

Abstract
Biosilicification is widespread across the eukaryotes and requires concentration of silicon in intracellular vesicles. Knowledge of the molecular mechanisms underlying this process remains limited, with unrelated silicon-transporting proteins found in the eukaryotic clades previously studied. Here, we report the identification of silicon transporter (SIT)-type genes from the siliceous loricate choanoflagellates Stephanoeca diplocostata and Diaphanoeca grandis. Until now, the SIT gene family has been identified only in diatoms and other siliceous stramenopiles, which are distantly related to choanoflagellates among the eukaryotes. This is the first evidence of similarity between SITs from different eukaryotic supergroups. Phylogenetic analysis indicates that choanoflagellate and stramenopile SITs form distinct monophyletic groups. The absence of putative SIT genes in any other eukaryotic groups, including non-siliceous choanoflagellates, leads us to propose that SIT genes underwent a lateral gene transfer event between stramenopiles and loricate choanoflagellates. We suggest that the incorporation of a foreign SIT gene into the stramenopile or choanoflagellate genome resulted in a major metabolic change: the acquisition of biomineralized silica structures. This hypothesis implies that biosilicification has evolved multiple times independently in the eukaryotes, and paves the way for a better understanding of the biochemical basis of silicon transport through identification of conserved sequence motifs.
Keywords: choanoflagellate, silicon transporter, lateral gene transfer, diatom, SIT transport model, biomineralization
1. Introduction
Biosilicification is the genetically controlled incorporation of amorphous silicon dioxide into the physical macrostructure of an organism. In eukaryotes, biosilica formation occurs within a silicon deposition vesicle (SDV) to isolate the polymerizing silica from the general cell metabolism [1].
Silicon is present in the environment primarily as silicic acid (Si(OH)4). Silicic acid autopolymerizes to silica at concentrations above 2 mM, so silica formation within the SDV is more efficient close to this concentration [2]. However, the low concentrations of silicic acid in modern oceans and freshwaters (10–180 µM, [3]) are insufficient to support silicon uptake by passive transport. Silicifying organisms, therefore, require an active transport mechanism to take up silicon from the external environment and to concentrate silicon within the SDV. Because silicic acid is a relatively inert species, with no known biochemical role outside that of biomineralization [4,5], silicon transport requires the evolution of transmembrane proteins with highly specialized interactions with silicon.
Silicon-transporting proteins are known from only a few eukaryotic groups. Diatoms possess the silicon transporter (SIT) family, whose activity and expression is tightly coupled to biosilicification [6,7]. SIT-like genes have been found in other siliceous stramenopiles (Synura and Ochromonas [8]), but in no other groups. Land plants do not possess SIT-like genes, instead possessing NOD26-like intrinsic protein (NIP)-type transporters for silicic acid (Lsi1, Lsi6) [9,10] and active silicon efflux pumps (Lsi2) [11]. No SITs are known from siliceous sponges, but a 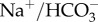 co-transporter has been postulated to have a role in silicon transport [12].
co-transporter has been postulated to have a role in silicon transport [12].
There is no obvious similarity or homology between the genes responsible for silicon transport from the different eukaryotic supergroups in the current literature. Within each transporter type, only a few silicon transport-related protein motifs have been identified, the GXQ amino acid motifs in SITs [13] and arginine/aromatic selectivity filter of silicon-related NIPs [14]. This lack of homology has hindered research into silicon biochemistry and prevented the identification of other silicon-interacting protein features.
Choanoflagellates are a group of heterotrophic aquatic protists that are the closest unicellular relatives of the animals [15,16]. Within choanoflagellates, the Acanthoecidae are a monophyletic group [17] characterized by the possession of an extracellular lorica constructed from a series of costal strips [18]. Each costal strip is a hollow tube of silica, formed individually within an SDV before being exocytosed from the cell [19]. Costal strip size and shape varies within individual loricae, and the overall lorica morphology varies between species [20].
Silicon-deprivation studies on loricate species revealed that costal strip formation and lorica assembly are affected incrementally in relation to silicon availability. Under silicon-depleted conditions, progressively thinner costal strips are produced, and under zero-silicon conditions no costal strips or SDVs are formed [21,22]. Upon silicon replenishment, costal strip production recommences, with silicon levels in the culture medium falling as new loricae are formed [23]. Electron microscopy reveals no evidence for vesicle-based pinocytotic uptake of silicic acid during lorica formation [24]. Collectively, this points towards choanoflagellates possessing a transporter-based system for the active uptake of silicon into the SDV for costal strip formation.
Here, we report the identification of genes from two loricate choanoflagellates, Stephanoeca diplocostata and Diaphanoeca grandis (figure 1) with significant sequence similarity to the SIT genes of diatoms. This is the first identification of homologous silicon-related genes between siliceous species from different eukaryotic supergroups. The limited taxonomic distribution of SIT homologues suggests that they originated by lateral gene transfer (LGT), transferring an important biochemical innovation between disparate eukaryotic groups.
Figure 1.

The loricate choanoflagellates (a,b) S. diplocostata and (c,d) D. grandis. Photographs taken using phase contrast at 100× magnification. Schematic figures are taken from the Micro*scope v. 6.0 website (http://starcentral.mbl.edu/microscope, drawings by Won-Je Lee) and used under a Creative Commons licence. Lor, Lorica; CB, cell body; Fl, flagellum; Col, collar. Bacteria in the photographs are annotated with asterisks (*). Scale bar, 5 μm. (Online version in colour.)
2. Material and methods
(a). Identification and cloning of silicon transporter sequences
RNA extracted from cultures of S. diplocostata was used to produce a cDNA library, which was sequenced using 454 pyrosequencing. The resulting transcriptome reads were assembled into contigs for bioinformatic analysis (see the electronic supplementary material for detailed description). Those contigs containing SIT domains were selected for further tBLASTx analysis [25] against the EMBL/Genbank non-redundant nucleotide databases (see the electronic supplementary material). The relevant open reading frames were used as queries in PSI-BLAST [25] searches against the EMBL/Genbank non-redundant protein database. ClustalX was used to generate a protein alignment of all the translated contigs containing the SIT domain.
The longest S. diplocostata SIT-like contig was used as query sequence for a tBLASTx search (significance threshold value = 1×10−10) against a partial genome sequence dataset from D. grandis (see the electronic supplementary material). A protein alignment of those contigs providing a significant hit was generated using ClustalX, and the longest contig analysed using InterProScan.
Stephanoeca diplocostata dTcDNA was made using Superscript III First-Strand Synthesis reverse transcriptase (Invitrogen) and 2 µl (approx. 1 µg) of cDNA used as RT-PCR template. A total of 200 ng of genomic DNA, extracted from D. grandis cultures using a cetyltrimethylammonium bromide (CTAB) buffer based method [26], was used as PCR template together with the DgSITa primers. The RT-PCR and PCR protocols were as follows: hot-start denaturing step of 94°C for 4 min; 35 cycles of 94°C (30 s), 50°C (30 s), 72°C (105 s); final elongation step of 72°C for 5 min (see the electronic supplementary material for primer sequences and combinations). Amplified sequences were cloned into the PGEM-T Easy Vector System (Promega) and Subcloning Efficiency DH5α Competent Cells (Invitrogen). Plasmids were extracted using a Qiaprep Spin Miniprep Kit (Qiagen) and sequenced by SourceBioScience (Cambridge, UK).
(b). Bioinformatic analyses and alignments
The tBLASTx, PSI-BLAST and InterProScan analyses were repeated for the consensus sequences for all cloned choanoflagellate SITs. The DgSITa protein sequence was identified by tBLASTx comparison to the SdSIT genes. The 5′ portion of the amplified sequence contained stop codons and a putative splice donor site, suggesting that it represents intronic sequence. The DgSITa sequence was trimmed to remove this putative intron. The remaining 554 bp exon, encoding a 185 amino acid protein sequence, was used for all further analyses.
WoLF PSORT analysis [27] was used to predict the subcellular location of the SdSIT proteins. The COILS prediction server (http://www.ch.embnet.org/software/COILS_form.html) was used to search for putative coiled-coil motifs. Topology predictions were carried out for all SdSITs and DgSITa to identify transmembrane domains (TMDs) using TMPred [28], HMMTop [29] and TMHMM [30].
Pairwise identity measurements between the choanoflagellate SITs were generated with ClustalW2 (www.ebi.ac.uk/Tools/msa/clustalw2/). A full alignment of the four SIT sequences was generated to ClustalX v. 2.0.9, using the default settings (gap opening = 10, gap extension = 0.2, delay divergent sequences = 30%) and the Gonnet Series matrix.
A second protein alignment incorporating non-choanoflagellate SITs was generated with ClustalX under the same settings. The 156 significant PSI-BLAST hits to SdSITa were aligned against the choanoflagellate SIT sequences (see the electronic supplementary material). Choanoflagellate and stramenopile SITs were compared to identify conserved motifs, charged residues and hydroxylated residues (following [13]). Features were noted if they were conserved across all SITs. A subset of this alignment was generated in order to display the results of the larger comparison and to include the predicted TMDs of SdSITs and Phaeodactylum tricornutum SITs [31].
(c). Phylogenetic analysis
Sequences were chosen from the EMBL/Genbank non-redundant protein database to cover a taxonomic range of SITs (see the electronic supplementary material) and aligned using ClustalX v. 2.0.9 (default settings and Gonnet Series matrix). The alignment was inspected and sequences manually trimmed to minimize missing data. The final alignment contained 20 sequences from 10 different species, a total of 769 positions.
Prottest [32] found that the LG + Г model provided the best fit (under the Akaike Information Criterion) to the data. Maximum-likelihood (ML) analysis was carried out using PhyML v. 3.0 [33] with the LG + Г model, the proportion of invariant sites and equilibrium frequencies of amino acids estimated from the data fixed according to the LG model. Starting trees were generated by BioNJ, with tree searching using SPR + NNI heuristic methods. Topology and branch lengths were optimized in ML calculations. A second ML analysis was performed using RaxML v. 7.2.8 [34]; the parameters used were identical to the PhyML analysis except using the WAG substitution matrix. One hundred bootstrapped datasets were analysed using the same respective model and method for each of the PhyML and RaxML analyses. Bootstrap proportions were added to the nodes of the ML tree. Bayesian MCMC analysis was done using MrBayes [35] running over 2 million generations, with four chains, sampling every 5000 generations, and with a burn-in of 100 000 generations; with the WAG substitution model, four γ-distributed rate categories with the α-value and proportion of invariant sites estimated. Bayesian posterior probabilities were added to the highest likelihood Bayesian analysis tree. Trees generated were viewed with FigTree v. 1.3.1 (A. Rambaut, Institute of Evolutionary Biology, University of Edinburgh 2006–2009).
3. Results
(a). Identification of silicon transporters in the Stephanoeca diplocostata transcriptome
Transcriptome sequencing of S. diplocostata produced 0.261 Gb of data assembled into 26 325 contigs. InterProScan analysis found six transcriptome contigs containing putative SIT protein domains. The most significant tBLASTx and PSI-BLAST search hits were to known diatom SIT sequences (see the electronic supplementary material). Searches excluding diatoms found significant similarity (e-value > 1 × 10−5) to SIT sequences from the siliceous stramenopiles Synura petersenii and Ochromonas ovalis. No significant similarity was found to any sequences from the non-loricate choanoflagellates Monosiga brevicollis, Salpingoeca rosetta or Monosiga ovata.
These six contigs appear to represent three full-length genes, with a shorter contig (08888) that may represent minor allelic variation, errors in sequencing that led to a distinct assembly or a partial or inactive gene. The three full-length genes were designated S. diplocostata silicon transporter (SdSIT) a, b and c.
The validity of the transcriptome assemblies for all three genes was confirmed by RT-PCR from S. diplocostata cDNA. SdSITa and SdSITb yielded products with 99 per cent nucleotide identity to the assembled contigs. In SdSITc, the gene-specific 5′ region was successfully amplified, while comparing the contigs showed that the 3′ region is identical to that of SdSITb. Repeating tBLASTx, PSI-BLAST and InterProScan analyses for the final sequences of each SdSIT gene produced the same results as the original contigs (see the electronic supplementary material).
(b). Identification of an incomplete silicon transporter in the Diaphanoeca grandis genome dataset
A tBLASTx search querying SdSITa against the partial D. grandis genome dataset found 25 hits at a significance threshold of 1 × 10−5 (data not shown). The longest contig is 931 bp and has potential to encode a protein with similarity to the S. diplocostata SITs over 193 amino acids. The remaining contigs, mostly shorter than 250 bp, encode protein fragments similar to short stretches within this region of the protein, but the fragmentary genome data precludes determination of how many genes they represent. We conclude that D. grandis also has a family of SIT-related genes.
The 5′ end of the 931 bp DgSITa contig has low sequence similarity to other SIT sequences and contains in-frame stop codons. There is a putative splice acceptor site just upstream of the conserved protein coding sequence, suggesting that this 5′ sequence represents an intron. This provides strong evidence against a bacterial origin for the SIT domain containing sequences in the D. grandis partial genome data. The conserved protein sequence runs to the 3′ end of the available contig.
PCR amplification of DgSITa from genomic DNA produced a sequence identical to the original contig, except that a 71 bp long tandem repeat in the putative intron was absent in the PCR-derived sequence. Attempts to extend the available DgSITa sequence using RT-PCR or 5′-RACE were unsuccessful. The results of the BLAST searches and InterProScan analysis using DgSITa are given in the electronic supplementary material.
(c). Bioinformatic analysis of Stephanoeca diplocostata silicon transporter
The SdSIT-deduced proteins show high levels of sequence similarity (89–95% nucleotide identity). WoLF PSORT was used to analyse the sequences for predicted subcellular localization. All three databases gave a majority prediction that each SdSIT protein is targeted to the plasma membrane. Topology prediction programmes identified nine, 10 or 11 TMDs (see the electronic supplementary material). As diatom SITs have been consistently predicted to have 10 TMDs (orange line, figure 2), we applied 10 TMD prediction from TMPred to the SdSITs (black line, figure 2). The TMDs predicted for choanoflagellate and diatom SITs overlap to some degree; however, the location of TMD 4 is considerably different. The COILS software found no evidence for putative coiled-coil motifs in the SdSIT sequences, unlike pennate diatom SITs which contain prominent C-terminal coiled-coil motifs [13].
Figure 2.

Alignment and identification of conserved residues and TM domains in stramenopile and choanoflagellate SITs. The black line is the TMPred SdSIT topology prediction. The orange line is the diatom SIT TMD prediction [30]. Red boxes indicate conserved motifs of three or more residues. The double blue circles mark the ‘CMLD’ motif in stramenopiles. Conserved charged or hydroxylated residues are noted as per [13,30]; purple arrows, negatively charged; red arrows, positively charged; green arrows, hydroxyl-containing. Oo, Ochromonas ovalis (chrysophyte, non-diatom stramenopile); Cf, Cylindrotheca fusiformis (pennate diatom); Pt, Phaeodactylum tricornutum (pennate diatom); Tp, Thalassiosira pseudonana (centric diatom); Sa, Synedra acus (pennate diatom).
(d). Identification of conserved motifs in silicon transporter proteins
Alignment of choanoflagellate, diatom and stramenopile SITs revealed five motifs in common across all SITs (figure 2). The two pairs of GXQ-containing motifs have been noted previously, the first in TMDs 2 and 3, the second located in TMDs 7 and 8, but symmetrically orientated with respect to the membrane polarity [13]. In our alignment, the C-terminal motifs of the pair contained GRQ, apart from one CRQ motif (Shionodiscus ritscheri). The N-terminal motif sequences in each pair were resolved as EGXQ, X being cysteine, glycine or glutamine in the first motif pair and methionine, leucine, isoleucine or threonine in the second. Variants were again only found at the second motif of the pair: EAMQ (in Navicula pelliculosa SIT4) and KGMQ (in Stephanodiscus yellowstonensis). In choanoflagellate SITs, all motifs were EGCQ–GRQ (first motif pair) and EGLQ–GRQ (second motif pair).
We also identified an additional conserved sequence between the EGXQ–GRQ motif pairs: this motif is (G/S)QL. GQL is by far the more common variant and is present in all choanoflagellate SIT sequences. The CMLD motif [36] was not found in any choanoflagellate SIT.
In addition to these motifs, three hydroxyl-containing residues (T115, T290 and S382 in SdSITa), three negatively charged residues (E133, E225 and D476 in SdSITa) and one positively charged residue (R134 in SdSITa) were identified as being conserved across all SITs. These residue classes have been proposed to function in silicic acid transport [13] (see §4).
The first GRQ motif (TMD 3), the second EGXQ motif (TMD 7) and the conserved residues are found in similar position with respect to membrane orientation in both choanoflagellates and stramenopiles. The first EGXQ motif is intracellular in diatoms but placed within TMD 2 in choanoflagellates. Conversely, the second GRQ motif is extracellular in choanoflagellates but is predicted to be within TMD 8 in diatoms. In choanoflagellates, the (G/S)QL motif is inside TMD 4, but is intracellular in diatom SIT structures. This relates to the major difference in the predicted location of TMD 4 between diatoms and choanoflagellates (figure 2).
(e). Evolution of silicon transporter genes
An ML tree produced from analysis of choanoflagellate, diatom and non-diatom stramenopile SIT protein sequences is shown in figure 3. This tree was obtained using the LG + Γ model implemented in PhyML; RaxML analysis and Bayesian analysis (see the electronic supplementary material) using the WAG substitution matrix produced identical topologies. Trees are unrooted because of the lack of any known SITs or SIT homologues, from outside the choanoflagellates or stramenopiles, to act as an outgroup.
Figure 3.

Phylogenetic tree demonstrates that choanoflagellate SITs are monophyletic within the stramenopile SITs. The tree was produced using PhyML ML analysis with the LG + Γ model from an alignment of 769 amino acid residues. The tree produced using RaxML and the WAG model had identical topology. Numbers at nodes are a percentage of 100 bootstrap replicates (PhyML bootstrap value/RaxML bootstrap value). Blue, pennate diatom; green, centric diatom; brown, non-diatom stramenopile; red, choanoflagellate. Dash (−/−) indicates a bootstrap value less than or equal to 70%. Scale bar indicates the average number of amino acid substitutions per site.
Choanoflagellate SITs (in red) form a distinct monophyletic clade (100% bootstrap support, posterior probability = 1). Within this, the S. diplocostata paralogues resolve as a clade, though with low bootstrap support. An exceptionally long branch marks the division between the choanoflagellate and stramenopile sequences.
The SITs from non-diatom stramenopiles (shown in brown) branch from within the diatom SITs with significant (94–100%) bootstrap support and posterior probability (=1). These synurophyte and chrysophyte SITs are sister to a subset of the pennate diatom SITs (blue). Centric diatom SITs branch together as a monophyletic clade (100% bootstrap support, posterior probability = 1) with pennate diatom SITs resolving as paraphyletic.
4. Discussion
The loricate choanoflagellates S. diplocostata and D. grandis possess a family of genes encoding the SIT protein domain, a domain previously found only in diatoms, synurophytes and chrysophytes. This is the first report of homology between putative SITs in two different eukaryotic supergroups, opisthokonts and stramenopiles [37]. These SIT domains are not present in any of the sequenced non-siliceous choanoflagellate species [15,17,38,39], implying a correlation between their presence and lorica biomineralization.
(a). Evidence against a contaminant origin
That distinct, but closely related, SIT genes are found in two different loricate choanoflagellate species is strong evidence against an origin from stramenopile contamination. This is further supported by phylogenetic analysis, which resolved choanoflagellate SITs as a strongly supported clade separate to the stramenopile SITs (figure 3). Furthermore, no housekeeping genes (rRNA, tubulins, etc.), were found among those transcriptome contigs with highest tBLASTx similarity to stramenopile sequences, and no non-choanoflagellate eukaryotes were observed in the cultures.
(b). Silicon transporters in choanoflagellates
The SIT gene family is best characterized from S. diplocostata, but the evidence indicates that an SIT gene family also exists in D. grandis. SdSITa, b and c show very high levels of sequence identity, up to 95 per cent (nucleotide) and 94 per cent (amino acid). This is similar to identity levels in diatom SITs [13,40]. Such a high degree of conservation implies either strong functional constraints [41] or that these genes are the product of a relatively recent gene duplication event.
Different SdSIT paralogues may be specialized to different subcellular locations (e.g. plasma membrane or SDV). This is supported by variation at the N-termini, where eukaryotic localization signal sequences often occur [42]. Alternatively, each SdSIT may have different transport activity, as suggested for diatom SIT3 [7,40]. Unlike diatoms [43], S. diplocostata continually produces biosilica under normal conditions [19]. This would require a steady, constant uptake of silicon, which may be reflected in the similar read numbers observed for SdSITa, b and c.
(c). Silicon transporter evolution
Phylogenetic reconstruction (figure 3) found that choanoflagellate and stramenopile SIT sequences are evolutionarily distinct, with a long and well-supported branch between the two groups. SdSITs appear to be monophyletic, indicating gene duplication within the loricate choanoflagellates.
The synurophyte and chrysophyte SIT sequences group together as a well-supported monophyletic clade, as do the centric diatom SITs. Both these clades branch from within the paraphyletic pennate diatom SITs. This branching order is incongruent with previous hypotheses of stramenopile evolution based on siliceous fossils [44], but this may be an artefact of taxon sampling that could be resolved by obtaining more SIT sequences from non-diatom stramenopiles. The phylogenetic analyses are in agreement with some aspects of previous analyses [13,31], such as the close relationship of C. fusiformis SIT genes and the highly divergent nature of TpSIT3 and PtSIT3.
The phylogenetic analysis clearly shows that the diatom SIT numerical classification system is incongruent with patterns of orthology between species and therefore choanoflagellate SIT genes were classified alphabetically, to avoid any statement of homology with individual members of the diatom SIT gene families.
(d). Proposed structure and function of choanoflagellate silicon transporter proteins
The similar TMD topology predictions and conserved residues between choanoflagellate and stramenopile proteins (figure 4) can be used to support a common mechanism for SIT transmembrane silicon transport in both these groups, similar to that proposed by Thamatrakoln et al. [13].
Figure 4.
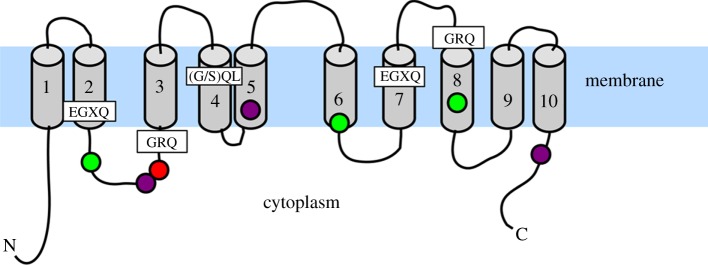
Schematic of the SdSIT protein showing transmembrane structure and orientations of conserved motifs and residues. Conservation was determined from the choanoflagellate and stramenopile SIT alignment (see figure 2 and electronic supplementary material). TMDs (grey cylinders) are taken from the SdSITa TMPred topology and numbered accordingly. Conserved motifs are in boxes, circles are conserved residues as per figure 2: red, positively charged; purple, negatively charged; green, hydroxyl group containing.
SITs possess an inverted symmetrical structure of 5 + 5 TMDs (figure 4). This is characteristic of the LeuT-fold-type sodium symporters, which all share a common inverted 5 + 5 TMD structural psuedosymmetry [45,46]. The degree of symmetry observable from the amino acid sequence of SITs is notable in comparison with other LeuT-fold transporters, whose inverted pseudosymmetry is only discernible from their three-dimensional structure and is not visible in the primary protein sequence [45]. This strongly supports the theory that SITs evolved by homodimerization of an ancestral transmembrane transporter by gene duplication followed by gene fusion [13,47]. The inverted symmetrical structure would allow bidirectional silicon transport, predicted for both diatom and choanoflagellate SITs. Silicic acid efflux transport is thought to have had a major role in the high-silicon pre-Cenozoic oceans [3], to prevent over-accumulation of silicic acid and subsequent uncontrolled auto-polymerization of silica in the cytoplasm [13,48].
Owing to the structural similarities, we applied the alternating access transporter mechanism developed for the LeuT-fold-type transporters (reviewed in [46,49]) to SITs. We suggest that the highly conserved TMDs 2, 3, 7 and 8 form the central helices (equivalent to TMDs 1, 2, 6 and 7 of LeuT), with the remaining TMDs forming the surrounding scaffold helices. The EGXQ and/or GRQ motifs are likely candidates to be the silicic acid-binding sites, as the charged residues could provide a localized polar environment characteristic of the unwound helix region found at binding sites [50]. The glutamine side-chains may interact with local negative charges in the hydroxyl groups of the silicic acid molecule [13]. The remaining EGXQ and/or GRQ motifs would lie at the point of substrate entry to or exit, where the charged residues could play a role in orientating the substrates into and out of the aqueous vestibule.
Sodium and silicon are transported in a 1 : 1 ratio in diatoms [51], and calculated Hill binding coefficients of SITs [48,52] indicate the presence of a second binding site. In LeuT-fold transporters, Na+ binding is required for correct substrate binding and to cause conformational change of the core helices for transport [46,49], and this must also be the case for SITs. The conserved hydroxylated residue (S382 in SdSITa) in the proposed central bundle region (figure 4) may act as a binding site for Na+, similar to the Na2 sodium-binding site LeuT and vSGLT transporters [45].
Marine diatom SITs require Na+, but in freshwater diatoms both Na+ and K+ are involved in silicon uptake, a feature related to salinity differences between the two environments [41]. Loricate choanoflagellates are primarily marine [53], with the important exception of Acanthocorbis mongolica [54]. In comparison, non-loricate choanoflagellates are found in a range of salinities [53]. It may be that most loricate choanoflagellate SITs cannot make use of K+ in the same way as freshwater diatom SITs, preventing biosilicification and limiting colonization of freshwaters. The SITs of A. mongolica will be of particular interest with regards Na+ and SIT activity.
At the cytoplasmic opening to the proposed binding vestibule (between TMD 2 and 3, figure 4), the intracellular loop contains conserved charged and hydroxylated amino acids. These residues may interact with the theorized cytoplasmic silicic acid-binding component that forms an organic complex for intracellular silicon transfer to the SDV [48]. The (G/S)QL motif and other conserved features lie outside the proposed central bundle helices. This hints that they may play a structural role, rather than having direct involvement in substrate binding.
Comparison of diatom and choanoflagellate SITs provides evidence against other proposed models. Choanoflagellate SITs lack the conserved extracellular cysteines previously suggested to play a role in diatom SIT activity [40], and the CMLD motif is absent in all choanoflagellate SITs, as is the possible YXXL-binding site, strongly arguing against a critical role for zinc-binding role in SIT transport [36].
(e). Lateral gene transfer and the origin of silicon transporters in loricate choanoflagellates
The genomes of choanoflagellates and some stramenopiles contain SIT genes encoding highly similar proteins. The absence of genes with any recognizable homology to SITs in eukaryotes other than loricate choanoflagellates and siliceous stramenopiles makes it extremely unlikely that such SIT genes were present in the last common ancestor of choanoflagellates and stramenopiles. This organism would be equivalent to the last common ancestor of all eukaryotic supergroups [37], and therefore this hypothesis would involve the loss of all SIT homologues from all other sequenced eukaryotes.
The convergent evolution of SITs in loricate choanoflagellates and siliceous stramenopiles would require not only parallel evolution of multiple SIT-specific features, but also the loss of all related or ancestral genes in all other opisthokonts and non-siliceous stramenopiles. The taxonomic coverage of available sequence data [55,56] makes such a conclusion highly unlikely.
We therefore believe that on the basis of current evidence, the best model for the evolution of SITs is through LGT. The most parsimonious hypothesis involves a single LGT event between a choanoflagellate and a stramenopile, in one direction or the other.
LGT is prevalent both in choanoflagellates [57] and diatoms [58]. In choanoflagellates, this may be related to their phagotrophic lifestyle [59]. In M. brevicollis, LGT from both eukaryotic and prokaryotic sources has been proposed to be responsible for the gain of stress-related genes [60]. Stramenopile-derived genes are thought to be unusually frequent in M. brevicollis [57], including genes linked with the evolution of multicellularity [61]. Diatoms are believed to have gained some of their biosilicification machinery (related to long-chain polyamine formation) from LGT [62] in addition to other diatom-specific metabolic pathways [58].
Though the genomic arrangement of choanoflagellate SIT genes is unknown, diatom biosilicification genes are often clustered [31]. These clusters possess their own promoter sequences, with a possible silicon-specific promoter suggested from T. pseudonana [63]. This spatial association of biosilicification genes allows for coordinated regulation in diatoms [31], but it may make SITs more amenable to LGT by having a self-regulating genomic unit that can interact with existing metabolic pathways with minimal disruption [60].
Major questions concern the direction and timing of the SIT-related LGT event. The earliest confirmed diatom fossils are from the Mesozoic [44], with putative Precambrian chrysophyte fossils [64]. Choanoflagellates lack a fossil record, though molecular clock estimates place the origin of the choanoflagellate branch to approximately 750 Ma [65]. Choanoflagellate biosilicification is believed to have evolved only once, in the ancestor of loricates [17], but further evidence is required to confirm this, or to determine whether SIT-related silica biomineralization is the ancestral condition of the choanoflagellates and was lost in non-loricate groups [17]. This ambiguity, together with the incomplete fossil record and phylogenetic results (figure 3), means that it remains inconclusive as to whether the LGT event was stramenopile-to-choanoflagellate, diatom-to-choanoflagellate or choanoflagellate-to-stramenopile.
(f). Evolutionary implications for biosilicification
In addition to SITs, there are several classes of genes involved in biosilicification in diatoms (e.g. silaffins [66] and silacidins [67]). None of these, other than SITs, were identified in our sequence data. Additionally, no evidence was found for choanoflagellate versions of the silica-related sponge enzyme silicatein [68]. This suggests that other components of the choanoflagellate biosilica metabolic machinery may have a choanoflagellate-specific origin.
Bulk organic analysis of the costal strips from S. diplocostata yielded a glycoprotein that has been proposed to be involved in costal strip silicification [69]. Multiple glycoprotein-encoding genes were identifiable in the S. diplocostata transcriptome dataset; however, no obvious candidates for a costal strip-specific glycoprotein could be identified (data not shown). These results support an origin of biosilicification in choanoflagellates that is independent both of sponges and diatoms, though the choanoflagellate and diatom cases may be related to each other by LGT.
In this scenario, biosilicification with a known mechanism arose a minimum of twice in the opisthokonts—once in the metazoans (siliceous sponges) and once in the choanoflagellates; and independently at least once in each of the Stramenopiles+Alveolates+Rhizaria supergroup (stramenopiles) and archaeplastids (in land plants). Biosilicification is found in isolated species or small groups in each of the eukaryotic supergroups [37]. However, current molecular evidence of differing mechanisms points towards multiple independent origins of biosilica, rather than biosilicification being present in the last common ancestor of all eukaryotes. In the case of the choanoflagellate and stramenopile SITs, and the land plant NIPs [70], the evolution of genes encoding silicon-transporting proteins appears to involve LGT events. Owing to the unusual biochemistry of silicon [71], the acquisition of silicon transport by LGT may be relatively more prevalent than their de novo evolution from existing transporter genes.
Biosilicification provides many selective benefits (e.g. protection, [72]), so once a foreign SIT gene was incorporated into the genome it would confer a strong selective advantage for that organism. Several common biomolecules, such as collagen [73], glycoproteins [69], polyamines [2] and proteases [74] are known to have the capacity to direct silica polymerization. It may be the case that many taxa possess the capacity for silicification but lack the means to concentrate sufficient silicon for polymerization to occur. This difficulty would be overcome by the acquisition of a laterally transferred gene for silicon transport, with important implications for the ecology [43], evolutionary diversity [53] and biogeochemistry [3] of the newly biomineralizing lineage.
Acknowledgements
We thank Barry Leadbeater (University of Birmingham) for providing cultures of S. diplocostata and D. grandis, and Dan Richter and Nicole King (University of California, Berkeley) for helpful discussions. Ken Siggens gave valuable technical advice. Sequencing work was supported by BBSRC Capacity and Capability Challenge Programme project no. CCC-1–10. This work was supported by the BBSRC Comparative Genomics training grant BB/E527604/1 to A.O.M. and a Leathersellers' Company Scholarship to A.O.M awarded by Fitzwilliam College, Cambridge, UK.
References
- 1.Simkiss K, Wilbur KM. 1989. Biomineralization: cell biology and mineral deposition. San Diego, CA: Academic Press Inc [Google Scholar]
- 2.Gröger C, Lutz K, Brunner E. 2008. Biomolecular self-assembly and its relevance in silica biomineralization. Cell Biochem. Biophys. 50, 23–39 10.1007/s12013-007-9003-2 (doi:10.1007/s12013-007-9003-2) [DOI] [PubMed] [Google Scholar]
- 3.Maldonado M, Carmona MC, Cruzado A. 1999. Decline in Mesozoic reef-building in sponges explained by silicon limitation. Nature 401, 785–788 10.1038/44560 (doi:10.1038/44560). [DOI] [Google Scholar]
- 4.Hildebrand M, Higgins DR, Busser K, Volcani BE. 1993. Silicon-responsive cDNA clones isolated from the marine diatom Cylindrotheca fusiformis. Gene 132, 213–218 10.1016/0378-1119(93)90198-C (doi:10.1016/0378-1119(93)90198-C) [DOI] [PubMed] [Google Scholar]
- 5.Jugdaohsingh R. 2009. Silicon and bone health. J. Nutr. Health Aging 11, 99–110 [PMC free article] [PubMed] [Google Scholar]
- 6.Hildebrand M, Volcani BE, Gassman W, Schroeder J. 1997. A gene family of silicon transporters. Nature 385, 688–689 10.1038/385688b0 (doi:10.1038/385688b0) [DOI] [PubMed] [Google Scholar]
- 7.Thamatrakoln K, Hildebrand M. 2007. Analysis of Thalassiosira pseudonana silicon transporters indicates distinct regulatory levels and transport activity through the cell cycle. Eukaryot. Cell 6, 271–279 10.1128/EC.00235-06 (doi:10.1128/EC.00235-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Likhoshway YV, Masyukova YA, Sherbakova TA, Petrova DP, Grachev AMA. 2006. Detection of the gene responsible for silicic acid transport in chrysophycean algae. Dokl. Biol. Sci. 408, 256–260 10.1134/S001249660603015X (doi:10.1134/S001249660603015X) [DOI] [PubMed] [Google Scholar]
- 9.Yamaji N, Ma JF. 2009. A transporter at the node responsible for intervascular transfer of silicon in rice. Plant Cell 21, 2878–2883 10.1105/tpc.109.069831 (doi:10.1105/tpc.109.069831) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma JF, Tamai K, Yamaji N, Mitani N, Konishi S, Katsuhara M. 2006. A silicon transporter in rice. Nature 440, 688–691 10.1038/nature04590 (doi:10.1038/nature04590) [DOI] [PubMed] [Google Scholar]
- 11.Ma JF, Yamaji N, Mitani N, Tamai K, Konishi S, Fujiwara T, Katsuhara M, Yano M. 2007. An efflux transporter of silicon in rice. Nature 448, 209–213 10.1038/nature05964 (doi:10.1038/nature05964) [DOI] [PubMed] [Google Scholar]
- 12.Schröder H-C, Perović-Ottstadt S, Rothenberger M, Wiens M, Schwertner H, Batel R, Korzhev M, Müller IM, Müller WEG. 2004. Silica transport in the demosponge Suberites domuncula: fluorescence emission analysis using the PDMPO probe and cloning of a potential transporter. Biochem. J. 381, 665–673 10.1042/BJ20040463 (doi:10.1042/BJ20040463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thamatrakoln K, Alverson AJ, Hildebrand M. 2006. Comparative sequence analysis of diatom silicon transporters: toward a mechanistic model of silicon transport. J. Phycol. 42, 822–834 10.1111/j.1529-8817.2006.00233.x (doi:10.1111/j.1529-8817.2006.00233.x) [DOI] [Google Scholar]
- 14.Mitani-Ueno N, Yamaji N, Zhao F-J, Ma JF. 2011. The aromatic/arginine selectivity filter of NIP aquaporins plays a critical role in substrate selectivity for silicon, boron, and arsenic. J. Exp. Bot. 62, 4391–4398 10.1093/jxb/err158 (doi:10.1093/jxb/err158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King N, et al. 2008. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451, 783–788 10.1038/nature06617 (doi:10.1038/nature06617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torruella G, Derelle R, Jordi P, Lang BF, Andrew R, Shalchian-Tabrizi K, Iñaki R-T. 2012. Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single copy protein domains. Mol. Biol. Evol. 29, 531–544 10.1093/molbev/msr185 (doi:10.1093/molbev/msr185). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitsche F, Carr M, Arndt H, Leadbeater BSC. 2011. Higher level taxonomy and molecular phylogenetics of the choanoflagellatea. J. Eukaryot. Microbiol. 58, 452–462 10.1111/j.1550-7408.2011.00572.x (doi:10.1111/j.1550-7408.2011.00572.x) [DOI] [PubMed] [Google Scholar]
- 18.Mann S, Williams RJP. 1982. High resolution electron microscopy studies of the silica lorica in the choanoflagellate Stephanoeca diplocostata Ellis. Proc. R. Soc. Lond. B 216, 137–146 10.1098/rspb.1982.0066 (doi:10.1098/rspb.1982.0066) [DOI] [Google Scholar]
- 19.Leadbeater BSC. 1979. Developmental studies on the loricate choanoflagellate Stephanoeca diplocostata Ellis. I. Ultrastructure of the non-dividing cell and costal strip production. Protoplasma 262, 241–262 10.1007/BF01281442 (doi:10.1007/BF01281442) [DOI] [Google Scholar]
- 20.Leadbeater BSC, Yu Q, Kent J, Stekel DJ. 2009. Three-dimensional images of choanoflagellate loricae. Proc. R. Soc. B 276, 3–11 10.1098/rspb.2008.0844 (doi:10.1098/rspb.2008.0844) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leadbeater BSC. 1985. Developmental studies on the loricate choanoflagellate Stephanoeca diplocostata Ellis. IV. Effects of silica deprivation on growth and loricate production. Protoplasma 127, 171–179 10.1007/BF01276261 (doi:10.1007/BF01276261) [DOI] [Google Scholar]
- 22.Leadbeater BSC, Cheng R. 2010. Costal strip production and lorica assembly in the large tectiform choanoflagellate Diaphanoeca grandis Ellis. Eur. J. Protistol. 46, 96–110 10.1016/j.ejop.2009.12.003 (doi:10.1016/j.ejop.2009.12.003) [DOI] [PubMed] [Google Scholar]
- 23.Leadbeater BSC. 1989. Developmental studies on the loricate choanoflagellate Stephanoeca diplocostata Ellis. VI. Effects of silica replenishment on silica impoverished cells. Protoplasma 153, 71–84 10.1007/BF01322467 (doi:10.1007/BF01322467) [DOI] [Google Scholar]
- 24.Leadbeater BSC. 1987. Developmental studies on the loricate choanoflagellate Stephanoeca diplocostata Ellis. V. The cytoskeleton and the effects of microtubule poisons. Protoplasma 136, 1–15 10.1007/BF01276313 (doi:10.1007/BF01276313) [DOI] [Google Scholar]
- 25.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 10.1093/nar/25.17.3389 (doi:10.1093/nar/25.17.3389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doyle J, Doyle J. 1987. A rapid DNA isolation method for small quantities of fresh tissues. Phytochem. Bull. 19, 11–15 [Google Scholar]
- 27.Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. 2007. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587 10.1093/nar/gkm259 (doi:10.1093/nar/gkm259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffman K, Stoffel W. 1993. TMbase: a database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seyler 374, 166 [Google Scholar]
- 29.Tusnády GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17, 849–850 10.1093/bioinformatics/17.9.849 (doi:10.1093/bioinformatics/17.9.849) [DOI] [PubMed] [Google Scholar]
- 30.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 10.1006/jmbi.2000.4315 (doi:10.1006/jmbi.2000.4315) [DOI] [PubMed] [Google Scholar]
- 31.Sapriel G, Quinet M, Heijde M, Jourdren L, Crom L, Lopez PJ. 2009. Genome-wide transcriptome analyses of silicon metabolism in Phaeodactylum tricornutum reveal the multilevel regulation of silicic acid transporters. PLoS ONE 4, e7458. 10.1371/journal.pone.0007458 (doi:10.1371/journal.pone.0007458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105 10.1093/bioinformatics/bti263 (doi:10.1093/bioinformatics/bti263) [DOI] [PubMed] [Google Scholar]
- 33.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 10.1093/sysbio/syq010 (doi:10.1093/sysbio/syq010) [DOI] [PubMed] [Google Scholar]
- 34.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771 10.1080/10635150802429642 (doi:10.1080/10635150802429642) [DOI] [PubMed] [Google Scholar]
- 35.Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 10.1093/bioinformatics/17.8.754 (doi:10.1093/bioinformatics/17.8.754) [DOI] [PubMed] [Google Scholar]
- 36.Sherbakova TA, et al. 2005. Conserved motif CMLD in silicic acid transport proteins of diatoms. Mol. Biol. 39, 269–280 10.1007/s11008-005-0038-4 (doi:10.1007/s11008-005-0038-4) [DOI] [PubMed] [Google Scholar]
- 37.Walker G, Dorrell RG, Schlacht A, Dacks JB. 2011. Eukaryotic systematics: a user's guide for cell biologists and parasitologists. Parasitology 138, 1638–1663 10.1017/S0031182010001708 (doi:10.1017/S0031182010001708) [DOI] [PubMed] [Google Scholar]
- 38.Dayel MJ, Alegado RA, Fairclough SR, Levin TC, Nichols SA, McDonald K, King N. 2011. Cell differentiation and morphogenesis in the colony-forming choanoflagellate Salpingoeca rosetta. Dev. Biol. 357, 73–82 10.1016/j.ydbio.2011.06.003 (doi:10.1016/j.ydbio.2011.06.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snell EA, Brooke NM, Taylor WR, Casane D, Philippe H, Holland PWH. 2006. An unusual choanoflagellate protein released by Hedgehog autocatalytic processing. Proc. R. Soc. B 273, 401–407 10.1098/rspb.2005.3263 (doi:10.1098/rspb.2005.3263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hildebrand M, Dahlin K, Volcani BE. 1998. Characterization of a silicon transporter gene family in Cylindrotheca fusiformis: sequences, expression analysis, and identification of homologs in other diatoms. Mol. Gen. Genet. 260, 480–486 10.1007/s004380050920 (doi:10.1007/s004380050920) [DOI] [PubMed] [Google Scholar]
- 41.Alverson AJ. 2007. Strong purifying selection in the silicon transporters of marine and freshwater diatoms. Plant Biol. 52, 1420–1429 [Google Scholar]
- 42.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. 2000. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016 10.1006/jmbi.2000.3903 (doi:10.1006/jmbi.2000.3903) [DOI] [PubMed] [Google Scholar]
- 43.Martin-Jézéquel V, Hildebrand M, Brzezinski MA. 2000. Silicon metabolism in diatoms: implications for growth. J. Phycol. 36, 821–840 10.1046/j.1529-8817.2000.00019.x (doi:10.1046/j.1529-8817.2000.00019.x) [DOI] [Google Scholar]
- 44.Finkel ZV, Kotrc B. 2010. Silica use through time: macroevolutionary change in the morphology of the diatom fustule. Geomicrobiol. J. 27, 596–608 10.1080/01490451003702941 (doi:10.1080/01490451003702941) [DOI] [Google Scholar]
- 45.Abramson J, Wright EM. 2009. Structure and function of Na+-symporters with inverted repeats. Curr. Opin. Struct. Biol. 19, 425–432 10.1016/j.sbi.2009.06.002 (doi:10.1016/j.sbi.2009.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boudker O, Verdon G. 2010. Structural perspectives on secondary active transporters. Trends Pharmacol. Sci. 31, 418–426 10.1016/j.tips.2010.06.004 (doi:10.1016/j.tips.2010.06.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khafizov K, Staritzbichler R, Stamm M, Forrest LR. 2010. A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry 49, 10 702–10 713 10.1021/bi101256x (doi:10.1021/bi101256x) [DOI] [PubMed] [Google Scholar]
- 48.Thamatrakoln K, Hildebrand M. 2008. Silicon uptake in diatoms revisited: a model for saturable and nonsaturable uptake kinetics and the role of silicon transporters. Plant Physiol. 146, 1397–1407 10.1104/pp.107.107094 (doi:10.1104/pp.107.107094) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krishnamurthy H, Piscitelli CL, Gouaux E. 2009. Unlocking the molecular secrets of sodium-coupled transporters. Nature 459, 347–355 10.1038/nature08143 (doi:10.1038/nature08143) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Screpanti E, Hunte C. 2007. Discontinuous membrane helices in transport proteins and their correlation with function. J. Struct. Biol. 159, 261–267 10.1016/j.jsb.2007.01.011 (doi:10.1016/j.jsb.2007.01.011) [DOI] [PubMed] [Google Scholar]
- 51.Bhattacharyya PEB, Volcani BE. 1980. Sodium-dependent silicate transport in the apochlorotic marine diatom Nitzschia alba. Proc. Natl Acad. Sci. USA 77, 6386–6390 10.1073/pnas.77.11.6386 (doi:10.1073/pnas.77.11.6386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curnow P, Senior L, Knight MJ, Thamatrakoln K, Hildebrand M, Booth PJ. 2012. Expression, purification, and reconstitution of a diatom silicon transporter. Biochemistry 51, 3776–3785 10.1021/bi3000484 (doi:10.1021/bi3000484) [DOI] [PubMed] [Google Scholar]
- 53.Leadbeater BSC. 2008. Choanoflagellate evolution: the morphological perspective. Protistology 5, 256–267 [Google Scholar]
- 54.Paul M. 2012. Acanthocorbis mongolica nov. spec.—description of the first freshwater loricate choanoflagellate (Acanthoecida) from a Mongolian lake. Eur. J. Protistol. 48, 1–8 10.1016/j.ejop.2011.07.001 (doi:10.1016/j.ejop.2011.07.001) [DOI] [PubMed] [Google Scholar]
- 55.Ruiz-Trillo I, Burger G, Holland PWH, King N, Lang BF, Roger AJ, Gray MW. 2007. The origins of multicellularity: a multi-taxon genome initiative. Trends Genet. 23, 113–118 10.1016/j.tig.2007.01.005 (doi:10.1016/j.tig.2007.01.005) [DOI] [PubMed] [Google Scholar]
- 56.Gobler CJ, et al. 2011. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc. Natl Acad. Sci. USA 108, 4352–4357 10.1073/pnas.1016106108 (doi:10.1073/pnas.1016106108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun G, Yang Z, Ishwar A, Huang J. 2010. Algal genes in the closest relatives of animals. Mol. Biol. Evol. 27, 2879–2889 10.1093/molbev/msq175 (doi:10.1093/molbev/msq175) [DOI] [PubMed] [Google Scholar]
- 58.Bowler C, et al. 2008. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456, 239–244 10.1038/nature07410 (doi:10.1038/nature07410) [DOI] [PubMed] [Google Scholar]
- 59.Keeling PJ, Palmer JD. 2008. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9, 605–618 10.1038/nrg2386 (doi:10.1038/nrg2386) [DOI] [PubMed] [Google Scholar]
- 60.Nedelcu AM, Blakney JC, Logue KD. 2009. Functional replacement of a primary metabolic pathway via multiple independent eukaryote-to-eukaryote gene transfers and selective retention. J. Evol. Biol. 22, 1882–1894 10.1111/j.1420-9101.2009.01797.x (doi:10.1111/j.1420-9101.2009.01797.x) [DOI] [PubMed] [Google Scholar]
- 61.Tucker RP, Beckmann J, Leachman NT, Schöler J. 2012. Phylogenetic analysis of the teneurins: conserved features and premetazoan ancestry. Mol. Biol. Evol. 29, 1019–1029 10.1093/molbev/msr271 (doi:10.1093/molbev/msr271). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michael AJ. 2011. Molecular machines encoded by bacterially-derived multi-domain gene fusions that potentially synthesize, N-methylate and transfer long chain polyamines in diatoms. FEBS Lett. 585, 2627–2634 10.1016/j.febslet.2011.07.038 (doi:10.1016/j.febslet.2011.07.038) [DOI] [PubMed] [Google Scholar]
- 63.Mock T, et al. 2008. Whole-genome expression profiling of the marine diatom Thalassiosira pseudonana identifies genes involved in silicon bioprocesses. Proc. Natl Acad. Sci. USA 105, 1579–1584 10.1073/pnas.0707946105 (doi:10.1073/pnas.0707946105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macdonald FA, Cohen PA, Dudas F, Schrag DP. 2010. Early Neoproterozoic scale microfossils in the Lower Tindir Group of Alaska and the Yukon Territory. Geology 38, 143–146 10.1130/G25637.1 (doi:10.1130/G25637.1) [DOI] [Google Scholar]
- 65.Parfrey LW, Lahr DJG, Knoll AH, Katz L. 2011. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl Acad. Sci. USA 108, 13 624–13 629 10.1073/pnas.1110633108 (doi:10.1073/pnas.1110633108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poulsen N, Sumper M, Kroger N. 2003. Biosilica formation in diatoms: characterization of native silaffin-2 and its role in silica morphogenesis. Proc. Natl Acad. Sci. USA 100, 12 075–12 080 10.1073/pnas.2035131100 (doi:10.1073/pnas.2035131100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wenzl S, Hett R, Richthammer P, Sumper M. 2008. Silacidins: highly acidic phosphopeptides from diatom shells assist in silica precipitation in vitro. Angew. Chem. 47, 1729–1732 10.1002/anie.200704994 (doi:10.1002/anie.200704994) [DOI] [PubMed] [Google Scholar]
- 68.Shimizu K, Cha J, Stucky GD, Morse DE. 1998. Silicatein alpha: cathepsin L-like protein in sponge biosilica. Proc. Natl Acad. Sci. USA 95, 6234–6238 10.1073/pnas.95.11.6234 (doi:10.1073/pnas.95.11.6234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gong N, Wiens M, Schröder HC, Mugnaioli E, Kolb U, Müller WEG. 2010. Biosilicification of loricate choanoflagellate: organic composition of the nanotubular siliceous costal strips of Stephanoeca diplocostata. J. Exp. Biol. 213, 3575–3585 10.1242/jeb.048496 (doi:10.1242/jeb.048496) [DOI] [PubMed] [Google Scholar]
- 70.Zardoya R, Ding X, Kitagawa Y, Chrispeels MJ. 2002. Origin of plant glycerol transporters by horizontal gene transfer and functional recruitment. Proc. Natl Acad. Sci. USA 99, 14 893–14 896 10.1073/pnas.192573799 (doi:10.1073/pnas.192573799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kinrade SD. 1999. Stable five- and six-coordinated silicate anions in aqueous solution. Science 285, 1542–1545 10.1126/science.285.5433.1542 (doi:10.1126/science.285.5433.1542) [DOI] [PubMed] [Google Scholar]
- 72.Hamm CE, Merkel R, Springer O, Jurkojc P. 2003. Architecture and material properties of diatom shells provide effective mechanical protection. Nature 421, 841–843 10.1038/nature01416 (doi:10.1038/nature01416). [DOI] [PubMed] [Google Scholar]
- 73.Ehrlich H, et al. 2010. Mineralization of the metre-long biosilica structures of glass sponges is templated on hydroxylated collagen. Nat. Chem. 2, 1084–1088 10.1038/nchem.899 (doi:10.1038/nchem.899) [DOI] [PubMed] [Google Scholar]
- 74.Coradin T, Livage J. 2003. Interactions of bovine serum albumin and lysozyme with sodium silicate solutions. Colloids Surf. 29, 189–196 10.1016/S0927-7765(02)00208-4 (doi:10.1016/S0927-7765(02)00208-4) [DOI] [Google Scholar]