

Abstract
To investigate the role of GRP78 in adipogenesis and metabolic homeostasis, we knocked down GRP78 in mouse embryonic fibroblasts and 3T3-L1 preadipocytes induced to undergo differentiation into adipocytes. We also created an adipose Grp78-knockout mouse utilizing the aP2 (fatty acid binding protein 4) promoter-driven Cre-recombinase. Adipogenesis was monitored by molecular markers and histology. Tissues were analyzed by micro-CT and electron microscopy. Glucose homeostasis and cytokine analysis were performed. Our results indicate that GRP78 is essential for adipocyte differentiation in vitro. aP2-cre-mediated GRP78 deletion leads to lipoatrophy with ∼90% reduction in gonadal and subcutaneous white adipose tissue and brown adipose tissue, severe growth retardation, and bone defects. Despite severe abnormality in adipose mass and function, adipose Grp78-knockout mice showed normal plasma triglyceride levels, and plasma glucose and insulin levels were reduced by 40-60% compared to wild-type mice, suggesting enhanced insulin sensitivity. The endoplasmic reticulum is grossly expanded in the residual mutant white adipose tissue. Thus, these studies establish that GRP78 is required for adipocyte differentiation, glucose homeostasis, and balanced secretion of adipokines. Unexpectedly, the phenotypes and metabolic parameters of the mutant mice, which showed early postnatal mortality, are uniquely distinct from previously characterized lipodystrophic mouse models.—Zhu, G., Ye, R., Jung, D. Y., Barron, E., Friedline, R. H., Benoit, V. M., Hinton, D. R., Kim, J. K., Lee, A. S. GRP78 plays an essential role in adipogenesis and postnatal growth in mice.
Keywords: conditional-knockout mouse model, aP2-Cre-recombinase, adipokine secretion
Adipose tissue plays a significant role in energy balance and glucose homeostasis, as it responds to nutrient, neural, and hormonal signals and secretes adipokines that influence feeding, thermogenesis, immunity, and neuroendocrine function (1). There are two types of adipose tissues, white adipose tissue (WAT), and brown adipose tissue (BAT). WAT acts mainly to store excess energy in the form of triglycerides (TGs) and serves as a major energy reservoir in mammals. In addition, WAT functions as an endocrine organ, as it integrates metabolic signals and regulates energy balance by secreting large amounts of peptides and inflammatory mediators, such as leptin, adiponectin, resistin, and tumor necrosis factor α (TNF-α) (1). In contrast, BAT plays a thermoregulatory role by generating nonshivering body heat. Obesity, which is rapidly becoming a major health disorder in the developed world, is characterized by increased mass of WAT due to imbalance between energy expenditure and uptake. Thus, understanding the molecular moieties required for adipogenesis will be important toward the development of effective preventive and therapeutic strategies to combat the obesity pandemic.
The endoplasmic reticulum (ER) is a specialized perinuclear organelle where secretory and membrane proteins, as well as lipids, are synthesized. Within the lumen of the ER, protein chaperones assist in folding of newly synthesized polypeptides and prevent aggregation of unfolded or misfolded protein (2). The most abundant ER chaperone is the 78-kDa glucose regulated protein (GRP78), which is also referred to as BiP/HSPA5. In addition to maintaining ER homeostasis, GRP78 is required for the integrity of the ER structure (3). GRP78 also serves as a master regulator of the unfolded protein response (UPR) that senses ER stress and mounts adaptive responses (4). During ER stress, when the protein load exceeds the protein-folding capacity of the ER, GRP78 is induced and assists in transporting misfolded proteins in the ER lumen to the cytosol for proteasome-mediated degradation (5). Cells that are specialized for high secretory capacity, such as pancreatic β cells and plasma cells, are known to expand their ER capacities to adapt to the increased demand in protein folding (6–8). Differentiated adipocytes, as potent endocrine cells, also exhibit high secretory activity (1, 9). Biosynthesis and secretion of large amounts of adipocytokines require molecular chaperones and folding enzymes in the ER. Thus, GRP78 may be a key determinant for adipocyte differentiation and a potential therapeutic target for obesity.
To directly investigate the function of GRP78 in vivo, we created mouse models with knockout and floxed alleles of Grp78. Homozygous knockout of Grp78 results in the reduction of embryonic cell proliferation and massive apoptosis of the inner cell mass, leading to embryonic lethality at d 3.5 (10). In adult tissues, conditional knockout of GRP78 in Purkinje cells in mice led to growth retardation and neuronal dysfunction (3). Interestingly, while specific deletion of GRP78 in the prostate epithelial cells potently suppressed prostate tumorigenesis, the prostate epithelium is phenotypically normal (11). Thus, these initial studies suggest that in adult mice, the requirement of GRP78 for cell survival appears to be cell-type specific and content dependent, although this remains to be determined. In the present study, to understand the role of GRP78 in adipogenesis, we knocked down GRP78 expression in mouse embryonic fibroblasts (MEFs) and 3T3-L1 cells induced to undergo adipocyte differentiation. We also created Grp78 floxed mice harboring the fatty acid binding protein 4 (aP2)-Cre-recombinase gene, which has been extensively used to generate conditional adipose selective inactivation of genes in both WAT and BAT during mouse development (12). Here, we report that GRP78 function is essential for adipogenesis and production and secretion of adipokines. Unexpectedly, we observed phenotypes and metabolic parameters of the aP2-Cre; Grp78f/f mice, referred to below as the c78f/f mice, uniquely distinct from previously characterized lipodystrophic mouse models. The implications of these results on the specificity of the aP2-Cre system and novel requirement of GRP78 function in the affected tissues will be discussed.
MATERIALS AND METHODS
Cell culture and induction of adipocyte differentiation
Primary MEFs were prepared from E12.5–E14.5 embryos of Grp78f/f and Grp78+/+ mice, as described previously (13). The primary Grp78f/f or wild-type Grp78+/+ MEFs of passage 3 were seeded into multiwell culture plates and infected with Ad5-CMV-Cre-GFP (Vector Development Laboratory, Baylor College of Medicine, Houston, TX, USA) at 95% confluency. The next day (d 0), the infected cells were induced to differentiate into adipocytes with the stimulation medium: DMEM (with 4.5 mg/ml glucose) supplemented with 10% FBS, 1% penicillin-streptomycin, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX; Sigma-Aldrich, St. Louis, MO, USA), 1 μM dexamethasone (Sigma-Aldrich), 10 μg/ml insulin (Sigma-Aldrich), and 5 μM rosiglitazone (Cayman Chemical, Ann Arbor, MI, USA). On d 2, the medium was replaced by poststimulation medium (DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, 5 μg/ml insulin, and 5 μM rosiglitazone). Fresh poststimulation medium was replaced every 2 d thereafter.
3T3-L1 preadipocytes (passage 7; gift of Dr. My Chouinard, University of Massachusetts Medical School, Worcester, MA, USA) were cultured in complete medium (DMEM with 10% FBS and 1% penicillin-streptomycin) and induced to differentiate into adipocytes, as described previously (14). Briefly, 3 d after 100% confluency (d 0), 3T3-L1 preadipocytes were cultured in complete medium supplemented with 0.5 mM IBMX, 0.25 μM dexamethasone, and 10 μg/ml insulin for 2 d. On d 2, the medium was replaced by insulin medium (complete medium supplemented with only 10 μg/ml insulin). Fresh insulin medium was replaced every 2 d thereafter.
Oil Red O staining
Cells were fixed with 10% buffered formalin for 1 h, washed 3 times by dH2O, and incubated with 60% isopropanol for 5 min. The cells were then air dried and stained with freshly prepared Oil Red O working solution [0.3% Oil Red O (Sigma-Aldrich) and 60% isopropanol] for 3 min, and washed with water 3 times.
Lentiviral shRNA infection
Nonsilencing-GIPZ lentiviral shRNAmir control (Open Biosystems, Lafayette, CO, USA) and GRP78 shRNA (h2) lentivirus [cat. no. sc-44261-V, Santa Cruz Biotechnology, Santa Cruz, CA, USA; gift of Dr. Yvonne Lin, Keck School of Medicine, University of Southern California (USC), Los Angeles, CA, USA] were used to infect 3T3-L1 cells. The GRP78 shRNA (h2) lentivirus particles consist of a pool of three sequences targeting human Grp78 mRNA, one of which crossreacts with mouse Grp78 mRNA. 3T3-L1 cells in passage 7 were seeded into 48-well plates at 50% confluency. The next day, the cells were pretreated with 250 μl DMEM with polybrene (4 μg/ml, Sigma-Aldrich) for 5 min. Then, the medium was replaced by fresh DMEM, followed by addition of lentivirus (MOI=2). After 24 h, the medium was replaced with complete medium. The infected cells were cultured in 48 wells for 2 d and reached 100% confluency. At 3 d after 100% confluency (d 0), 3T3-L1 preadipocytes were induced to differentiate as described above.
Real-time PCR
Total RNA was isolated from MEFs using TRIzol reagent (Invitrogen, Burlington, ON, Canada) following the manufacturer's instructions. First-strand cDNA was synthesized with SuperScript II (Invitrogen), as described previously (15). Primer sequences are as follows: sterol regulatory element binding protein 1c (SREBP-1c) F, 5′-GTTACTCGAGCCTGCCTTCAGG-3′; SREBP-1c R, 5′-CAAGCTTTGGACCTGGGTGTG-3′; peroxisome proliferator-activated receptor γ2 (PPAR-γ2) F, 5′-TCTGGGAGATTCTCCTGTTGA-3′; PPAR-γ2 R, 5′-GGTGGGCCAGAATGGCATCT-3′; aP2 F, 5′-GAATTCGATGAAATCACCGCA-3′; aP2 R, 5′-CTCTTTATTGTGGTCGACTTTCCA-3′; 18S rRNA F, 5′-ACGGCCGGTACAGTGAAAC-3′; and 18S rRNA R, 5′-GAGGGAGCTCACCGGG-3′.
Generation of adipose conditional-knockout mice
The generation of Grp78f/f mice was described previously (10, 11). Male aP2-Cre transgenic mice (C57BL/6 background; Jackson Laboratory, Bar Harbor, ME, USA), were mated with female Grp78f/f mice (C57/129 background) to generate aP2-Cre; Grp78f/+ offspring. Breeding between Grp78f/f mice and aP2-Cre; Grp78f/+ mice yielded aP2-Cre; Grp78f/f and Grp78f/f mice used in the experiments. Both male and female mice were analyzed. All protocols for animal use and dissection were reviewed and approved by the University of Southern California Institutional Animal Care and Use Committee. Genotyping of Grp78 WT, floxed, and KO alleles was performed with genomic DNA extracted from mouse tails, adipose tissue, and liver as described previously (11).
Tissue processing and histological analysis
Mouse tissues or organs were isolated and fixed in 10% buffered formalin for ≥24 h before embedding. Paraffin-embedded tissues were sectioned and stained with hematoxylin and eosin (H&E) for histological evaluation. Tissues for immunoblotting were put into liquid nitrogen immediately after isolation and stored at −80°C.
Immunoblotting
Tissues or cells were homogenized in ice-cold RIPA buffer containing cocktails of proteinase inhibitors and phosphatase inhibitors (Pierce, Rockford, IL, USA), following protocol, as described previously (16). Western blot was performed by standard protocol (16). Primary antibodies used were mouse monoclonal anti-GRP78 antibody (1:1000; gift of Dr. Parkash Gill, Keck School of Medicine, USC), mouse monoclonal anti-β-actin antibody (1:5000; Sigma-Aldrich) and rabbit polyclonal anti-XBP-1 antibody (1:500; Santa Cruz Biotechnology).
Electron microscopy and analysis
Freshly dissected WAT was subjected to electron microscopy, as described previously (8). Briefly, the tissue was fixed in half-strength Karnovsky's fixative for 24 h at 4°C, then postfixed in 1% osmium tetroxide for 2 h on ice. The samples were dehydrated in serially graded ethanol and then infiltrated in Eponate prior to embedding. Ultrathin sections were cut at a thickness of 70 nm and stained with uranyl acetate and lead citrate. The sections were examined on a Jeol JEM 2100 (Jeol USA, Peabody, MA, USA) and photographed with the Orius SC1000B digital camera (Gatan, Pleasanton, CA, USA).
Micro-CT imaging of femurs and fat
The preparation of femurs and scanning procedure have been described previously (17). Micro-CT scanning of femurs was performed with Inveon CT scanner (Siemens, Los Angeles, CA, USA). For fat imaging, mice under deep anesthesia were scanned in the micro-CT scanner with optimized scan energy and voxel size, as described previously (18, 19). Scanning freshly harvested WAT and BAT identified the upper and lower thresholds that separated adipose tissue from tissues with higher or lower density and fluids (18). Fat compartments in the abdominal region (lumbar 1 to lumbar 5) were quantified using Amira software (18, 19).
Measurement of body composition and plasma parameters
The following studies were performed at the University of Massachusetts Medical School and approved by the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. Blood was collected from retroorbital bleeding in Microtainer plasma separator tubes with lithium heparin (Becton Dickinson, Franklin Lakes, NJ, USA), and plasma was prepared. Plasma hormones, cytokines, and lipid levels were measured using Luminex and Cobas Clinical Chemistry Analyzer (Roche Diagnostics, Indianapolis, IN, USA).
Statistical analysis
A 2-tailed Student's t test was applied for all pairwise comparisons. Data are expressed as mean ± se. Cochran-Armitage test for trend was performed to examine the association between the percentage of embryos and postnatal progenies carrying the aP2-Cre; Grp78f/f genotype and age.
RESULTS
GRP78 is required for adipocyte differentiation in vitro
To assess the role of GRP78 in adipogenesis in vitro, several approaches were used. First, we compared the ability of GRP78-knockout MEFs and wild-type MEFs to differentiate into adipocytes. To achieve this, we infected MEFs carrying either the wild-type Grp78+/+ alleles or the Grp78 floxed alleles with adenovirus containing the Cre-recombinase gene (Adeno-Cre). MEFs carrying the Grp78+/+ alleles were not affected by the Adeno-Cre, and as the cells differentiated into adipocytes on hormonal stimulation, a gradual increase in GRP78 protein level was observed (Fig. 1A, B). In contrast, when the Grp78 alleles were deleted from the genome of the Grp78f/f MEFs by Adeno-Cre, the GRP78 protein level was nearly depleted and the GRP78-deficient MEFs showed significant repression of adipocyte differentiation under the same conditions (Fig. 1A, B). In parallel with GRP78 induction, Grp78+/+ MEFs showed gradual increase in a transcription factor, the spliced X-box binding protein 1 (XBP-1s) protein level, consistent with the previous finding that XBP-1s is essential for adipocyte differentiation (20). Correspondingly, XBP-1s level was reduced in the GRP78-deficient MEFs with impaired adipogenesis (Fig. 1A). We noted that after d 10 of Adeno-Cre treatment, a low level of GRP78 and more XBP-1s were detected in the Grp78f/f MEFs, likely due to proliferation of cells that escaped Cre-recombination.
Figure 1.
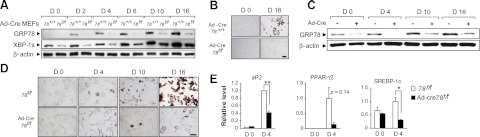
GRP78 is required for adipogenesis from embryonic fibroblasts. A) Grp78+/+ and Grp78f/f MEFs were infected with adenovirus-Cre (Ad-Cre) and induced to differentiate into adipocytes at d 0. Western blots were performed to detect the levels of GRP78 and XBP-1s, with β-actin serving as loading control. B) Oil Red O staining of MEFs treated in A on d 0 and 16. C) Western blot detection of GRP7 and β-actin levels in Grp78f/f MEFs either noninfected or infected with Ad-Cre. D) Oil Red O staining of MEFs treated in C on the days indicated. E) Real-time-PCR analysis of adipocyte marker genes aP2, PPAR-γ2, and SREBP-1c in MEFs treated in C. Expression levels of each gene were normalized to the levels of 18S rRNA. The experiment was repeated twice, in duplicate each time. Data are presented as means ± se. Scale bars = 50 μm. *P < 0.05; **P < 0.01.
Second, to exclude the possibility that the differences in adipocyte differentiation may be due to intrinsic differences between the Grp78+/+ and Grp78f/f MEFs, the induction experiment was repeated by comparing Grp78f/f MEFs with and without infection of Adeno-Cre, or the same MEFs infected with an inactive Adeno-Cre. As shown in Fig. 1C, D, Grp78 f/f MEFs with intact GRP78 were able to differentiate into adipocytes, in parallel with an increased GRP78 level. In the Adeno-Cre-infected cells, GRP78 was efficiently knocked out, and as observed above, by d 10, a low level of GRP78 reappeared. The GRP78-deficient MEFs showed greatly attenuated differentiation into adipocytes, as compared to noninfected cells or cells infected with inactive Adeno-Cre (Fig. 1D and data not shown). In agreement with impairment of adipogenesis, the transcript levels of adipogenesis markers, such as aP2, PPAR-γ2, and SREBP-1c, were all reduced in the GRP78-deficient MEFs at d 4 following hormonal stimulation (Fig. 1E). Thus, GRP78 is required for the induction of adipocyte differentiation in MEFs.
Third, we examined the effect of GRP78 knockdown on adipocyte differentiation in cells already committed to the adipocyte lineage. To test this, 3T3-L1 preadipocytes were infected with either lentivirus targeting GRP78 (sh-GRP78) or lentivirus sh-control (sh-CON), followed by induction of adipocyte differentiation. As shown in Fig. 2A, GRP78 level was gradually upregulated with the differentiation of 3T3-L1 preadipocytes into adipocytes. 3T3-L1 infected with sh-GRP78 exhibited around 70% lower level of GRP78 compared to sh-CON treated cells (Fig. 2B, C). We observed prominent attenuation of adipogenesis with reduced number of both small and large fat droplets on d 4 in the sh-GRP78-treated cells; and the reduction in large fat droplets persisted through d 8 (Fig. 2D, E). Thus, these experiments demonstrate that knockdown of GRP78 significantly delayed adipocyte differentiation from preadipocytes in vitro.
Figure 2.
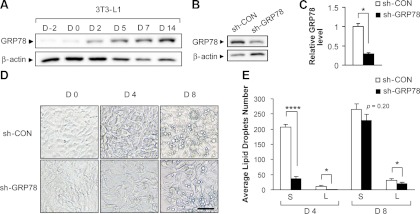
Knockdown of GRP78 suppresses adipocyte differentiation in 3T3-L1 preadipocytes. A) 3T3-L1 cells were induced to differentiate into adipocytes on d 0, and GRP78 and β-actin levels were analyzed by Western blot for the days indicated. B) 3T3-L1 cells infected with either lentivirus sh-control (sh-CON) or sh-GRP78. The levels of GRP78 and β-actin were detected by Western blot on d 8 after induction of differentiation. C) Quantitation of GRP78 level after normalization to the β-actin level in the lentivirus-infected 3T3-L1 cells in B. Data are presented as means ± se. *P < 0.05. D) Morphology of the lentivirus-infected 3T3-L1 cells undergoing adipogenesis was examined by light microscopy (Nikon Eclipse TS100) on the days indicated. Fat droplets were visible starting from d 4. Scale bar = 100 μm. E) Quantitation of number of small (S) and large (L) lipid droplets in differentiating 3T3-L1 cells at d 4 and 8. Mean level in each group was determined by 4 randomly selected areas on the image. Data are presented as means ± se. *P < 0.05; ****P < 0.0001.
Adipose deletion of Grp78 gene leads to lipodystrophy
To examine the role of GRP78 in adipogenesis in vivo, we created an adipose-knockout mouse model of GRP78. This was achieved by crossing the Grp78 floxed mice with the aP2-Cre; Grp78f/+ mice to generate the aP2-Cre; Grp78f/f (c78f/f) mice, with sibling Grp78f/f (78f/f) mice without the Cre-transgene serving as wild-type controls. The Cre recombinase-mediated GRP78 knockout in adipose tissue was confirmed by genotyping. Using the genomic DNA extracted from tissues of 78f/f and c78f/f mice, we detected the Grp78 knockout alleles in WAT and BAT of the c78f/f mice, but not in the liver of the same mice or the same tissues in the 78f/f mice (Fig. 3A). Compared to the 78f/f mice, the c78f/f mice displayed ∼50% smaller body size and 90% reduction in the amount of gonadal WAT and BAT (Fig. 3B). Micro-CT scan analysis further revealed severe reduction of WAT located in the abdomen and beneath the skin and BAT located in the lower neck (Fig. 3C). Quantification of volume of WAT between abdominal L1 to L5 of the c78f/f and 78f/f mice (69 and 724 mm3, respectively) showed a 90% reduction in the c78f/f mice (Fig. 3C).
Figure 3.
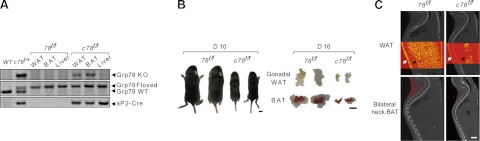
Adipose deletion of Grp78 leads to lipoatrophy. A) Representative PCR genotyping results from WAT (gonadal) and BAT and liver isolated from the 78f/f and c78f/f mice, using the tail DNA from the indicated genotypes as controls. B) Representative images of 78f/f and c78f/f mice at postnatal d 10 (left panel) and gonadal WAT and BAT from the 78f/f and c78f/f mice at postnatal d 16 (right panel). C) Micro-CT scout views comparing the adiposity of 78f/f and c78f/f mice at postnatal d 23. WAT was visualized as yellow dots (top panels) and BAT as red dots (bottom panels). White arrows indicate the locations of subcutaneous WAT and black arrows locations of abdominal WAT. Scale bars = 5 mm (B); 3 mm (C).
Residual WAT from c78f/f mice shows reduced lipid accumulation and grossly dilated ER
As noted above, postnatal c78f/f mice only had ∼10% the amount of WAT and BAT compared to the 78f/f mice. Western blot analysis of the residual WAT and BAT showed GRP78 was knocked down in the c78f/f mice, and the reduction was ∼60% for both types of adipose tissues (Fig. 4A). Histological examination using H&E staining revealed that the subcutaneous WAT between skin and muscle layer was ∼70 to 80% thinner in the c78f/f mice compared to the 78f/f control (Fig. 4B). Histological analysis of gonadal WAT showed a decrease in lipid accumulation in the residual adipocytes from the c78f/f mice compared to the 78f/f mice, suggesting that GRP78 depletion blunted lipogenic capacity in adipose tissue (Fig. 4C). Since GRP78 is a major ER chaperone protein, we next examined the morphology of the ER in the residual gonadal WAT of c78f/f mice. We observed that the WAT of the c78f/f mice showed grossly expanded ER lumen structure, indicative that GRP78 is required for the integrity of ER in WAT (Fig. 4D).
Figure 4.
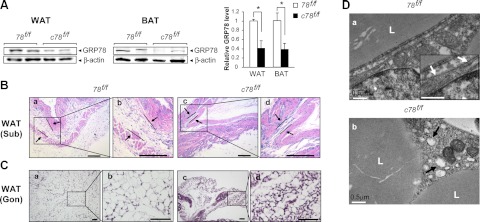
Residual white adipocytes from c78f/f mice display reduced lipid accumulation and dilated endoplasmic reticulum. A) Left panel: representative Western blot detection of GRP78 level in residual WAT and BAT isolated from 78f/f and c78f/f mice, with β-actin serving as loading control. Right panel: quantitation of GRP78 level after normalization to the β-actin level (n=3/genotype). Data are presented as means ± se. *P < 0.05. B) H&E staining of subcutaneous WAT tissues in 78f/f and c78f/f mice at postnatal d 23 at low magnification (a, c) and at high magnification (b, d) for the boxed regions. Arrows denote the boundaries of subcutaneous WAT. C) H&E staining of gonadal WAT from 78f/f and c78f/f mice at postnatal d 16 at low magnification (a, c) and at high magnification (b, d) for the boxed regions. D) Representative electron micrographs of white adipocytes from WAT in 78f/f mice (a) and c78f/f mice (b) at postnatal d 16. Inset (a): enlarged view showing normal ER structure, indicated by white arrows. Black arrows (b) indicate examples of expanded ER lumen in the adipocytes of c78f/f mice. L, lipid droplet. Scale bars = 200 μm (B); 50 μm (C); 0.5 μm (D).
aP2-mediated inactivation of the Grp78 gene results in growth retardation, bone reduction, and early mortality
While the aP2-Cre transgene is highly expressed in both WAT and BAT, selective expression in nonadipogenic tissues has been reported (21, 22). In the course of generating the c78f/f mice, we noted that the birth ratio of the c78f/f mice was ∼20% (Table 1), which is only slightly lower than the expected 25% from the breeding scheme. These data suggest that deletion of the Grp78 gene by aP2-Cre recombination only had a minor effect, if any, on mouse embryonic survival. The weight at birth of the c78f/f mice was ∼90% of the 78f/f mice; however, the c78f/f pups showed minimal gain in body weight after birth and early mortality (Table 1 and Fig. 5A). By postnatal days P3 and P15, the percentage of live c78f/f mice dropped to 10.5 and 5.0%, respectively, and by P28, none of the c78f/f mice was viable (Table 1 and Fig. 5A). Biostatistics analysis confirmed that the percentage of embryos and postnatal progenies carrying the aP2-Cre; Grp78f/f genotype decreased with age (Cochran-Armitage Trend Test P<0.001).
Table 1.
Percentage of embryos/mice with the c78f/f genotype from E9.5 to P28
| Age | Progeny (n) | c78f/f mice (n) | c78f/f mice (%) |
|---|---|---|---|
| E9.5–E18.5 | 61 | 12 | 19.7 |
| P2 | 41 | 7 | 17.1 |
| P3 | 38 | 4 | 10.5 |
| P4–P7 | 68 | 8 | 11.8 |
| P8–P14 | 72 | 8 | 11.1 |
| P15–P22 | 181 | 9 | 5.0 |
| P23–P25 | 163 | 6 | 3.7 |
| P26 | 154 | 3 | 1.9 |
| P27 | 147 | 1 | 0.7 |
| After P28 | 160 | 0 | 0 |
E, embryonic day; P, postnatal day. Expected percentage according to the breeding scheme was 25%.
Figure 5.
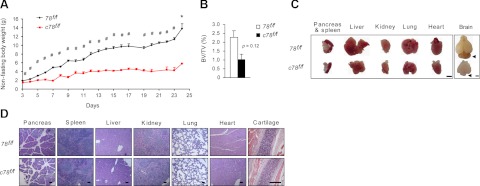
aP2-Cre mediated knockout of Grp78 results in developmental defects. A) Growth curves of 78f/f mice (n=3–7) and c78f/f mice (n=2–9) mice from postnatal d 3 to 24. Data are expressed as means ± se. *P < 0.05; #P < 0.01. B) Quantitation of micro-CT scan of femurs from 78f/f and c78f/f mice at postnatal d 16. Percentage of bone volume (BV) over total volume (TV); data are presented as means ± se. C) Comparison of various organs isolated from 78f/f and c78f/f mice at postnatal d 10. Arrowheads indicate cerebellum in the brain. D) Representative H&E staining of various organs and the cartilage from 78f/f and c78f/f mice at postnatal d 10. Scale bars = 2 mm (C); 200 μm (D).
In view of the reported gene inactivation in bone and cartilage by the aP2-Cre recombinase (23), we performed micro-CT scan of the mouse femurs and observed a 60% reduction in the bone volume of the c78f/f mice compared to the 78f/f mice (Fig. 5B). The pancreas, spleen, liver, kidney, lung, and heart exhibited reduced organ size in proportion to the reduced overall body size of the c78f/f mice (Fig. 5C). These organs showed similar histological morphology in both genotypes (Fig. 5D). Western blot analysis of GRP78 level showed minimal difference in the liver and lung; however, we observed ∼30% reduction (P=0.04) in the kidney of the c78f/f mice (Supplemental Fig. S1A). The cerebellum was disproportionally reduced in the brain of the c78f/f mice (Fig. 5C), corresponding with a reduction (∼50%) of GRP78 in brain and some Purkinje cells in the c78f/f mice (Supplemental Fig. S1B, C). Thus, in addition to severe lipoatrophy, c78f/f mice exhibit other developmental defects that could be related to ectopic aP2-Cre-recombination. These growth abnormalities, coupled with severe lipoatrophy, may be contributing factors to the early mortality of the c78f/f mice.
Conditional knockout of the Grp78 gene by aP2-Cre recombinase alters glucose homeostasis and adipokine secretion
Adipose tissue is an important endocrine organ, which secretes multiple hormones and cytokines that regulate nutrient environment. We examined whether aP2-Cre-mediated GRP78 knockout leads to perturbations in the plasma profile of metabolic parameters. Plasma glucose and insulin levels were significantly lower in c78f/f mice compared to 78f/f mice, suggesting that c78f/f mice are more insulin sensitive (Fig. 6A, B). Circulating C-peptide levels were also reduced by ∼70% in c78f/f mice, suggesting that lower insulin levels are due to reduced insulin secretion, as opposed to altered insulin clearance, in c78f/f mice (Fig. 6C). Plasma glucagon levels were significantly lower in c78f/f mice, consistent with lower basal glucose levels and enhanced insulin sensitivity in these mice (Fig. 6D). Plasma triglyceride levels were normal in c78f/f mice, contrary to other lipodystrophic mouse models that showed elevated plasma lipid profiles (Fig. 6E). Plasma leptin levels were very low in c78f/f mice, which was consistent with their lipodystrophic phenotypes (Fig. 6F). However, leptin level normalized by the volume of WAT showed minimal difference between c78f/f and 78f/f mice, consistent with the notion that the reduced leptin level was mostly due to less WAT (Fig. 6F). Plasma adiponectin level was not altered in c78f/f mice, but resistin level was markedly lower in c78f/f mice (Fig. 6F, G). In that regard, resistin is a well-studied adipokine that negatively regulates insulin action (24). These data indicate that lower resistin levels might be responsible for increased insulin sensitivity in c78f/f mice. Circulating levels of inflammatory cytokines, such as TNF-α and monocyte chemotactic protein-1 (MCP-1), were not affected in the c78f/f mice (Fig. 6G). Plasma glucagon-like peptide 1 (GLP-1) levels were not affected in c78f/f mice (Fig. 6G).
Figure 6.

aP2-Cre mediated knockout of Grp78 alters glucose homeostasis and adipokine secretion. Blood parameters of 78f/f (n=5–9) and c78f/f mice (n=4–11) at postnatal d 16 to 26. A) Feeding glucose level of 78f/f and c78f/f mice. B) Insulin level. C) C-peptide 2 level. D) Glucagon level. E) Triglyceride level. F) Leptin and adiponectin levels, both of which are hormones secreted by adipose tissue. Normalization of leptin to the WAT volume showed minimal difference between the two genotypes. G) Resistin, TNF-α, monocyte chemotactic protein-1 (MCP-1), and glucagon-like peptide-1 (GLP-1) levels. Data are presented as means ± se. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
DISCUSSION
GRP78 is a major chaperone in the ER and is also a regulator of ER stress signaling (2, 25–27); however, its role in adipogenesis is not known. In the present study, we used both cell culture and conditional-knockout mouse models to directly investigate the requirement of GRP78 in adipogenesis. In the tissue culture systems, we observed that depletion of GRP78 in MEFs through deletion of the Grp78 floxed alleles by Adeno-Cre led to impaired adipocyte differentiation and lower expression level of adipogenic genes. This, coupled with the attenuation of adipogenesis in 3T3-L1 preadipocytes treated with lenti-shGRP78 particles, demonstrate that GRP78 is required for adipocyte differentiation from MEFs, as well as 3T3-L1 preadipocytes, which have already committed to be adipocytes. Recently, it was reported that a reduction in the expression of activating transcription factor 6α (ATF6α) in the adipogenic cell line (C3H10T1/2) resulted in impaired expression of key adipogenic genes and reduced lipid accumulation following the induction of adipogenesis (28). Since GRP78 is a major downstream target of ATF6α (29), GRP78 reduction may be a contributing factor. Depletion of GRP78 in MEFs induced to differentiate into adipocytes also correlated with a reduction of the expression of transcription factor XBP-1s. In agreement, XBP1-deficient MEFs and 3T3-L1 cells exhibited defects in adipogenesis (20). To further examine the role of GRP78 in adipogenesis and whole organism metabolism homeostasis, we created an adipose knockout mouse model of GRP78, utilizing the aP2-Cre transgene, which is highly expressed in WAT and BAT and has been widely used for creation of adipose-specific knockout mouse models (12).
The novel mouse model reveals that conditional knockout of the Grp78 alleles using the aP2-Cre-recombinase system caused severe lipoatrophy, growth retardation, and early mortality. These effects were associated with reduced adipocyte size and lipid accumulation, and dilated ER lumen in adipocytes of c78f/f mice. Growth retardation was accompanied by overall developmental defects, and severe hypoglycemia in some c78f/f mice that appeared sickly (data not shown) could lead to early mortality. Despite severe lipoatrophy, c78f/f mice maintained normal plasma triglyceride levels that contrast with other lipodystrophic or lipoatrophic mouse models (30–32). Human and animal models of lipodystrophy have consistently shown profound alterations in lipid metabolism and glucose homeostasis. Highly active antiretroviral therapy (HAART)-associated lipodystrophic subjects develop ectopic fat accumulation and type 2 diabetes (33). Lipodystrophic mice also develop marked hypertriglyceridemia, fatty liver, insulin resistance, and type 2 diabetes (30–32). Mice with targeted expression of an attenuated diphtheria toxin A chain progressively develop adipocyte atrophy and necrosis from 5 mo old and generalized lipodystrophy, insulin resistance, hyperglycemia, dyslipidemia, low leptin levels, and hyperphagia by the age of 9 mo (30). A-ZIP/F-1 transgenic mice express a dominant-negative protein that prevents the DNA binding of B-ZIP transcription factors of both the CCAAT/enhancer binding protein (C/EBP) and Jun families. As a result, these mice displayed dramatically reduced WAT from birth and developed severe hyperglycemia, hyperlipidemia, and hepatomegaly as early as at 3-4 wk of age (31). In aP2-SREBP-1c transgenic mice, which overexpress nSREBP-1c in adipose tissue under the control of the aP2 promoter, showed immature and smaller white adipocytes, marked insulin resistance, hyperglycemia, hyperlipidemia, and fatty liver (32).
In contrast, in the present study, lipoatrophic c78f/f mice did not develop hypertriglyceridemia or hyperglycemia. In fact, c78f/f mice maintained normal plasma lipid levels and lower glucose levels. Lower insulin levels indicate enhanced insulin sensitivity in c78f/f mice. These findings suggest that ectopic accumulation of lipids observed in previous lipodystrophic mouse models may not be simply due to lack of adipose tissue, but other factors/hormones might be involved in lipid partitioning to nonadipose organs in these mice. Since ectopic fat accumulation in liver was largely responsible for insulin resistance in these lipodystrophic mouse models, our findings of normal liver and plasma lipid profile are consistent with insulin-sensitive phenotypes of c78f/f mice. Alternatively, ectopic fat accumulation in other lipodystrophic mice may be partly due to elevated insulin levels and their role in lipogenesis. Consistent with this notion, c78f/f mice showed markedly lower insulin levels, which might suppress de novo lipogenesis in liver. Lower glucagon levels in c78f/f mice further suggest that ablation of GRP78 in adipose tissues may also affect pancreatic islets, thereby reducing both insulin and glucagon levels.
There are a number of tissue or organ defects outside of WAT and BAT that likely contribute to the early death of our mouse model. While it is well-established that the aP2-Cre transgene is highly expressed in both WAT and BAT, it has also been demonstrated to be active in the trigeminal ganglia, dorsal root ganglia, cartilage primordia, and vertebrae of developing embryos (21). Further, a recent study demonstrated that the aP2 promoter drives gene inactivation in the ganglia of the peripheral nervous system, adrenal medulla, and neurons throughout the central nervous system, including Purkinje cells in the cerebellum of 4-wk-old mice (22). Consistent with these reports, there was a moderate but detectable reduction of GRP78 level in the kidney in the c78f/f mice. GRP78 is required for Purkinje cell survival and function and integrity of the cerebellum (3). Interestingly, we observed reduction of GRP78 expression in the brain and some Purkinje cells in the c78f/f mice. Another mouse model in which aP2-Cre was used to knockout acetyl-CoA carboxylase 1 exhibited growth retardation and bone defects (23). Thus, it is possible that ectopic expression of aP2 in nonadipose tissues and the critical role of GRP78 for survival and development of those tissues contributed to the bone defects and early mortality of our mouse model. In view of this, ectopic expression of aP2-Cre should be taken into consideration when it is used to establish conditional knockout models to study potential regulators of adipose tissue. A novel 5.4-kb adiponectin promoter fragment conveying adipocyte-specific expression of passenger genes has recently been developed, and this may be useful in generating greater adipose tissue specificity in transgenic and knockout mouse models (34).
In summary, depletion of GRP78 blocks adipocyte differentiation in vitro and leads to dramatic decrease in adipose tissues in the conditional-knockout model. Despite severe lipoatrophy, the mutant mice do not exhibit dyslipidemia, fatty liver, or type 2 diabetes phenotypes. Rather, they exhibit normal plasma triglyceride levels with lower glucose and insulin levels, which contrast with other lipodystrophic mouse models. Because of the pleiotrophic expression of the aP2-driven Cre-recombinase in the bone, we uncovered a novel role of GRP78 in bone development, which warrants further investigation. The identification of GRP78 as a critical player for adipogenesis suggests that it could be targeted in adipocyte tissues to reduce adiposity to combat obesity.
Supplementary Material
Acknowledgments
The authors thank Dr. My Chouinard (University of Massachusetts Medical School, Worcester, MA, USA) and Drs. Mike Gray, Yvonne Lin, and Parkash Gill [Keck School of Medicine, University of Southern California (USC), Los Angeles, CA, USA] for the gifts of cell lines and reagents. The authors thank Ms. Archana Tank (USC Comprehensive Cancer Center Small Animal Imaging Core and Translational Pathology Core) and Dr. Dongyun Yang (USC Biostatistics Core) for assistance. The authors also thank the USC Comprehensive Cancer Center Cell and Tissue Imaging Core for assistance in electron microscopy.
This work is supported in part by U.S. National Institutes of Health (NIH) grants R01 DK 079999 to A.S.L and R01 DK 080756 to J.K.K. The USC cores described were supported by NIH grant P30 CA014089 from the National Cancer Institute.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- aP2
- fatty acid binding protein 4
- ATF6α
- activating transcription factor 6α
- BAT
- brown adipose tissue
- ER
- endoplasmic reticulum
- GLP-1
- glucagon-like peptide 1
- GRP78
- 78-kDa glucose-regulated protein
- H&E
- hematoxylin and eosin
- IBMX
- 3-isobutyl-1-methylxanthine
- MCP-1
- monocyte chemotactic protein 1
- MEF
- mouse embryonic fibroblast
- PPAR-γ2
- peroxisome proliferator-activated receptor γ2
- SREBP-1c
- sterol regulatory element binding protein 1c
- TG
- triglyceride
- TNF-α
- tumor necrosis factor α
- UPR
- unfolded protein response
- WAT
- white adipose tissue
- XBP-1s
- spliced X-box binding protein 1
REFERENCES
- 1. Rosen E. D., Spiegelman B. M. (2006) Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444, 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ni M., Lee A. S. (2007) ER chaperones in mammalian development and human diseases. FEBS Lett. 581, 3641–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang M., Ye R., Barron E., Baumeister P., Mao C., Luo S., Fu Y., Luo B., Dubeau L., Hinton D. R., Lee A. S. (2010) Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 17, 488–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang M., Wey S., Zhang Y., Ye R., Lee A. S. (2009) Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 11, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D. Y., Nagata K. (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572 [DOI] [PubMed] [Google Scholar]
- 6. Reimold A. M., Iwakoshi N. N., Manis J., Vallabhajosyula P., Szomolanyi-Tsuda E., Gravallese E. M., Friend D., Grusby M. J., Alt F., Glimcher L. H. (2001) Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307 [DOI] [PubMed] [Google Scholar]
- 7. Federovitch C. M., Ron D., Hampton R. Y. (2005) The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409–414 [DOI] [PubMed] [Google Scholar]
- 8. Ye R., Mareninova O. A., Barron E., Wang M., Hinton D. R., Pandol S. J., Lee A. S. (2010) Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am. J. Pathol. 177, 2827–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gregor M. F., Hotamisligil G. S. (2007) Thematic review series: Adipocyte biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J. Lipid Res. 48, 1905–1914 [DOI] [PubMed] [Google Scholar]
- 10. Luo S., Mao C., Lee B., Lee A. S. (2006) GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 26, 5688–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fu Y., Wey S., Wang M., Ye R., Liao C. P., Roy-Burman P., Lee A. S. (2008) Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc. Natl. Acad. Sci. U. S. A. 105, 19444–19449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He W., Barak Y., Hevener A., Olson P., Liao D., Le J., Nelson M., Ong E., Olefsky J. M., Evans R. M. (2003) Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. U. S. A. 100, 15712–15717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baerga R., Zhang Y., Chen P. H., Goldman S., Jin S. (2009) Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 5, 1118–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burkart A., Shi X., Chouinard M., Corvera S. (2011) Adenylate kinase 2 links mitochondrial energy metabolism to the induction of the unfolded protein response. J. Biol. Chem. 286, 4081–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ni M., Zhou H., Wey S., Baumeister P., Lee A. S. (2009) Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS One 4, e6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye R., Jung D. Y., Jun J. Y., Li J., Luo S., Ko H. J., Kim J. K., Lee A. S. (2010) Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 59, 6–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verdelis K., Lukashova L., Atti E., Mayer-Kuckuk P., Peterson M. G., Tetradis S., Boskey A. L., van der Meulen M. C. (2011) MicroCT morphometry analysis of mouse cancellous bone: intra- and inter-system reproducibility. Bone 49, 580–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bastie C. C., Zong H., Xu J., Busa B., Judex S., Kurland I. J., Pessin J. E. (2007) Integrative metabolic regulation of peripheral tissue fatty acid oxidation by the SRC kinase family member Fyn. Cell Metab. 5, 371–381 [DOI] [PubMed] [Google Scholar]
- 19. Luu Y. K., Lublinsky S., Ozcivici E., Capilla E., Pessin J. E., Rubin C. T., Judex S. (2009) In vivo quantification of subcutaneous and visceral adiposity by micro-computed tomography in a small animal model. Med. Eng. Phys. 31, 34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sha H., He Y., Chen H., Wang C., Zenno A., Shi H., Yang X., Zhang X., Qi L. (2009) The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 9, 556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urs S., Harrington A., Liaw L., Small D. (2006) Selective expression of an aP2/fatty acid binding protein 4-Cre transgene in non-adipogenic tissues during embryonic development. Transgenic Res. 15, 647–653 [DOI] [PubMed] [Google Scholar]
- 22. Martens K., Bottelbergs A., Baes M. (2010) Ectopic recombination in the central and peripheral nervous system by aP2/FABP4-Cre mice: implications for metabolism research. FEBS Lett. 584, 1054–1058 [DOI] [PubMed] [Google Scholar]
- 23. Mao J., Yang T., Gu Z., Heird W. C., Finegold M. J., Lee B., Wakil S. J. (2009) aP2-Cre-mediated inactivation of acetyl-CoA carboxylase 1 causes growth retardation and reduced lipid accumulation in adipose tissues. Proc. Natl. Acad. Sci. U. S. A. 106, 17576–17581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Steppan C. M., Bailey S. T., Bhat S., Brown E. J., Banerjee R. R., Wright C. M., Patel H. R., Ahima R. S., Lazar M. A. (2001) The hormone resistin links obesity to diabetes. Nature 409, 307–312 [DOI] [PubMed] [Google Scholar]
- 25. Rutkowski D. T., Kaufman R. J. (2004) A trip to the ER: coping with stress. Trends Cell Biol. 14, 20–28 [DOI] [PubMed] [Google Scholar]
- 26. Pfaffenbach K. T., Lee A. S. (2011) The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 23, 150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo B., Lee A. S. (2012) The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anti-cancer therapies. [E-pub ahead of print] Oncogene doi: 10.1038/onc.2012.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lowe C. E., Dennis R. J., Obi U., O'Rahilly S., Rochford J. J. (2012) Investigating the involvement of the ATF6alpha pathway of the unfolded protein response in adipogenesis. Int. J. Obes. (Lond.) 36, 1248–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo S., Lee A. S. (2002) Requirement of the p38 MAPK signaling pathway for the induction of Grp78/BiP by azetidine stress: ATF6 as a target for stress-induced phosphorylation. Biochem. J. 366, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ross S. R., Graves R. A., Spiegelman B. M. (1993) Targeted expression of a toxin gene to adipose tissue: transgenic mice resistant to obesity. Genes Dev. 7, 1318–1324 [DOI] [PubMed] [Google Scholar]
- 31. Moitra J., Mason M. M., Olive M., Krylov D., Gavrilova O., Marcus-Samuels B., Feigenbaum L., Lee E., Aoyama T., Eckhaus M., Reitman M. L., Vinson C. (1998) Life without white fat: a transgenic mouse. Genes Dev. 12, 3168–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shimomura I., Hammer R. E., Richardson J. A., Ikemoto S., Bashmakov Y., Goldstein J. L., Brown M. S. (1998) Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. 12, 3182–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flint O. P., Noor M. A., Hruz P. W., Hylemon P. B., Yarasheski K., Kotler D. P., Parker R. A., Bellamine A. (2009) The role of protease inhibitors in the pathogenesis of HIV-associated lipodystrophy: cellular mechanisms and clinical implications. Toxicol. Pathol. 37, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Z. V., Deng Y., Wang Q. A., Sun K., Scherer P. E. (2010) Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology 151, 2933–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.