

Abstract
Evolutionary and acclimatory responses require functional variability, but in contrast with mRNA and protein abundance data, most physiological measurements cannot be obtained in a high-throughput manner. Consequently, one must either rely on high-throughput transcriptomic or proteomic data with only predicted functional information, or accept the limitation that most physiological measurements can give fewer data than those provided by transcriptomics or proteomics. We evaluated how transcriptional and redox enzyme activity data agreed with regard to population differentiation (i.e. a system in steady state in which any time lag between transcription, translation and post-translational effects would be irrelevant) and in response to an acute 6°C increase in temperature (i.e. a disequilibrium state wherein translation could not have caught up with transcription) in the three-spined stickleback (Gasterosteus aculeatus). Transcriptional and enzyme activity data corresponded well with regard to population differentiation, but less so with regard to acute temperature increase. The data thus suggest that transcriptional and functional measurements can lead to similar conclusions when a biological system is in a steady state. The responses to acute changes must, as has been demonstrated earlier, be based on changes in cellular conditions or properties of existing proteins without significant de novo synthesis of new gene products.
Keywords: mRNA–protein correlation, temperature, population differentiation
1. Introduction
A drawback of most genomic studies is that functional information is not combined with genomic information [1]. Yet, studies often discuss functional implications (e.g. enzyme activity changes) on the basis of transcript data (as given by quantitative PCR or cDNA microarrays) without information about how the protein levels and their activities change. In part, genomic data translate to information of gene product activity via protein abundance. In the past 10 years, there have been numerous studies examining the relationship between protein abundance and mRNA abundance (reviewed, e.g. in Abreu et al. [2]). About 40 per cent of the variance in protein expression is explained by mRNA expression [2,3]. A complicating factor is that not only does the relationship vary among organisms and among genes within an organism, but also with a given gene in different conditions. The relationship between genomic data and overall metabolic activity becomes even more complex, as the same physiological response can be a result of different cellular pathways [4]. Because of this, network-based analyses are often used. The idea behind network-based analysis is that by grouping differentially expressed transcripts into networks of related predicted functions, the biological function related to the observed transcriptional changes can be inferred. This, however, is not the full story as regulation can also occur translationally and post-translationally. Both of these regulatory steps influence the final outcome of any metabolic network. Also, information on the genome-to-gene product activity relationship is confined mostly to a small number of model organisms. This biases functional predictions based on gene sequences—they can give accurate results as long as the function of the gene product is similar to that in model organisms, but in the case of divergent pathways or neofunctionalization, such predictions may be erroneous. Consequently, there is a need for investigations simultaneously assessing transcriptional changes and protein activities for the same animals with the ultimate goal of evaluating conditions under which conclusions of functional or structural changes can be drawn from transcript data and those in which it cannot.
Self-evidently, the ecological and evolutionary success of organisms depends both on their functions and the adaptability of these functions. However, a full understanding of the evolutionary potential and constraints of such functions remains a challenge since inference based strictly on gene sequences or protein abundances rather than actual protein activities gives us an incomplete picture. For example, Oleksiak et al. [5] have estimated the relationship between transcriptional differences of genes coding for enzymes involved in cardiac energy metabolism and protein activity changes. They found that there was marked inter-individual variation in the relationship between mRNA expression and activity of enzymes coded for by the genes. The individuals could be divided into different groups with regard to major metabolic substrate type used. These results strengthen the conclusion that information regarding gene product activity is required to get a complete picture about the ecologically and evolutionarily important organismal activities and their relationship to genomic data.
It is now possible to use high-throughput methods to obtain transcriptomic [6] and protein abundance [7] data. Similar high-throughput methodology is not available for most protein activity measurements. As an example, while it is possible to determine how a treatment affects the transcription of thousands of genes with microarrays, one cannot measure the biological activities of the thousands of gene products even if rapid measurements of one or a few gene product activities are possible. It has become common to carry out microarray and quantitative reverse transcription PCR measurements also in evolutionary studies, and use thus obtained transcript data to infer functions, or use variations in protein abundance to infer the overall activity of the protein. To increase their usefulness in evolutionary studies, the relationship of such genomic and proteomic data to gene product activity data should be evaluated.
In the present study, we evaluated the correspondence of genomic and enzyme activity data in both steady-state and disequilibrium conditions using the three-spined stickleback as model. This species was selected for the following reasons: first, its genome has been sequenced [8] which has facilitated development of genomic resources, such as a high density custom microarray [9]; second, it is a major model in evolutionary studies [10]; and third, physiological and genomic measurements in this species have been integrated [11]. Two different types of data were examined: the relationship between transcription and redox enzyme activities at the steady state was studied by evaluating the differences between populations, and the relationship in disequilibrium conditions by looking at the responses to acute temperature change. The acute temperature increase was of such a short duration that transcriptional and translational responses could be uncoupled [12,13]. We used redox balance and redox enzyme activities to estimate some physiological features, as the redox balance is a major evolutionarily adjusted parameter [14] integrating both temperature and oxygen-level changes [15–17].
2. Material and methods
(a). Fish
Mature sticklebacks from Lake Pulmankijärvi in Finnish Lapland (69°58′5″ N, 27°59′35″ E), the Baltic Sea in the vicinity of Helsinki, Finland (59°49′17″ N, 22°58′14″ E) and Lake Vättern (58°38′00″ N, 14°51′00″ E) from south-central Sweden were spawned, and fertilized eggs were transported to the laboratory at the University of Helsinki where the fish hatched. After hatching, the fish were maintained at 17±1°C, with a photoperiod of 18 L : 6 D cycle for six months, whereafter their environmental conditions were gradually changed to simulate wintering conditions (24 h darkness; temperature 9±1°C). After five months, fish were stimulated to breed by gradually reverting the photoperiod back to an 18 L : 6 D cycle and the water temperature to 17±1°C. The generated F2 offspring from each population was used in the experiments at approximately 20 months of age. Although sexually mature, all experimental fish were reproductively inactive. Ten F2 fish from each population were housed in one of two identical tanks (20 fish per population altogether). Fish were acclimated overnight. One tank was maintained as a control at 17°C, and water in the second tank was heated approximately 1°C per hour for 6 h to a final temperature of 23°C. After 1 h at the final temperature, every fish was euthanized with a lethal amount of MS-222 in water, and its liver removed and immediately frozen in liquid nitrogen and stored at −80°C. In total, sufficient quantities of RNA and protein could be extracted from 50 fish and used to measure enzyme activities; a random subset of these same individuals (n = 35) were also used for transcriptome-wide (microarray) analysis of transcript expression.
(b). Sample preparation
For transcriptome work, total RNA was isolated from liver tissue by means of Tri Reagent (Sigma; St Louis, MO, USA), following the manufacturer's protocol. RNA was treated with DNase (Promega; Madison, WI, USA) and re-isolated using Tri Reagent. RNA concentration was quantified using a Nanodrop ND-1000 (Thermo Scientific; Waltham, MA, USA), and RNA quality was assessed using an Experion automated electrophoresis system (Bio-Rad; Hercules, CA, USA).
For glutathione (GSH) concentration and enzyme activity measurements, the frozen liver samples were homogenized using TissueLyser II Bead mill (Qiagen, Hamburg, Germany) in 0.1 M K2HPO4+0.15 M KCl-buffer (pH 7.4). The homogenate was centrifuged for 15 min at 10 000g at +4°C. The supernatant was divided into several aliquots of which one was used for the preparation of the GSH determination sample and the rest were frozen in liquid nitrogen and stored at −80°C until further measurements.
(c). Microarray analysis
A short oligo microarray was custom-designed as earlier described [9]. Each sample (1.65 μg) was hybridized to the custom-designed stickleback array at 65°C overnight (17 h) using Agilent's GE Hybridization Kit. Washes were carried out as recommended by the manufacturer using Agilent's Gene Expression Wash Pack. Arrays were scanned with Agilent Technologies Scanner, model G2505B. Spot intensities and other quality control features were extracted with Agilent's Feature Extraction Software v. 9.5.3.1. Array quality was assessed through the use of Agilent control features, as well as spike-in controls (Agilent One-Color Spike-in Kit for RNA experiment). Feature Extraction Software was used to flag features above background at the 99%; however, to further filter the data within each population, only probes with a background-corrected median intensity value of greater than 50 across their respective groups were retained. Because probes with very low expression levels may not be reliable, this is similar to keeping only the probes that are more than two times as intense as background levels. Post-processed signals were further normalized within and across arrays using the quantile method, implemented in the R/Bioconductor package ‘limma’ [18]. Probes with missing values were removed from further analyses, which were thus based on data for 23 946 unique probes, representing 11 203 predicted/projected genes (Ensembl Stickleback Genome v. 65.1, updated May 2010). The microarray data are available at http://www.ebi.ac.uk/microarray-as/ae/ with accession no. A-MEXP-1443.
Differentially expressed transcripts between populations were identified using the empirical Bayes procedure [19], as implemented in the R/Bioconductor package ‘siggenes’ [20]. This was performed within each temperature treatment separately, and by iteratively selecting a Z-score correction factor ‘a0’ parameter to achieve an acceptable posterior false discovery rate (less than or equal to 0.10). Transcript lists were then pooled prior to enrichment analyses. Similarly, to identify probes differentially expressed between temperature treatments, data were analysed via the empirical Bayes procedure (populations pooled) contrasting control and temperature treatment groups.
(d). Functional annotation analysis
The database for annotation, visualization and integrated discovery (DAVID) [21] was used to determine if differentially expressed probes were significantly over-represented by genes of particular functional categories [22]. Although DAVID will accept several probe identifiers, Entrez Gene identifiers generally produce the most comprehensive annotation data [23,24]. Stickleback genes were initially matched to their human orthologues using Biomart [25,26], and further supplemented via BLAST search to increase inferred annotations. Of the 23 946 probes, 75 per cent (18 060) were assigned an Entrez GeneID number. Approximately the same proportions (1874) of the 2509 probes differentially expressed among populations, and (2698) of the 3516 probes differentially expressed between temperature treatments were also assigned ENTREZ identifiers. Input for analysis consisted of the list of differentially expressed probes with Entrez GeneID, contrasted with a customized ‘background’ set, including only those genes represented on the custom array. Though DAVID can integrate non-redundant functional annotations across multiple databases, we focused on gene ontology (GO) annotations at the ‘biological process’ level [27]. We further restricted output by grouping enriched terms into functional annotation clusters, using default settings [28].
(e). Glutathione concentration determinations
Samples for GSH determination were deproteinized with 5 per cent sulfosalicylic acid. Reduced and oxidized GSH species (GSH/GSSG) were measured with ThioStar GSH detection reagent (Arbor Assays, MI, USA) using reduced GSH as the standard (Sigma Chemicals, St. Louis, MO, USA) as described by Lilley et al. [29].
(f). Determinations of enzyme activities
The enzyme activity determinations were carried out mainly as described by Vuori et al. [30] at 25°C. Briefly, glutathione reductase (GSR) activity was measured according to Smith et al. [31]. Glutathione S-transferase (GST) activity was measured according to Habig et al. [32] with the exception of using 2 mM GSH instead of 1 mM. Glutathione peroxidase (GPX) activity was measured using a Sigma kit (Sigma Chemicals) with 2 mM H2O2 as a substrate. The inhibition rate of superoxide dismutase (SOD) was measured using a Fluka kit (Fluka, Buchs, Germany). The measurement of catalase (CAT) activity was modified to microplate from the Catalase Assay kit (Sigma Chemicals), where a reaction of CAT and H2O2 is stopped with NaN3 [33], an aliquot is pipetted to a microplate and the leftover H2O2 is detected with colorimetric reaction [34]. The protein contents of the samples were determined using Pierce BCA Reagents (Thermo Fisher Scientific, Rockford, IL, USA) with bovine serum albumin (Sigma Chemicals) as the standard.
Both GSH and enzyme activity analyses were conducted with EnSpire plate reader (Wallac, PerkinElmer Life Sciences, Turku, Finland) using either 96-well (protein and CAT assays) or 384-well plates (GSH, GSR, GST, GPX and SOD).
(g). Analysis of trait divergence in enzyme and glutathione concentration data
To infer if between population differences in enzyme data were best explained by neutral or putatively selective processes, we contrasted an index of quantitative genetic differentiation (QST) with that of neutral genetic divergence [35]. Among and within-group phenotypic variance were estimated as random effects via mixed effects modelling, after first controlling for temperature effects as a fixed model term. Since family groups were unavailable within each population—the standard source of within-group variance—we first created five pseudo-replicate samples within each population-by-treatment group by random sampling with replacement. Within-group variance was estimated as variation among replicate samples, nested within each population. Confidence intervals (95%) were obtained by parametric bootstrapping (10 000 simulations). The range of neutral expectation was defined based on a previously published dataset of 17 microsatellite markers [36], using data pertaining only to the three populations in the current study. We evaluated the dataset via outlier analysis to ensure that all markers fell within a simulated range of neutral expectation [37], and used the observed range of FST values for comparison against QST estimates.
(h). Multivariate association between mRNA expression and enzyme activity
Associations between multivariate descriptors of enzymatic and transcriptional variation were inferred via co-inertia analysis (CoIA). In brief, CoIA finds common sets of principal axes for two datasets pertaining to the same cases—in this instance cases were individual fish, and datasets were an individual's transcriptional profile at all 23 946 probes detected above background and activity data for four enzymes and total GSH concentration—such that principal components have maximum covariance, while also accounting for partitioning between and co-variation within discrete classes/groups of individual cases [38,39]. Only individuals for which both transcriptomic and enzyme activity data were available were used for CoIA. Between-class (i.e. among treatment-specific population groups) PCA and CoIA of both data projections were performed using the ‘ade4’ package [40] implemented in R. Significant contributions of transcriptional probes to among-group discrimination was ascertained by evaluating their respective loadings on the dominant principal axes: probes whose squared score exceeded the 90th quantile of a distribution describing all squared loading scores for a focal axis were deemed to be significant.
3. Results and discussion
With regard to population differentiation, transcriptional and enzyme activity data lead to the same conclusion. There is a marked difference in the activities of GPX and SOD enzymes between the lake Vättern population on one the hand, and lake Pulmanki and Helsinki populations on the other hand (figure 1). In the case of GPX, the difference also exceeds neutral expectation (i.e. QST > FST; figure 2). This divergence cannot be explained either by genetic salinity adaptation, as the two lake populations are different, or by latitude-related adaptation, as the populations from similar latitudes are also different. Thus, the result shows that there are some physiological differences between populations which may reflect divergent selection pressures. The populations are different, although northern European populations of three-spined stickleback share a relatively recent and common evolutionary origin [41]. However, only a small number of amino acid changes (i.e. only few nucleotide changes in a gene) are required to change the function of a protein markedly [42]. On the other hand, although CAT activity showed a tendency for population differentiation, and SOD activity differed significantly among populations, the tail of their bootstrapped QST distributions overlapped with the range of FST estimated from neutral microsatellite markers (figure 2). Neither GSR activity nor total GSH concentration (the level of which depends on the function of several gene products) differed significantly from neutrality (figure 2).
Figure 1.

GPX, GSR, SOD and CAT activities and total GSH concentration in Helsinki (HEL, n = 18), Vättern (VAT, n = 13) and Pulmanki (PUL, n = 18) populations. The statistical significance of the difference between the means was tested using two-way ANOVA with population and sex as explanatory factors. An ad hoc test to indicate differences between individual groups was carried out using the Holm–Sidak method. Asterisks indicate that the population with asterisks differs significantly from the other populations. *p < 0.05; ***p < 0.001.
Figure 2.
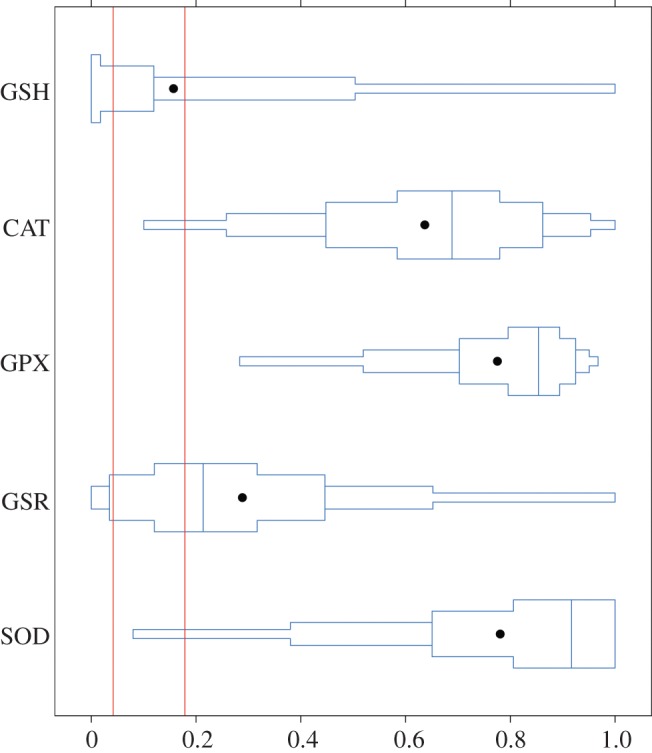
Box-percentile plots showing the bootstrap distribution (10 000 iterations) for estimates of the index of quantitative trait differentiation (QST) for enzyme data. Vertical lines denote the putative range of neutral differentiation (FST), as inferred from variation in 17 assumedly neutral microsatellite loci. (Online version in colour.)
The differentiation between populations was also seen in the transcriptome data. In the microarray analysis, we identified 1698 genes (1834 splice variants in total) which were differentially transcribed among populations. This reduced probe-set was largely divided into transcripts that were differentially expressed in the lake Vättern population when compared with the other two populations. Enrichment analysis grouped the differentially transcribed genes into 164 functional clusters. We excluded those clusters with an enrichment score less than 0.5, and for which the mean fold enrichment of constituent GO biological processes was significantly less than 1. Ultimately, we retained 27 functional annotation clusters which met these criteria (table 1). Of these, most inferred functions relate to basic cellular function and maintenance. However, of the retained groups, ‘responses to oxidative stress’ and closely related ‘GSH, peptide and sulphur metabolism’ were in line with trends in independent enzyme activity data, suggesting inferential correspondence between datasets.
Table 1.
Functional enrichment analysis of differentially transcribed genes. (Functional clusters are based on inter-relationships and redundancies among GO biological processes, implemented in DAVID. Ad hoc descriptions of retained clusters are based on common themes/functions of constituent GO annotations. Clusters are ranked by overall enrichment score (enrich. score). Data pertaining to the number of GO terms (no. BP), the number of differentially expressed gene transcripts (no. genes) and the mean (95% quantiles) fold enrichment score (fold enrich.) of constituent GO terms are also presented. Functional clusters relevant to enzymatic data are italicized.)
| functional cluster | enrich. score | no. BP | no. genes | fold enrich. |
|---|---|---|---|---|
| regulation of protein localization transport and secretion | 2.34 | 14 | 63 | 2.50 (1.50–4.20) |
| detection of external stimuli | 1.66 | 4 | 13 | 2.95 (2.05–4.28) |
| response to steroidal stimuli | 1.36 | 4 | 12 | 2.83 (2.02–3.60) |
| regulation of cellular growth | 1.11 | 10 | 24 | 1.76 (1.19–2.40) |
| regulation of GTPase activity | 1.11 | 12 | 40 | 2.23 (1.10–3.90) |
| regulation of cell adhesion | 1.08 | 3 | 12 | 2.17 (1.83–2.55) |
| glucose and carbohydrate homeostasis | 1.08 | 3 | 9 | 2.37 (2.20–2.65) |
| regulation of ion transport | 1.08 | 6 | 12 | 2.40 (1.83–3.70) |
| nuclear organization | 1.03 | 6 | 11 | 2.18 (1.55–2.68) |
| regulation of protein signalling | 0.99 | 3 | 9 | 2.70 (2.70–2.70) |
| SMAD protein localization | 0.91 | 3 | 11 | 2.50 (1.61–3.95) |
| glucose and carbohydrate metabolism | 0.87 | 27 | 26 | 2.37 (1.63–3.87) |
| membrane protein proteolysis | 0.87 | 4 | 8 | 3.23 (1.44–5.82) |
| regulation of immune response | 0.86 | 13 | 28 | 2.31 (1.04–4.80) |
| response to intra-cellular pathogens | 0.85 | 7 | 10 | 2.49 (1.33–3.80) |
| mitochondrial organization | 0.83 | 3 | 7 | 3.70 (2.00–4.70) |
| regulation of intra-cellular protein transport | 0.81 | 8 | 19 | 1.68 (1.24–2.03) |
| water homeostasis | 0.81 | 6 | 10 | 3.22 (1.65–4.70) |
| response to oxidative stress | 0.75 | 9 | 25 | 1.92 (1.22–2.62) |
| regulation of lipid metabolism | 0.74 | 14 | 17 | 2.33 (1.23–3.80) |
| regulation of macromolecular secretion | 0.64 | 7 | 7 | 2.29 (1.76–2.70) |
| exocytosis | 0.63 | 6 | 26 | 1.53 (1.13–2.03) |
| GSH, peptide and sulphur metabolism | 0.60 | 3 | 17 | 1.57 (1.24–1.87) |
| regulation of cellular development | 0.59 | 7 | 25 | 1.90 (1.22–2.70) |
| transport of organic acids | 0.55 | 5 | 16 | 1.60 (1.14–2.52) |
| regulation of muscle development | 0.53 | 10 | 10 | 1.86 (1.19–3.33) |
| DNA catabolism | 0.51 | 7 | 14 | 1.71 (1.36–1.97) |
Even though the number of parameters and the number of data points in the enzyme activity measurements were small, they revealed the same clusters as microarray determinations based on several thousand genes with regard to the different populations. Common ordination in multivariate space revealed a significant co-variation (35.7%; p = 0.002) between transcriptional and enzymatic data. A total of 65.7% co-inertia was captured by a single axis which separated Vättern samples from the other populations (figure 3). A second co-inertia axis captured 22 per cent of total (co)variation in the data, and described common differences attributable to thermal treatments. This finding is very reassuring for environmentally oriented physiologists, as it suggests that evolutionarily relevant findings can be made with datasets that are manageable in terms of measuring. The observation is also in line with the recent result that there was more divergence in markers linked to physiologically important genes than to neutral genes, indicating that physiological processes are important targets of selection underlying adaptive divergence in this species [43]. The present study goes further than the previous one, as we have looked into both transcriptional and physiological (i.e. enzyme activity) data. The observation also lends support for the use of using transcriptional findings for suggesting functional pathways in cases where a priori function is not evident when population divergence is studied.
Figure 3.

CoIA of between-group ordinations of log2 transcript abundance (triangles) and GSH enzymatics (circles). Global similarity (co-inertia) between transcription and enzyme datasets is 35.7% (p = 0.002). A total of 65.7% of co-inertia is described by the first axis (screeplot; inset lower right) which appears to describe differentiation between Lake Vättern samples from the other populations. Loadings on this axis (correlation circle; inset upper right) suggest that these differences are largely driven by SOD and GPX activity. The second co-inertia axis capture 22% of total (co)variation in the data, and describes common differences attributable to thermal treatment—this ‘effect’ seems more pronounced in Helsinki and Lake Pulmankijärvi populations. Loadings on axis two suggest that thermally induced patterns of (co)variation are mostly observed with respect to GSR and CAT activity. (Online version in colour.)
While transcriptional and enzyme activity data led to similar conclusions with regard to population differentiation, the responses to acute temperature increase were somewhat different. With regard to enzyme activity data, no significant differences between populations in their temperature response were observed. Also, the acute temperature increase did not affect any of the measured parameters significantly (figure 4). Only 199 genes (215 splice variants) were identified as potentially differentially expressed in both population and temperature contrasts. By contrast, 1925 genes (2141 unique transcripts) responded transcriptionally to the acute temperature increase in the three populations. Among the transcripts differentially regulated, only 11 were in the genes coding for redox-relevant proteins. Although two of them were transcripts of a gene coding for GPX (GPX4), post hoc analysis of probe-specific false discovery rate-corrected significance levels (q-values) indicated that the fold change in response to the acute temperature treatment (0.85 in treatment relative to control) was not statistically significant. It should be noted that one of the main clusters in GO, based on transcriptional responses to acute temperature increase, is regulation of metabolism suggesting that acute temperature changes may be more associated with metabolism than redox changes.
Figure 4.

The effects of temperature on GPX, GSR, SOD and CAT activities, and total GSH concentration in the different populations. n = 9 for the Helsinki population, six for the Lake Vättern population and nine for the Lake Pulmankijärvi population at 23°C, and nine for the Helsinki population, seven for the Lake Vättern population and nine for the Lake Pulmankijärvi population at 17°C . The statistical significance of the differences between means was determined using two-way ANOVA with population and temperature as explanatory factors. An ad hoc test to indicate differences between individual groups was carried out using the Holm–Sidak method. No statistically significant effects were observed.
There are a couple of possible explanations as to why there can be significant transcriptional changes but enzyme activities remain unchanged after an acute temperature increase. The first is that transcription and translation have different time courses. Even if transcription of a gene would later lead to a change in protein activity, the different time courses [12] would result in transcriptional change immediately upon an increase in temperature without a measurable change in protein level. The second possibility is that cellular and tissue conditions are such that increased transcription and further production of a protein is needed to maintain a constant activity [4]. This can be caused by the facts that temperature increase speeds up the breakdown of the proteins, and that the activity of unit amount of protein is reduced by increased temperature [44,45]. Notably, while the former could be investigated using a proteomics approach, the latter requires protein activity measurements. Overall, as a response to acute temperature change, any physiological change must occur either with pre-existing proteins or as a result of changes in protein–lipid interactions (for reviews see [46–48]). On the other hand, the population differentiation has occurred over many generations, whereby the system is in the steady state regarding transcription and translation which is also reflected in the concordance of transcriptional and enzyme activity data.
In conclusion, the results show a very good agreement between transcriptional and enzyme activity data for a biological system in steady state as is the case for population differentiation, but less so for a system in disequilibrium as when an acute environmental stress, temperature increase, is applied. Because of this, and as any fitness-related effect has to be functional, conclusions of environmental effects on populations should be based on functional data whenever possible. Predictions of functional effects can be made with high-throughput transcriptional or proteomics observations when population differentiation is studied, but, if possible, they should be supported with functional data to ascertain that the predicted function is actually affected. The use of high-throughput transcriptional data requires no pre-conceived notions of which ecological and/or selective factors may have been important in driving functional differences. Another possibility is to make an a priori guess of possibly important environmental differences, and measure traits that are likely to be associated with physiological responses to such environmental variations.
Acknowledgements
The work with stickleback was approved by the National Authority on Animal Experimentation (Finland).
The authors' work is supported by grants from the Academy of Finland (grant nos. 129662, 250435, 258078, 133875, 136464 and 134728). We thank the following people for help: Jose Cano Arias (animal breeding), Wolfgang Waser and Anti Vasemagi (sampling) and Heidi Viitaniemi (sample processing). In addition, several other persons helped us during different stages of the study, for which we extend our gratitude.
References
- 1.Furlong EEM. 2011. Molecular biology: a fly in the face of genomics. Nature 471, 458–459 10.1038/471458a (doi:10.1038/471458a) [DOI] [PubMed] [Google Scholar]
- 2.Abreu RD, Penalva LO, Marcotte EM, Vogel C. 2009. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5, 1512–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473, 337–342 10.1038/nature10098 (doi:10.1038/nature10098) [DOI] [PubMed] [Google Scholar]
- 4.Nikinmaa M, Waser W. 2007. Molecular and cellular studies in evolutionary physiology of natural vertebrate populations: influences of individual variation and genetic components on sampling and measurements. J. Exp. Biol. 210, 1847–1857 10.1242/jeb.002717 (doi:10.1242/jeb.002717) [DOI] [PubMed] [Google Scholar]
- 5.Oleksiak MF, Roach JL, Crawford DL. 2005. Natural variation in cardiac metabolism and gene expression in Fundulus heteroclitus. Nat. Genet. 37, 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu N, Drinnenberg I, Kelso J, Wu JR, Paabo S, Zeng R, Khaitovich P. 2007. Comparison of protein and mRNA expression evolution in humans and chimpanzees. PLoS ONE 2, e216. 10.1371/journal.pone.0000216 (doi:10.1371/journal.pone.0000216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shim YH, Paik YK. 2010. Caenorhabditis elegans proteomics comes of age. Proteomics 10, 846–857 10.1002/pmic.200900542 (doi:10.1002/pmic.200900542) [DOI] [PubMed] [Google Scholar]
- 8.Jones FC, et al. 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61 10.1038/nature10944 (doi:10.1038/nature10944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leder EH, Merila J, Primmer CR. 2009. A flexible whole-genome microarray for transcriptomics in three-spine stickleback (Gasterosteus aculeatus). BMC Genomics 10, 426. 10.1186/1471-2164-10-426 (doi:10.1186/1471-2164-10-426) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Östlund-Nilsson S, Mayer I, Huntingford F. 2007. The biology of three-spined stickleback. Boca Raton, FL: CRC Press [Google Scholar]
- 11.Leveelahti L, Leskinen P, Leder EH, Waser W, Nikinmaa M. 2011. Responses of threespine stickleback (Gasterosteus aculeatus, L) transcriptome to hypoxia. Comp. Biochem. Physiol. D Genomics Proteomics 6, 370–381 10.1016/j.cbd.2011.08.001 (doi:10.1016/j.cbd.2011.08.001) [DOI] [PubMed] [Google Scholar]
- 12.Buckley BA, Gracey AY, Somero GN. 2006. The cellular response to heat stress in the goby Gillichthys mirabilis: a cDNA microarray and protein-level analysis. J. Exp. Biol. 209, 2660–2677 10.1242/jeb.02292 (doi:10.1242/jeb.02292) [DOI] [PubMed] [Google Scholar]
- 13.Nikinmaa M, Rytkonen KT. 2011. Functional genomics in aquatic toxicology: do not forget the function. Aquat. Toxicol. 105, 16–24 10.1016/j.aquatox.2011.05.019 (doi:10.1016/j.aquatox.2011.05.019) [DOI] [PubMed] [Google Scholar]
- 14.Rytkonen KT, Vuori KAM, Primmer CR, Nikinmaa M. 2007. Comparison of hypoxia-inducible factor-1 alpha in hypoxia-sensitive and hypoxia-tolerant fish species. Comp. Biochem. Physiol. D-Genomics Proteomics 2, 177–186 10.1016/j.cbd.2007.03.001 (doi:10.1016/j.cbd.2007.03.001) [DOI] [PubMed] [Google Scholar]
- 15.Heise K, Puntarulo S, Nikinmaa M, Abele D, Portner HO. 2006. Oxidative stress during stressful heat exposure and recovery in the North Sea eelpout Zoarces viviparus L. J. Exp. Biol. 209, 353–363 10.1242/jeb.01977 (doi:10.1242/jeb.01977) [DOI] [PubMed] [Google Scholar]
- 16.Heise K, Puntarulo S, Nikinmaa M, Lucassen M, Portner HO, Abele D. 2006. Oxidative stress and HIF-1 DNA binding during stressful cold exposure and recovery in the North Sea eelpout (Zoarces viviparus). Comp. Biochem. Physiol. A-Mol. Integr. Physiol. 143, 494–503 10.1016/j.cbpa.2006.01.014 (doi:10.1016/j.cbpa.2006.01.014) [DOI] [PubMed] [Google Scholar]
- 17.Lesser MP. 2006. Oxidative stress in marine environments: biochemistry and physiological ecology. Annu. Rev. Physiol. 68, 253–278 10.1146/annurev.physiol.68.040104.110001 (doi:10.1146/annurev.physiol.68.040104.110001) [DOI] [PubMed] [Google Scholar]
- 18.Smyth GK, Speed T. 2003. Normalization of cDNA microarray data. Methods 31, 265–273 10.1016/S1046-2023(03)00155-5 (doi:10.1016/S1046-2023(03)00155-5) [DOI] [PubMed] [Google Scholar]
- 19.Efron B, Tibshirani R, Storey JD, Tusher V. 2001. Empirical Bayes analysis of a microarray experiment. J. Am. Stat. Assoc. 96, 1151–1160 10.1198/016214501753382129 (doi:10.1198/016214501753382129) [DOI] [Google Scholar]
- 20.Schwender H, Krause A, Ickstadt K. 2006. Identifying interesting genes with siggenes. RNews 6, 45–50 [Google Scholar]
- 21.Debat V, David P. 2001. Mapping phenotypes: canalization, plasticity and developmental stability. Trends Ecol. Evol. 16, 555–561 10.1016/S0169-5347(01)02266-2 (doi:10.1016/S0169-5347(01)02266-2) [DOI] [Google Scholar]
- 22.Huang DW, et al. 2007. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8, R183. 10.1186/gb-2007-8-9-r183 (doi:10.1186/gb-2007-8-9-r183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maglott D, Ostell J, Pruitt KD, Tatusova T. 2005. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 33, D54–D58 10.1093/nar/gki031 (doi:10.1093/nar/gki031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherman BT, et al. 2007. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinform. 8, 426. 10.1186/1471-2105-8-426 (doi:10.1186/1471-2105-8-426) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. 2005. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 10.1093/bioinformatics/bti525 (doi:10.1093/bioinformatics/bti525) [DOI] [PubMed] [Google Scholar]
- 26.Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A. 2009. BioMart—biological queries made easy. BMC Genomics 10, 22. 10.1186/1471-2164-10-22 (doi:10.1186/1471-2164-10-22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris MA, et al. 2004. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 10.1093/nar/gkh036 (doi:10.1093/nar/gkh036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 10.1038/nprot.2008.211 (doi:10.1038/nprot.2008.211) [DOI] [PubMed] [Google Scholar]
- 29.Lilley TM, Ruokolainen L, Meierjohann A, Kanerva M, Stauffer J, Laine VN, Nikinmaa M. In press. Impacts of organic tin compounds on redox regulation and complement reaction in natural populations of Daubenton's bats (Myotis daubentonii). Comp. Biochem. Physiol. C Toxicol. Pharmacol . [DOI] [PubMed] [Google Scholar]
- 30.Vuori KA, Kanerva M, Ikonen E, Nikinmaa M. 2008. Oxidative stress during Baltic salmon feeding migration may be associated with yolk-sac fry mortality. Environ. Sci. Technol. 42, 2668–2673 10.1021/es702632c (doi:10.1021/es702632c) [DOI] [PubMed] [Google Scholar]
- 31.Smith IK, Vierheller TL, Thorne CA. 1988. Assay of glutathione reductase in crude tissue homogenates using 5,5′-dithiobis(2-nitrobenzoic acid). Anal. Biochem. 175, 408–413 10.1016/0003-2697(88)90564-7 (doi:10.1016/0003-2697(88)90564-7) [DOI] [PubMed] [Google Scholar]
- 32.Habig WH, Pabst MJ, Jakoby WB. 1974. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 249, 7130–7139 [PubMed] [Google Scholar]
- 33.Deisseroth A, Dounce AL. 1970. Catalase: physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 50, 319–375 [DOI] [PubMed] [Google Scholar]
- 34.Fossati P, Prencipe L, Berti G. 1980. Use of 3,5-dichloro-2-hydroxybenzenesulfonic acid/4-aminophenazone chromogenic system in direct enzymic assay of uric acid in serum and urine. Clin. Chem. 26, 227–231 [PubMed] [Google Scholar]
- 35.Spitze K. 1993. Population—structure in Daphnia-obtusa—quantitative genetic and allozymic variation. Genetics 135, 367–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cano JM, Matsuba C, Makinen H, Merila J. 2006. The utility of QTL-Linked markers to detect selective sweeps in natural populations: a case study of the EDA gene and a linked marker in threespine stickleback. Mol. Ecol. 15, 4613–4621 10.1111/j.1365-294X.2006.03099.x (doi:10.1111/j.1365-294X.2006.03099.x) [DOI] [PubMed] [Google Scholar]
- 37.Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G. 2008. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform. 9, 323. 10.1186/1471-2105-9-323 (doi:10.1186/1471-2105-9-323) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dray S, Chessel D, Thioulouse J. 2003. Co-inertia analysis and the linking of ecological data tables. Ecology 84, 3078–3089 10.1890/03-0178 (doi:10.1890/03-0178) [DOI] [Google Scholar]
- 39.Jombart T, Pontier D, Dufour AB. 2009. Genetic markers in the playground of multivariate analysis. Heredity 102, 330–341 10.1038/hdy.2008.130 (doi:10.1038/hdy.2008.130) [DOI] [PubMed] [Google Scholar]
- 40.Dray S, Dufour AB. 2007. The ade4 package: implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20 [Google Scholar]
- 41.Makinen HS, Cano M, Merila J. 2008. Identifying footprints of directional and balancing selection in marine and freshwater three-spined stickleback (Gasterosteus aculeatus) populations. Mol. Ecol. 17, 3565–3582 10.1111/j.1365-294X.2008.03714.x (doi:10.1111/j.1365-294X.2008.03714.x) [DOI] [PubMed] [Google Scholar]
- 42.Weber RE, Hiebl I, Braunitzer G. 1988. High altitude and hemoglobin function in the vultures Gyps rueppellii and Aegypius monachus. Biol. Chem. Hoppe-Seyler 369, 233–240 10.1515/bchm3.1988.369.1.233 (doi:10.1515/bchm3.1988.369.1.233) [DOI] [PubMed] [Google Scholar]
- 43.Shimada Y, Shikano T, Merila J. 2011. A high incidence of selection on physiologically important genes in the three-spined stickleback, Gasterosteus aculeatus. Mol. Biol. Evol. 28, 181–193 10.1093/molbev/msq181 (doi:10.1093/molbev/msq181) [DOI] [PubMed] [Google Scholar]
- 44.Coquelle N, Fioravanti E, Weik M, Vellieux F, Madern D. 2007. Activity, stability and structural studies of lactate dehydrogenases adapted to extreme thermal environments. J. Mol. Biol. 374, 547–562 10.1016/j.jmb.2007.09.049 (doi:10.1016/j.jmb.2007.09.049) [DOI] [PubMed] [Google Scholar]
- 45.Gatt S. 1969. Thermal lability of beta galactosidase from pink salmon liver. Science 164, 1422–1423 10.1126/science.164.3886.1422 (doi:10.1126/science.164.3886.1422) [DOI] [PubMed] [Google Scholar]
- 46.Cossins AR, Raynard RS. 1987. Adaptive responses of animal cell membranes to temperature. In Temperature and animal cells (eds Bowler K, Fuller BJ.), pp. 95–111 Cambridge, UK: Company of Biologists; [PubMed] [Google Scholar]
- 47.Hazel JR. 1984. Effects of temperature on the structure and metabolism of cell membranes in fish. Am. J. Physiol. 246, R460–R470 [DOI] [PubMed] [Google Scholar]
- 48.Hochachka PW, Somero GN. 2002. Biochemical adaptation: mechanism and process in physiological evolution. Oxford, UK: Oxford University Press [Google Scholar]