified by silica gel column chromatography (0–6% MeOH in CH2Cl2, v/v) to afford nucleoside 32 (10 mg, 65%). Rf = 0.5 (8% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 488.1466 ([M + H] + (C19H27N5O6ClP, calcd 488.1466). 1H NMR (MeOD-d4) δ 8.14 (s, 1H), 5.35 (d, 1H, J = 7.2 Hz), 4.96 (s, 1H), 4.79 (d, 1H, J = 7.2 Hz), 4.58–4.62 (m, 1H), 4.12–4.21 (m, 4H), 4.02–4.08 (m, 2H), 1.75–1.79 (m, 1H), 1.53 (s, 3H), 1.29–1.39 (m, 6H), 1.27–1.33 (m, 4H), 1.11–1.16 (m, 1H). 31P NMR (MeOD-d4) δ −1.34.
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[(diisopropylphosphate) methyl]-2,3-(O-isopropylidene)-bicyclo[3.1.0]hexane (33)
Nucleoside 24 (15 mg, 38 µmol) was coevaporated with anhydrous toluene (3 × 10 mL), dissolved in anhydrous pyridine (2 mL) and diisopropyl chlorophosphate (90 µL, 190 µmol) was added to this. After stirring at rt for 17 h, the reaction mixture was diluted with EtOAc (25 mL) and washed with aqueous NaHCO3 (2 × 10 mL). The aqueous phase was back extracted with EtOAc (1 × 10 mL) and combined organic phase was evaporated to dryness. The resulting crude residue was purified by silica gel column chromatography (0–4% MeOH in CH2Cl2, v/v) to afford nucleoside 33 (9.6 mg, 49%) as a white solid. Rf = 0.3 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 516.1790 ([M + H] + (C21H32N5O6ClP, calcd 516.1779). 1H NMR (CDCl3) δ 8.08 (s, 1H), 6.16 (s, 2H), 5.33 (d, 1H, J = 6.9 Hz), 5.01 (s, 1H), 4.65–4.73 (m, 2H), 4.60 (d, 1H, J = 7.2 Hz), 4.42–4.48 (m, 1H), 4.09–4.15 (m, 5H), 1.69–1.75 (m, 1H), 1.57 (s, 3H), 1.33–1.42 (m, 12H), 1.22–1.29 (m, 4H), 1.07–1.12 (m, 1H). 31P NMR (CDCl3) δ −2.65.
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(6-chloro-2-iodo-purin-9-yl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane phosphonate (37)
Diisopropyl azodicarboxylate (86 µL, 0.44 mmol) was added at rt to a mixture of triphenylphosphine (115 mg, 0.44 mmol) and 6-chloro-2-iodopurine (122 mg, 0.44 mmol) in anhydrous THF (4 mL). After stirring for 45 min, a solution of the compound 36 (70 mg, 0.22 mmol) in THF (4 mL) was added to the mixture. After stirring for 36 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–3% MeOH in CH2Cl2, v/v) to afford nucleoside 37 (89 mg, 70%) as a white solid. Rf = 0.5 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 583.0374 [M + H]+, C19H26ClIN4O5P +: Calcd. 583.0373); 1H NMR (CDCl3) δ 8.64 (s, 1H), 5.39 (d, 1H, J = 7.5 Hz), 5.07 (s, 1H), 4.64 (d, 1H, J = 7.5 Hz), 4.15–4.21 (m, 4H), 2.41 (t, 1H, J = 16.0 Hz), 2.11–2.22 (m, 1H), 1.73–1.79 (m, 1H), 1.59–1.64 (m, 1H), 1.54 (s, 3H), 1.35 (t, 3H, J = 7.1 Hz), 1.28 (t, 3H, J = 7.1 Hz), 1.25 (s, 3H), 1.04–1.09 (m, 1H).
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-iodopurin-9-yl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane phosphonate (38)
Nucleoside 37 (80 mg, 0.14 mmol) was treated with 2M NH3 in i-PrOH (8 mL) and the mixture was heated to 70 °C and stirred for 17 h. The reaction mixture was evaporated to dryness, and the resulting residue was purified by silica gel column chromatography (0–6% MeOH in CH2Cl2, v/v) to afford nucleoside 38 (48 mg, 64%) as a white solid. Rf = 0.4 (7% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 564.0873 [M + H]+, C19H28IN5O5P+: Calcd. 564.0856); 1H NMR (CDCl3) δ 8.15(s, 1H), 5.74 (s, 2H), 5.34 (d, 1H, J = 6.1 Hz), 4.94 (s, 1H), 4.64 (d, 1H, J = 6.1 Hz), 4.12–4.19 (m, 4H), 2.19–2.42 (m, 2H), 1.69–1.74 (m, 1H), 1.55 (s, 3H), 1.33 (t, 3H, J = 7.1 Hz), 1.26 (t, 3H, J = 7.1 Hz), 1.22 (s, 3H), 1.18–1.21 (m, 1H), 1.03–1.09 (m, 1H).
Determination of aqueous solibility of selected phosphonate triesters
To 5 mg of solid compound 14 was added water in a drop-wise fashion with agitation at room temperature to prepare a saturated solution, i.e. with solid remaining. Total volume of water added was 50 µL. The solution was allowed to stand at room temperature for 1 h, and then it was centrifuged. The supernatant was removed and lyophilized. The amount of solid obtained was 1.3 mg, providing an aqueous solubility of 60 mM. A similar procedure was used with 35 mg of compound 16 (2 mL water added, with 6.75 mg remaining after lyophilization) to provide an aqueous solubility of 7.25 mM.
Biological evaluation
Post-infarction ischemic heart failure model
An ischemic heart failure model was carried out by permanent ligation of the LAD in mice using a procedure similar to that described previously.17 Adult 12-week old WT (BL6) mice or transgenic mice with cardiac-specific overexpression of the P2X4R were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) intraperitoneally. After endotracheal intubation and left intercostal thoracotomy, myocardial Infarction was produced by ligating the LAD with an 8-0 nylon suture within 2 mm below the edge of left atrium near the origin of the artery. All animal procedures were performed according to guidelines of and approved by the University of Connecticut School of Medicine Review Board.
Measurements of intact heart function by in vivo echocardiography or by ex vivo working heart model and infarct size
Transthoracic echocardiography was performed using a linear 30-MHz transducer according to the manufacturer’s instructions (Vevo 660 High Resolution Imaging System from VisualSonics, Toronto, Canada) similar to previously described methods.5 Two-dimensional-targeted M-mode echocardiographic measurements were carried out at mid-papillary muscle level after anesthesia with 1% isoflurane. FS was determined as LVEDD - LVESD/LVEDD where LVEDD and LVESD represented left ventricular end diastolic and end systolic dimensions in mm respectively. Measurements were averaged from more than three cardiac cycles. Another measure of intact heart function was the left ventricular rate of pressure development (+dP/dt) using an ex vivo working heart preparation as previously described10. The afterload against which buffered perfusate solution was ejected from left ventricle was constant at 56 mmHg of hydrostatic pressure from a column of buffer connected to the aorta. Hemodynamic parameters from the various groups of mice were obtained under the same afterload. Data were analyzed by computer software (WorkBench for Windows+, Kent Scientific Corp., Torrington, CT).
The infarct size was quantified as previously described.17 After fixing in 10% formalin, sections were embedded in paraffin, cut into 4-µm slices, and stained with Masson’s trichrome to measure area of infarcted myocardium as fibrosis. Infarct size was calculated as the ratio of infarct length to the circumference of both the endocardium and the epicardium after tracing with a planimeter image analyzer (ImageProPlus). Contractile response was measured in adult WT murine cardiac ventricular myocytes in which contraction shortening in the presence of drugs was determined as previously described18.
Drug administration
Compound 1 and its analogues were dissolved in phosphate-buffered saline, pH=7.4 at 10 µM (200 µL total volume), filtered for sterility for in vivo administration at 6 µL per d for 28 d via a mini-osmotic pump (Alzet) in mice. Intact heart function in vivo was assessed by echocardiography following infusion of nucleotide or vehicle.
Data analysis
Unless otherwise indicated, data were provided as mean ± standard error of the mean. For analysis of multiple groups, one-way ANOVA and Newman Keuls post-test comparison were used. Student’s t-test for paired or unpaired samples was used to evaluate the effects of experimental interventions; P<0.05 was taken as statistically significant.
Supplementary Material
Acknowledgments
Mass spectral measurements were carried out by Dr. John Lloyd and Dr. Noel Whittaker (NIDDK). This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. This work was supported in part by RO1-HL48225 and Ray Neag Distinguished Professorship to Bruce T. Liang.
Abbreviations
- 5’-AMP
adenosine 5’-monophosphate
- CS
contraction shortening
- CSQ
calsequestrin
- DIBAL-H
diisobutylaluminum hydride
- DMEM
Dulbecco’s modified Eagle medium
- DMF
dimethylformamide
- FS
fractional shortening
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectroscopy
- LAD
left anterior descending coronary artery
- LV
left ventricular
- NS
normal saline
- rt
room temperature
- SAR
structure activity relationship
- TBAP
tributylammonium phosphate
- THF
tetrahydrofuran
- TEAA
triethylammonium acetate
Footnotes
Supporting Information Available: NMR spectral data and HPLC traces of selected nucleotide derivatives, stability data for selected phosphonates, in vivo assay methods and results for the CSQ model are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev. 2011;63:641–683. doi: 10.1124/pr.110.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen J-B, Pappano A, Liang BT. Extracellular ATP-stimulated current in wild type and P2X4 receptor transgenic mouse ventricular myocytes. implication for a cardiac physiologic role of P2X4 receptors. FASEB J. 2006;20:277–284. doi: 10.1096/fj.05-4749com. [DOI] [PubMed] [Google Scholar]
- 3.Yang A, Sonin D, Jones L, Liang BT. A beneficial role of cardiac P2X4 receptors in heart failure: Rescuing the calsequestrin-overexpression model of cardiomyopathy. Am. J. Physiol. 2004;287:H1096–H1103. doi: 10.1152/ajpheart.00079.2004. [DOI] [PubMed] [Google Scholar]
- 4.Shen JB, Cronin C, Sonin D, Joshi BV, Carolina M, Nieto G, Harrison D, Jacobson KA, Liang BT. P2X purinergic receptor-mediated ionic current in cardiac myocytes of calsequestrin model of cardiomyopathy. Implications for the treatment of heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1077–H1084. doi: 10.1152/ajpheart.00515.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar TS, Zhou SY, Joshi BV, Balasubramanian R, Yang T, Liang BT, Jacobson KA. Structure activity relationship of (N)-methanocarba phosphonate analogues of 5’-AMP as cardioprotective agents acting through a cardiac P2X receptor. J. Med. Chem. 2010;53:2562–2576. doi: 10.1021/jm9018542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, Boyer JL, Harden TK, Jacobson KA. Adenine nucleotides analogues locked in a Northern methanocarba conformation: Enhanced stability and potency as P2Y1 receptor agonists. J. Med. Chem. 2002;45:2090–2100. doi: 10.1021/jm010538v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Joshi BV, Melman A, Mackman RL, Jacobson KA. Synthesis of ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: A versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleos. Nucleot. Nucleic Acids. 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn PM, Kim HS, Jacobson KA, Burnstock G. Northern ring conformation of methanocarba-adenosine 5′-triphosphate required for activation of P2X receptors. Drug Devel. Res. 2004;61:227–232. doi: 10.1002/ddr.10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Moody EC, Hecht SS, Sturla SJ. Deoxygenated phosphorothioate inositol phosphate analogs: synthesis, phosphatase stability, and binding affinity. Bioorg Med Chem. 2008;16:3419–3427. doi: 10.1016/j.bmc.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 9.Shao L, Abolin C, Hewitt MC, Koch P, Varney M. Derivatives of tramadol for increased duration of effect. Bioorg Med Chem Lett. 2006;16:691–694. doi: 10.1016/j.bmcl.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 10.Zhou SY, Mamdani M, Qanud K, Shen JB, Pappano A, Kumar TS, Jacobson KA, Hintze T, Recchia FA, Liang BT. Treatment of heart failure by a methanocarba derivative of adenosine monophosphate. Implication for a role of cardiac P2X purinergic receptors. J. Pharm. Exp. Therap. 2010;333:920–928. doi: 10.1124/jpet.109.164376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGuigan C, Bellevergue P, Jones BCNM, Mahmood N, Hay AJ, Petrik J, Karpas A. Alkyl hydrogen phosphonate derivatives of the anti-HIV agent AZT may be less toxic than the parent nucleoside analogue. Antiviral Chem. Chemother. 1994;5:271–277. 1994. [Google Scholar]
- 12.Khan SR, Nowak B, Plunkett W, Farquhar D. Bis(pivaloyloxymethyl) thymidine 5′-phosphate is a cell membrane-permeable precursor of thymidine 5′-phosphate in thymidine kinase deficient CCRF CEM cells. Biochem. Pharmacol. 2005;69:1307–1313. doi: 10.1016/j.bcp.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Peyrottes S, Egron D, Lefebvre I, Gosselin G, Imbach JL, Perigaud C. SATE pronucleotide approaches: an overview. Mini-Rev. Med. Chem. 2004;4:395–408. doi: 10.2174/1389557043404007. [DOI] [PubMed] [Google Scholar]
- 14.(a) Hecker SJ, Erion MD. Prodrugs of Phosphates and Phosphonates. J. Med. Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]; (b) Schultz C. Prodrugs of biologically active phosphate esters. Bioorg. Med. Chem. 2003;11:885–898. doi: 10.1016/s0968-0896(02)00552-7. [DOI] [PubMed] [Google Scholar]
- 15.Rosowsky A, Kim S, Ross J, Wick MM. Lipophilic 5’-(alkyl phosphate) esters of l-beta-arabinofuranosylcytosine and its N-acyl derivatives as potential prodrugs. J. Med. Chem. 1982;25:171–178. doi: 10.1021/jm00344a016. [DOI] [PubMed] [Google Scholar]
- 16.Moss GP. Extension and revision of the von Baeyer system for naming polycyclic compounds (including bicyclic compounds) Pure Appl. Chem. 1999;71:513–529. [Google Scholar]
- 17.Sonin D, Zhou SY, Cronin C, Sonina T, Wu J, Jacobson KA, Pappano A, Liang BT. Role of P2X purinergic receptors in the rescue of ischemic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H1191–H1197. doi: 10.1152/ajpheart.00577.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen JB, Shutt R, Pappano A, Liang BT. Characterization and mechanism of P2X receptor-mediated increase in cardiac myocyte contractility. Am. J Physiol. Heart Circ. Physiol. 2007;293:H3056–H3062. doi: 10.1152/ajpheart.00515.2007. [DOI] [PubMed] [Google Scholar]
- 19.Silvestri MA, Nagarajan M, De Clercq E, Pannecouque C, Cushman M. Design, Synthesis, Anti-HIV Activities, and Metabolic Stabilities of Alkenyldiarylmethane (ADAM) Non-nucleoside Reverse Transcriptase Inhibitors. J. Med. Chem. 2004;47:3149–3162. doi: 10.1021/jm049916x. [DOI] [PubMed] [Google Scholar]
- 20.Sun H. Capture hydrolysis signals in the microsomal stability assay: Molecular mechanisms of the alkyl ester drug and prodrug metabolism. Bioorg. Med. Chem. Lett. 2012;22:989–995. doi: 10.1016/j.bmcl.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Singh JK, Solanki A, Shirsath VS. Comparative in-vitro Intrinsic clearance of imipramine in multiple species liver microsomes: Human, Rat, Mouse and Dog. J. Drug Metab. Toxicol. 2012;3:4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.