

Abstract
Differential coexistence among species underlies geographical patterns of biodiversity. Understanding such patterns has relied either on ecological or historical approaches applied separately. Recently, macroecology and community phylogenetics have tried to integrate both ecological and historical approaches. However, macroecology is mostly non-phylogenetic, whereas community phylogenetics is largely focused on local scales. Here, we propose a conceptual framework to link macroecology and community phylogenetics by exploring the evolutionary context of large-scale species coexistence, introducing the phylogenetic field concept. This is defined as the phylogenetic structure of species co-occurrence within a focal species' geographical range. We developed concepts and methods for analysing phylogenetic fields and applied them to study coexistence patterns of the bat family Phyllostomidae. Our analyses showed that phyllostomid bats coexist mostly with closely related species, revealing a north–south gradient from overdispersed to clustered phylogenetic fields. Patterns at different phylogenetic levels (i.e. all species versus close relatives only) presented the same gradient. Results support the tropical niche conservatism hypothesis, potentially mediated by higher speciation rates in the region of origin coupled with shared environmental preferences among species. The phylogenetic field approach enables species-based community phylogenetics, instead of those that are site-based, allowing the description of historical processes at more appropriate macroecological and biogeographic scales.
Keywords: biodiversity, macroecology, community phylogenetics, niche conservatism, historical processes, bats
1. Introduction
Differential coexistence among species in distinct regions of the globe results in species richness varying geographically [1,2]. One of the groups that exemplify this phenomenon is the New World bat family Phyllostomidae, which shows a strong latitudinal gradient in species richness, as well as in phenetic and functional diversity [3,4]. Geographical patterns of phyllostomid biodiversity are thought to result in part from historical processes [5], among which those related to tropical niche conservatism (TNC; e.g. shared environmental preferences among related species and higher speciation at region of origin [6]) have gained recent support [7]. Studies supporting historical processes as major determinants of phyllostomid assemblage patterns have been based on species coexisting at particular assemblages or sites (i.e. site-based; [8–10]). However, such scale of analysis does not consider complete areas of distribution of species throughout which different assemblages are formed, as it only represents particular spatial instances of species coexistence patterns [11]. What remains to be determined is how species coexistence is reflected at the level of species' ranges (i.e. species-based) and the relative influence of ecological and evolutionary processes on those patterns.
Currently, phylogenetic approaches are being widely used to study community assembly and the resulting patterns of species coexistence [12]. This integration of ecology and evolution under a community phylogenetics approach [13] is based on the assumption that species interact through traits that are non-randomly distributed within the phylogeny [14]. Accordingly, genealogical relationships among species inhabiting an assemblage (i.e. its phylogenetic structure) are interpreted as a product of either ecological processes such as biotic interactions and habitat filters acting at local spatial scales [13], or historical processes like in situ speciation [15–17] and niche conservatism [6,7,18] acting at larger spatial and temporal scales [15,19]. The community phylogenetics approach has concentrated mainly on local to regional spatial scales and focusing on sites, whether single, multiple sites or entire regions [12,15,19]). Nonetheless, the phylogenetic structure of a set of sites is not sufficient to reveal species-level patterns because this may differ from the overall phylogenetic structure within a species' range, unless all sites and species within it are considered.
A recently developed framework [11,20] allows the analysis of species coexistence patterns as depicted within individual species' ranges. Such framework is based on the inherent relationship between species richness and spatial distribution, especially at broad spatial scales where richness is usually measured as the overlap of species' ranges within a gridded domain [21]. This relationship can be analysed through a frequency distribution of grid cells with different richness values (species richness frequency distribution (SRFD)) and interpreted as a function of species' coexistence [11]. Furthermore, an SRFD can be built independently for each individual species to describe its diversity field [11]. The diversity field of a species characterizes the assemblages occupied throughout its range, reflecting its tendency to occur in species-rich or species-poor regions. For instance, depending on its co-occurring species (e.g. all species within a clade versus specific sub-lineages or ecological guilds), a species may coexist with a higher or lower number of species. In fact, a community phylogenetics approach might predict lower or higher coexistence among closely related species depending on the processes involved [13]. Consequently, a phylogenetic component of the diversity field is to be expected and could be used to infer potential processes determining geographical coexistence among species.
Phylogenetic patterns of diversity fields can be investigated with standard methods applied in community phylogenetics together with macroecological analyses of their geographical structure. For instance, given a ‘focal’ species, the shape of its SRFD can describe coexistence patterns throughout its geographical range [11]. In addition, phylogenetic relationships between co-occurring and focal species characterize the evolutionary component of the coexistence patterns, which can be scrutinized from a phylogenetic point-of-view (e.g. in terms of clustering or overdispersion). Furthermore, specific predictions for such coexistence can be derived from biogeographic and evolutionary theory. For instance, processes such as higher in situ speciation [16,17] and niche conservatism [6] would predict coexistence among closely related species (i.e. phylogenetic clustering). By contrast, niche evolution, evolutionary convergence and colonization would promote coexistence among distantly related species (i.e. phylogenetic overdispersion).
Here, we introduce the concept of phylogenetic field, defined as the phylogenetic structure of species co-occurrence within a focal species' geographical range, to study species-level patterns of coexistence and phylogenetic structure. A phylogenetic field can be viewed as a species' attribute describing the overall phylogenetic relatedness with its coexisting species. Although it depends on species co-occurrence, which is defined geographically, a species' phylogenetic field is best depicted in the phylogenetic tree (i.e. showing the phylogenetic position of species co-occurring within its range, as in figure 1). We focus on species' ranges, instead of sites or local assemblages, as the observational unit to study phylogenetic structure at geographical scales and infer historical processes involved in species coexistence. In doing so, we extend the traditional site-based perspective of phylogenetic structure to a broad-scale biogeographic and species-based setting, proposing a conceptual link between macroecological methods and phylogenetic approaches to study large-scale biodiversity patterns.
Figure 1.
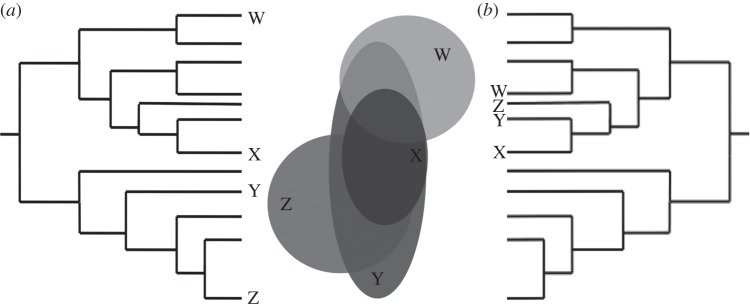
Schematic of the phylogenetic field, showing (a) overdispersed and (b) clustered phylogenetic fields of species X, determined by its phylogenetic relationship with species co-occurring within its range (species W, Y and Z).
Based on previous studies that suggest an important role of historical processes in the origin and maintenance of phyllostomid assemblages previously discussed [5,7–10], we expect that phylogenetic fields of phyllostomid bats should also reveal the effect of such historical processes. According to TNC, we could expect species to coexist with a high number of related species, especially towards the equator, whereas at subtropical/temperate latitudes, we may expect coexistence among less related species [5,7]. Thus, we make two specific predictions about the phylogenetic fields of phyllostomid bats: (i) a positive relationship between species richness and clustering of phylogenetic fields, in which species with richer diversity fields and negatively skewed SRFDs will show clustered phylogenetic fields; and (ii) a geographical gradient from clustered to overdispersed phylogenetic fields as we move away from the equator. We believe that using phyllostomid bats as an example reveals how studying phylogenetic fields can help to infer potential processes responsible for geographical patterns of coexistence.
2. Material and methods
(a). Distributional data and diversity fields
A distributional database was compiled from previous studies [11], following the taxonomical arrangement of Simmons [22]. This database was used to map the geographical distribution (i.e. extent of occurrence) of 126 species of phyllostomid bats for which we had phylogenetic information (see below). Range maps were built using ArcGIS with data from the primary literature up to 2004 for North American species and augmented with data from the Nature Serve database [23] for South American species [11]. A presence–absence matrix was built from overlying a regular grid of 0.5° resolution onto the distributional maps, obtaining a 126 species × 6489 cells matrix.
From the presence–absence matrix, we extracted information on diversity fields of species by an Rq-mode approach, in which data from a row (i.e. species) is gathered by considering data in the columns (i.e. cells) it intersects [20]. The SRFD that emerges from this procedure can be described by its standard statistical moments (e.g. mean, s.d., skewness and kurtosis). We estimated the total number of co-occurring species (i.e. species richness) within each focal range as well as the mean and skewness of its diversity field's SRFD. Mean and skewness of a SRFD describe the within-range richness structure present over all sites occupied by a focal species, representing the average and variance in species richness across its geographical range, respectively [11]. In addition, we also recorded the latitudinal midpoint of each species' range to describe its geographical location.
(b). Phylogenetic data and phylogenetic fields
Phylogenetic information for phyllostomid bats was obtained from a time-calibrated species-level supertree encompassing all of Chiroptera [24]. The subtree containing the family Phyllostomidae was pruned from the supertree using Mesquite v. 2.72 [25], keeping branch length information. This tree was then used as the global phylogeny to estimate phylogenetic fields of species. We were interested in describing the phylogenetic field of each phyllostomid species, as a way to characterize the overall phylogenetic structure contained within a range including the focal species. This allows assessing if individual species coexist with either closely or distantly related or a random set of phyllostomid bats. Thus, each phylogenetic field is described by a single value of phylogenetic structure representing the total set of coexisting species within a focal species' range, rather than an aggregate measure (e.g. mean, variance) of phylogenetic diversity or any other metrics from individual sites composing that range.
Phylogenetic structure of a set of species can be described by different metrics, which aim to inform about the degree of clustering (i.e. species are, on average, more closely related than expected by chance), overdispersion (i.e. species are, on average, more distantly related than expected by chance) or randomness in the phylogenetic structure of an assemblage [13]. We used here the phylogenetic species variability (PSV) and phylogenetic species clustering (PSC) indices [26] that summarize the degree of phylogenetic relatedness among species in an assemblage, considering all species (i.e. deep phylogenetic level) or closest relatives only (i.e. shallow phylogenetic level), respectively. Thus, results from both indices may differ if distinct processes determine the assembly of lineages at different phylogenetic levels, making their interpretations complementary.
PSV and PSC values can be interpreted in terms of variance of a neutral trait among species in an assemblage, varying between 0 (reduced variability, clustering) and 1 (maximum variability, overdispersion), with unity being the maximum attainable value under a ‘star phylogeny’ representing species' phylogenetic independence [26]. In addition, these indices are not affected by species richness and abundance, allowing the estimation of pure phylogenetic structure [26]. We used both PSV and PSC to statistically describe and compare the phylogenetic fields of species. Calculations of both indices included the focal species and are represented by an ‘sp’ subscript (e.g. PSVsp/PSCsp) to differentiate them from common PSV/PSC indices used for local assemblages. Also, we used the K-statistic [27] to test for phylogenetic signal (in terms of deviations from a Brownian expectation) in phylogenetic field (i.e. PSVsp and PSCsp) and range attributes (i.e. range size and skewness of SRFD) of species, and to evaluate if species' variation for these attributes was phylogenetically correlated.
The particular phylogenetic structure of an assemblage depends on the clade under consideration [28,29]. For instance, one could either consider all species within a clade or only species within particular subclades or ecological groupings (e.g. feeding guilds). In phyllostomid bats, the inclusion of a species within a particular feeding guild has a direct correspondence with the delimitation of subfamilies that are phylogenetically well established [24,30–33]. To evaluate if different phylogenetic scales could be responsible for observed patterns in Phyllostomidae, we followed a pattern deconstruction approach [34] by estimating restricted phylogenetic fields defined only by species within each of the three most diverse phyllostomid subfamilies (Stenodermatinae, Phyllostominae and Glossophaginae) as the coexistence set. In this case, a species' phylogenetic field would only consider co-occurring species belonging to the same subfamily.
For comparison and to help interpret geographical components of species' phylogenetic fields, we also mapped the PSV and PSC values of phyllostomid assemblages throughout the New World, calculating these metrics for the grid cells of the domain (see earlier). Note that, in this case, both PSV and PSC indices represent the phylogenetic structure of species occurring at each individual cell rather than whole species' ranges, as proposed for phylogenetic fields (i.e. PSV/PSC ≠ PSVsp/PSCsp).
(c). Statistical analyses
As a species' property, the phylogenetic field can be related to other species' attributes relevant to its coexistence patterns. To address if there was a relationship between phylogenetic fields and within-range species richness and structure, we correlated PSVsp and PSCsp values of phylogenetic fields with total and mean species richness within ranges and with skewness of their SRFDs. In addition, to explore if there was a geographical gradient of phylogenetic field structure, we correlated their PSVsp and PSCsp values against ranges' latitudinal midpoints. Because previous analyses showed no phylogenetic signal in the studied traits (i.e. phylogenetic field's PSVsp and PSCsp, range size, skewness of SRFD), we did not consider necessary to include phylogenetic structure to avoid biased type I error estimates in these correlation analyses.
We developed two null models to determine if observed phylogenetic field patterns differed from those expected by chance. This was done by comparing each phylogenetic field against a distribution of 1000 randomly generated phylogenetic fields per null model. In the first null model (I), species identity is shuffled to entertain the possibility of independent distributional patterns among species in which any species may coexist with any other species. Thus, for each model replicate, the null phylogenetic field of each species is generated by randomly sampling, without replacement, the observed number of co-occurring species from the global phylogeny. However, this first null model does not take into account the variation in species' geographical range sizes, which makes coexistence with large-ranged species more likely. Thus, we designed a second null model (II) following a similar procedure, but with the probability of sampling a co-existing species being proportional to its range size. In this second model, distributional patterns of species are still independent of each other, but large-ranged species are more likely to coexist with other species than small-ranged species.
In addition, we conducted partial randomizations [28] restricted by subfamily membership. Partial randomizations shuffle species only within a defined clade, allowing testing of phylogenetic structure independently within each clade and informing which parts of the phylogenetic tree contribute to the observed patterns [28]. We followed the same procedure mentioned above, differing only in the choice of global phylogeny and species pool. In this case, only species belonging to the same subfamily were considered in the null models. In addition to reporting the number of species with significant phylogenetic field values, we also conducted a Fisher's test of combined probabilities [35] to evaluate the overall significance pattern of species from each null model. Null models were constructed and run in the R statistical language [36], using the APE [37] and PICANTE [38] packages. Phylogenetic analyses were performed in the new software Phylogenetic Analysis in Macroecology (T. F. L. V. B. Rangel & J. A. F. Diniz-Filho 2012, unpublished data).
3. Results
(a). Geographical coexistence and phylogenetic structure
On average, phyllostomid bats coexist geographically with a high number of other phyllostomids. Total species richness within individual ranges varied from a few species (15) to all of the species studied (126), averaging high richness for all species' ranges (mean = 93.43 species). In fact, the majority of species (62%) coexisted with a higher number of phyllostomids than the overall mean for all ranges (median = 101 species). The majority of species showed highly structured diversity fields with negatively skewed SRFDs (see the electronic supplementary material, table S1), implying that species richness within a species' range is variable with most sites holding more species than the range's average.
Phylogenetic structure of co-occurring species within individual phyllostomid ranges revealed a pattern of high PSVsp and PSCsp values (see the electronic supplementary material, table S1), varying from 0.732 to 0.809 for PSVsp and 0.677 to 0.799 for PSCsp. Hence, observed values of phyllostomids' phylogenetic fields would seem to be close to the maximum value (1.0), suggesting an overdispersed phylogenetic structure determined by coexistence among distantly related species. In addition, differences among individual species' values suggest that some species have clustered phylogenetic fields, defined by coexistence among comparatively more related species. However, such values need to be compared with theoretical expectations before reaching satisfying conclusions (see below).
(b). Diversity fields versus phylogenetic fields
Species ranges' attributes, such as size, diversity field structure (i.e. total and mean richness and skewness) and phylogenetic field values did not exhibit a phylogenetic signal; hence, relationships among them were evaluated with standard statistical methods. Phylogenetic fields showed significant relationships with other coexistence attributes of species' ranges. These relationships varied depending on the index used to describe phylogenetic fields, PSVsp or PSCsp. For instance, a negative relationship was found between phylogenetic fields' PSVsp and total species richness within ranges (r = −0.552) as well as with mean richness (results not shown; figure 2a). Instead, these relationships were inverted when phylogenetic fields were described by PSCsp. Positive relationships were found between phylogenetic fields' PSCsp and total species richness within ranges (r = 0.829) as well as with mean richness (results not shown; figure 2c). Opposite relationships were found between SRFDs' skewness and PSVsp/PSCsp. A positive relationship was found between PSVsp and SRFDs' skewness (r = 0.355), whereas a negative relationship was found between PSCsp and SRFDs' skewness (r = −0.284) (figure 2b,d). The relationship between the latitudinal midpoint of species' ranges and their phylogenetic fields also showed different patterns for PSVsp and PSCsp. For PSVsp, there was no clear geographical patterning, whereas a significant quadratic relationship was observed between latitudinal midpoints and PSCsp (p < 0.001) (see the electronic supplementary material, figure S1).
Figure 2.

Statistical relationships between phylogenetic and diversity fields. (a,b) Illustrate relationships between phylogenetic fields' PSVsp and total within-range richness and SRFDs' skewness, respectively. (c,d) Illustrate relationships between phylogenetic fields' PSCsp and total within-range richness and SRFDs' skewness, respectively. Grey triangles indicate significantly clustered (point down) and overdispersed (point up) phylogenetic fields from null model II comparisons. Fitted lines represent adjusted least-squares regressions for all data points.
(c). Single-site phylogenetic structure
At local geographical scales, the phylogenetic structure of assemblages within each cell of the domain showed clear geographical structuring. Bat assemblages located at higher latitudes had lower values of PSV and PSC than those around the equator (figure 3b,c). For PSV, the pattern was less obvious and some assemblages at higher latitudes also showed higher values. Moreover, for both PSV and PSC, some northern assemblages, namely along the Chihuahuan desert in northern Mexico, showed a reversed pattern from adjacent regions: lower PSV and higher PSC values, respectively (figure 3b,c).
Figure 3.
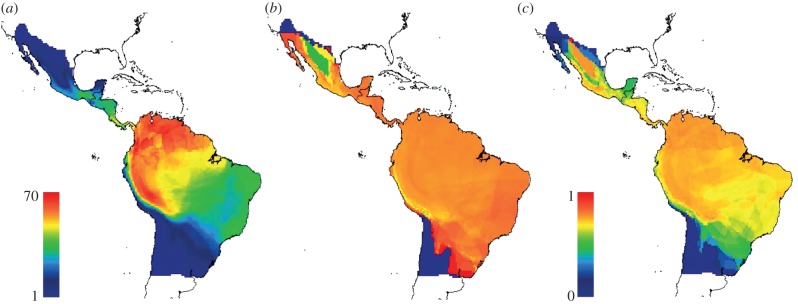
Geographic variation of (a) species richness, (b) PSV and (c) PSC for phyllostomid assemblages' present at each cell of the gridded domain. Scaled colour ramp for (b) and (c) is the same (0,1). (Online version in colour.)
(d). Null models and pattern deconstruction
Comparison between observed patterns and null model simulations showed similar results for PSVsp and PSCsp, revealing no significant phylogenetic field structure for the vast majority of species when considering the complete phylogeny (see the electronic supplementary material, table S1). Indeed, when compared with null model I results, none of the species showed significant PSVsp values and only few species showed significant PSCsp values (13 species with clustered and four species with overdispersed phylogenetic fields). Significant species patterns for both PSVsp and PSCsp were found when compared with null model II results (range size biased). Still, few species showed significantly clustered (17 and 32 species for PSVsp and PSCsp, respectively), or overdispersed (four species for both PSVsp and PSCsp) phylogenetic fields. Significant phylogenetic fields described by either PSVsp or PSCsp confirmed the statistical relationships between phylogenetic structure and coexistence patterns. Species with clustered phylogenetic fields coexisted with more species and had latitudinal midpoints around the equator, whereas species with overdispersed phylogenetic fields coexisted with less species and had northern latitudinal midpoints (figure 4).
Figure 4.

(a,c) Null model II simulation results depicting the latitudinal midpoint and (b,d) total within-range richness of significantly overdispersed and clustered, (a,b) PSVsp and (c,d) PSCsp, phylogenetic fields.
Deconstructing patterns by subfamily membership showed differences among subfamilies and between PSVsp and PSCsp phylogenetic fields. For PSVsp, the subfamily Stenodermatinae, with 55 primarily frugivore species, had few species with significantly overdispersed phylogenetic fields when contrasted against simulations of null models I and II (one and 13 species, respectively). The subfamily Phyllostominae, with 33 mostly insectivore species, also had few species with significant phylogenetic field structure when compared with null model I and II results (five and three species, respectively). By contrast, none of the species within the subfamily Glossophaginae (with 19 primarily nectarivore species) showed significant phylogenetic fields when compared with either null model.
Conversely, PSCsp phylogenetic fields of several species within all three subfamilies showed significantly clustered phylogenetic fields when compared with null model II results and for the first two subfamilies also for null model I results, whereas none had species with significantly overdispersed phylogenetic fields (see the electronic supplementary material, table S2). The Fisher's combined probability tests showed support for the observed patterns' overall significance when compared with null models simulations, mainly when comparing PSCsp values with null model II results. Nonetheless, from 16 comparisons between observed and simulated patterns, considering the family and subfamily analyses and both PSVsp and PSCsp indices, only nine showed overall significance. The overall non-significant patterns corresponded mainly to comparisons with null model I and using the PSVsp index (see the electronic supplementary material, table S3).
4. Discussion
(a). Phylogenetic fields and geographical coexistence
Species coexistence mediated by their overlapping ranges ultimately determines broad-scale biodiversity patterns [21]. This broad-scale coexistence results mainly from underlying historical (i.e. evolutionary) processes [39]. Thus, relative importance of these processes may be inferred from the evolutionary information contained within species' ranges resulting from coexistence with other species. Our phylogenetic field approach (describing the phylogenetic structure of species co-occurrence within species' ranges) revealed the effect of such historical processes on the coexistence among New World leaf-nosed bats.
Current biogeographic theory predicts geographical variation in the phylogenetic characteristics of species assemblages, mainly resulting from niche conservatism [6,13] and differential speciation [16,17,40]. Support for these predictions has been found in different taxa (reviewed in Wiens et al. [41]). For phyllostomid bats, Stevens [5,7] found that local assemblages showed evidence in agreement with TNC, under which the proportion of derived and least variable taxa increased from the centre to the periphery of the family's range. Extending predictions of TNC and differential speciation to a larger scale based on species' ranges, we also found support for historical processes. For instance, processes related to range size variation (e.g. geographical speciation, range dynamics, range size inheritance or traits related to it [42,43]) and niche conservatism (e.g. higher speciation rates at region of origin and shared environmental preferences among species [5–7]), as well as other potential processes (e.g. niche evolution and convergence) determine phylogenetic field patterns and their geographical arrangement in phyllostomids. Our results suggest a strong pattern of coexistence with high numbers of closely related species within the geographical ranges of phyllostomid bats.
Disentangling the relative importance of different historical processes remains challenging, as these processes drive biodiversity patterns throughout large spatial and temporal scales [42]. At best, we can differentiate among potential processes producing distinct outcomes. For instance, in concert with our first prediction, our finding that statistically significant phylogenetic field clustering increases with species richness and coexistence structure (i.e. SRFD negatively skewed: most sites hold more species than the ranges' mean) is in broad agreement with TNC predictions. On the other hand, the presence of significantly overdispersed phylogenetic fields suggests that niche evolution and convergence of less related species may also play a role determining geographical coexistence among phyllostomid bats.
Local phyllostomid assemblages at different latitudes are composed by different species, probably owing to distinct processes acting at different locations [5,11]. We found a geographical pattern of phyllostomids' phylogenetic fields, clustered at equatorial versus overdispersed at northern latitudes, agreeing with our second prediction and revealing the action of distinct processes shaping the composition of phyllostomid faunas at the periphery of the family's range. At high latitudes, processes such as historically recent dispersal and colonization of those subtropical and temperate environments by a few species could have resulted in lower richness and coexistence among less related species. Accordingly, if TNC acted equally on the distribution of different phyllostomid lineages, this would explain phylogenetic overdispersion at the northern edge of the family's range. For instance, some members of each major phyllostomid lineage have extensive ranges and may have evolved the ability to face seasonality and freezing temperatures. Thus, the presence of one or a few of these distantly related phyllostomids (e.g. species from different genera and subfamilies such as Artibeus, Glossophaga and Macrotus) at northern latitudes could certainly give rise to phylogenetic overdispersion of species' coexistence patterns at the periphery of the family's range (R. D. Stevens 2012, personal communication).
By contrast, at equatorial latitudes, processes such as higher speciation or lower extinction (or both) coupled with a tropical origin of the clade, could have produced higher species richness and coexistence among closely related species. At local scales, assemblages within individual grid cells showed a different gradient from that of phylogenetic fields. However, these local-scale patterns cannot be equated and interpreted exactly like species range patterns (i.e. phylogenetic fields) because they only represent specific sites within complete ranges. That is, local assemblages do not contain all species with which a particular species coexists and fail to represent the complete evolutionary context of species coexistence patterns, which is the main advantage of the phylogenetic field concept as proposed here.
(b). Deconstructing patterns and phylogenetic levels
As a species' trait, the phylogenetic field of a species can be influenced by other such traits determining the particular coexistence patterns and resulting phylogenetic structure. For instance, different ecological or morphological traits (e.g. diet, body size, etc.), may be related to specific phylogenetic field patterns. However, complete information on such traits is still lacking for many species for which the only available information may be their geographical distribution. Alternatively, exploring patterns within homogeneous groups of organisms defined ecologically or phylogenetically (i.e. guilds or families) can facilitate interpretation of observed patterns [34]. Indeed, our closer inspection within phyllostomid subfamilies highlighted the importance of evaluating patterns within such homogeneous groups of organisms. Species within the Phyllostomidae exhibit extraordinary feeding specializations ranging from insectivory to nectarivory and blood feeding, which are remarkably embedded within particular subfamilies [30–33] suggesting phylogenetic conservatism of these ecological characteristics, and providing more informative means to evaluate coexistence patterns.
Our findings of within-subfamily phylogenetic fields revealed idiosyncrasies among and within subfamilies, suggesting the action of different processes or timing of events. For instance, only species within Stenodermatinae showed overdispersed phylogenetic fields when analysed as a group. Some stenodermatine species coexist with less related species. This could potentially result from either a dominant role of allopatric speciation followed by dispersal and convergent environmental preference among distantly related stenodermatines [44,45] or even geographical exclusion of closely related species [9]. Interestingly, the latter possibility may suggest that ecological sorting processes could be acting at larger scales [19]. In fact, at local scales, different studies have found that the structure of frugivore ensembles (i.e. stenodermatines) involves ecological sorting processes, whereas other functional groups show random patterns suggesting historical processes [8–10].
A common expectation for organisms within the same guild or lineage is that negative interactions may be stronger and result in similar species not coexisting together [13,19]. In contrast to this common expectation, we found an opposite pattern within phyllostomid subfamilies. At shallower phylogenetic levels within subfamilies, more species tended to coexist with closest relatives, suggesting similar historical processes (i.e. in situ speciation and shared environmental preferences) driving their coexistence. For instance, clustered or randomly structured phylogenetic fields were observed for frugivore, insectivore and nectarivore species alike (subfamilies Stenodermatinae, Phyllostominae and Glossophaginae, respectively). Thus, similar processes seem to be equally important for geographical coexistence within subfamilies regardless of such different ecological and phylogenetic groupings. In contrast to family level patterns, no species within subfamilies had an overdispersed phylogenetic field among closest relatives. Hence, there is no direct evidence for potential niche evolution and convergence within subfamilies. Instead, niche conservatism seems to be more important among closest relatives within subfamilies. For instance, the Mexican long-nosed bat (Leptonycteris nivalis) located at northern latitudes showed a clustered phylogenetic field among closest relatives within the subfamily Glossophaginae contrasting with its overdispersed phylogenetic field at the family level.
(c). Generality of patterns and multiplicity of factors
Contrasting observed patterns against theoretical or null expectations represents the main strategy for assessing pattern significance and relating them to underlying theory [21,46]. Non-random patterns are particularly informative, providing evidence that actual mechanisms act beyond stochasticity [47] and, in our case, beyond species richness and range size variation. Yet, depending on the specified null hypothesis or null model expectations, interpretations can still be made when observed patterns do not differ from such expectations [48]. We tested two null models with different constraints preserving some features of empirical data; observed species richness and range sizes. In null model I, keeping observed species richness within focal species' ranges and assuming any species could occur anywhere; most results did not differ from random. These results imply that phylogenetic fields could be determined by random collections of species or, alternatively, that coexistence among species is phylogenetically varied without evidence of overdispersion or clustering among species. In fact, this is in agreement with empirical patterns of coexistence among phyllostomids where most species coexist with a high number of other phyllostomids [11], thus making it difficult to distinguish from random patterns under our null model I assumptions.
Alternatively, our null model II further included the observed species' range sizes and significant patterns were identified, though most species still showed no differences from random expectations. These results coincide with those from null model I where species may occur anywhere. Indeed, phyllostomid bats have relatively large ranges compared with other mammalian orders and show ample variation in range sizes [49,50]. Moreover, previous studies found that range size variation of phyllostomids is higher at the species than at supraspecific levels (e.g. genera or families) [50]. Accordingly, we did not find a phylogenetic signal in phyllostomid range sizes. Therefore, species from different phyllostomid subfamilies presenting large geographical ranges are bound to coexist with many phyllostomids at some point within their ranges. Thus, for many phyllostomid species, phylogenetic fields are not distinguishable from randomizations based on within-range species richness and range size. Consequently, observed relationships between phylogenetic fields and coexistence and geographical patterns could be considered as null expectations derived from species richness and range size variation alone. Nonetheless, as in classical null model analyses, deviations from null expectations allowed us the interpretation of potential processes not included in our models (i.e. historical processes mentioned above [47,48]).
Another potential explanation for random phylogenetic fields within phyllostomids may also be related to niche conservatism. It is possible that most species share similar environmental preferences owing to their tropical origin and diversification within the same geographical domain [5]. This would allow them to occur at the same regions and coexist with closely and distantly related species, producing phylogenetic fields indistinguishable from range size variation alone. In fact, recent phylogenetic analyses suggest phenotypic and ecological stasis after early species' differentiation within Phyllostomidae followed by increasing speciation rates [44,51,52], which may account for the high number of closely related coexisting species. Finally, we cannot discard the potential effect of evolutionary range dynamics on current coexistence patterns [43,53], which may explain the lability and lack of phylogenetic signal in phyllostomid range sizes.
(d). Concluding remarks
Our study provides a conceptual and methodological framework to evaluate geographical coexistence patterns under a phylogenetic perspective. The phylogenetic field approach has the advantage of extending traditional community phylogenetics based on multiple sites or local communities into a species-based, biogeographic setting. Thus, it enables the discussion of broad-scale historical processes (i.e. speciation, extinction, dispersal and phylogenetic conservatism) under the appropriate arena where they actually occur: complete geographical distributions of species as study units and whole regions as domains. More importantly, phylogenetic fields reveal the actual evolutionary information contained in the phylogeny resulting from geographical coexistence among species, which ultimately determined large-scale biodiversity patterns.
Phylogenetic field can be further investigated by relating it to other species' traits potentially influencing species' coexistence (e.g. dispersal ability, body size). Also, future analysis of phylogenetic fields could also be evaluated through a pattern-oriented modelling approach to test the influence of macro-evolutionary processes (speciation/extinction) and the effect of deep-time climate dynamics on niche conservatism/evolution [54]. Alternatively, comparisons among different taxa with distinct evolutionary histories and similar (or dissimilar) geographical patterns [55] could also be investigated under a phylogenetic field approach to reveal potential driving processes. With these further developments in mind, we suggest that the novel approach proposed here might be important to integrate large spatial and temporal scales towards a more comprehensive understanding of broad-scale biodiversity patterns.
Acknowledgements
We thank Irby Lovette, Gavin Thomas and Richard D. Stevens for helpful comments that greatly improved this manuscript, and D.B. Provete, S. F. Gouveia and R. Dobrovolski for discussions. F.V. thanks H. T. Arita, M. E. Olson and J. Soberón for continuous support. F.V. was supported by grants from CONACYT (no. 172694), and CNPq. TFR and JAFD-F have been continuously supported by CNPq and CAPES grants. We thank Rebecca Crosthwait for kindly reviewing the English.
References
- 1.Hillebrand H. 2004. On the generality of the latitudinal diversity gradient. Am. Nat. 163, 192–211 10.1086/381004 (doi:10.1086/381004) [DOI] [PubMed] [Google Scholar]
- 2.Willig MR, Kaufman DM, Stevens RD. 2003. Latitudinal gradients of biodiversity: pattern, process, scale, and synthesis. Ann. Rev. Ecol. Evol. Syst. 34, 273–309 10.1146/annurev.ecolsys.34.012103.144032 (doi:10.1146/annurev.ecolsys.34.012103.144032) [DOI] [Google Scholar]
- 3.Stevens RD, Cox SB, Strauss RE, Willig MR. 2003. Patterns of functional diversity across an extensive environmental gradient: vertebrate consumers, hidden treatments and latitudinal trends. Ecol. Lett. 6, 1099–1108 10.1046/j.1461-0248.2003.00541.x (doi:10.1046/j.1461-0248.2003.00541.x) [DOI] [Google Scholar]
- 4.Stevens RD, Willig MR, Strauss RE. 2006. Latitudinal gradients in the phenetic diversity of New World bat communities. Oikos 112, 41–50 10.1111/j.0030-1299.2006.13167.x (doi:10.1111/j.0030-1299.2006.13167.x) [DOI] [Google Scholar]
- 5.Stevens RD. 2006. Historical processes enhance patterns of diversity along latitudinal gradients. Proc. R. Soc. B 273, 2283–2289 10.1098/rspb.2006.3596 (10.1098/rspb.2006.3596) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiens JJ, Donoghue MJ. 2004. Historical biogeography, ecology and species richness. Trends Ecol. Evol. 19, 639–644 10.1016/j.tree.2004.09.011 (doi:10.1016/j.tree.2004.09.011) [DOI] [PubMed] [Google Scholar]
- 7.Stevens RD. 2011. Relative effects of time for speciation and tropical niche conservatism on the latitudinal diversity gradient of phyllostomid bats. Proc. R. Soc. B 278, 2528–2536 10.1098/rspb.2010.2341 (doi:10.1098/rspb.2010.2341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arita HT. 1997. Species composition and morphological structure of the bat fauna of Yucatan, Mexico. J. Anim. Ecol. 66, 83–97 10.2307/5967 (doi:10.2307/5967) [DOI] [Google Scholar]
- 9.Moreno CE, Arita HT, Solis L. 2006. Morphological assembly mechanisms in Neotropical bat assemblages and ensembles within a landscape. Oecologia 149, 133–140 10.1007/s00442-006-0417-0 (doi:10.1007/s00442-006-0417-0) [DOI] [PubMed] [Google Scholar]
- 10.Stevens RD, Willig MR. 1999. Size assortment in New World bat communities. J. Mammal 80, 644–658 10.2307/1383309 (doi:10.2307/1383309) [DOI] [Google Scholar]
- 11.Villalobos F, Arita HT. 2010. The diversity field of New World leaf-nosed bats (Phyllostomidae). Glob. Ecol. Biogeogr. 19, 200–211 10.1111/j.1466-8238.2009.00503.x (doi:10.1111/j.1466-8238.2009.00503.x) [DOI] [Google Scholar]
- 12.Cavender-Bares J, Ackerly DD, Kozak KH. 2012. Integrating ecology and phylogenetics: the footprint of history in modern-day communities. Ecology 93, S1–S3 10.1890/12-0092.1 (doi:10.1890/12-0092.1) [DOI] [Google Scholar]
- 13.Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. 2002. Phylogenies and community ecology. Ann. Rev. Ecol. Evol. Syst. 33, 475–505 10.1146/annurev.ecolsys.33.010802.150448 (doi:10.1146/annurev.ecolsys.33.010802.150448) [DOI] [Google Scholar]
- 14.Vamosi SM, Heard SB, Vamosi JC, Webb CO. 2009. Emerging patterns in the comparative analysis of phylogenetic community structure. Mol. Ecol. 18, 572–592 10.1111/j.1365-294X.2008.04001.x (doi:10.1111/j.1365-294X.2008.04001.x) [DOI] [PubMed] [Google Scholar]
- 15.Kissling WD, Eiserhardt WL, Baker WJ, Borchsenius F, Couvreur TLP, Balslev H, Svenning JC. 2012. Cenozoic imprints on the phylogenetic structure of palm species assemblages worldwide. Proc. Natl Acad. Sci. USA 109, 7379–7384 10.1073/pnas.1120467109 (doi:10.1073/pnas.1120467109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen AP, Gillooly JF. 2006. Assessing latitudinal gradients in speciation rates and biodiversity at the global scale. Ecol. Lett. 9, 947–954 10.1111/j.1461-0248.2006.00946.x (doi:10.1111/j.1461-0248.2006.00946.x) [DOI] [PubMed] [Google Scholar]
- 17.Cardillo M. 1999. Latitude and rates of diversification in birds and butterflies. Proc. R. Soc. Lond. B 266, 1221–1225 10.1098/rspb.1999.0766 (doi:10.1098/rspb.1999.0766) [DOI] [Google Scholar]
- 18.Lovette IJ, Hochachka WM. 2006. Simultaneous effects of phylogenetic niche conservatism and competition on avian community structure. Ecology 87, S14–S28 10.1890/0012-9658(2006)87[14:SEOPNC]2.0.CO;2 (doi:10.1890/0012-9658(2006)87[14:SEOPNC]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 19.Cardillo M. 2011. Phylogenetic structure of mammal assemblages at large geographical scales: linking phylogenetic community ecology with macroecology. Phil. Trans. R. Soc. B 366, 2545–2553 10.1098/rstb.2011.0021 (doi:10.1098/rstb.2011.0021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arita HT, Christen JA, Rodríguez P, Soberón J. 2008. Species diversity and distribution in presence–absence matrices: mathematical relationships and biological implications. Am. Nat. 172, 519–532 10.1086/590954 (doi:10.1086/590954) [DOI] [PubMed] [Google Scholar]
- 21.Gotelli NJ, et al. 2009. Patterns and causes of species richness: a general simulation model for macroecology. Ecol. Lett. 12, 873–886 10.1111/j.1461-0248.2009.01353.x (doi:10.1111/j.1461-0248.2009.01353.x) [DOI] [PubMed] [Google Scholar]
- 22.Simmons NB. 2005. Order Chiroptera. In Mammal species of the World (eds Wilson DE, Reeder DM.), pp. 312–529 Baltimore, MD: The Johns Hopkins University Press [Google Scholar]
- 23.Patterson BD, Ceballos G, Sechrest W, Tognelli MF, Brooks TM, Luna L, Ortega P, Salazar I, Young B. 2007. Digital distribution maps of the mammals of the western hemisphere, version 3. Arlington, VA: NatureServe [Google Scholar]
- 24.Jones KE, Purvis A, MacLarnon A, Bininda-Emonds ORP, Simmons NB. 2002. A phylogenetic supertree of the bats (Mammalia: Chiroptera). Biol. Rev. 77, 223–259 10.1017/S1464793101005899 (doi:10.1017/S1464793101005899) [DOI] [PubMed] [Google Scholar]
- 25.Maddison W, Maddison D. 2009. Mesquite: a modular system for evolutionary analysis, v. 2.72. See http://mesquiteproject.org [Google Scholar]
- 26.Helmus MR, Bland TJ, Williams CK, Ives AR. 2007. Phylogenetic measures of biodiversity. Am. Nat. 169, E68–E83 10.1086/511334 (doi:10.1086/511334) [DOI] [PubMed] [Google Scholar]
- 27.Blomberg SP, Garland J, Ives AR. 2003. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745 [DOI] [PubMed] [Google Scholar]
- 28.Hardy OJ, Senterre B. 2007. Characterizing the phylogenetic structure of communities by an additive partitioning of phylogenetic diversity. J. Ecol. 95, 493–506 10.1111/j.1365-2745.2007.01222.x (doi:10.1111/j.1365-2745.2007.01222.x) [DOI] [Google Scholar]
- 29.Parra JL, McGuire JA, Graham CH. 2010. Incorporating clade identity in analyses of phylogenetic community structure: an example with hummingbirds. Am. Nat. 176, 573–587 10.1086/656619 (doi:10.1086/656619) [DOI] [PubMed] [Google Scholar]
- 30.Baker RJ, Hoofer SR, Porter CA, Van Den Bussche RA. 2003. Diversification among New World leaf-nosed bats: an evolutionary hypothesis and classification inferred from digenomic congruence of DNA sequence. Occas. Pap. Mus. Texas Tech. Univ. 230, 1–32 [Google Scholar]
- 31.Datzmann T, Von Helversen O, Mayer F. 2010. Evolution of nectarivory in phyllostomid bats (Phyllostomidae Gray, 1825, Chiroptera: Mammalia). BMC Evol. Biol. 10, 165. 10.1186/1471-2148-10-165 (doi:10.1186/1471-2148-10-165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freeman P. 2000. Macroevolution in Microchiroptera: recoupling morphology and ecology with phylogeny. Evol. Ecol. Res. 2, 317–335 [Google Scholar]
- 33.Wetterer AL, Rockman MV, Simmons NB. 2000. Phylogeny of phyllostomid bats (Mammalia: Chiroptera): data from diverse morphological systems, sex chromosomes, and restriction sites. B. Am. Mus. Nat. Hist. 1–200 (doi:10.1206/0003-0090(2000)248<0001:POPBMC>2.0.CO;2) [DOI] [Google Scholar]
- 34.Marquet PA, Fernández M, Navarrete SA, Valdovinos C. 2004. Diversity emerging: toward a deconstruction of biodiversity patterns. In Frontiers of biogeography: new directions in the geography of nature (eds Lomolino MV, Heaney LR.), pp. 192–209 Sunderland, MA: Sinauer [Google Scholar]
- 35.Sokal RR, Rohlf FJ. 1995. Biometry: the principles and practice of statistics in biological research, 3rd edn New York, NY: W. H. Freeman [Google Scholar]
- 36.R Development Core Team 2011. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See http://www.R-project.org/ [Google Scholar]
- 37.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 10.1093/bioinformatics/btg412 (doi:10.1093/bioinformatics/btg412) [DOI] [PubMed] [Google Scholar]
- 38.Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO. 2010. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464 10.1093/bioinformatics/btq166 (doi:10.1093/bioinformatics/btq166) [DOI] [PubMed] [Google Scholar]
- 39.Ricklefs RE. 2008. Disintegration of the ecological community. Am. Nat. 172, 741–750 10.1086/593002 (doi:10.1086/593002) [DOI] [PubMed] [Google Scholar]
- 40.Stephens PR, Wiens JJ. 2003. Explaining species richness from continents to communities: the time-for-speciation effect in emydid turtles. Am. Nat. 161, 112–128 10.1086/345091 (doi:10.1086/345091) [DOI] [PubMed] [Google Scholar]
- 41.Wiens JJ, et al. 2010. Niche conservatism as an emerging principle in ecology and conservation biology. Ecol. Lett. 13, 1310–1324 10.1111/j.1461-0248.2010.01515.x (doi:10.1111/j.1461-0248.2010.01515.x) [DOI] [PubMed] [Google Scholar]
- 42.Borregaard MK, Gotelli NJ, Rahbek C. 2012. Are range-size distributions consistent with species-level heritability? Evolution 66, 2216–2226 10.1111/j.1558-5646.2012.01581.x (doi:10.1111/j.1558-5646.2012.01581.x) [DOI] [PubMed] [Google Scholar]
- 43.Pigot AL, Phillimore AB, Owens IPF, Orme CDL. 2010. The shape and temporal dynamics of phylogenetic trees arising from geographic speciation. Syst. Biol. 59, 660–673 10.1093/sysbio/syq058 (doi:10.1093/sysbio/syq058) [DOI] [PubMed] [Google Scholar]
- 44.Monteiro LR, Nogueira MR. 2011. Evolutionary patterns and processes in the radiation of phyllostomid bats. BMC Evol. Biol. 11, 137. 10.1186/1471-2148-11-137 (doi:10.1186/1471-2148-11-137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rojas D, Vale A, Ferrero V, Navarro L. 2012. The role of frugivory in the diversification of bats in the Neotropics. J. Biogeogr. 39, 1948–1960 10.1111/j.1365-2699.2012.02709.x (doi:10.1111/j.1365-2699.2012.02709.x) [DOI] [Google Scholar]
- 46.Harte J. 2004. The value of null theories in ecology. Ecology 85, 1792–1794 10.1890/03-0681 (doi:10.1890/03-0681) [DOI] [Google Scholar]
- 47.Gotelli NJ, Graves GR. 1996. Null models in ecology, 368 p Washington, DC: Smithsonian Institution Press [Google Scholar]
- 48.Gotelli NJ, Ulrich W. 2012. Statistical challenges in null model analysis. Oikos 121, 171–180 10.1111/j.1600-0706.2011.20301.x (doi:10.1111/j.1600-0706.2011.20301.x) [DOI] [Google Scholar]
- 49.Lyons SK, Willig MR. 1997. Latitudinal patterns of range size: methodological concerns and empirical evaluations for New World bats and marsupials. Oikos 79, 568–580 10.2307/3546901 (doi:10.2307/3546901) [DOI] [Google Scholar]
- 50.Willig MR, Patterson BD, Stevens RD. 2003. Patterns of range size, richness, and body size in the Chiroptera. In Bat ecology (eds Kunz TH, Fenton MB.), pp. 580–621 Chicago, IL: University of Chicago Press [Google Scholar]
- 51.Rojas D, Vale A, Ferrero V, Navarro L. 2011. When did plants become important to leaf-nosed bats? Diversification of feeding habits in the family Phyllostomidae. Mol. Ecol. 20, 2217–2228 10.1111/j.1365-294X.2011.05082.x (doi:10.1111/j.1365-294X.2011.05082.x) [DOI] [PubMed] [Google Scholar]
- 52.Dumont ER, Dávalos LM, Goldberg A, Santana SE, Rex K, Voigt CC. 2011. Morphological innovation, diversification and invasion of a new adaptive zone. Proc. R. Soc. B 279, 1797–1805 10.1098/rspb.2011.2005 (doi:10.1098/rspb.2011.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arita HT, Vázquez-Domínguez E. 2008. The tropics: cradle, museum or casino? A dynamic null model for latitudinal gradients of species diversity. Ecol. Lett. 11, 653–663 10.1111/j.1461-0248.2008.01197.x (doi:10.1111/j.1461-0248.2008.01197.x) [DOI] [PubMed] [Google Scholar]
- 54.Rangel TFLVB, Diniz-Filho JAF, Colwell RK. 2007. Species richness and evolutionary niche dynamics: a spatial pattern-oriented simulation experiment. Am. Nat. 170, 602–616 10.1086/521315 (doi:10.1086/521315) [DOI] [PubMed] [Google Scholar]
- 55.Hawkins BA, et al. 2012. Different evolutionary histories underlie congruent species richness gradients of birds and mammals. J. Biogeogr. 39, 825–841 10.1111/j.1365-2699.2011.02655.x (doi:10.1111/j.1365-2699.2011.02655.x) [DOI] [Google Scholar]