

Abstract
The Human Microbiome Project used rigorous good clinical practice standards to complete comprehensive body site sampling in healthy 18- to 40-yr-old adults, creating an unparalleled reference set of microbiome specimens. To ensure that specimens represented minimally perturbed microbiomes, we first screened potential participants using exclusion criteria based on health history, including the presence of systemic diseases (e.g., hypertension, cancer, or immunodeficiency or autoimmune disorders), use of potential immunomodulators, and recent use of antibiotics or probiotics. Subsequent physical examinations excluded individuals based on body mass index (BMI), cutaneous lesions, and oral health. We screened 554 individuals to enroll 300 (149 men and 151 women, mean age 26 yr, mean BMI 24 kg/m2, 20.0% racial minority, and 10.7% Hispanic). We obtained specimens from the oral cavity, nares, skin, gastrointestinal tract, and vagina (15 specimens from men and 18 from women). The study evaluated longitudinal changes in an individual's microbiome by sampling 279 participants twice (mean 212 d after the first sampling; range 30-359 d) and 100 individuals 3 times (mean 72 d after the second sampling; range 30-224 d). This sampling strategy yielded 11,174 primary specimens, from which 12,479 DNA samples were submitted to 4 centers for metagenomic sequencing. Our clinical design and well-defined reference cohort has laid a foundation for microbiome research.—Aagaard, K., Petrosino, J., Keitel, W., Watson, M., Katancik, J., Garcia, N., Patel, S., Cutting, M., Madden, T., Hamilton, H., Harris, E., Gevers, D., Simone, G., McInnes, P., Versalovic, J. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters.
Keywords: metagenomics, HMP clinical data, clinical metadata, clinical research studies, metagenomic medicine
Homo sapiens and its microbiomes have probably coevolved to form a physiological community comprising distinct body site niches (1–4). The convergence and divergence of these niches results in metabolically and antigenically vibrant diverse communities of microbes including mutualists (symbiotically beneficial microbes), commensals (microbes that are neither harmful nor beneficial to the host), and pathogens (microbes that are detrimental to the host; refs. 5, 6). What is unique about our so-called second genome (or the “metagenome” of the human microbiome) is its incredible diversity and plasticity (7, 8). When we define the diversity of microbes within a given body habitat by the number and abundance of organisms of distinct genus and species, it is evident that microbial diversity varies among individuals. Our recently published work with the Human Microbiome Project Consortium (9–12) provided the first reliable estimates of the structure, function, and diversity of the healthy (“reference”) human microbiome. Before this work, suppositions of human microbiome diversity and lack of plasticity in disease states were based on studies in relatively small subject cohorts or within a limited set of body sites (8, 13–18).
There are numerous biological illustrations of the plasticity and fluidity of the human microbiome. For example, studies have shown that neonates typically acquire complex microbial communities in the gut within the first week of life, with fluctuations in bacterial representation during the first year until a relative equilibrium is reached (19–24). Compared with vaginally delivered infants, those delivered by cesarean have higher rates of colonization by horizontally transmitted, environmentally acquired microbes such as Clostridium difficile and Klebsiella and Enterobacter species (19, 21, 24–26). Transmission of taxa may be altered during vaginal birth by the presence of vertically transmitted symbionts, and cesarean delivery may enhance colonization of the oral cavity and intestine by components of the skin microbiome (26). In studies of adult female monozygotic and dizygotic twins and their mothers, in which the twin pairs were concordant for leanness or obesity, obesity was associated with phylum-level changes in the gut microbiota and reduced bacterial diversity (27, 28). Low diversity in the gut has also been associated with inflammatory bowel disease (17). The composition of the vaginal microbiota in reproductive-aged women is affected by several host factors, including age, menarche, sexual activity, pregnancy, and exogenous exposures such as contraceptives; at this body site, greater microbial diversity has been associated with bacterial vaginosis (29–32).
Characterizing the processes that govern the structure and dynamics of human microbial communities is essential for understanding human development and physiology. This characterization is also crucial for advancing the field of metagenomic medicine, which currently ranges from treatments targeting individual bacterial strains to reported clinical success with fecal transplantation (33–36). Metagenomic medicine is predicated on the premise that some of the host factors and environmental exposures affecting the microbiome composition may be modifiable, providing opportunities to reshape our plastic metagenome. Assessment of clinical outcomes in metagenomic medicine will rely on robust body sampling protocols and strong analytical approaches for health disease intervention data comparisons.
The U.S. National Institutes of Health (NIH) Roadmap Human Microbiome Project (HMP) is the first microbiome study to combine three essential elements critical to advancing the field: comparisons across multiple body sites in many different individuals, comparisons of longitudinal samples, and efforts to minimize exogenous and environmental exposures that may influence species abundance in core microbiomes at different body sites. The HMP used new technologies for metagenomic analysis to establish an unparalleled reference data set characterizing the microbiomes of accessible body sites of healthy adults. To achieve a combination of organismal and functional data, the HMP combined multiple analytical tools including taxonomic profiling by 16S ribosomal RNA gene sequences, metagenomic profiling by whole-genome shotgun (WGS) sequencing, and alignment of assembled sequences to reference microbial genomes. Recent publications describe the sequencing and analytical methods developed under the HMP initiative, and the findings on structure, function, and diversity of the human microbiome, which included the demonstration of distinct microbiomes at different body sites (9–11).
This article expands on the HMP consortium articles by describing the carefully designed and executed HMP specimen collection study and providing detailed data on the characteristics of the study population. To produce the high-quality clinical specimens required for metagenomic analysis, we sampled 15 body sites in 149 men and 18 body sites in 151 women. We describe here the rigorous design standards for subject screening and recruitment, the standardized procedures for collection of blood and specimens from the oral cavity, nares, epidermis, vagina, and distal gut (via fecal specimens), and the longitudinal sampling, which permitted an initial assessment of short-term microbiome population dynamics in healthy individuals. We present the detailed demographic and clinical data from the full 300-subject HMP cohort, which demonstrate our ability to correlate metagenomic data with the broad host phenotype (i.e., sex) in our healthy adult population.
MATERIALS AND METHODS
Protocol development
A national panel of experts representing different clinical specialties as well as experts in ethics and informed consent contributed to the study development. An HMP clinical working group developed and refined the study protocol, consent forms, and the manual of procedures (MOP), which provided detailed, standardized instructions to ensure consistency in screening, enrollment, body site sampling, and specimen processing and compliance with regulatory and data management requirements. The final study protocol and the MOP are publicly available through the NIH Database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000228.v3.p1).
Protection of human subjects
Specimen collection procedures involved minimal risk to participants. As defined in 45 U.S. Code of Federal Regulations 46.102 (i), “Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.” As part of the consent process, we discussed with participants the potential risks of participation, including the physical risks associated with specimen collection, the possibility that traces of human DNA sequence data could be contained in the metagenomic sequence data and the possibility that protected health information or deidentified project data stored in a public repository could be accidentally released. The protocol and consent form described precautions taken to reduce these risks. The study sites created consent documents using a template developed specifically for the project, based on National Human Genome Research Institute (NHGRI) guidelines for genomics studies that address the ethical, legal, and social implications of such research (see sample consent document at http://www.genome.gov/27526660). The institutional review boards at each study site reviewed and approved the protocol and informed consent and other study documents. We obtained certificates of confidentiality intended to protect against the compelled disclosure of participants' identities in legal proceedings. Additional protections for participants included coding genomic and metagenomic specimens and sequence data, using controlled-access repositories for extracted metagenomic nucleic acids and for deidentified medical and human genome sequence data and making efforts to remove any human sequence traces from the microbial data deposited in open access databases. If a participant withdrew consent after providing specimens, the remaining specimens and extracted nucleic acids were to be destroyed; however, any metagenomic sequence data that were already published in open access databases could not be retracted.
Study oversight
An independent clinical study oversight committee (CSOC) was convened to review safety data, unanticipated problems, study conduct, and progress. The CSOC considered background information on specimen collection procedures and study-specific data and provided recommendations to NIH and the study investigators.
Protocol compliance
The complex logistics of the longitudinal, multisite body sampling in this study required months of close coordination of health care professionals in primary care, dentistry, and obstetrics and gynecology. Understanding of and adherence to presampling instructions for promotion of an “antimicrobe-free” state at the time of sampling required careful planning and education. We identified 51 protocol deviations, none of which compromised the study or the protection of human subjects. Deviations included specimen collections outside of defined time windows, participant noncompliance with presampling instructions, eligibility issues, and laboratory errors. Of the participants, 21 participants terminated the protocol early for reasons including loss to follow-up, relocation, and pregnancy. No serious adverse events were reported.
Adaptability of the study and protocol refinement
The regular meetings of the HMP clinical working group permitted ongoing assessment of study issues and the ability to adapt as needed. The clinical protocol and accompanying MOP were revised on multiple occasions based on refinements in clinical practice, yielding 9 versions of the protocol and 12 versions of the MOP.
Early in the pilot phase, we changed the upper limit for blood pressure (BP) from 140/90 to 160/100 mmHg to avoid exclusion of subjects who had a single elevated BP determination but no history of hypertension. When influenza vaccination rates rose in response to the 2009 H1N1 influenza outbreak, we added an exclusion criterion to avoid introducing the variable of a perturbed nasal cavity microbiome after administration of live, attenuated, nasally delivered vaccine. Preliminary 16S metagenomic sequencing data indicated that the extent of intraindividual variation between visits 1 and 2 was body site-dependent and generated interest in additional sampling efforts. We then expanded the study by 50 participants (from 250 to 300 participants) and added a third sampling of 100 participants (50/study site). These enhancements were intended to make compositional data from multiple body sites more robust and enable investigators to study intraindividual variation more comprehensively across body sites and over an extended time.
Processing of body site specimens for microbial DNA extraction began while the specimen collection portion of the study was ongoing. The clinical site laboratories provided feedback on specimen quality, indicating that most specimens reproducibly yielded DNA samples with average concentrations between 0.1 and 21 ng/ml. Although some specimens (palate, buccal mucosa, gingiva, and throat) yielded relatively low quantities of total nucleic acid and required post-PCR assessment with 16S recombinant RNA (rRNA) gene primer sets to ascertain adequacy of samples for metagenomic sequencing, the clinical site processing laboratories deemed DNA quantity and quality adequate for all body sites except skin, for which quality was inconsistent. Based on earlier studies of the human skin microbiome (37), we initially used scalpel blades to obtain microbial DNA by scraping the retroauricular creases and antecubital fossae. After sampling 56 participants, we paused enrollment because of concerns about human DNA contamination, reproducibility of the skin scraping method, and skin trauma. Human DNA could confound metagenomic analyses or affect the ability to amplify target 16S rRNA gene regions. It also presented risks of participant identifiability. We adopted a skin swabbing approach, key aspects of which were application of a moist swab with sufficient vigor, for a sufficient period of time, and on a sufficient area of skin (38). Specimens obtained with the swab method showed an improved ratio of microbial to human DNA.
Participant recruitment and screening
The intent of the JumpStart phase of HMP was to study healthy young adults whose core microbiomes were not likely to be perturbed by infectious comorbidities and exogenous exposures. In a cohort of 300 individuals, we aimed for equal sex distribution and ∼20% minority (racial and ethnic) participants. We recruited subjects in two ethnically, racially, and socioeconomically diverse U.S. cities, with study sites at Baylor College of Medicine (BCM) in Houston, Texas, and Washington University in St. Louis (WUSTL) and St. Louis University (SLU) in St. Louis, Missouri. The age range of 18 to 40 yr was established to minimize variability due to childhood growth and development, aging, and hormonal influences during adolescence and menopause. We recruited participants from the general population and telephone-screened using a general health questionnaire. Exclusion criteria included a body mass index (BMI) outside the range of 18 to 35 kg/m2, a history of cancer, a compromised immune status, a history of specified chronic diseases, or exposure to some medications or dietary supplements within the last 6 mo (antibiotics, corticosteroids, cytokines, immunosuppressive agents, or large doses of probiotics). We excluded individuals who reported recent major dietary changes or a recent history of moderate to high alcohol intake. We excluded individuals who had more than 8 missing teeth, unless they were missing because of third molar extractions, orthodontic- or trauma-related extractions, or congenital absence. Women were required to have a regular 21- to 35-d menstrual cycle or a history of regular cycles before beginning contraception. We excluded women who used the combination hormone vaginal ring contraceptive device, but other forms of hormonal contraception were not exclusionary.
Individuals who passed telephone screening were invited for further screening in the research clinic. After participants signed a consent form, investigators completed a detailed medical history and review of systems, documented concomitant medications, measured vital signs, collected urine for pregnancy testing, performed targeted physical examinations with attention to body site-specific exclusion criteria (oral cavity, skin, and nasal cavity), and measured vaginal pH in women (pH>4.5 at the posterior fornix was exclusionary). If all criteria were met, blood was collected for serum human immunodeficiency virus, hepatitis B virus, and hepatitis C virus testing (all required to be negative). A complete listing of 35 exclusion criteria is found in Supplemental Table S1.
The microbiome of a healthy person may be altered qualitatively and quantitatively by host actions. To reduce the affect of these environmental factors, we instructed participants to avoid certain activities and the use of some medications and personal care products for specified periods before sampling. The restrictions included use of topical antibiotics and vaginal antifungal agents (7 d); use of antimicrobial soaps, mouthwashes, and hand sanitizers; vaginal, oral, and anal sexual activity; and swimming in chlorinated pools or hot tubs (48 h); and bathing, washing, and teeth brushing or flossing (12 h). To encourage compliance, we distributed kits containing acceptable personal hygiene components, such as soap and shampoo that did not contain residue-producing antimicrobial agents (see study MOP, Appendices D and E).
Clinical data collection
The EMMES Corp. (Rockville, Maryland, USA; http://www.emmes.com) provided operational support and a secure Internet-based data entry system for the study. Clinical data elements included sex, race, ethnicity, age, birthplace, occupation, BMI, vital signs, vaginal pH, date of last menstrual period, tobacco use, and dental and health insurance status. We recorded a medical history, targeted physical examination findings, and medication history for each participant. After completion of data checks, the clinical data were entered into dbGaP (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/about.cgi) to facilitate access by investigators approved by a NIH Data Access Committee.
Body site sampling
Supplemental Table S2 lists the study specimens, which were collected as follows.
Oral cavity
We collected 9 spatially distinct specimens from the oral cavity (saliva, swabs from 6 soft tissue sites, and supra- and subgingival dental plaque). Participants drooled into a 50-ml collection tube after allowing saliva to collect in the mouth for ≥1 min. For individuals who had difficulty forming saliva, flow was stimulated with a sugar- and flavor-free chewing gum base product. We collected soft tissue specimens from the tongue dorsum, hard palate, buccal mucosa, keratinized gingiva, palatine tonsils, and throat by swabbing with sterile Catch-All specimen collection swabs (Epicenter Technologies, Madison, WI, USA). We used sterile Gracey dental curettes to collect supra- and subgingival plaque from 6 index teeth: 2 molar teeth (upper right and lower left first molars, nos. 3 and 19), 2 premolar teeth (upper left and lower right first premolars, nos. 12 and 28), and 2 incisor teeth (upper left and lower right central incisors, nos. 9 and 25). If the index teeth did not have adequate plaque to sample, adjacent teeth were sampled, and those teeth numbers were recorded in the participant's dental record. Specimens collected with swabs and curettes were placed immediately into tubes containing MO BIO buffer (MO BIO Laboratories, Carlsbad, CA, USA), which aids in subsequent bacterial lysis.
Skin
We collected 4 separate skin specimens from the two retroauricular creases and the two antecubital fossae. Specimens from the first 56 participants were collected by scraping the skin with a sterile no. 15 scalpel blade. Concerns about the amount of human DNA in the specimens and other technical issues prompted a change to use of a sterile Catch-All swab. The swab was premoistened with specimen collection fluid (Tris-EDTA and 0.5% Tween 20), and the areas to be sampled were swiped vigorously for 30 s. Swab specimens were inserted into a tube containing MO BIO buffer.
Nasal cavity
We collected specimens from the nasal cavity by gently rubbing the mucosal surfaces of the anterior nares with a sterile Catch-All swab. The right and left sides were pooled into one tube containing MO BIO buffer.
Gastrointestinal tract
The stool specimen was self-collected by participants. We provided participants with a commercial “toilet hat” stool specimen collection kit (specimen container, shipping box, and cold packs; Fisherbrand Commode Specimen Collection System; Thermo Fisher Scientific, Waltham, MA, USA) at the screening visit and as needed for resampling. We instructed participants to collect stool specimens within 24 h before each sampling visit and bring them to the visits.
Vagina
We collected separate specimens from the vaginal introitus, the posterior fornix, and the midpoint of the vagina. All specimens were collected by applying a sterile Catch-All swab to a single site, swirling it 5 times, withdrawing the swab without contamination from other sites, and transferring the specimen into a tube with MO BIO buffer. We first used a digital display pH meter with accuracy to ±0.01 (Waterproof BigDisplay pH Spear; Oakton, Vernon Hills, IL, USA) to measure the pH at the vaginal introitus and collected the vaginal introitus specimen. A clear small or medium Pederson speculum (McKesson, San Francisco, CA, USA) was then introduced in the absence of lubrication and turned to an approximate 45° angle to enable placement of the pH spear for determination of posterior fornix pH. We collected the posterior fornix specimen and then slowly withdrew the speculum to obtain the specimen from the midpoint of the vagina. The vaginal pH for screening was measured at least 48 h after menstrual blood flow ceased, and participants were not menstruating on the days of specimen collection and must have abstained from sexual intercourse, douching, tampon use, or vaginal cream use for the preceding 48 h.
We sampled study-eligible individuals at baseline within 30 d of the screening visit. To collect information on the variation in an individual's microbiome over time, we asked participants to provide a second set of specimens within 1 yr after the first specimen collection, and 279 of the 300 participated. Participants were eligible for second sampling even if exclusionary medications were used in the interval between samplings. Whenever possible, we delayed resampling for at least 6 mo after exposure to exclusionary medications. Intercurrent medications were noted as part of the clinical data. Based on early data analyses, we amended the protocol to allow collection of a third set of specimens from 100 participants, obtained within 6 mo after the second specimen collection.
Blood collection
At the first sampling visit, we collected blood specimens (30 ml) from each participant. We sent blood to the NHGRI Sample Repository for Human Genetic Research at Coriell Institute for Medical Research (Camden, NJ, USA) for human DNA extraction and cryopreservation of lymphocytes for possible creation of lymphoblastoid cell lines. Coded human DNA was stored at the repository for possible future approved human microbiome studies. We separated and stored serum in clinical site repositories for possible future studies of humoral immune responses to microbiome organisms.
Specimen processing and microbiome analyses
After collection, body site specimens were immediately placed on ice, transported to the laboratory, and then stored at −80°C until DNA extraction. Genomic DNA was isolated using the standard protocol from the MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories). Preprocessing steps were performed on saliva and stool specimens. For saliva, polypropylene tubes (Falcon; BD Biosciences, San Jose, CA, USA) containing collected saliva were centrifuged at 2600 g for 15 min at room temperature. If the solids were not sufficiently separated, the specimen was centrifuged again for 20 min. The supernatant was transferred to two tubes containing 750 μl of lysis buffer and processed according to MO BIO PowerSoil Kit instructions. For stool, ∼2 ml of specimen was transferred into one 50-ml Falcon tube using a sterile spatula. Then 5ml of MO BIO lysis buffer (PowerLyzer PowerSoil Bead Solution, 12855-50-BS) was added to the Falcon tube, and the specimen was homogenized by vortexing for 30–40 s. The Falcon tube was centrifuged for 5 min at 1500 relative centrifugal force, and 1 ml of supernatant was pipetted into 4 barcoded MO BIO Garnet Bead tubes containing 750 μl of MO BIO buffer. The specimens were heated at 65°C for 10 min, incubated at 95°C for 10 min, and processed per PowerSoil instructions. All extracted DNA samples were stored in kit solution C6 at −20°C. DNA was quantified by NanoDrop (all samples) and PicoGreen Assay (>75% of samples; Thermo Fisher Scientific). We obtained sufficient quantities of DNA from the majority of specimens. Time between specimen collection and extraction was examined as a possible confounder, and no bias was observed (9).
Before study specimens were processed, the laboratory created a mock microbial community (simulated microbiome) to test and validate the DNA extraction methods. The mock community contained 22 bacterial strains associated with the human microbiome. The strains were cultured, and cells from confluent growth were mixed in equal ratios at 108 colony-forming units/ml. Sequencing of cloned 16S recombinant DNA amplicons was performed, and sequences were analyzed by Basic Local Area Search Tool (BLASTN; version 2.2.23) vs. the U.S. National Center for Biotechnology Information (Bethesda, MD, USA) Nucleotide database (download April 2010). Sequence-based identifications were compared with the known mock community composition. The two specimen-processing laboratories (WUSTL and BCM) compared two DNA extraction kits for extraction yields of DNA from the mock microbial community. The MO BIO PowerSoil Kit was selected for use in the study because it yielded superior reproducibility within and between the two laboratories and because it included steps to remove potentially inhibitory compounds that could impede PCR or sequencing steps.
Site clinical laboratories coded, stored, and processed body site specimens for nucleic acid extraction, using a common protocol to reduce variability between samples and processing centers. The clinical site repositories distributed coded nucleic acid samples to sequencing centers for metagenomic sequencing. The EMMES Global Trace system tracked all specimens from collection through shipment for sequencing.
A full description of analysis methods, including 16S data processing, generation of operational taxonomic unit (OTU) tables, association testing, and standardization of protocols across sequencing centers is available in our HMP Consortium publications (9, 10, 12).
RESULTS
Supplemental Figure S1 shows the number of screening and sampling visits completed over the study period. We completed screening visits for 554 adults to enroll 300, including 30 who failed initial screening but passed a second screening (e.g., after receiving dental care). An unmeasured number of initial contacts self-excluded after telephone screening and did not attend screening visits. Among recorded reasons for screen failure were 128 instances of periodontal pockets >4 mm or >10% of sites with bleeding on probing, 88 instances of untreated cavitated carious lesions or oral abscesses, 22 instances of vaginal pH outside allowed limits or vaginal irritation, and 21 instances of corticosteroid, antibiotic (antifungal, antiviral, and antiparasitic drugs) or probiotic use during the protocol-specified exclusionary time frame. Reasons also included unrecognized hypertension, history of medication usage, underweight (BMI≤18 kg/m2) or overweight (BMI>35 kg/m2) status, acne or other cutaneous lesions, or inability to comply with the protocol. For example, because of their role in the health care profession, some potential participants could not refrain from using bactericidal agents for the required >24 h interval.
The mean age of study participants was 26 yr, and the mean BMI was 24 kg/m2. Study enrollment included 149 men, 151 women, 60 self-identified members of racial minorities (20.0%), and 32 participants who self-identified their ethnicity as Hispanic (10.7%). The time between screening and baseline sampling was less than 30 d for nearly all participants. For those who participated in repeat collections, average intervals between the first and second and second and third samplings were 212 d (range 30–359 d) and 72 d (range 30–224 d), respectively (Fig. 1). The average time between the first and third (final) samplings was 296 d (range 83–444 d). Overall, we collected 11,174 primary specimens, which served as the source of 12,479 DNA samples submitted for 16S and WGS sequencing.
Figure 1.
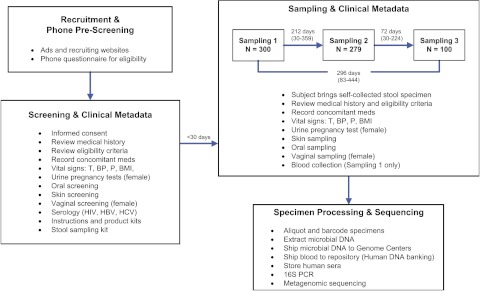
Timeline and flow during the screening and sampling process. The basic processes of human screening and sampling (including repeat sampling) are outlined in this figure. Individual steps in the screening and sampling processes are listed, and the average intervals (and ranges; in days) between the steps are shown.
With revision of the BP inclusion criterion, 25 of the 300 subjects enrolled had a single elevated screening BP determination, with elevation of systolic only (19 subjects), diastolic only (2 subjects), or both (4 subjects). The majority of values in those with elevated systolic BP were in the range of 140–145 mm Hg; 6 individuals had elevated diastolic BP in the range of 92–98 mm Hg.
After sampling was complete, the HMP Data Analysis Working Group requested additional clinical data to further characterize the study population. We succeeded in reaching 277 of the 300 participants (92%). Of these, 244 (88.1%) consumed meat/fish/poultry at least 3 d/wk, 17 (6.1%) consumed meat/fish/poultry 1 or 2 d/wk, 15 (5.4%) consumed eggs and dairy products but no meat, and 1 (0.4%) consumed no animal products and at least 70% raw food. A majority of these 277 participants (n=198, 71.5%) reported being breastfed in infancy. Among the 144 women contacted, 26 (18.1%) were parous; of these, 18 (69.2%) had only vaginal deliveries, 7 (26.9%) had only cesarean deliveries, and 1 (3.8%) had both vaginal and cesarean deliveries.
Because genetic and environmental differences may affect body site microbial composition and function, we compared participant characteristics at the two clinical sites. Table 1 summarizes subject characteristics and shows the relationship between clinical site and subject demographic determinants for categorical variables (P value derived from bivariate analysis with Fisher's exact test or Fisher-Freeman-Halton, as appropriate). Whereas there were no significant differences between clinical sites for the subject's birthplace, with 86.7% from the United States or Canada, the sites differed significantly in the birth countries/regions of the subjects' parents (P<0.0001). For the total study population, 76% had parents born in the United States or Canada, but BCM enrolled only 42% of the total number of subjects whose parents were U.S. or Canadian natives. Subjects whose father was born in Asia were more common at BCM (n=26) than at WUSTL (n=3). The sites also differed significantly with respect to subjects' self-reported education levels, occupations, and dental insurance coverage.
Table 1.
Analysis of relationship of clinical site of enrollment and subject demographics
| Subject characteristic | Clinical enrollment site |
Total, n = 300 |
P | ||||
|---|---|---|---|---|---|---|---|
| BCM, n = 150 |
WUSTL, n = 150 |
||||||
| n | % | n | % | n | % total study population | ||
| Birth region or countrya,b | 0.12 | ||||||
| Canada/United States | 125 | 48.1 | 135 | 51.9 | 260 | 86.7 | |
| Asia (South and Southeast) | 9 | 75.0 | 3 | 25.0 | 12 | 4.0 | |
| Africa (Central and South Africa) | 2 | 100.0 | 0 | 2 | 0.7 | ||
| Europe | 2 | 20.0 | 8 | 80.0 | 10 | 3.3 | |
| Middle East | 2 | 100.0 | 0 | 2 | 0.7 | ||
| South America | 2 | 50.0 | 2 | 50.0 | 4 | 1.3 | |
| Central America/Caribbean | 4 | 80.0 | 1 | 20.0 | 5 | 1.7 | |
| Mexico | 4 | 80.0 | 1 | 20.0 | 5 | 1.7 | |
| Educationb | <0.0001 | ||||||
| Some college or associate degree | 12 | 23.1 | 40 | 76.9 | 52 | 17.3 | |
| Bachelor or baccalaureate degree | 64 | 49.2 | 66 | 50.8 | 130 | 43.3 | |
| Some college or professional school (postcollege graduation) | 20 | 71.4 | 8 | 28.6 | 28 | 9.3 | |
| Master's degree | 19 | 57.6 | 14 | 42.4 | 33 | 11.0 | |
| Doctoral degree | 12 | 63.2 | 7 | 36.8 | 19 | 6.3 | |
| Occupation (top three reported)b | <0.0001 | ||||||
| Health care | 16 | 43.2 | 21 | 56.8 | 37 | 12.3 | |
| Research | 28 | 73.7 | 10 | 26.3 | 38 | 12.7 | |
| Student | 83 | 55.3 | 67 | 44.7 | 150 | 50.0 | |
| Insurancec | |||||||
| Has dental insurance | 93 | 66.9 | 124 | 85.5 | 217 | 76.4 | <0.0001 |
| Does not have dental insurance | 46 | 33.1 | 21 | 14.5 | 67 | 23.6 | |
| Has medical insurance | 129 | 92.8 | 136 | 93.8 | 265 | 93.3 | ns |
| Does not have medical Insurance | 10 | 7.2 | 9 | 6.2 | 19 | 6.7 | |
| Tobacco usec | ns | ||||||
| Does not use tobacco products | 143 | 95.3 | 138 | 92.0 | 281 | 93.7 | |
| Uses tobacco products | 7 | 4.7 | 12 | 8.0 | 19 | 6.3 | |
ns, not significant.
Significant difference existed between subject's mother's and father's birth country in association with site of enrollment (data not shown).
For birth region/country, education, and occupation, the table shows the distribution by clinical site of each characteristic (%) and the representation of each characteristic in the total study population (% total study population).
For insurance and tobacco use, the table shows the percentage of respondents at each site who reported the characteristic (%) and the percentage of respondents study-wide who reported the characteristic (% total study population).
Tables 2 and 3 display demographic characteristics and physical findings in additional subjects, indicating significant differences between clinical sites associated with race (P<0.0001, Fisher's exact test), ethnicity (P=0.0002, χ2 test), and diastolic BP (P=0.0002, t test or Kruskal-Wallis test), and the suggestion of a difference associated with age (P=0.04, Kruskal-Wallis test). Given our initial observations on potential bias by bivariate analyses, we used multivariate analyses and a logistic regression model to assess a possible association between clinical site (BCM or WUSTL) and selected participant characteristics: age, race, ethnicity, BMI, and diastolic and systolic BP. The logistic regression model (data not shown) used forward and backward selection with P = 0.20 for covariate entry or retention into the model. In both logistic regression approaches, race (all five categories), ethnicity, and diastolic BP entered or stayed in the model, with final P ≤ 0.003; age was not retained in the model. Taken together, these analyses suggest that the participant populations at the two clinical sites differed with respect to race, ethnicity, and diastolic BP.
Table 2.
Bivariate analysis of relationship of clinical site of enrollment and subject race and ethnicity
| Subject characteristic | Clinical enrollment site |
Total, n = 300 |
P | ||||
|---|---|---|---|---|---|---|---|
| BCM, n = 150 |
WUSTL, n = 150 |
||||||
| n | % | n | % | n | % total study population | ||
| Race | <0.0001 | ||||||
| Asian | 26 | 17.3 | 4 | 2.7 | 30 | 10.0 | |
| Black/African American | 9 | 6.0 | 10 | 6.7 | 19 | 6.3 | |
| White | 99 | 66.0 | 133 | 88.7 | 232 | 77.3 | |
| Multiracial | 9 | 6.0 | 2 | 1.3 | 11 | 3.7 | |
| Other/unknown | 7 | 4.7 | 1 | 0.7 | 8 | 2.7 | |
| Ethnicity | 0.0002 | ||||||
| Non-Hispanic | 124 | 82.7 | 144 | 96.0 | 268 | 89.3 | |
| Hispanic | 26 | 17.3 | 6 | 4.0 | 32 | 10.7 | |
ns, not significant.
Table 3.
Bivariate analysis of relationship of clinical site of enrollment and subject physical findings
| Subject physical finding | Clinical enrollment site |
Total, n = 300 | Kruskal-Wallis | |
|---|---|---|---|---|
| BCM, n = 150 | WUSTL, n = 150 | |||
| BMI (kg/m2) | 24.3 | 24.5 | 24.4 | ns |
| Diastolic BP (mmHg) | 73.7 | 69.9 | 71.4 | 0.0002 |
| Systolic BP (mmHg) | 121.0 | 118.6 | 119.8 | ns |
| Age (yr) | 26.8 | 25.8 | 26.3 | 0.04 |
Values are means. ns, not significant.
To objectively measure the strength of our protocol in generating highly concordant subject cohorts (with minimal bias based on factors such as study site), we analyzed microbial community data for each separate site within the oral cavity, nares, skin, and gastrointestinal tract, calculating significant associations of sex with microbial clades (Fig. 2). Fifteen linear models were built to associate each genus or higher taxonomic clade (total features=981) in the hierarchically binned 16S V3-5 OTU table (phylotypes “pty”; http://hmpdacc.org/HMQCP) with 5 potential confounders (clinical recruitment site, sequencing center, input DNA concentration, sequencing depth, and study day processed) and with sex. Based on criteria recommended for handling sequence artifacts (39), relative abundance of features that were ≥0.01% in ≥10% of samples per body site were arcsin square root-transformed and tested for associations, and the resulting P values were false discovery rate (FDR)-corrected (before prefiltering) as Benjamini-Hochberg q values. As demonstrated in Fig. 2, we observe that microbial communities were both body site- and sex-specific, but variance in our data is not significantly associated with clinical recruitment site, sequencing center, input DNA concentration, sequencing depth, or study day processed. Further extensive evidence of data robustness is presented in the HMP Consortium manuscripts (9, 10).
Figure 2.

Host-associated microbial organisms correlate with sex. A selection of taxonomic clade abundances (raw relative abundance in percentage) that are most significantly (all FDR q<0.2) associated with sex across several different body sites is presented. A total of 981 features (genus, family, order, class, and phylum) were tested for each body site. At the top, those body sites with significantly different α diversity values between men and women are shown. HP, hard palate; TD, tongue dorsum; RC, retroauricular crease; BM, buccal mucosa). A complete list of significant associations is included in ref. 9.
DISCUSSION
We describe herein how we completed the most comprehensive clinical study to date for evaluating the human microbiome. We further identified logistic challenges with specimen collection, such as human DNA contamination, and addressed these by optimizing our protocols. Our study's emphasis on minimal perturbation of the microbial environment was important in determining the composition of the human microbiome in relatively healthy adults and in maximizing the ability to detect diversity of the microbial population between individuals and between different body sites in and on a single person (Tables 1–3 and Fig. 2). Collection of specimens from different sites in the oral cavity and different locations in the vagina presented the opportunity to detect diversity within a single anatomic site and allowed more analyses per study participant. The study's repeat sample collections provide the foundation for limited analysis of temporal variation in an individual's microbiome.
The aim of our study was to create a reference cohort using rigorous screening, enrollment, and sampling criteria applied consistently across study sites (Tables 1–3). Our intent was not to characterize the entire human microbiome from a population-wide perspective but rather to gain a glimpse of the composition of minimally perturbed microbiomes in a reference collection of human specimens. The data presented here and in our companion articles (9–11) support the validity of our cohort as the source of a reference metagenomic data set for this and future studies. Consistent with early evidence from others (29, 40–42), our data show that differences may be correlated with human phenotypic differences such as sex (Fig. 2) and race or ethnicity (9). Although the small sample size and the differences between the Houston and St. Louis cohorts with respect to these characteristics may confound the analysis (Tables 1–3), variance in our data was not significantly associated with potential confounders related to the multisite study structure. We observed distinct differences in microbial communities associated with body site and sex, but minimal variance within subjects of a single sex (Fig. 2), suggesting that future metagenomic studies should, at a minimum, incorporate sex stratification in study analyses.
We completed in-person screening for nearly twice as many individuals as were ultimately enrolled (Supplemental Fig. S1). This outcome emphasizes the stringent screening criteria and the significant resources required for this study and for future metagenomic studies. Exclusions for oral health issues were common in the young, otherwise healthy, adults targeted in this study. Recent data indicate that only 67 and 43% of young adult whites and Hispanics, respectively, have visited a dentist within 1 yr, and lack of insurance correlates with infrequent dental visits (43). A small percentage of subjects with no history of hypertension had mild elevations in screening BP. It is possible that some of these subjects were truly hypertensive, but a single BP measurement is not adequate to determine this diagnosis.
Environmental, genetic, and physiological factors may affect the composition of the human microbiome, and future studies should address these influences. In this study, a postsampling questionnaire enabled investigators to acquire basic dietary information. In the future, it may be useful to prospectively collect more detailed dietary histories, using food frequency questionnaires or 3-d food records, to allow habitual diet or dietary changes to be correlated with microbial composition and function. Physiological and immune effects due to routine surgery should be considered in future studies. Although prior appendectomy was not an exclusion criterion for our study's healthy population, some research has indicated that the appendix may influence the composition of the microbiome (44). The current study excluded pregnant women, but body site sampling completed before and during pregnancy and during the postpartum interval could add to our understanding of this profound alteration in the physiology of a healthy adult. Future studies could assess the affect of exogenous hormones on the microbiome by sampling postmenopausal women who receive hormone replacement therapy. In future research, changes over time in basic physiological features such as weight and body metabolism could be studied to assess their effects on the microbiome.
In the future, human microbiomes will be defined at many body sites and during different periods in the human life span. A metagenomic survey alone can describe the microbial communities present, but clinical data are required to understand the factors that affect community composition. Studies must consider the role of sex, diet, race/ethnicity, age, residence location, use of medications, dietary supplements, and hygiene products, and many other factors that shape and cause fluctuations in individual microbiomes. Current NIH-funded demonstration projects are exploring differences in microbial communities and whole metagenomes in disease states. Our study was designed and executed using rigorous quality standards typical of randomized controlled trials, setting the standard for future human microbiome research and providing access to a solid reference data set for the health-associated microbiome.
Supplementary Material
Acknowledgments
The authors thank the staff directly involved in clinical evaluation of subjects, human body sampling, and specimen processing. At Baylor College of Medicine (BCM), the staff included Chanei Henry, Michelle Rubio-Gonzales, Janet Wells, V. A. Mancha, Joanna Allaire, Kavitha Parthasarathy, Jayne McWherter, Antone Opekun, Coni Cheesman, Shannon Hawkins, Matthew Ross, Tulin Ayvaz, Bonnie Youmans, and Yue Shang. At Washington University in St. Louis (WUSTL), the staff included Sally Anderson, Shea Roesel, Linda Ventimiglia, Debra Kemp, Teresa Arb, Arlyn Pittler, Cindy Neske, Laura Granderson, Beth Reagan, Amy Brink, Nicole Gaudin, Emily Rozycki, and Amanda Alyatim. At St Louis University, the staff included Mary Signorino and Diane Collier. The authors are grateful to Dr. Sarah Highlander (BCM) for assisting with protocol development for DNA isolation and sample processing, and to Dr. Curtis Huttenhower (Harvard School of Public Health) for generating essential data used in Fig. 2.
The authors gratefully acknowledge the support of the Human Microbiome Project funded through the U.S. National Instiutes of Health (NIH) Director's Common Fund (as part of NIH RoadMap 1.5). The genome centers participating in this study were funded by direct support from the National Human Genome Research Institute at NIH (U54HG004973, BCM; and U54HG004968, WUSTL).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BCM
- Baylor College of Medicine
- BMI
- body mass index
- BP
- blood pressure
- CSOC
- clinical study oversight committee
- dbGaP
- Database of Genotypes and Phenotypes
- FDR
- false discovery rate
- HMP
- Human Microbiome Project
- MOP
- manual of procedures
- NHGRI
- National Human Genome Research Institute
- NIH
- U.S. National Institutes of Health
- OTU
- operational taxonomic unit
- rRNA
- recombinant RNA
- WGS
- whole-genome shotgun
- WUSTL
- Washington University in St. Louis
REFERENCES
- 1. Ley R. E., Peterson D. A., Gordon J. I. (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848 [DOI] [PubMed] [Google Scholar]
- 2. Turnbaugh P. J., Ley R. E., Hamady M., Fraser-Liggett C. M., Knight R., Gordon J. I. (2007) The human microbiome project. Nature 449, 804–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ley R. E., Hamady M., Lozupone C., Turnbaugh P. J., Ramey R. R., Bircher J. S., Schlegel M. L., Tucker T. A., Schrenzel M. D., Knight R., Gordon J. I. (2008) Evolution of mammals and their gut microbes. Science 320, 1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ley R. E., Lozupone C. A., Hamady M., Knight R., Gordon J. I. (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pamer E. G. (2007) Immune responses to commensal and environmental microbes. Nat. Immunol. 8, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 6. Hooper L. V., Wong M. H., Thelin A., Hansson L., Falk P. G., Gordon J. I. (2001) Molecular analysis of commensal host-microbial relationships in the intestine. Science 291, 881–884 [DOI] [PubMed] [Google Scholar]
- 7. Petrosino J. F., Highlander S., Luna R. A., Gibbs R. A., Versalovic J. (2009) Metagenomic pyrosequencing and microbial identification. Clin. Chem. 55, 856–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gill S. R., Pop M., Deboy R. T., Eckburg P. B., Turnbaugh P. J., Samuel B. S., Gordon J. I., Relman D. A., Fraser-Liggett C. M., Nelson K. E. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. The Human Microbiome Project Consortium (2012) Structure, function and diversity of the human microbiome in an adult reference population. Nature 486, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. The Human Microbiome Project Consortium (2012) A framework for human microbiome research. Nature 486, 215–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aagaard K. M., Riehle K., Ma J., Segata N., Mistretta T. A., Coarfa C., Raza S., Rosenbaum S., Van den Veyver I., Milosavljevic A., Gevers D., Huttenhower C., Petrosino J., Versalovic J. (2012) A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS One 7, e36466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jumpstart Consortium Human Microbiome Project Data Generation Working Group (2012) Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One 7, e39315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zoetendal E. G., Akkermans A. D., De Vos W. M. (1998) Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl. Environ. Microbiol. 64, 3854–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eckburg P. B., Bik E. M., Bernstein C. N., Purdom E., Dethlefsen L., Sargent M., Gill S. R., Nelson K. E., Relman D. A. (2005) Diversity of the human intestinal microbial flora. Science 308, 1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown C. J., Wong M., Davis C. C., Kanti A., Zhou X., Forney L. J. (2007) Preliminary characterization of the normal microbiota of the human vulva using cultivation-independent methods. J. Med. Microbiol. 56, 271–276 [DOI] [PubMed] [Google Scholar]
- 16. Costello E. K., Lauber C. L., Hamady M., Fierer N., Gordon J. I., Knight R. (2009) Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tap J., Mondot S., Levenez F., Pelletier E., Caron C., Furet J. P., Ugarte E., Muñoz-Tamayo R., Paslier D. L., Nalin R., Dore J., Leclerc M. (2009) Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 11, 2574–2584 [DOI] [PubMed] [Google Scholar]
- 18. Qin J., Li R., Raes J., Arumugam M., Burgdorf K. S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., Mende D. R., Li J., Xu J., Li S., Li D., Cao J., Wang B., Liang H., Zheng H., Xie Y., Tap J., Lepage P., Bertalan M., Batto J. M., Hansen T., Le Paslier D., Linneberg A., Nielsen H. B., Pelletier E., Renault P., Sicheritz-Ponten T., Turner K., Zhu H., Yu C., Li S., Jian M., Zhou Y., Li Y., Zhang X., Li S., Qin N., Yang H., Wang J., Brunak S., Doré J., Guarner F., Kristiansen K., Pedersen O., Parkhill J., Weissenbach J., MetaHIT Consortium, Bork P., Ehrlich S. D., Wang J. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mackie R. I., Sghir A., Gaskins H. R. (1999) Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 69, 1035S–1045S [DOI] [PubMed] [Google Scholar]
- 20. Favier C. F., Vaughan E. E., De Vos W. M., Akkermans A. D. (2002) Molecular monitoring of succession of bacterial communities in human neonates. Appl. Environ. Microbiol. 68, 219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Penders J., Thijs C., Vink C., Stelma F. F., Snijders B., Kummeling I., van den Brandt P. A., Stobberingh E. E. (2006) Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521 [DOI] [PubMed] [Google Scholar]
- 22. Kurokawa K., Itoh T., Kuwahara T., Oshima K., Toh H., Toyoda A., Takami H., Morita H., Sharma V. K., Srivastava T. P., Taylor T. D., Noguchi H., Mori H., Ogura Y., Ehrlich D. S., Itoh K., Takagi T., Sakaki Y., Hayashi T., Hattori M. (2007) Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 14, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Palmer C., Bik E. M., DiGiulio D. B., Relman D. A., Brown P. O. (2007) Development of the human infant intestinal microbiota. PLoS Biol. 5, e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reinhardt C., Reigstad C. S., Backhed F. (2009) Intestinal microbiota during infancy and its implications for obesity. J. Pediatr. Gastroenterol. Nutr. 48, 249–256 [DOI] [PubMed] [Google Scholar]
- 25. Gronlund M. M., Lehtonen O. P., Eerola E., Kero P. (1999) Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. J. Pediatr. Gastroenterol. Nutr. 28, 19–25 [DOI] [PubMed] [Google Scholar]
- 26. Dominguez-Bello M. G., Costello E. K., Contreras M., Contreras M., Magris M., Hidalgo G., Fierer N., Knight R. (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U. S. A. 107, 11971–11975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Turnbaugh P. J., Gordon J. I. (2009) The core gut microbiome, energy balance and obesity. J. Physiol. 587, 4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turnbaugh P. J., Hamady M., Yatsunenko T., Cantarel B. L., Duncan A., Ley R. E., Sogin M. L., Jones W. J., Roe B. A., Affourtit J. P., Egholm M., Henrissat B., Heath A. C., Knight R., Gordon J. I. (2009) A core gut microbiome in obese and lean twins. Nature 457, 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou X., Brown C. J., Abdo Z., Davis C. C., Hansmann M. A., Joyce P., Foster J. A., Forney L. J. (2007) Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 1, 121–133 [DOI] [PubMed] [Google Scholar]
- 30. Forney L. J., Foster J. A., Ledger W. (2006) The vaginal flora of healthy women is not always dominated by Lactobacillus species. J. Infect. Dis. 194, 1468–1469; author reply 1469–1470 [DOI] [PubMed] [Google Scholar]
- 31. Kim T. K., Thomas S. M., Ho M., Sharma S., Reich C. I., Frank J. A., Yeater K. M., Biggs D. R., Nakamura N., Stumpf R., Leigh S. R., Tapping R. I., Blanke S. R., Slauch J. M., Gaskins H. R., Weisbaum J. S., Olsen G. J., Hoyer L. L., Wilson B. A. (2009) Heterogeneity of vaginal microbial communities within individuals. J. Clin. Microbiol. 47, 1181–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ravel J., Gajer P., Abdo Z., Schneider G. M., Koenig S. S., McCulle S. L., Karlebach S., Gorle R., Russell J., Tacket C. O., Brotman R. M., Davis C. C., Ault K., Peralta L., Forney L. J. (2011) Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl. 1), 4680–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao L., Shen J. (2010) Whole-body systems approaches for gut microbiota-targeted, preventive healthcare. J. Biotechnol. 149, 183–190 [DOI] [PubMed] [Google Scholar]
- 34. Morowitz M. J., Denef V. J., Costello E. K., Thomas B. C., Poroyko V., Relman D. A., Banfield J. F. (2011) Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc. Natl. Acad. Sci. U. S. A. 108, 1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lemon K. P., Armitage G. C., Relman D. A., Fischbach M. A. (2012) Microbiota-targeted therapies: an ecological perspective. Sci. Transl. Med. 4, 137rv5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jorup-Rönström C., Håkanson A., Sandell S., Edvinsson O., Midtvedt T., Persson A. K., Norin E. (2012) Fecal transplant against relapsing Clostridium difficile-associated diarrhea in 32 patients. Scand. J. Gastroenterol. 47, 548–552 [DOI] [PubMed] [Google Scholar]
- 37. Grice E. A., Kong H. H., Renaud G., Young A. C., NISC Comparative Sequencing Program, Bouffard G. G., Blakesley R. W., Wolfsberg T. G., Turner M. L., Segre J. A. (2008) A diversity profile of the human skin microbiota. Genome Res. 18, 1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paulino L. C., Tseng C. H., Strober B. E., Blaser M. J. (2006) Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. J. Clin. Microbiol. 44, 2933–2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schloss P. D., Gevers D., Westcott S. L. (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6, e27310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grice E. A., Kong H. H., Conlan S., Deming C. B., Davis J., Young AC, NISC Comparative Sequencing Program, Bouffard G. G., Blakesley R. W., Murray P. R., Green E. D., Turner M. L., Segre J. A. (2009) Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Y., Zhang C., Zhao L., Nardini C. (2010) Adapting functional genomic tools to metagenomic analyses: investigating the role of gut bacteria in relation to obesity. Brief Funct. Genomics 9, 355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arumugam M., Raes J., Pelletier E., Le Paslier D., Yamada T., Mende D. R., Fernandes G. R., Tap J., Bruls T., Batto J. M., Bertalan M., Borruel N., Casellas F., Fernandez L., Gautier L., Hansen T., Hattori M., Hayashi T., Kleerebezem M., Kurokawa K., Leclerc M., Levenez F., Manichanh C., Nielsen H. B., Nielsen T., Pons N., Poulain J., Qin J., Sicheritz-Ponten T., Tims S., Torrents D., Ugarte E., Zoetendal E. G., Wang J., Guarner F., Pedersen O., de Vos W. M., Brunak S., Doré J., and MetaHIT Consortium. (2011) Enterotypes of the human gut microbiome. Nature 473, 174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mulye T. P., Park M. J., Nelson C. D., Adams S. H., Irwin C. E., Jr., Brindis C. D. (2009) Trends in adolescent and young adult health in the United States. J. Adolesc. Health 45, 8–24 [DOI] [PubMed] [Google Scholar]
- 44. Randal Bollinger R., Barbas A. S., Bush E. L., Lin S. S., Parker W. (2007) Biofilms in the large bowel suggest an apparent function of the human vermiform appendix. J. Theor. Biol. 249, 826–831 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.