

Abstract
Like other cancers, uveal melanomas (UM) are characterised by an uncontrolled, clonal, cellular proliferation, occurring as a result of numerous genetic, and epigenetic aberrations. Signalling pathways known to be disrupted in UM include: (1) the retinoblastoma pathway, probably as a result of cyclin D1 overexpression; p53 signalling, possibly as a consequence of MDM2 overexpression; and the P13K/AKT and mitogen-activated protein kinase/extracellular signal-related kinase pathway pathways that are disturbed as a result of PTEN and GNAQ/11 mutations, respectively. Characteristic chromosomal abnormalities are common and include 6p gain, associated with a good prognosis, as well as 1p loss, 3 loss, and 8q gain, which correlate with high mortality. These are identified by techniques such as fluorescence in situ hybridisation, comparative genomic hybridisation, microsatellite analysis, multiplex ligation-dependent probe amplification, and single-nucleotide polymorphisms. UM can also be categorised by their gene expression profiles as class 1 or class 2, the latter correlating with poor survival, as do BRCA1-associated protein-1 (BAP1) inactivating mutations. Genetic testing of UM has enhanced prognostication, especially when results are integrated with histological and clinical data. The identification of abnormal signalling pathways, genes and proteins in UM opens the way for target-based therapies, improving prospects for conserving vision and prolonging life.
Keywords: uveal melanoma, cytogenetics, molecular alterations, signalling pathways, MLPA, GEP
Introduction
Uveal melanoma (UM) is the most common primary adult intraocular cancer. More than 90% involve the choroid, the remainder being confined to iris and ciliary body. UM affects approximately six individuals per million per year in the United Kingdom, with the age at diagnosis peaking at 50–60 years. It is markedly different to cutaneous melanoma in its clinical and molecular genetic features. Concerning predisposition to UM, rare reports of families with an excess of UM cases have been published.1, 2, 3 Recent evidence suggests that patients with a cancer susceptibility may have higher frequencies of UM compared with the normal population.4 Other risk factors are congenital ocular melanocytosis, melanocytoma, and neurofibromatosis.
There is a wide range of therapeutic options for the treatment of primary UM. These include various forms of radiotherapy, surgical resection, and phototherapy.5 The 5-year local-tumour control rates in most specialised treatment centres exceed 90%. Despite this, almost 50% of patients with UM will develop disseminated disease, predominantly in the liver, but also in the lungs (24% of patients) and bone (16%).6 Early surgical removal of metastases has improved patient survival in some cases;7, 8 however, in general, the prognosis of UM patients with metastatic disease is currently poor because of the lack of effective chemotherapeutic agents.
Intense efforts have been made in the last decades to understand the molecular genetics involved in the development and the progression of UM, to recognise those that are likely to metastasise, and to identify signalling pathways and possible ‘druggable' molecules in the neoplastic melanocytes, which can be targeted using systemic therapies. Some of the early genetic events causing disruption of the cell cycle and apoptotic control in uveal melanocytes have been determined, as well as those leading to their malignant transformation and metastasis promotion.
This review summarises the current insights into the molecular mechanisms underlying UM pathogenesis, prognostic tests, and potential therapeutic strategies.
The hallmarks of cancer
Cancer is defined as an uncontrolled, clonal proliferation of cells, which progressively acquire most if not all of the six ‘hallmarks' of neoplasia, as defined by Hanahan and Weinberg in 2000, and then updated in 2011.9, 10 These hallmarks are considered to be absolute biological prerequisites of neoplastic cells for their survival and proliferation capacity at the primary site, and for their ability to invade and metastasise (Table 1). These features are acquired by a multistep process and include: (1) insensitivity to anti-growth signals; (2) self-sufficiency in growth signals; (3) avoiding apoptosis; (4) limitless replicative potential; (5) sustained angiogenesis; and (6) tissue invasion and metastasis.
Table 1. The classic and emerging ‘hallmarks of cancer' and their application to UM.
| Hallmarks of cancer | Example of gene/mechanism affected | Mechanism(s) in UM | References |
|---|---|---|---|
| Classical hallmarks | |||
| 1. Insensitivity to anti-growth signals | Loss of cell cycle inhibitor, such as retinoblastoma (Rb) suppressor | The retinoblastoma tumour suppressor pathway is disrupted in most UM either through: hyperphosphorylation of Rb; elevated expression of cyclin D1; or through methylation and inactivation of the INK4A gene | 11, 12, 13, 14, 15, 16 |
| 2. Self-sufficiency in growth signals | Gain of cell cycle stimulator—activation of pathways | The PI3K–AKT prosurvival pathway is constitutively activated in UM. LOH of the PTEN locus occurs in 76% of UM | 17, 18, 19, 20, 21 |
| The RAF/MEK/ERK pathway is constitutively activated: activating mutations in GNAQ or GNA11 occur in >80% of UM and can activate the RAF/MEK/ERK pathway | 11, 22, 23, 24, 25, 26, 27, 28 | ||
| 3. Avoid apoptosis | p53 pathway alterations | The p53 pathway is functionally blocked by its inhibitor MDM2 | 13, 14, 15, 29, 30 |
| BCL-2 | Defects in the Bcl2 pathway in UM contribute to apoptosis resistance | 11, 14, 29 | |
| PTEN downregulation | The PI3K–AKT prosurvival pathway is constitutively activated in UM to avoid apoptosis | 19, 20 | |
| Produce insulin-like growth factor (IGF-1) survival factors | IGF1R is often upregulated and can activate the PI3K–AKT pathway | 31, 32 | |
| 4. Limitless replicative potential | Turn on telomerase | Upregulated telomerase activity in UM | 33, 34 |
| 5. Sustained angiogenesis | Production of vascular endothelial growth factor (VEGF) inducer either by the tumour cells or by accompanying inflammatory cells | Upregulated expression of VEGF in UM; association with macrophage densities in UM | 35, 36, 37 |
| Increased expression of IGF-1 and IGF-1R in UM | 32, 38 | ||
| Raised levels of hypoxia-inducible factor 1 alpha | 39, 40, 41 | ||
| 6. Tissue invasion and metastasis | Activation of E-cadherin | Upregulation of E-cadherin and Wnt/beta-catenin signalling pathways | 42, 43 |
| Downregulation of the helix-loop-helix inhibitor ID2 | 39, 40 | ||
| Increased expression of matrix metalloproteinases (MMPs) and downregulation of their tissue inhibitors (TIMPs) by UM | 44, 45, 46, 47 | ||
| ALCAM expression | 48 | ||
| NOTCH pathway activation | 49 | ||
| Biallelic methylation of EFS | 50 | ||
| Emerging hallmarks | |||
| 1. Avoid immune destruction | Reduced tumour cell immunogenicity | Expression of PD-L1 by UM regulates T-cell function by suppressing IL-2 production and impairing T-cell function | 51 |
| Downregulation of HLA class 1 and 2 expression | Downregulation of HLA class 1 expression on UM cells | 52, 53 | |
| T-cell exhaustion | 51 | ||
| 2. Deregulating cellular energetics | Upregulating glucose transporters, for example, GLUT1, resulting in substantial increases in glucose import into cytoplasm. | Indirect evidence through the increased levels of the HIF1α and HIF2α transcription factors in UM | 39, 40, 41 |
| Hypoxia response of tumours acts by upregulating glucose transporters and multiple enzymes of the glycolytic pathway | |||
| Enabling characteristics | |||
| 1. Genome instability and mutation | See Tables 2 and 3 | See Tables 2 and 3 | |
| 2. Tumour-promoting inflammation | Lymphocytes Macrophages Dendritic cells | Varying densities of tumour infiltrating lymphocytes and macrophages; both associated with worse prognoses | 54, 55, 56, 57, 58, 59 |
These six core characteristics of the malignant cell are acquired in different tumour types via distinct mechanisms and at various times during the course of multistep tumourigenesis. An increasing body of evidence suggests that two additional ‘emerging' hallmarks of cancer and two ‘enabling characteristics' are involved in the pathogenesis of some and perhaps all cancers. These were added in the revised article of Hanahan and Weinberg in 2011, and include: (1) deregulating cellular energetics; (2) avoidance of immune destruction; (3) tumour-promoting inflammation and (4) genome instability and mutation (Table 1). As these capabilities have yet to be generalised to all tumour types and yet to be fully validated, they are still considered to be provisional.10
How do these hallmarks of cancer relate to UM?
UM is considered to be a cancer arising from uveal melanocytes. The precursors of melanocytes are non-pigmented melanoblasts derived from the neural crest, which bypass natural tissue barriers and basement membranes of the eye when migrating during embryogenesis. As with processes occurring in the skin, the melanoblasts mature into melanocytes within the uvea, and/or give rise to melanocytic stem cells, which maintain the ocular melanocytic ‘system'. Although it is known that melanocytic stem cells reside in the hair bulge in the skin,60 the location of uveal melanocytic stem cells is still unknown.
It is hypothesised that various genetic and epigenetic alterations occur along the ‘melanoblast–melanocyte–naevus–UM' pathway, resulting in their malignant transformation and propensity to spread. It is unclear whether the ‘melanoma-initiating' or ‘cancer stem-like' cells, recently shown to be present in UM cell lines,61 are derived directly from ocular melanocyte progenitors or from more mature melanocytes that have de-differentiated. Recent analysis of gene expression data of UM would suggest that de-differentiation indeed does occur during UM development, but this requires further investigation.11, 39 It is also unclear whether the naevus stage is a prerequisite in UM development: it has been estimated that <1 in 8000 naevi undergo malignant transformation to form UM.62 Histologically, it is exceptionally rare for a residual naevus to be evident adjacent to or within a choroidal melanoma, supporting this observation.
Despite this large gap in our knowledge regarding initial UM development, it has been demonstrated by several groups that most (if not all) of the ‘hallmarks of cancer' can be applied to UM pathogenesis (Table 1). The genetic and epigenetic events involved in UM development and dissemination enable malignant uveal melanocytes to proliferate and survive autonomously. These events include: mutation or amplification of proto-oncogenes; inactivating mutations or deletions of tumour (and metastasis) suppressor genes; and chromosomal aberrations (Tables 1, 2 and 3).11 Some of these genetic alterations are considered to occur in the early stages of UM development; others, at later stages (ie, prior to or at haematogeneous dissemination). It has been proposed that the genetic developmental pathway of UM bifurcates at an early stage to result in two very distinct genetic signatures: (1) disomy 3 with chromosome 6p gain and class 1 gene expression profile, and (2) monosomy 3 with class 2 molecular signature and a high metastatic propensity.40, 78, 80, 81 Later genetic events in UM development are suggested to be increasing aneuploidy and alterations in chromosome 8 (eg, gains in 8q and loss of 8p; see below for further details).
Table 2. Known genetic and epigenetic alterations in UM.
| Proto-oncogenes affected | Mechanism | Chromosome | Frequency (%) | References |
|---|---|---|---|---|
| NRAS | Mutation | 1p13 | a | 63, 64, 65, 66 |
| BRAF | Mutation | 7q34 | a | 67, 68 |
| NSB1 | Amplification | 8q21 | 50 | 69 |
| MYC | Amplification | 8q24 | 43 | 70 |
| DDEF1 (ASAP1) | Amplification | 8q24 | 50 | 71 |
| GNAQ/GNA11 | Mutation | 9p21 | >80 | 25, 26, 27 |
| CCND1 | Amplification | 11q13 | 65 | 12, 14, 15, 16 |
| MDM2 | Amplification | 12q15 | 65 | 13, 14, 15, 29, 30 |
| BCL-2 | Amplification | 18q21 | >95 | 11, 14, 29 |
| Tumour-suppressor genes | ||||
| LZTS1 | Deletion | 1p13 | — | 72 |
| CDKN2A-sporadic | Deletion, mutation | 9p21 | a | 1, 2, 3 |
| CDKN2A-familial | Deletion, mutation | 9p21 | a | 1, 2, 3 |
| PTEN | Deletion, mutation | 10q23 | 15 | 20 |
| BAP1 | Inactivating mutation | 0b–84% | 31 | |
| Epigenetic alterations | ||||
| CDKN2A | Hypermethylation | 9p21 | 4–33% | 1, 2, 3, 48, 73, 74 |
| RASSF1 | Hypermethylation | 3p21.3 | 13–70% | 28, 75 |
| hTERT | Hypermethylation | 52% | 74 | |
| MicroRNA alterations | ||||
| let-7b | Overexpression | NA | c | 76, 77 |
| miR18a | Overexpression | NA | c | 76, 77 |
| miR-199a | Overexpression | NA | c | 76, 77 |
| miR495 | Overexpression | NA | c | 76, 77 |
| miR549 | Overexpression | NA | c | 76, 77 |
Abbreviation: NA, not applicable.
Rare.
Data from Lake et al, unpublished results.
Not documented.
Table 3. Most common chromosomal aberrations in UM.
| Chromosome | Frequencya |
|---|---|
| 1p loss | 28–34% |
| 1q gain | 24% |
| 3 loss | 50–61% |
| 6p gain | 28–54% |
| 6q loss | 35–37% |
| 8p loss | 17–28% |
| 8q gain | 36–63% |
Although this dichotomous model is helpful in ‘classifying' UM into two risk groups with respect to the development of metastasis, it essentially discounts any concept of clonal heterogeneity within UM, which has been demonstrated by several groups using differing methods.82, 83, 84, 85, 86 Furthermore, it does not fit well with the concept of clonal evolution.87, 88 Recent evidence does indeed suggest that this model may be too simple, and that different clones of malignant melanocytes may evolve and co-exist within UM, with some having the potential to override or dominate others.79 This has been observed in longitudinal studies using next-generation sequencing in paired primary and metastatic carcinomas,89 and warrants further investigation using such techniques in UM.
In the following paragraphs, the major molecular pathway defects and the most common chromosomal alterations in UM development will be summarised.
Molecular pathway defects in primary UM
In most UM, the retinoblastoma (Rb) and p53 pathways are functionally inhibited, although actual mutations in the RB1 and TP53 genes are rare.11, 12, 13, 29, 30, 90 The Rb protein is constitutively hyperphosphorylated and functionally inactivated in most UM, probably as a result of cyclin D1 overexpression in about 65% of cases.12, 14, 15 (Figure 1; Table 1) The role of CDKN2A promoter hypermethylation, as an additional mechanism of Rb inactivation, is controversial as the frequency of hypermethylation of this gene varies between 4 and 33% of UM.12, 14, 15, 16, 48, 74 Increased cyclin D1 protein expression has been associated with larger tumour basal diameter, epithelioid cell type, and poor prognosis.15 The p53 pathway is inhibited downstream to p53 in many UM,30 and this may be a consequence of MDM2 overexpression, which is also common in UM and associated with a poor outcome.13, 15
Figure 1.
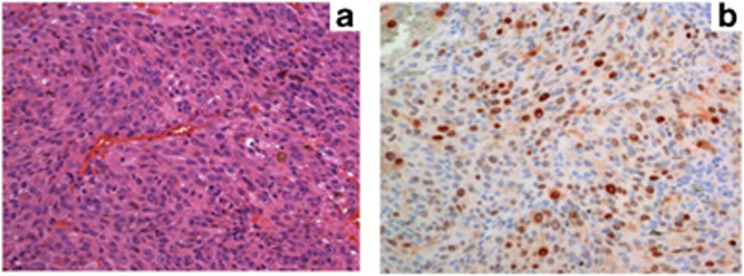
(a) Histological section of a UM of mixed cellularity (HE, × 20 magnification). (b) Immunostaining of the same tumour with clear nuclear cyclin D1 staining (APAAP, × 20 magnification).
The PI3K/AKT pathway is constitutively activated in most UM17 (Table 1). It has been demonstrated using immunohistochemistry that phosphorylated AKT correlates with poor prognosis in UM.18 While a study of 9 UM cell lines did not observe mutations in PTEN,19 a much larger study of 75 primary UM demonstrated loss of heterozygosity of the PTEN locus in 76% of tumours and actual mutations within the PTEN coding region in 11% of tumours.20 PTEN inactivation is also associated with increased aneuploidy and decreased survival in UM.20, 21 Taken together, these findings implicate a role for PTEN in UM progression, indicating scope for further investigation in this area.
The mitogen-activated protein kinase/extracellular signal-related kinase pathway (MAPK/ERK) pathway is essential for mediating cell-cycle progression, and in several types of cancer mutations in this pathway result in it being constitutively activated, producing inappropriate and autonomous proliferation of neoplastic cells.91 Most UM demonstrate constitutive activation of the MAPK pathway, suggesting the presence of upstream activating mutations.11, 22, 23 However, a systematic interrogation of candidate oncogenes in the MAPK pathway undertaken by several groups was initially disappointing.24 For example, mutations in KIT and the three RAS family members, which can activate the MAPK pathway, were shown to be exceedingly rare in UM.23, 63, 64, 65, 66, 92 Similarly, BRAF mutations, well described in cutaneous melanomas, have been reported in choroidal melanomas,67 but are very rare.22, 23, 65, 68, 93 Further, mutations in the other two members of the RAF family, ARAF and CRAF, are not found in UM.24
Guanine nucleotide-binding protein G(q) subunit (GNAQ, or G-alpha-q) and GNA11 mutations
This puzzle concerning the MAPK pathway, and the mechanisms by which it was constitutively activated, persisted until the recent discovery of mutations in GNAQ in almost half of UM.24, 25 GNAQ is one of a subfamily of G protein α subunit encoding genes, comprising GNAQ, GNA11, GNA14 and GNA15/16. The GNAQ mutation is somatically acquired, arising almost exclusively in exon 5 at codon 209 and resulting in substitution of the original glutamine at this point. There are at least five known variants, with GNAQQ209L or GNAQQ209P being the most frequent.25 More recently, mutations of codon 209 were also discovered in GNA11, and both GNAQ and GNA11 can also have mutations of exon 4 affecting codon 183.26 Thus, in a recent study, over 80% of UM were found to have GNAQ or GNA11 mutations affecting either Q209 or R183 in a mutually exclusive pattern.26
GNAQ and GNA11 mutations are also found in uveal naevi and in most UM regardless of their tumour stage, chromosomal constellation or other outcome predictors (see below).27 These mutations appear to be necessary but not sufficient for complete malignant transformation to melanoma.25 These data suggest that GNAQ and GNA11 mutations are early events in the molecular pathogenesis of UM. Therefore, although the timing of the alterations in the MAPK pathway during UM development have been clarified to some extent, the temporal sequences of those affecting the Rb, p53 and PI3K/AKT pathways remain to be determined.
Chromosomal alterations in primary UM
It has been known for almost 20 years that UM show specific chromosomal alterations, which are quite distinct from melanomas at other sites, particularly those of the skin (Table 3). The most striking abnormality in UM is the complete or partial loss of chromosome 3. Other common genetic abnormalities of UM include loss on 1p, 6q, 8, and 9p as well as gain on 1q, 6p, and 8q (Table 3). These alterations were initially identified by standard karyotypic analyses.94, 95, 96, 97, 98, 99, 100, 101, 102, 103 They have subsequently been confirmed by several groups using differing technologies, including: fluorescence in situ hybridisation (FISH);104, 105, 106, 107, 108, 109 comparative genomic hybridisation (CGH);21, 110, 111, 112, 113, 114, 115, 116 spectral karyotyping;117, 118 microsatellite analysis (MSA);119, 120, 121, 122, 123, 124 multiplex ligation-dependent probe amplification (MLPA),79, 125, 126, 127 and single-nucleotide polymorphisms (SNPs).109, 128, 129, 130, 131, 132
The above-mentioned chromosomal alterations in primary UM are clinically relevant because of their correlation with the risk of metastatic death. Chromosome 3 loss is associated with a reduction of the 5-year survival probability from approximately 100% to <50%.98, 101 Similarly, chromosome 8 gains and loss of chromosome 1 significantly correlate with reduced survival.79, 104, 108 Both chromosome 3 loss and polysomy 8q are also associated with other poor prognostic factors, including increasing tumour basal diameter, ciliary body involvement, presence of epithelioid cells, high mitotic count, and closed connective tissue loops.79 Conversely, gains in chromosome 6p correlate with a good prognosis, suggesting this aberration has a functionally protective effect.133
Molecular techniques used for prognostication in primary UM
The dramatic finding of the adverse prognostic significance of chromosome 3 loss in UM was confirmed by several groups, but was only translated from the ‘bench to the bedside' by a very few ocular oncology centres initially for use in the clinical management and counselling of patients.134 Since 1999, the cytogenetic status of UM cells has been assessed in consented UM patients undergoing either enucleation or local tumour resection at the Liverpool Ocular Oncology Centre using FISH.108 As a result of low specificity with respect to detecting partial deletions of chromosome 3 (also associated with a poorer prognosis79), the Liverpool team, in late 2007, replaced FISH with a polymerase chain reaction-based technique, MLPA, which examines 38 loci across chromosomes 1p, 3, 6, and 8. This was done after validation of this technique using a cohort of UM with a median follow-up of over 6 years125 and following comparative analysis with other techniques.135 MLPA, or MSA in selected cases where limited DNA is generated for analysis, is currently provided on a routine basis to all UM patients requesting genetic tumour typing. Those receiving radiotherapy or phototherapy undergo a tumour biopsy. The costs are modest and covered by the UK National Health Service (NHS). It is important to note that the genetic testing is performed in an accredited molecular pathology laboratory, and is undertaken with both regular internal and external quality assessment in accordance with European and UK Genetic Testing Network guidelines (http://www.ukgtn.nhs.uk/gtn). This is also the case for the co-author (MZ) performing MLPA and MSA at the Institute of Human Genetics in Essen, Germany.124 Consequently, the results of these tests can be considered as robust: they are certainly not ‘investigational' or ‘unethical', as has been unjustly suggested elsewhere.136
In the meantime, other ocular oncology centres are also offering molecular prognostication testing for their UM patients, using other techniques (Table 4), which are both DNA and RNA based. There are advantages and disadvantages to all methods (Table 4). The most commonly used tests are FISH, MLPA, MSA, SNP array (aSNP) and a PCR-based 12-gene assay based on gene expression profiling (GEP).137 The latter technique divides UM into two ‘classes' on the basis of an mRNA expression signature:138 class 1 and class 2. Class 1 UM often show 6p and 8q gain.40 Class 2 UM, tend to show more aneuploidy with 1p loss, 3 loss, 8p loss, and 8q gain. Class 2 UM are also strongly associated with inactivating mutations of ‘BRCA1-associated protein-1' (BAP1), located at 3p21 (see below).139 The GEP-based test has been patented (DecisionDx-UM; www.castlebiosciences.com/test_UM.html) and has received a considerable amount of publicity in the lay press, as it is claimed by Harbour and associates, its originators, to be superior to all other testing methods.137, 138 A recent study was performed to compare the prognostic accuracy of the GEP assay and monosomy 3, detected by a “multi-SNP” assay, which was designed by the authors as a research tool and still requires external validation as a clinical molecular test.128, 138 Although the GEP-based assay is marketed as a ‘stand-alone' assay for prognostication, MLPA has been demonstrated to provide as accurate prognostication when considered with clinical and histomorphological features of UM. Consequently, the study by Onken et al, that assessed monosomy 3 alone, fails to compare the most robust approach to genetic prognostic testing available. In addition, preliminary studies comparing the GEP-based assay with MLPA-based prognostication incorporating clinical and histomorphological tumour features in formalin-fixed paraffin-embedded material, show high concordance between these two techniques, with respect to the tumours examined and in their correlations with predictors of metastasis (Coupland et al, unpublished data). It is also worth noting that survival data in the literature suggest that as many as 15% of class 1 UM metastasise, and these have belatedly been reclassified as class 1B (http://talkabouthealth.com/what-are-the-potential-results-from-the-decisiondx-um-gene-expression-profiling-assay). Furthermore, class 1 profiles are observed in some liver metastases, an observation that also needs further investigation.129
Table 4. Most commonly used techniques for genetic typing of UM.
| Type of analysis | Molecular basis | Quantitative dosage analysis | Degree of automation | Cost | Remarks |
|---|---|---|---|---|---|
| Karyotype | Gross gain, loss, or alteration of chromosomes | + | + | + | Low-resolution copy number variation, cannot use FFPE |
| FISH | Gain or loss of small number of chromosome segments labelled with specific probes | + | + | ++ | Low-resolution copy number variation, can use FFPE |
| CGH | Gain or loss of large number of chromosome segments labelled with specific probes | ++ | ++ | +++ | c-CGH: low-resolution a-CGH: high-resolution copy number variation both can use FFPE |
| MSA | LOH of a small number of highly polymorphic DNA segments | ++ | ++ | + | Low-resolution copy number variation/LOH, can use FFPE |
| SNP | LOH of moderately polymorphic DNA segments | + | +++ | ++ | High-resolution copy number variation/LOH, can use FFPE |
| MLPA | Gain or loss of multiple chromosome segments | +++ | +++ | ++ | High-resolution copy number variation. 50 targets in one reaction can use FFPE |
| UM-GEP (PCR based) | Simultaneous measurement of mRNA expression of multiple genes | Not applicable | +++ | ++++ | Gene expression Can use FFPE, but fresh material preferred |
Abbreviations: CGH, comparative genomic hybridisation; microsatellite analysis; multiplex ligation-dependent probe amplification; FFPE, formalin-fixed paraffin-embedded material; FISH, fluorescence in situ hybridisation; LOH, loss of heterozygosity; SNP, single-nucleotide polymorphisms; UM-GEP, uveal melanoma gene expression profiling.
Finally, it is worth considering that molecular genetic diagnostics is a very rapidly advancing field with the costs of technologies plummeting at astounding rates, previously not experienced by the industry. Tests now considered to be ‘state of the art' are therefore likely to have a short ‘shelf life' and will be soon outdated by more accurate and less costly methods.
Molecular alterations proposed to be involved in UM metastasis
Aberrations of several genes and signalling pathways appear to promote dissemination of UM cells: these include LZTS1 (located on chromosome 8p22),72 DDEF1 (‘development and differentiation enhancing factor 1', also known as ASAP1, mapping to chromosome 8q24.21),71 PTP4A3 (‘protein tyrosine phosphatase type IV A member 3', located on chromosome 8q24.3),140 TCEB1 (chromosome 8q21.11), and NOTCH signalling.49 At present, the most convincing metastasis-regulatory gene in UM is BAP1, identified by exome sequencing.139 The BAP1 gene encodes a deubiquitinating enzyme that binds to BRCA1 and BARD1 to form a tumour suppressor heterodimeric complex.140 It is mapped to chromosome 3p21.31-p21.2, a region previously noted by Trolet et al129 to be deleted in UM. BAP1 is implicated in other cancers, which include mesothelioma, lung-, breast- and renal cell carcinomas.141 Inactivating BAP1 somatic mutations have been described in up to 84% of class 2 UM. Interestingly, germline BAP1 mutations have been described in families with a high risk for hereditary cancer and a novel ‘BAP1 cancer syndrome' including UM has been since been described by several groups.142, 143, 144, 145, 146
Besides BAP1, other genes are likely to be involved in the metastatic process, as indicated by our aSNP and whole exome sequencing of clinically and cytogenetically well-defined cohorts of UM with long-term follow-up (Lake et al, in press).147
Molecular alterations in UM metastases
Few cytogenetic or molecular genetic data are available on UM metastases. Trolet et al129 examined 63 liver metastases using array CGH and showed these to be clustered in the same groups as the primary tumours but in differing proportions, with the ‘class 2' gene signature as defined by this group. This is confirmed by studies examining UM metastases using FISH, demonstrating that the disseminated tumour cells are characterised by chromosome 3 loss and 8q gain.109, 113 We are currently examining paired primary and metastatic UM using aSNP and functional assays with the aim of identifying signalling pathways that are aberrant within the metastases and, consequently, potential ‘druggable targets'.
As outlined by Hanahan and Weinberg,9, 10 the metastatic process is multi-stepped and complex, and depends on numerous alterations occurring both within the tumour and its microenvironment. The latter is also of significance at the metastatic site, and often determines the location of metastases. This concept of ‘seed and soil' has long been accredited to Stephen Paget;148 it should actually be attributed to Ernst Fuchs, who interestingly described the notion of metastatic tropism 7 years earlier when discussing metastases of uveal tract ‘sarcomas' (ie, melanomas).149 This hepatic tropism of UM metastases remains unexplained but may be due to several factors including the chemokine receptor-ligand axis (eg, CXCR4 and CXCL12),150 interactions between FAS and FAS-ligand,151 IGF1 and IGF1-R, as well as C-Met and hepatocyte growth factor/scatter factor.32, 38
Strategies for targeted therapy in UM metastases
It is beyond the scope of this article to summarise the various chemotherapeutic regimens applied in metastatic melanoma, which have been reviewed elsewhere.8, 152, 153, 154 It is, however, worth mentioning the potential targets for UM therapy, which have been revealed by efforts unravelling key signalling cascades implicated in the development and progression of UM. These include the inhibitors of the: MAPK/MEK signalling pathway; PI3K/AKT pathway at the level of AKT; mTOR; mTOR blockade combined with an IGF-1R antibody; tyrosine kinases; c-Met pathway; CXCR4 and histone deacetylase.154 The progress of these varied approaches will be followed with enthusiasm by clinicians, researchers, and patients alike.
Conclusions
Although considerable work has demonstrated that the ‘hallmarks of cancer' are applicable to UM, the identification of the genetic pathways associated with UM oncogenesis and particularly those with metastasis is still at a preliminary stage. Characteristic copy number alterations and DNA expression profiles have been identified in UM, which are strongly correlated with prognosis. When these data are incorporated with the clinical and histomorphological features of UM in prediction models, a high degree of accuracy can be achieved for the individual patient, enhancing patient management.134, 155 It can be anticipated that the rapidly developing field of molecular genetics will shed further light on key signalling pathways involved in UM oncogenesis and progression, opening the way for target-based therapies.
The authors declare no conflict of interest.
References
- Jay M, McCartney AC. Familial malignant melanoma of the uvea and p53: a Victorian detective story. Surv Ophthalmol. 1993;37 (6:457–462. doi: 10.1016/0039-6257(93)90142-t. [DOI] [PubMed] [Google Scholar]
- Easton DF, Steele L, Fields P, Ormiston W, Averill D, Daly PA, et al. Cancer risks in two large breast cancer families linked to BRCA2 on chromosome 13q12-13. Am J Hum Genet. 1997;61 (1:120–128. doi: 10.1086/513891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlston RS, Damato BE. Genetic predisposition to ocular melanoma. Eye (Lond) 1999;13 (Part 1:43–46. doi: 10.1038/eye.1999.9. [DOI] [PubMed] [Google Scholar]
- Abdel-Rahman MH, Pilarski R, Ezzat S, Sexton J, Davidorf FH. Cancer family history characterization in an unselected cohort of 121 patients with uveal melanoma. Fam Cancer. 2010;9 (3:431–438. doi: 10.1007/s10689-010-9328-7. [DOI] [PubMed] [Google Scholar]
- Damato B, Heimann H.Personalized treatment of uveal melanoma Eye (Lond) 2012. e-pub ahead of print 23 November 2012; doi: 10.1038/eye.2012.242 [DOI] [PMC free article] [PubMed]
- Einhorn LH, Burgess MA, Gottlieb JA. Metastatic patterns of choroidal melanoma. Cancer. 1974;34 (4:1001–1004. doi: 10.1002/1097-0142(197410)34:4<1001::aid-cncr2820340406>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Frenkel S, Nir I, Hendler K, Lotem M, Eid A, Jurim O, et al. Long-term survival of uveal melanoma patients after surgery for liver metastases. Br J Ophthalmol. 2009;93 (8:1042–1046. doi: 10.1136/bjo.2008.153684. [DOI] [PubMed] [Google Scholar]
- Mariani P, Servois V, Piperno-Neumann S. Therapeutic options in metastatic uveal melanoma. Dev Ophthalmol. 2011;49:166–181. doi: 10.1159/000328333. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100 (1:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144 (5:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- An J, Wan H, Zhou X, Hu DN, Wang L, Hao L, et al. A comparative transcriptomic analysis of uveal melanoma and normal uveal melanocyte. PLoS One. 2011;6 (1:e16516. doi: 10.1371/journal.pone.0016516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley Jr MA, Harbour JW. Inactivation of retinoblastoma protein in uveal melanoma by phosphorylation of sites in the COOH-terminal region. Cancer Res. 2000;60 (16:4320–4323. [PMC free article] [PubMed] [Google Scholar]
- Brantley Jr MA, Harbour JW. Deregulation of the Rb and p53 pathways in uveal melanoma. Am J Pathol. 2000;157 (6:1795–1801. doi: 10.1016/s0002-9440(10)64817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupland SE, Bechrakis N, Schuler A, Anagnostopoulos I, Hummel M, Bornfeld N, et al. Expression patterns of cyclin D1 and related proteins regulating G1-S phase transition in uveal melanoma and retinoblastoma. Br J Ophthalmol. 1998;82 (8:961–970. doi: 10.1136/bjo.82.8.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupland SE, Anastassiou G, Stang A, Schilling H, Anagnostopoulos I, Bornfeld N, et al. The prognostic value of cyclin D1, p53, and MDM2 protein expression in uveal melanoma. J Pathol. 2000;191 (2:120–126. doi: 10.1002/(SICI)1096-9896(200006)191:2<120::AID-PATH591>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- van der Velden PA, Metzelaar-Blok JA, Bergman W, Monique H, Hurks H, Frants RR, et al. Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001;61 (13:5303–5306. [PubMed] [Google Scholar]
- Babchia N, Calipel A, Mouriaux F, Faussat AM, Mascarelli F. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: interaction with B-Raf/ERK. Invest Ophthalmol Vis Sci. 2010;51 (1:421–429. doi: 10.1167/iovs.09-3974. [DOI] [PubMed] [Google Scholar]
- Saraiva VS, Caissie AL, Segal L, Edelstein C, Burnier Jr MN. Immunohistochemical expression of phospho-Akt in uveal melanoma. Melanoma Res. 2005;15 (4:245–250. doi: 10.1097/00008390-200508000-00003. [DOI] [PubMed] [Google Scholar]
- Naus NC, Zuidervaart W, Rayman N, Slater R, van Drunen E, Ksander B, et al. Mutation analysis of the PTEN gene in uveal melanoma cell lines. Int J Cancer. 2000;87 (1:151–153. doi: 10.1002/1097-0215(20000701)87:1<151::aid-ijc23>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Abdel-Rahman MH, Yang Y, Zhou XP, Craig EL, Davidorf FH, Eng C. High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J Clin Oncol Jan. 2006;1024 (2:288–295. doi: 10.1200/JCO.2005.02.2418. [DOI] [PubMed] [Google Scholar]
- Ehlers JP, Worley L, Onken MD, Harbour JW. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res. 2008;14 (1:115–122. doi: 10.1158/1078-0432.CCR-07-1825. [DOI] [PubMed] [Google Scholar]
- Weber A, Hengge UR, Urbanik D, Markwart A, Mirmohammadsaegh A, Reichel MB, et al. Absence of mutations of the BRAF gene and constitutive activation of extracellular-regulated kinase in malignant melanomas of the uvea. Lab Invest. 2003;83 (12:1771–1776. doi: 10.1097/01.lab.0000101732.89463.29. [DOI] [PubMed] [Google Scholar]
- Zuidervaart W, van Nieuwpoort F, Stark M, Dijkman R, Packer L, Borgstein AM, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92 (11:2032–2038. doi: 10.1038/sj.bjc.6602598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Long MD, Duan S, Council ML, Bowcock AM, et al. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49 (12:5230–5234. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457 (7229:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363 (23:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Kilic E, Vaarwater J, Bastian BC, Garbe C, de Klein A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br J Cancer. 2009;101 (5:813–815. doi: 10.1038/sj.bjc.6605226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipel A, Abonnet V, Nicole O, Mascarelli F, Coupland SE, Damato B, et al. Status of RASSF1A in uveal melanocytes and melanoma cells. Mol Cancer Res. 2011;9 (9:1187–1198. doi: 10.1158/1541-7786.MCR-10-0437. [DOI] [PubMed] [Google Scholar]
- Chana JS, Wilson GD, Cree IA, Alexander RA, Myatt N, Neale M, et al. c-myc, p53, and Bcl-2 expression and clinical outcome in uveal melanoma. Br J Ophthalmol. 1999;83 (1:110–114. doi: 10.1136/bjo.83.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Tran BN, Worley LA, Delston RB, Harbour JW. Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005;46 (5:1561–1564. doi: 10.1167/iovs.04-1362. [DOI] [PubMed] [Google Scholar]
- All-Ericsson C, Girnita L, Seregard S, Bartolazzi A, Jager MJ, Larsson O. Insulin-like growth factor-1 receptor in uveal melanoma: a predictor for metastatic disease and a potential therapeutic target. Invest Ophthalmol Vis Sci. 2002;43 (1:1–8. [PubMed] [Google Scholar]
- Wu X, Zhou J, Rogers AM, Jänne PA, Benedettini E, Loda M, et al. c-Met, epidermal growth factor receptor, and insulin-like growth factor-1 receptor are important for growth in uveal melanoma and independently contribute to migration and metastatic potential. Melanoma Res. 2012;22 (2:123–132. doi: 10.1097/CMR.0b013e3283507ffd. [DOI] [PubMed] [Google Scholar]
- Heine B, Coupland SE, Kneiff S, Demel G, Bornfeld N, Hummel M, et al. Telomerase expression in uveal melanoma. Br J Ophthalmol. 2000;84 (2:217–223. doi: 10.1136/bjo.84.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach JM, Riedinger C, Wild M, Partsch M. [Telomerase activity in uveal melanomas] Ophthalmologe. 2000;97 (5:359–363. doi: 10.1007/s003470050537. [DOI] [PubMed] [Google Scholar]
- Abdel-Rahman MH, Craig EL, Davidorf FH, Eng C. Expression of vascular endothelial growth factor in uveal melanoma is independent of 6p21-region copy number. Clin Cancer Res. 2005;11 (1:73–78. [PubMed] [Google Scholar]
- Cools-Lartigue J, Marshall JC, Caissie AL, Saraiva VS, Burnier MN. Secretion of hepatocyte growth factor and vascular endothelial growth factor during uveal melanoma-monocyte in vitro interactions. Melanoma Res. 2005;15 (3:141–145. doi: 10.1097/00008390-200506000-00001. [DOI] [PubMed] [Google Scholar]
- Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18 (10:2726–2732. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakalian S, Marshall JC, Logan P, Faingold D, Maloney S, Di Cesare S, et al. Molecular pathways mediating liver metastasis in patients with uveal melanoma. Clin Cancer Res. 2008;14 (4:951–956. doi: 10.1158/1078-0432.CCR-06-2630. [DOI] [PubMed] [Google Scholar]
- Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res. 2006;66 (9:4602–4609. doi: 10.1158/0008-5472.CAN-05-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res. 2012;25 (2:171–181. doi: 10.1111/j.1755-148X.2012.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor N, Ivy A, Jiang BH, Agani FH. Involvement of HIF-1 in invasion of Mum2B uveal melanoma cells. Clin Exp Metastasis. 2006;23 (1:87–96. doi: 10.1007/s10585-006-9024-z. [DOI] [PubMed] [Google Scholar]
- Zuidervaart W, Pavey S, van Nieuwpoort FA, Packer L, Out C, Maat W, et al. Expression of Wnt5a and its downstream effector beta-catenin in uveal melanoma. Melanoma Res. 2007;17 (6:380–386. doi: 10.1097/CMR.0b013e3282f1d302. [DOI] [PubMed] [Google Scholar]
- Anastassiou G, Tschentscher F, Zeschnigk M, Bornfeld N. Cadherin expression in uveal melanoma. Exp Eye Res. 2002;74 (3:423–425. doi: 10.1006/exer.2002.1140. [DOI] [PubMed] [Google Scholar]
- Baker JK, Elshaw SR, Mathewman GE, Nichols CE, Murray AK, Parsons MA, et al. Expression of integrins, degradative enzymes and their inhibitors in uveal melanoma: differences between in vitro and in vivo expression. Melanoma Res. 2001;11 (3:265–273. doi: 10.1097/00008390-200106000-00008. [DOI] [PubMed] [Google Scholar]
- Canovas D, Rennie IG, Nichols CE, Sisley K. Local environmental influences on uveal melanoma: vitreous humor promotes uveal melanoma invasion, whereas the aqueous can be inhibitory. Cancer. 2008;112 (8:1787–1794. doi: 10.1002/cncr.23358. [DOI] [PubMed] [Google Scholar]
- Nareyeck G, Zeschnigk M, von der Haar D, Schilling H, Bornfeld N, Anastassiou G. Differential expression of tissue inhibitor of matrix metalloproteinases 3 in uveal melanoma. Ophthalmic Res. 2005;37 (1:23–28. doi: 10.1159/000082940. [DOI] [PubMed] [Google Scholar]
- van der Velden PA, Zuidervaart W, Hurks MH, Pavey S, Ksander BR, Krijgsman E, et al. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int J Cancer. 2003;106 (4:472–479. doi: 10.1002/ijc.11262. [DOI] [PubMed] [Google Scholar]
- Zeschnigk M, Tschentscher F, Lich C, Brandt B, Horsthemke B, Lohmann DR. Methylation analysis of several tumour suppressor genes shows a low frequency of methylation of CDKN2A and RARB in uveal melanomas. Comp Funct Genomics. 2003;4 (3:329–336. doi: 10.1002/cfg.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnaghi L, Ebrahimi KB, Schreck KC, Bar EE, Coonfield ML, Bell WR, et al. Notch signaling promotes growth and invasion in uveal melanoma. Clin Cancer Res. 2012;18 (3:654–665. doi: 10.1158/1078-0432.CCR-11-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann LC, Weinhausel A, Thomas S, Horsthemke B, Lohmann DR, Zeschnigk M. EFS shows biallelic methylation in uveal melanoma with poor prognosis as well as tissue-specific methylation. BMC Cancer. 2011;11:380. doi: 10.1186/1471-2407-11-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chen PW, Li H, Alizadeh H, Niederkorn JY. PD-L1: PD-1 interaction contributes to the functional suppression of T-cell responses to human uveal melanoma cells in vitro. Invest Ophthalmol Vis Sci. 2008;49 (6:2518–2525. doi: 10.1167/iovs.07-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurks HM, Metzelaar-Blok JA, Mulder A, Claas FH, Jager MJ. High frequency of allele-specific down-regulation of HLA class I expression in uveal melanoma cell lines. Int J Cancer. 2000;85 (5:697–702. doi: 10.1002/(sici)1097-0215(20000301)85:5<697::aid-ijc16>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- de Waard-Siebinga I, Hilders CG, Hansen BE, van Delft JL, Jager MJ. HLA expression and tumor-infiltrating immune cells in uveal melanoma. Graefes Arch Clin Exp Ophthalmol. 1996;234 (1:34–42. doi: 10.1007/BF00186516. [DOI] [PubMed] [Google Scholar]
- Whelchel JC, Farah SE, McLean IW, Burnier MN. Immunohistochemistry of infiltrating lymphocytes in uveal malignant melanoma. Invest Ophthalmol Vis Sci. 1993;34 (8:2603–2606. [PubMed] [Google Scholar]
- Makitie T, Summanen P, Tarkkanen A, Kivela T. Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42 (7:1414–1421. [PubMed] [Google Scholar]
- Toivonen P, Makitie T, Kujala E, Kivela T. Microcirculation and tumor-infiltrating macrophages in choroidal and ciliary body melanoma and corresponding metastases. Invest Ophthalmol Vis Sci. 2004;45 (1:1–6. doi: 10.1167/iovs.03-0622. [DOI] [PubMed] [Google Scholar]
- Jager MJ, Ly LV, El Filali M, Madigan MC. Macrophages in uveal melanoma and in experimental ocular tumor models: Friends or foes. Prog Retin Eye Res. 2010;30 (2:129–146. doi: 10.1016/j.preteyeres.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Herwig MC, Grossniklaus HE. Role of macrophages in uveal melanoma. Expert Rev Ophthalmol. 2011;6 (4:405–407. doi: 10.1586/eop.11.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronkhorst IH, Jager MJ. Uveal melanoma: the inflammatory microenvironment. J Innate Immun. 2012;4 (5-6:454–462. doi: 10.1159/000334576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Fang D, Kumar SM, Li L, Nguyen TK, Acs G, et al. Isolation of a novel population of multipotent adult stem cells from human hair follicles. Am J Pathol. 2006;168 (6:1879–1888. doi: 10.2353/ajpath.2006.051170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalirai H, Damato BE, Coupland SE. Uveal melanoma cell lines contain stem-like cells that self-renew, produce differentiated progeny, and survive chemotherapy. Invest Ophthalmol Vis Sci. 2011;52 (11:8458–8466. doi: 10.1167/iovs.11-7379. [DOI] [PubMed] [Google Scholar]
- Singh AD, Kalyani P, Topham A. Estimating the risk of malignant transformation of a choroidal nevus. Ophthalmology. 2005;112 (10:1784–1789. doi: 10.1016/j.ophtha.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Mooy CM, Van der Helm MJ, Van der Kwast TH, De Jong PT, Ruiter DJ, Zwarthoff EC. No N-ras mutations in human uveal melanoma: the role of ultraviolet light revisited. Br J Cancer. 1991;64 (2:411–413. doi: 10.1038/bjc.1991.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soparker CN, O'Brien JM, Albert DM. Investigation of the role of the ras protooncogene point mutation in human uveal melanomas. Invest Ophthalmol Vis Sci. 1993;34 (7:2203–2209. [PubMed] [Google Scholar]
- Cruz F, Rubin BP, Wilson D, Town A, Schroeder A, Haley A, et al. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 2003;63 (18:5761–5766. [PubMed] [Google Scholar]
- Kilic E, Bruggenwirth HT, Verbiest MM, Zwarthoff EC, Mooy NM, Luyten GP, et al. The RAS-BRAF kinase pathway is not involved in uveal melanoma. Melanoma Res. 2004;14 (3:203–205. doi: 10.1097/01.cmr.0000130006.46885.a0. [DOI] [PubMed] [Google Scholar]
- Malaponte G, Libra M, Gangemi P, Bevelacqua V, Mangano K, D'Amico F, et al. Detection of BRAF gene mutation in primary choroidal melanoma tissue. Cancer Biol Ther. 2006;5 (2:225–227. doi: 10.4161/cbt.5.2.2429. [DOI] [PubMed] [Google Scholar]
- Cohen Y, Goldenberg-Cohen N, Parrella P, Chowers I, Merbs SL, Pe'er J, et al. Lack of BRAF mutation in primary uveal melanoma. Invest Ophthalmol Vis Sci. 2003;44 (7:2876–2878. doi: 10.1167/iovs.02-1329. [DOI] [PubMed] [Google Scholar]
- Ehlers JP, Harbour JW. NBS1 expression as a prognostic marker in uveal melanoma. Clin Cancer Res. 2005;11 (5:1849–1853. doi: 10.1158/1078-0432.CCR-04-2054. [DOI] [PubMed] [Google Scholar]
- Parrella P, Caballero OL, Sidransky D, Merbs SL. Detection of c-myc amplification in uveal melanoma by fluorescent in situ hybridization. Invest Ophthalmol Vis Sci. 2001;42 (8:1679–1684. [PubMed] [Google Scholar]
- Ehlers JP, Worley L, Onken MD, Harbour JW. DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res. 2005;11 (10:3609–3613. doi: 10.1158/1078-0432.CCR-04-1941. [DOI] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Harbour JW. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res. 2008;14 (12:3737–3745. doi: 10.1158/1078-0432.CCR-07-5144. [DOI] [PubMed] [Google Scholar]
- Merbs SL, Sidransky D. Analysis of p16 (CDKN2/MTS-1/INK4A) alterations in primary sporadic uveal melanoma. Invest Ophthalmol Vis Sci. 1999;40 (3:779–783. [PubMed] [Google Scholar]
- Moulin AP, Clement G, Bosman FT, Zografos L, Benhattar J. Methylation of CpG island promoters in uveal melanoma. Br J Ophthalmol. 2008;92 (2:281–285. doi: 10.1136/bjo.2007.127035. [DOI] [PubMed] [Google Scholar]
- Maat W, Beiboer SH, Jager MJ, Luyten GP, Gruis NA, van der Velden PA. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49 (4:1291–1298. doi: 10.1167/iovs.07-1135. [DOI] [PubMed] [Google Scholar]
- Worley LA, Long MD, Onken MD, Harbour JW. Micro-RNAs associated with metastasis in uveal melanoma identified by multiplexed microarray profiling. Melanoma Res. 2008;18 (3:184–190. doi: 10.1097/CMR.0b013e3282feeac6. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Badhrinarayanan N, Biswas J, Krishnakumar S. Analysis of chromosomal aberration (1, 3, and 8) and association of microRNAs in uveal melanoma. Mol Vis. 2009;15:2146–2154. [PMC free article] [PubMed] [Google Scholar]
- Hoglund M, Gisselsson D, Hansen GB, White VA, Säll T, Mitelman F, et al. Dissecting karyotypic patterns in malignant melanomas: temporal clustering of losses and gains in melanoma karyotypic evolution. Int J Cancer. 2004;108 (1:57–65. doi: 10.1002/ijc.11558. [DOI] [PubMed] [Google Scholar]
- Damato B, Dopierala JA, Coupland SE. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res. 2010;16 (24:6083–6092. doi: 10.1158/1078-0432.CCR-10-2076. [DOI] [PubMed] [Google Scholar]
- Parrella P, Sidransky D, Merbs SL. Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999;59 (13:3032–3037. [PubMed] [Google Scholar]
- Tschentscher F, Husing J, Holter T, Kruse E, Dresen IG, Jöckel KH, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63 (10:2578–2584. [PubMed] [Google Scholar]
- Mensink HW, Vaarwater J, Kilic E, Naus NC, Mooy N, Luyten G, et al. Chromosome 3 intratumor heterogeneity in uveal melanoma. Invest Ophthalmol Vis Sci. 2009;50 (2:500–504. doi: 10.1167/iovs.08-2279. [DOI] [PubMed] [Google Scholar]
- Maat W, Jordanova ES, van Zelderen-Bhola SL, Barthen ER, Wessels HW, Schalij-Delfos NE, et al. The heterogeneous distribution of monosomy 3 in uveal melanomas: implications for prognostication based on fine-needle aspiration biopsies. Arch Pathol Lab Med. 2007;131 (1:91–96. doi: 10.5858/2007-131-91-THDOMI. [DOI] [PubMed] [Google Scholar]
- Dopierala J, Damato BE, Lake SL, Taktak AF, Coupland SE. Genetic heterogeneity in uveal melanoma assessed by multiplex ligation-dependent probe amplification. Invest Ophthalmol Vis Sci. 2010;51 (10:4898–4905. doi: 10.1167/iovs.09-5004. [DOI] [PubMed] [Google Scholar]
- Bronkhorst IH, Maat W, Jordanova ES, Kroes WG, Schalij-Delfos NE, Luyten GP, et al. Effect of heterogeneous distribution of monosomy 3 on prognosis in uveal melanoma. Arch Pathol Lab Med. 2011;135 (8:1042–1047. doi: 10.5858/2010-0477-OAR1. [DOI] [PubMed] [Google Scholar]
- Aronow M, Sun Y, Saunthararajah Y, Biscotti C, Tubbs R, Triozzi P, et al. Monosomy 3 by FISH in uveal melanoma: variability in techniques and results. Surv Ophthalmol. 2012;57 (5:463–473. doi: 10.1016/j.survophthal.2011.12.004. [DOI] [PubMed] [Google Scholar]
- White VA, McNeil BK, Thiberville L, Horsman DE. Acquired homozygosity (isodisomy) of chromosome 3 during clonal evolution of a uveal melanoma: association with morphologic heterogeneity. Genes Chromosomes Cancer. 1996;15 (2:138–143. doi: 10.1002/(SICI)1098-2264(199602)15:2<138::AID-GCC10>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Callejo SA, Dopierala J, Coupland SE, Damato B. Sudden growth of a choroidal melanoma and multiplex ligation-dependent probe amplification findings suggesting late transformation to monosomy 3 type. Arch Ophthalmol. 2011;129 (7:958–960. doi: 10.1001/archophthalmol.2011.181. [DOI] [PubMed] [Google Scholar]
- Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72 (19:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes AG, Liloglou T, Maloney P, Hagan S, Nunn J, Hiscott P, et al. Loss of heterozygosity on chromosomes 3, 9, 13, and 17, including the retinoblastoma locus, in uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42 (11:2472–2477. [PubMed] [Google Scholar]
- Denhardt DT. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J. 1996;318 (Pt 3:729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pache M, Glatz K, Bosch D, Dirnhofer S, Mirlacher M, Simon R, et al. Sequence analysis and high-throughput immunohistochemical profiling of KIT (CD 117) expression in uveal melanoma using tissue microarrays. Virchows Arch. 2003;443 (6:741–744. doi: 10.1007/s00428-003-0883-2. [DOI] [PubMed] [Google Scholar]
- Rimoldi D, Salvi S, Lienard D, Lejeune FJ, Speiser D, Zografos L, et al. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003;63 (18:5712–5715. [PubMed] [Google Scholar]
- Griffin CA, Long PP, Schachat AP. Trisomy 6p in an ocular melanoma. Cancer Genet Cytogenet. 1988;32 (1:129–132. doi: 10.1016/0165-4608(88)90319-6. [DOI] [PubMed] [Google Scholar]
- Horsman DE, Sroka H, Rootman J, White VA. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet Cytogenet. 1990;45 (2:249–253. doi: 10.1016/0165-4608(90)90090-w. [DOI] [PubMed] [Google Scholar]
- Horsman DE, White VA. Cytogenetic analysis of uveal melanoma. Consistent occurrence of monosomy 3 and trisomy 8q. Cancer. 1993;71 (3:811–819. doi: 10.1002/1097-0142(19930201)71:3<811::aid-cncr2820710325>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Becher R. Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst. 1990;82 (22:1765–1769. doi: 10.1093/jnci/82.22.1765. [DOI] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Horsthemke B, Becher R. Chromosomal aberrations defining uveal melanoma of poor prognosis. Lancet. 1992;339 (8794:691–692. [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Becher R. Two subclones in a case of uveal melanoma. Relevance of monosomy 3 and multiplication of chromosome 8q. Cancer Genet Cytogenet. 1994;77 (2:144–146. doi: 10.1016/0165-4608(94)90230-5. [DOI] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Friedrichs W, Seeber S, Becher R. Cytogenetics of twelve cases of uveal melanoma and patterns of nonrandom anomalies and isochromosome formation. Cancer Genet Cytogenet. 1995;80 (1:40–46. doi: 10.1016/0165-4608(94)00165-8. [DOI] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347 (9010:1222–1225. doi: 10.1016/s0140-6736(96)90736-9. [DOI] [PubMed] [Google Scholar]
- Sisley K, Rennie IG, Cottam DW, Potter AM, Potter CW, Rees RC. Cytogenetic findings in six posterior uveal melanomas: involvement of chromosomes 3, 6, and 8. Genes Chromosomes Cancer. 1990;2 (3:205–209. doi: 10.1002/gcc.2870020307. [DOI] [PubMed] [Google Scholar]
- Wiltshire RN, Elner VM, Dennis T, Vine AK, Trent JM. Cytogenetic analysis of posterior uveal melanoma. Cancer Genet Cytogenet. 1993;66 (1:47–53. doi: 10.1016/0165-4608(93)90148-f. [DOI] [PubMed] [Google Scholar]
- Sisley K, Rennie IG, Parsons MA, Jacques R, Hammond DW, Bell SM, et al. Abnormalities of chromosomes 3 and 8 in posterior uveal melanoma correlate with prognosis. Genes Chromosomes Cancer. 1997;19 (1:22–28. doi: 10.1002/(sici)1098-2264(199705)19:1<22::aid-gcc4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- McNamara M, Felix C, Davison EV, Fenton M, Kennedy SM. Assessment of chromosome 3 copy number in ocular melanoma using fluorescence in situ hybridization. Cancer Genet Cytogenet. 1997;98 (1:4–8. doi: 10.1016/s0165-4608(96)00405-0. [DOI] [PubMed] [Google Scholar]
- Patel KA, Edmondson ND, Talbot F, Parsons MA, Rennie IG, Sisley K. Prediction of prognosis in patients with uveal melanoma using fluorescence in situ hybridisation. Br J Ophthalmol. 2001;85 (12:1440–1444. doi: 10.1136/bjo.85.12.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisley K, Tattersall N, Dyson M, Smith K, Mudhar HS, Rennie IG. Multiplex fluorescence in situ hybridization identifies novel rearrangements of chromosomes 6, 15, and 18 in primary uveal melanoma. Exp Eye Res. 2006;83 (3:554–559. doi: 10.1016/j.exer.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Damato B, Duke C, Coupland SE, Hiscott P, Smith PA, Campbell I, et al. Cytogenetics of uveal melanoma: a 7-year clinical experience. Ophthalmology. 2007;114 (10:1925–1931. doi: 10.1016/j.ophtha.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Singh AD, Aronow ME, Sun Y, Bebek G, Saunthararajah Y, Schoenfield LR, et al. Chromosome 3 status in uveal melanoma: a comparison of fluorescence in situ hybridization and single-nucleotide polymorphism array. Invest Ophthalmol Vis Sci. 2012;53 (7:3331–3339. doi: 10.1167/iovs.11-9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon KB, Thompson CT, Char DH, O'Brien JM, Kroll S, Ghazvini S, et al. Comparative genomic hybridization in the detection of DNA copy number abnormalities in uveal melanoma. Cancer Res. 1994;54 (17:4764–4768. [PubMed] [Google Scholar]
- Speicher MR, Prescher G, du Manoir S, Jauch A, Horsthemke B, Bornfeld N, et al. Chromosomal gains and losses in uveal melanomas detected by comparative genomic hybridization. Cancer Res. 1994;54 (14:3817–3823. [PubMed] [Google Scholar]
- Ghazvini S, Char DH, Kroll S, Waldman FM, Pinkel D. Comparative genomic hybridization analysis of archival formalin-fixed paraffin-embedded uveal melanomas. Cancer Genet Cytogenet. 1996;90 (2:95–101. doi: 10.1016/s0165-4608(96)00076-3. [DOI] [PubMed] [Google Scholar]
- Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42 (2:313–317. [PubMed] [Google Scholar]
- Hughes S, Damato BE, Giddings I, Hiscott PS, Humphreys J, Houlston RS. Microarray comparative genomic hybridisation analysis of intraocular uveal melanomas identifies distinctive imbalances associated with loss of chromosome 3. Br J Cancer. 2005;93 (10:1191–1196. doi: 10.1038/sj.bjc.6602834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic E, van Gils W, Lodder E, Beverloo HB, van Til ME, Mooy CM, et al. Clinical and cytogenetic analyses in uveal melanoma. Invest Ophthalmol Vis Sci. 2006;47 (9:3703–3707. doi: 10.1167/iovs.06-0101. [DOI] [PubMed] [Google Scholar]
- Petrausch U, Martus P, Tonnies H, Bechrakis NE, Lenze D, Wansel S, et al. Significance of gene expression analysis in uveal melanoma in comparison to standard risk factors for risk assessment of subsequent metastases. Eye (Lond) 2008;22 (8:997–1007. doi: 10.1038/sj.eye.6702779. [DOI] [PubMed] [Google Scholar]
- Naus NC, van Drunen E, de Klein A, Luyten GP, Paridaens DA, Alers JC, et al. Characterization of complex chromosomal abnormalities in uveal melanoma by fluorescence in situ hybridization, spectral karyotyping, and comparative genomic hybridization. Genes Chromosomes Cancer. 2001;30 (3:267–273. [PubMed] [Google Scholar]
- White JS, Becker RL, McLean IW, Director-Myska AE, Nath J. Molecular cytogenetic evaluation of 10 uveal melanoma cell lines. Cancer Genet Cytogenet. 2006;168 (1:11–21. doi: 10.1016/j.cancergencyto.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Tschentscher F, Prescher G, Zeschnigk M, Horsthemke B, Lohmann DR. Identification of chromosomes 3, 6, and 8 aberrations in uveal melanoma by microsatellite analysis in comparison to comparative genomic hybridization. Cancer Genet Cytogenet. 2000;122 (1:13–17. doi: 10.1016/s0165-4608(00)00266-1. [DOI] [PubMed] [Google Scholar]
- Scholes AG, Damato BE, Nunn J, Hiscott P, Grierson I, Field JK. Monosomy 3 in uveal melanoma: correlation with clinical and histologic predictors of survival. Invest Ophthalmol Vis Sci. 2003;44 (3:1008–1011. doi: 10.1167/iovs.02-0159. [DOI] [PubMed] [Google Scholar]
- Hausler T, Stang A, Anastassiou G, Jöckel KH, Mrzyk S, Horsthemke B, et al. Loss of heterozygosity of 1p in uveal melanomas with monosomy 3. Int J Cancer. 2005;116 (6:909–913. doi: 10.1002/ijc.21086. [DOI] [PubMed] [Google Scholar]
- Shields CL, Ganguly A, Materin MA, Teixeira L, Mashayekhi A, Swanson LA, et al. Chromosome 3 analysis of uveal melanoma using fine-needle aspiration biopsy at the time of plaque radiotherapy in 140 consecutive cases: the Deborah Iverson, MD, Lectureship. Arch Ophthalmol. 2007;125 (8:1017–1024. doi: 10.1001/archopht.125.8.1017. [DOI] [PubMed] [Google Scholar]
- Shields CL, Ganguly A, Bianciotto CG, Turaka K, Tavallali A, Shields JA. Prognosis of uveal melanoma in 500 cases using genetic testing of fine-needle aspiration biopsy specimens. Ophthalmology. 2010;118 (2:396–401. doi: 10.1016/j.ophtha.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Thomas S, Putter C, Weber S, Bornfeld N, Lohmann DR, Zeschnigk M. Prognostic significance of chromosome 3 alterations determined by microsatellite analysis in uveal melanoma: a long-term follow-up study. Br J Cancer. 2012;106 (6:1171–1176. doi: 10.1038/bjc.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damato B, Dopierala J, Klaasen A, van Dijk M, Sibbring J, Coupland SE. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Invest Ophthalmol Vis Sci. 2009;50 (7:3048–3055. doi: 10.1167/iovs.08-3165. [DOI] [PubMed] [Google Scholar]
- Damato B. Progress in the management of patients with uveal melanoma. The 2012 Ashton Lecture. Eye (Lond) 2012;26 (9:1157–1172. doi: 10.1038/eye.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaarwater J, van den Bosch T, Mensink HW, van Kempen C, Verdijk RM, Naus NC, et al. Multiplex ligation-dependent probe amplification equals fluorescence in-situ hybridization for the identification of patients at risk for metastatic disease in uveal melanoma. Melanoma Res. 2011;22 (1:30–37. doi: 10.1097/CMR.0b013e32834e6a67. [DOI] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW. Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma. Clin Cancer Res. 2007;13 (10:2923–2927. doi: 10.1158/1078-0432.CCR-06-2383. [DOI] [PubMed] [Google Scholar]
- Trolet J, Hupe P, Huon I, Lebigot I, Decraene C, Delattre O, et al. Genomic profiling and identification of high-risk uveal melanoma by array CGH analysis of primary tumors and liver metastases. Invest Ophthalmol Vis Sci. 2009;50 (6:2572–2580. doi: 10.1167/iovs.08-2296. [DOI] [PubMed] [Google Scholar]
- Lake SL, Coupland SE, Taktak AF, Damato BE. Whole-genome microarray detects deletions and loss of heterozygosity of chromosome 3 occurring exclusively in metastasizing uveal melanoma. Invest Ophthalmol Vis Sci. 2010;51 (10:4884–4891. doi: 10.1167/iovs.09-5083. [DOI] [PubMed] [Google Scholar]
- McCannel TA, Burgess BL, Nelson SF, Eskin A, Straatsma BR. Genomic identification of significant targets in ciliochoroidal melanoma. Invest Ophthalmol Vis Sci. 2010;52 (6:3018–3022. doi: 10.1167/iovs.10-5864. [DOI] [PubMed] [Google Scholar]
- Abi-Ayad N, Kodjikian L, Couturier J. [Genomic techniques used in uveal melanoma: a literature review] J Fr Ophtalmol. 2011;34 (4:259–264. doi: 10.1016/j.jfo.2010.11.012. [DOI] [PubMed] [Google Scholar]
- White VA, Chambers JD, Courtright PD, Chang WY, Horsman DE. Correlation of cytogenetic abnormalities with the outcome of patients with uveal melanoma. Cancer. 1998;83 (2:354–359. [PubMed] [Google Scholar]
- Damato B, Eleuteri A, Taktak AF, Coupland SE. Estimating prognosis for survival after treatment of choroidal melanoma. Prog Retin Eye Res. 2011;30 (5:285–295. doi: 10.1016/j.preteyeres.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Coupland SE, Kalirai H, Baudo M, Maye U, Zeschnigk M, Damato B. Quality assessment of multiplex ligation-dependent probe amplification (MLPA) in uveal melanoma by comparison with array comparative genomic hybridization (aCGH) and microsatellite analysis (MSA) IOVS. 2012.
- Harbour JW. Molecular prognostic testing and individualized patient care in uveal melanoma. Am J Ophthalmol. 2009;148 (6:823–829 e821. doi: 10.1016/j.ajo.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010;12 (4:461–468. doi: 10.2353/jmoldx.2010.090220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Char DH, Augsburger JJ, Correa ZM, Nudleman E, et al. Collaborative Ocular Oncology Group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology. 2012;119 (8:1596–1603. doi: 10.1016/j.ophtha.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330 (6009:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent C, Valet F, Planque N, Silveri L, Maacha S, Anezo O, et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2010;71 (3:666–674. doi: 10.1158/0008-5472.CAN-10-0605. [DOI] [PubMed] [Google Scholar]
- Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16 (9:1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48 (12:856–859. doi: 10.1136/jmedgenet-2011-100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43 (10:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43 (10:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njauw CN, Kim I, Piris A, Gabree M, Taylor M, Lane AM, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One. 2012;7 (4:e35295. doi: 10.1371/journal.pone.0035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone M, Korb Ferris L, Baumann F, Napolitano A, Lum CA, Flores EG, et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med. 2012;10 (1:179. doi: 10.1186/1479-5876-10-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake SL, Damato BE, Taktak AFG, Lloyd BH, Coupland SE.Copy number alteration of genes key to metastatic progression in uveal melanoma Am J Patholin press.
- Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;133 (3421:571–573. [PubMed] [Google Scholar]
- Fuchs E. Das Sarkom des Uvealtractus. Graefe's Archiv für Ophthalmologie. 1882;XII (2:233. [Google Scholar]
- Scala S, Ierano C, Ottaiano A, Franco R, La Mura A, Liguori G, et al. CXC chemokine receptor 4 is expressed in uveal malignant melanoma and correlates with the epithelioid-mixed cell type. Cancer Immunol Immunother. 2007;56 (10:1589–1595. doi: 10.1007/s00262-007-0303-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiou G, Coupland SE, Stang A, Boeloeni R, Schilling H, Bornfeld N. Expression of Fas and Fas ligand in uveal melanoma: biological implication and prognostic value. J Pathol. 2001;194 (4:466–472. doi: 10.1002/path.926. [DOI] [PubMed] [Google Scholar]
- Patel M, Smyth E, Chapman PB, Wolchok JD, Schwartz GK, Abramson DH, et al. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clin Cancer Res. 2011;17 (8:2087–2100. doi: 10.1158/1078-0432.CCR-10-3169. [DOI] [PubMed] [Google Scholar]
- Woodman SE. Metastatic uveal melanoma: biology and emerging treatments. Cancer J. 2012;18 (2:148–152. doi: 10.1097/PPO.0b013e31824bd256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreville S, Agapova OA, Matatall KA, Kneass ZT, Onken MD, Lee RS, et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2011;18 (2:408–416. doi: 10.1158/1078-0432.CCR-11-0946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SA, Damato B, Marshall E, Salmon P. Psychological aspects of cytogenetic testing of uveal melanoma: preliminary findings and directions for future research. Eye (Lond) 2009;23 (3:581–585. doi: 10.1038/eye.2008.54. [DOI] [PubMed] [Google Scholar]