

Summary
Background
Factor VII-activating protease (FSAP) is a serine protease that circulates in plasma in its inactive single-chain form and can be activated upon contact with dead cells. When activated by apoptotic cells, FSAP leads to the release of nucleosomes. The serpins C1-inhibitor and α2-antiplasmin are reported to be the major inhibitors of FSAP. However, regulation of FSAP activity by Kunitz-type inhibitors is not well studied.
Objectives
To compare the inhibition of FSAP activity and FSAP-induced nucleosome release from apoptotic cells by tissue factor pathway inhibitor (TFPI) with that of C1-inhibitor and α2-antiplasmin.
Methods
Apoptotic cells were incubated with plasma or FSAP in presence of the inhibitor, and nucleosome release was analyzed with flow cytometry. Monoclonal antibodies against TFPI and altered forms of TFPI were used to investigate which domains of TFPI contribute to FSAP inhibition.
Results and Conclusions
We show that TFPI abrogates FSAP activity and nucleosome release from apoptotic cells. TFPI is a much more efficient inhibitor than C1-inhibitor or α2-antiplasmin. The active site of K2 is required for inhibition of FSAP. A direct binding interaction between FSAP and the C-terminal domain of TFPI is also required for efficient inhibition. Inhibition of FSAP-induced nucleosome release by recombinant TFPI might, in part, explain the anti-inflammatory effects of recombinant TFPI infusion observed in animal and human sepsis.
Keywords: apoptosis, factor VII-activating protease, hyaluronic acid-binding protein 2, inflammation, nucleosomes, tissue factor pathway inhibitor
Introduction
Nucleosomes and DNA-binding proteins are endogenous mediators of inflammation and lethality in sepsis [1–4]. Levels of circulating nucleosomes correlate with disease severity and outcome in sepsis patients [5,6]. Histone release contributes to death following inflammatory injury or chemical-induced cellular injury in mouse models, mediated in part through Toll-like receptors [7]. Although the cellular source of nucleosomes is unclear, it is likely that they are derived from apoptotic cells. Nucleosomes stay associated with the apoptotic cell under serum-free or plasma-free conditions [8], and are released after contact of apoptotic cells with serum or plasma [9]. Activation of factor VII-activating protease (FSAP) is essential for nucleosome release [10]. FSAP, also known as plasma hyaluronic acid-binding protein 2, is a serine protease consisting of three epidermal growth factor domains, a kringle domain, and a serine protease domain at its C-terminus. In plasma, FSAP circulates as an inactive single-chain molecule of 78 kDa that can be converted in its active two-chain form consisting of a 50-kDa heavy chain and a 28-kDa light chain connected by a disulfide bond [11]. In purified systems, several serpins, such as C1-inhibitor (C1inh), α2-antiplasmin (AP), antithrombin III, and plasminogen activator inhibitor-1 [11–15], have been reported to inhibit the amidolytic activity of FSAP. In plasma, C1inh has been reported to be the main inhibitor of activated FSAP [12]. We demonstrated that when FSAP is activated in plasma, complexes with AP and C1inh are formed [16]. Incubation of plasma or serum with apoptotic cells leads to activation of FSAP, release of nucleosomes, and the formation of FSAP–C1inh and FSAP–AP complexes. We observed FSAP activation in postsurgery patients, and patients suffering from severe sepsis, septic shock, and meningococcal sepsis, and the levels of FSAP activation correlated with nucleosome levels, disease severity and mortality in these patients [16].
In a search for inhibitors of FSAP, we found that aprotinin, a synthetic Kunitz-type inhibitor, inhibits nucleosome release from apoptotic cells by FSAP in plasma [10]. Tissue factor pathway inhibitor (TFPI) is a Kunitz-type inhibitor present on endothelium [17], within platelets [18,19] and in plasma [20] that has anti-inflammatory properties [21–23]. The primary physiologic function of TFPI is to inhibit coagulation through inhibition of FXa and tissue factor (TF)–FVIIa [24]. Here, we report that TFPI is an efficient inhibitor of FSAP.
Materials and methods
Materials
Mouse mAbs against FSAP (anti-FSAP4), complexed C1inh (KOK-12) and AP (AAP-20) were prepared at our department (all IgG1κ) [10,25]. The mAbs against the various domains of TFPI, plasma purified human C1inh (Cetor), high-performance ELISA buffer and poly-horseradish peroxidase (HRP)-labeled streptavidin were obtained from Sanquin Reagents (Amsterdam, The Netherlands). Iscove's modified Dulbecco's medium (IMDM) was obtained from BioWhittaker Europe (Verviers, Belgium). Fetal bovine serum (FBS) was obtained from Bodinco BV (Alkmaar, The Netherlands). Penicillin and streptomycin were obtained from Gibco/Invitrogen (Groningen, The Netherlands). Etoposide, β-mercaptoethanol, RNase, mineral oil and AP were obtained from Sigma (Zwijndrecht, The Netherlands). 3,5,3’,5’-Tetramethylbenzidine (TMB) was obtained from Merck (Darmstadt, Germany). Chromogenic substrate S2288 was obtained from Chromogenix (Milan, Italy). Recombinant full-length TFPI, TFPI-K1M (Ile36) and TFPI-K2M (Leu107) expressed in Escherichia coli were a kind gift from A. Creasey (Chiron Corporation, Emeryville, CA, USA). In these altered forms of TFPI, the residue at the active-site cleft of Kunitz domain 1 (K1) or Kunitz domain 2 (K2) has been individually changed, leading to a dysfunctional Kunitz domain [24]. TFPI-160 was obtained as described by Warshawsky et al. [26,27].
Cell culture and induction of apoptosis
Jurkat cells were cultured in IMDM containing 5% (v/v) FBS, penicillin (100 IU mL)1), streptomycin (100 lg mL–1), and 50 μm β-mercaptoethanol. Before apoptosis induction, cells were washed three times with culture medium without FBS by centrifugation at 360 ×g for 10 min, and resuspended in culture medium without FBS. Cells (1 × 106 cells mL–1) were incubated for 48 h with etoposide at a final concentration of 200 μm to induce apoptosis.
Recalcified plasma
Serum clotted in the presence of cells contains microparticles that obscure fluorescence-activated cell sorting (FACS) analysis. Therefore, we used recalcified citrated plasma. It removed nucleosomes from apoptotic cells as efficiently as serum, and the clotting did not lead to FSAP activation [9]. In the text, recalcified citrated plasma is denoted as serum.
Blood was obtained from healthy donors in vials containing a final concentration of 10 mm sodium citrate, and centrifuged twice at 1300 ×g. Citrated plasma was recalcified with 20 mm CaCl2 in a glass vial, and incubated at 37 °C for 30 min. Subsequently, the recalcified plasma was incubated at 4 °C for 30 min, and the formed clot was removed. The serum was stored at – 20 °C until use. All donors were homozygous for the wild-type form of FSAP.
Nucleosome-releasing factor (NRF) assay
Active two-chain FSAP (tcFSAP) was purified as described previously [10]. Apoptotic Jurkat cells were washed in HN buffer (10 mm Hepes, 140 mm NaCl, pH 7.2) and 1% (w/v) bovine serum albumin (BSA), and resuspended in HN/1% BSA to a final concentration of 2 × 106 cells mL–1. Cells were incubated with RNase (40 μg mL–1) for 30 min at 37 °C. After incubation of 100 μL of sample (either plasma or tcFSAP diluted in HN) with 100 μL of cells for 30 min at 37 °C in a glass vial, 150 μL was removed and added to a microtiter plate (96 wells, round bottom). After three washes with FACS buffer (10 mm Hepes, 150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 0.5% BSA), cells were resuspended in 100 μL of FACS buffer and stained with propidium iodide at a final concentration of 4 μg mL–1. The median fluorescence intensity was measured with flow cytometry.
FSAP–C1inh and FSAP–AP complex ELISA
Complexes of FSAP with C1inh and AP were determined as described previously [16]. Briefly, the mAbs KOK-12 against C1inh in complex or AAP-20 against AP were used for capture of the FSAP–inhibitor complexes. Biotinylated mAb anti-FSAP4, recognizing the light chain of FSAP in combination with poly-HRP-labeled streptavidin, was used for detection. Results were expressed in AU mL–1 by reference to a standard, which was recalcified citrated plasma activated with apoptotic cells (1 × 106 cells mL–1) in the presence of 20 mm EDTA. This standard was arbitrarily set to 50 AU mL–1.
FSAP inhibition in chromogenic assay
Increasing concentrations of TFPI, C1inh, AP, TFPI-160, TFPI-K1M or TFPI-K2M were added to an excess of chromogenic substrate S2288 (1 mm) [13] in HN/0.1% Tween-20 in a 96-well plate. In addition, increasing concentrations of TFPI were preincubated with mAbs against Kunitz domain 1 (K1), Kunitz domain 2 (K2), Kunitz domain 3 (K3) or the C-terminus (Cter) of TFPI, or an irrelevant antibody (50 μg mL–1), and were added to an excess of chromogenic substrate S2288 (1 mm) in HN/0.1% Tween-20 in a 96-well plate. Subsequently, a fixed concentration of purified tcFSAP (10 nm) was added to the plate. To prevent evaporation, the samples were covered with a layer of mineral oil. The absorbance at 405 nm was recorded for 60 min at 37 °C with a Multiskan Spectrum Reader (Thermo Labsystems; Waltham, MA, USA). The change in absorbance perminute (velocity) was calculated. Data were plotted as Vi/Vo against the inhibitor concentration, where Vi and Vo are the velocities of substrate hydrolysis by 10 nm tcFSAP in the presence and absence of inhibitor, respectively. The kinetic parameters were calculated from the Michaelis–Menten regression model. The IC50 values for purified tcFSAP inhibition by TFPI, C1inh and AP were determined by plotting Vi/Vo against the inhibitor concentration (Hill slope model). The Ki was calculated with the Cheng–Prusoff equation [28].
TFPI binding to FSAP
A 96-well microtiter plate (Maxisorp; Nunc, Roskilde, Denmark) was coated with purified tcFSAP (2 μg mL–1 in phosphate-buffered saline [PBS]) overnight at room temperature. All further incubation steps were performed in PBS/0.1% (v/v) Tween-20/0.2% (w/v) gelatin (PTG), and after each step the wells were washed five times with washing fluid (PBS, 0.02% Tween-20) witha Microplate Autowasher (Bio-tek Instruments, Inc. Winooski, VT, USA). After coating, wells were incubated with 1% (w/v) BSA overnight to prevent non-specific binding to the plate. Biotinylated TFPI was preincubated for 60 min with anti-K1, anti-K2, anti-K3, anti-Cter, or an irrelevantanti body of the same isotype (50 μg mL–1). Subsequently, the TFPI samples were added to the plate and incubated for 60 min at room temperature. Finally streptavidin-polymerized HRP (poly-HRP) conjugate (dilution 1 : 10 000 v/v) diluted in PTG was added to each well and incubated for 20 min. Plates were developed by the addition of 100 μg mL–1 TMB and 0.003% hydrogen peroxide in 0.11 m sodium acetate buffer (pH 5.5). The coloring reaction was stopped by addition of 100 μL of 2 m H2SO4. The absorbance was measured at 450–540 nm with an ELISA plate reader. Results were expressed as percentage binding of TFPI to FSAP, where binding of TFPI, preincubated with an irrelevant antibody, to FSAP was set at 100%.
Results
Inhibition of FSAP activity by TFPI
To test whether TFPI is able to inhibit the nucleosome-releasing activity of FSAP, we incubated serum (Fig. 1A) or purified FSAP (Fig. 1B) with TFPI prior to incubation with apoptotic cells. C1inh and AP served as controls. Surprisingly, C1inh and AP were significantly less efficient in the inhibition of nucleosome release than TFPI. Inhibition of purified FSAP required lower levels of C1inh and AP, whereas TFPI was equally efficient. To determine whether the apoptotic cells play a role in the inhibition of FSAP by TFPI, we also investigated whether TFPI inhibits the amidolytic activity of plasma-derived tcFSAP in the absence of cells. Again, TFPI turned out to be a very efficient inhibitor of the amidolytic activity of purified tcFSAP (Fig. 1C). The estimated inhibition constant (Ki) of the inhibition of purified FSAP by recombinant TFPI was 0.63 nm. As it is known that physiologic concentrations of calcium reduce the inhibition of FXa by TFPI, we tested the inhibition of FSAP by TFPI in the presence of calcium. However, addition of physiologic concentrations of calcium with TFPI did not alter the inhibition of FSAP by TFPI (data not shown).
Fig. 1.

Inhibition of factor VII-activating protease (FSAP) activity by α2-antiplasmin (AP), C1-inhibitor (C1inh), and tissue factor pathway inhibitor (TFPI). Serum 10% (A) or purified FSAP 10 AU mL–1 (B) was preincubated with increasing concentrations of AP, C1inh or TFPI prior to incubation with apoptotic cells for 30 min at 37 °C. Cells were stained with propidium iodide, and median fluorescence intensity (MFI) was measured by fluorescence-activated cell sorting analysis. Nucleosome-releasing factor (NRF) activity is expressed as percentage of the decrease in MFI after incubation with 10% serum (which contains ~ 18.5 nm FSAP) or 10 AU mL–1 purified two-chain FSAP (tcFSAP) (~ 18.5 nm). Increasing concentrations of TFPI, C1inh and AP were added to the chromogenic substrate S2288 (1 mm). Purified tcFSAP (10 nm) was added, and the absorbance at 405 nm was measured (C). Data are plotted as Vi/Vo against the inhibitor concentration. Vi and Vo indicate the velocities of substrate hydrolysis by 10 nm tcFSAP in the presence and absence of inhibitor, respectively. The potency in inhibiting 50% of the reference amidolytic activity (IC50) was determined. Results are given as mean ± standard error of the mean (n = 3).
Inhibition of FSAP–inhibitor complex formation by TFPI
Quantification of FSAP–C1inh and FSAP–AP complexes can be used to monitor both in vitro and in vivo FSAP activation [16]. Upon incubation with apoptotic cells, FSAP is activated and FSAP–AP and FSAP–C1inh complexes are formed. To confirm the results of the nucleosome-releasing assay, FSAP–inhibitor complexes were measured after serum incubation with apoptotic cells in the presence of TFPI. TFPI at a concentration of 125 nm was sufficient to inhibit the formation of complexes with AP (~ 0.5 μm in 50% plasma) (Fig. 2A). This was also true for C1inh with an estimated concentration of 1.2 μm (Fig. 2B). These results support the data obtained in the nucleosome-releasing assay and the chromogenic assay, indicating TFPI to be a more efficient inhibitor than the plasma inhibitors AP and C1inh.
Fig. 2.

Inhibition of factor VII-activating protease (FSAP)–α2-antiplasmin (AP) and FSAP–C1-inhibitor (C1inh) complex formation by tissue factor pathway inhibitor (TFPI). Serum (50%) was preincubated with increasing concentrations of TFPI prior to incubation with apoptotic cells for 30 min at 37 °C. FSAP–AP (A) and FSAP–C1inh (B) complexes were measured by ELISA. Results are given as mean ± standard error of the mean (n = 3).
K2, K3 and Cter of TFPI inhibit FSAP activity
Full-length TFPI consists of three Kunitz-type domains and a basic C-terminal end. We tested which domain of TFPI is involved in the inhibition of FSAP activity by using mAbs directed against the various domains of TFPI. TFPI was preincubated with antibodies, added to serum, and incubated with apoptotic cells. Anti-K2 reversed the inhibitory effect of TFPI on FSAP-mediated nucleosome release (Fig. 3A). Anti-Cter and, to a lesser extent, anti-K3 and anti-K1 had a partial effect. Similar results were obtained when FSAP activation was monitored via formation of complexes of FSAP with AP and C1inh (data not shown). To determine whether the involvement of the various domains of TFPI is related to the presence of cells, we tested the effect of anti-TFPI antibodies in a chromogenic assay in the absence of cells. Again, anti-K2 was the most efficient inhibitor of TFPI, followed by anti-Cter and anti-K3. In contrast to the plasma system, anti-K1 had no effect on FSAP inhibition in the chromogenic assay (Fig 3B).
Fig. 3.

Role of Kunitz domains and C-terminus (Cter) of tissue factor pathway inhibitor (TFPI) in inhibition of factor VII-activating protease (FSAP) activity. TFPI was preincubated with blocking antibodies against Kunitz domain 1 (K1), Kunitz domain 2 (K2), Kunitz domain 3 (K3), or Cter (50 μg mL–1). Serum was then incubated with TFPI and antibodies prior to incubation with apoptotic cells for 30 min at 37 °C. Cells were stained with propidium iodide, and median fluorescence intensity (MFI) was measured by flow cytometry (A). Nucleosome-releasing factor (NRF) is expressed as a percentage of the decrease in MFI after incubation with 10% serum (which contains ~ 18.5 nm FSAP). TFPI was preincubated with blocking antibodies against K1, K2, K3, or Cter (50 μg mL–1), and added to the chromogenic substrate S2288 (1 mm). Purified two-chain FSAP (tcFSAP) (10 nm) was added, and the absorbance at 405 nm was measured (B). Data are plotted as Vi/Vo against the inhibitor concentration. Vi and Vo indicate the velocities of substrate hydrolysis by 10 nm tcFSAP in the presence and absense of TFPI, respectively. Results are given as mean ± standard error of the mean (n = 3).
To confirm the involvement of the various domains of TFPI in FSAP inhibition, we analyzed the effects of various TFPI mutants. TFPI-160, lacking K3 and Cter, was not able to inhibit FSAP activity (Fig. 4A), although TFPI-160 was still able to inhibit FXa activity (data not shown). TFPI-K1M, with an inactive K1, was still able to inhibit FSAP activity, whereas TFPI-K2M, with an inactive K2, completely lacked this inhibitory capacity (Fig. 4B).
Fig. 4.

Inhibition of factor VII-activating protease (FSAP) activity by tissue factor pathway inhibitor (TFPI), TFPI-160, TFPI-K1M, and TFPI-K2M. Increasing concentrations of TFPI, TFPI-160 (A), TFPI-K1M or TFPI-K2M (B) were added to the chromogenic substrate S2288 (1 mm). Purified two-chain (tcFSAP) (10 nm) was added, and the absorbance at 405 nm was measured. Data are plotted as Vi/Vo against the inhibitor concentration. Vi and Vo indicate the velocities of substrate hydrolysis by 10 nm tcFSAP in the presence and absence of inhibitor, respectively. Results are given as mean ± standard error of the mean (n = 3).
Binding of TFPI to FSAP
As the basic Cter tail of TFPI is involved in FSAP inhibition, and has been reported to bind to cell surfaces and heparin-like structures, we tested whether FSAP binds to TFPI and whether Cter is involved in this binding. Full-length TFPI bound to FSAP dose-dependently. Preincubation with anti-K1, anti-K2 or anti-K3 inhibited binding to FSAP by ~ 50%. When TFPI was preincubated with anti-Cter, the binding of TFPI to FSAP was reduced to 10% of its original level (Fig. 5). This indicates that Cter is important for TFPI binding to FSAP (Fig. 6).
Fig. 5.

Binding of tissue factor pathway inhibitor (TFPI) to factor VII-activating protease (FSAP). To measure binding of TFPI to FSAP, microtiter plates were coated with purified two-chain FSAP (tcFSAP). Biotinylated TFPI was preincubated with anti-Kunitz domain 1 (K1), anti-Kunitz domain 2 (K2), anti-Kunitz domain 3 (K3), or anti-C-terminus (Cter) of TFPI, or an irrelevant antibody (50 μg mL–1), before addition to the plate. Results are expressed as percentage binding of TFPI to FSAP, where binding of TFPI, preincubated with an irrelevant antibody, to FSAP was set at 100%. Results are given as mean ± standard error of the mean (n = 4).
Fig. 6.
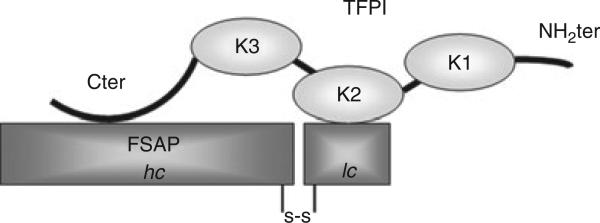
Schematic model of factor VII-activating protease (FSAP) inhibition by tissue factor pathway inhibitor (TFPI). TFPI binds directly to FSAP with its C-terminal domain (Cter). Upon binding of TFPI to FSAP, the Kunitz domain 2 of TFPI (K2) is able to inhibit FSAP activity. hc, heavy chain; K1, Kunitz domain 1; K3, Kunitz domain 3; lc, light chain; NH2ter, N-terminal domain; s-s, disulfide bond.
Discussion
We recently reported that activated tcFSAP in plasma can release nucleosomes from late apoptotic cells [9,10]. This nucleosome-releasing activity of FSAP can be efficiently inhibited by TFPI. Surprisingly, C1inh and AP, serpins that have been reported to be efficient inhibitors of FSAP in purified systems [13], were significantly less efficient. Inhibition of purified tcFSAP required lower levels of C1inh and AP, whereas TFPI was equally efficient. Most likely, this is because, during preincubation of (partially activated) FSAP with C1inh or AP, complex formation was achieved before apoptotic cells were added. The difference might be attributable to the fact that TFPI is a Kunitz-type protease inhibitor that has a different inhibitory mechanism from the serpins AP and C1inh. The efficacy of TFPI in inhibiting FSAP activity was also demonstrated in the absence of apoptotic cells. The amidolytic activity of plasma-derived tcFSAP was inhibited more efficiently than by C1inh and AP. Why TFPI is much more efficient in inhibiting FSAP than C1inh and AP is not clear. TFPI binds to surfaces, and is therefore considered to be an efficient surface-bound inhibitor of proteases. However, also in a cell-free system, as shown by the results of the chomogenic assay, TFPI was efficient in inhibiting FSAP activity. TFPI turned out to be 300 times more efficient than C1inh and AP in inhibiting the chromogenic activity of purified FSAP.
Full-length TFPI consists of three Kunitz-type domains and a basic C-terminal end. K1 and K2 inhibit TF–FVIIa and FXa, respectively, resulting in a quaternary complex TFPI–FXa– TF–FVIIa [24]. Cter has been reported to bind to cell surfaces and heparin-like structures [29,30]. By using mAbs against the various domains of TFPI, we demonstrated that inhibition of FSAP activity by TFPI is completely abrogated by anti-K2 and attenuated by anti-Cter and anti-K3. It is unlikely that the binding of TFPI to the surface of apoptotic cells is important for inhibition of FSAP activity, as the effect of the antibodies was also observed in a chromogenic assay in the absence of cells. To confirm the role of K2 in FSAP inhibition, TFPI-K1M and TFPI-K2M mutants were tested as well. In these point mutants of TFPI, the residue at the active-site cleft of K1 and K2 has been individually mutated, leading to inactive Kunitz domains [24]. TFPI-K1M was still able to inhibit FSAP activity, whereas TFPI-K2M completely lacked this inhibitory capacity. This confirms the results obtained with the anti-TFPI antibodies showing that K2 is indeed essential for the inhibition of FSAP. Anti-K3 and anti-Cter partially reversed the inhibition of NRF activity and the amidolytic activity of FSAP. When TFPI-160, which contains the first two Kunitz domains, was used [26,27], FSAP activity was not inhibited. This suggested that Cter and/or K3 are important for FSAP inhibition. Cter has been reported to bind to cell surfaces and heparin-like structures [29,30]. As Cter is involved in the inhibition of FSAP in the absence of a cell surface, we tested whether TFPI directly binds FSAP. By means of ELISA, we showed that, in the absence of cells, TFPI did indeed bind directly to FSAP, and that this interaction was partially abrogated by addition of anti-Cter. This suggests that FSAP binding to TFPI is a prerequisite for efficient inhibition of protease activity by K2 (Fig. 6). Interestingly, this mechanism is similar to FXa inhibition by TFPI, where binding of Cter to FXa and direct interaction of FXa with K2 were shown to be crucial for FXa inhibition [24,31]. As shown by the use of an altered form of TFPI lacking K3 but containing Cter, FXa interacts with Cter directly [31]. A recent paper reported that FSAP increased FXa generation by proteolytic degradation of TFPI [32]. Proteolytic degradation of TFPI was strongly dependent on FSAP binding to Cter. This is in agreement with our results showing that inhibition is dependent on binding of FSAP to Cter. Proteolytic degradation of TFPI, as suggested by Kanse et al., might be a mechanism by which FSAP is inhibited by TFPI in our system. TFPI would thereby serve as a substrate for FSAP, and hence compete with the chromogenic substrate or target proteases of FSAP. However, the inhibition curves of the amidolytic activity after 30 and 60 min of incubation are similar (data not shown), indicating no substantial loss of TFPI activity, which one would expect after proteolytic degradation of TFPI.
The TFPI concentrations used in the present study are much higher than the human plasma concentration of TFPI, which is 1.0–2.5 nm [30,33]. Moreover, we used full-length recombinant TFPI for our experiments. In contrast, only a minor fraction of the TFPI measured in plasma is considered to be full-length [34]. TFPI is mainly produced by and bound to vascular endothelial cells [20,35]. Therefore, the plasma concentration of TFPI does not reflect the local concentration of full-length TFPI on cellular surfaces. Administration of recombinant full-length TFPI was shown to attenuate the inflammatory response in animal and human sepsis [21–23]. The plasma concentrations reached in the human studies after therapeutic administration are in a similar range as the concentrations used in the present study in vitro [23,36,37]. Circulating histones and nucleosomes were shown to induce a potential fatal inflammatory response in sepsis [4,5]. FSAP was shown to induce nucleosome release from apoptotic cells and to be activated in sepsis [10,16]. We have now demonstrated that recombinant TFPI efficiently inhibits FSAP-mediated nucleosome release from apoptotic cells. Inhibition of FSAP-induced nucleosome release might therefore prevent the organism from the harmful effects of circulating nucleosomes and histones during inflammation. Inhibition of FSAP-induced nucleosome release might, in part, explain the anti-inflammatory effects of high-dose administration of recombinant TFPI observed in animal and human sepsis [21–23].
Acknowledgements
We thank K. Mertens for his helpful comments on this paper. This work was supported by a grant of the Landsteiner Foundation for Blood Transfusion Research (LSBR 0817).
Footnotes
Disclosure of Conflict of Interests
S. Zeerleder is in receipt of an unrestricted grant from Viropharma. A. E. Mast is in receipt of a grant from Novo Nordisk. The other authors state that they have no conflict of interest.
References
- 1.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 2.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164:1768–73. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 3.Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–96. doi: 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 4.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeerleder S, Zwart B, Wuillemin WA, Aarden LA, Groeneveld AB, Caliezi C, van Nieuwenhuijze AE, van Mierlo GJ, Eerenberg AJ, Lammle B, Hack CE. Elevated nucleosome levels in systemic inflammation and sepsis. Crit Care Med. 2003;31:1947–51. doi: 10.1097/01.CCM.0000074719.40109.95. [DOI] [PubMed] [Google Scholar]
- 6.van Till JW, van Veen SQ, den Broeder V, Bresser P, Lutter R, Out TA, Schultz MJ, Gouma DJ, Boermeester MA. Compartmental apoptosis and neutrophil accumulation in severe peritonitis. J Surg Res. 2010;164:321–8. doi: 10.1016/j.jss.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–31. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Nieuwenhuijze AE, van Lopik T, Smeenk RJ, Aarden LA. Time between onset of apoptosis and release of nucleosomes from apoptotic cells: putative implications for systemic lupus erythematosus. Ann Rheum Dis. 2003;62:10–14. doi: 10.1136/ard.62.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeerleder S, Zwart B, te Velthuis H, Manoe R, Bulder I, Rensink I, Aarden LA. A plasma nucleosome releasing factor (NRF) with serine protease activity is instrumental in removal of nucleosomes from secondary necrotic cells. FEBS Lett. 2007;581:5382–8. doi: 10.1016/j.febslet.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 10.Zeerleder S, Zwart B, te Velthuis H, Stephan F, Manoe R, Rensink I, Aarden LA. Nucleosome-releasing factor: a new role for factor VII-activating protease (FSAP). FASEB J. 2008;22:4077–84. doi: 10.1096/fj.08-110429. [DOI] [PubMed] [Google Scholar]
- 11.Etscheid M, Hunfeld A, Konig H, Seitz R, Dodt J. Activation of proPHBSP, the zymogen of a plasma hyaluronan binding serine protease, by an intermolecular autocatalytic mechanism. Biol Chem. 2000;381:1223–31. doi: 10.1515/BC.2000.150. [DOI] [PubMed] [Google Scholar]
- 12.Choi-Miura NH, Saito K, Takahashi K, Yoda M, Tomita M. Regulation mechanism of the serine protease activity of plasma hyaluronan binding protein. Biol Pharm Bull. 2001;24:221–5. doi: 10.1248/bpb.24.221. [DOI] [PubMed] [Google Scholar]
- 13.Romisch J, Vermohlen S, Feussner A, Stohr H. The FVII activating protease cleaves single-chain plasminogen activators. Haemostasis. 1999;29:292–9. doi: 10.1159/000022515. [DOI] [PubMed] [Google Scholar]
- 14.Hunfeld A, Etscheid M, Konig H, Seitz R, Dodt J. Detection of a novel plasma serine protease during purification of vitamin K-dependent coagulation factors. FEBS Lett. 1999;456:290–4. doi: 10.1016/s0014-5793(99)00959-x. [DOI] [PubMed] [Google Scholar]
- 15.Wygrecka M, Morty RE, Markart P, Kanse SM, Andreasen PA, Wind T, Guenther A, Preissner KT. Plasminogen activator inhibitor-1 is an inhibitor of factor VII-activating protease in patients with acute respiratory distress syndrome. J Biol Chem. 2007;282:21671–82. doi: 10.1074/jbc.M610748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stephan F, Hazelzet JA, Bulder I, Boermeester MA, van Till JO, van der Poll T, Wuillemin WA, Aarden LA, Zeerleder S. Activation of factor VII-activating protease in human inflammation: a sensor for cell death. Crit Care. 2011;15:R110. doi: 10.1186/cc10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajaj MS, Kuppuswamy MN, Saito H, Spitzer SG, Bajaj SP. Cultured normal human hepatocytes do not synthesize lipoprotein-associated coagulation inhibitor: evidence that endothelium is the principal site of its synthesis. Proc Natl Acad Sci USA. 1990;87:8869–73. doi: 10.1073/pnas.87.22.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Novotny WF, Girard TJ, Miletich JP, Broze GJ., Jr Platelets secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood. 1988;72:2020–5. [PubMed] [Google Scholar]
- 19.Maroney SA, Haberichter SL, Friese P, Collins ML, Ferrel JP, Dale GL, Mast AE. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007;109:1931–7. doi: 10.1182/blood-2006-07-037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novotny WF, Girard TJ, Miletich JP, Broze GJ., Jr Purification and characterization of the lipoprotein-associated coagulation inhibitor from human plasma. J Biol Chem. 1989;264:18832–7. [PubMed] [Google Scholar]
- 21.Creasey AA, Chang AC, Feigen L, Wun TC, Taylor FB, Jr, Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–60. doi: 10.1172/JCI116529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carr C, Bild GS, Chang AC, Peer GT, Palmier MO, Frazier RB, Gustafson ME, Wun TC, Creasey AA, Hinshaw LB. Recombinant E. coli-derived tissue factor pathway inhibitor reduces coagulopathic and lethal effects in the baboon gram-negative model of septic shock. Circ Shock. 1994;44:126–37. [PubMed] [Google Scholar]
- 23.Abraham E, Reinhart K, Svoboda P, Seibert A, Olthoff D, Dal NA, Postier R, Hempelmann G, Butler T, Martin E, Zwingelstein C, Percell S, Shu V, Leighton A, Creasey AA. Assessment of the safety of recombinant tissue factor pathway inhibitor in patients with severe sepsis: a multicenter, randomized, placebo-controlled, single-blind, dose escalation study. Crit Care Med. 2001;29:2081–9. doi: 10.1097/00003246-200111000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Girard TJ, Warren LA, Novotny WF, Likert KM, Brown SG, Mile-tich JP, Broze GJ., Jr Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989;338:518–20. doi: 10.1038/338518a0. [DOI] [PubMed] [Google Scholar]
- 25.Nuijens JH, Huijbregts CC, Eerenberg-Belmer AJ, Abbink JJ, Strack van Schijndel RJ, Felt-Bersma RJ, Thijs LG, Hack CE. Quantification of plasma factor XIIa-Cl(-)-inhibitor and kallikrein-Cl(-)-inhibitor complexes in sepsis. Blood. 1988;72:1841–8. [PubMed] [Google Scholar]
- 26.Warshawsky I, Bu G, Mast A, Saffitz JE, Broze GJ, Jr, Schwartz AL. The carboxy terminus of tissue factor pathway inhibitor is required for interacting with hepatoma cells in vitro and in vivo. J Clin Invest. 1995;95:1773–81. doi: 10.1172/JCI117855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lockett JM, Mast AE. Contribution of regions distal to glycine-160 to the anticoagulant activity of tissue factor pathway inhibitor. Biochemistry. 2002;41:4989–97. doi: 10.1021/bi016058n. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Harenberg J, Malsch R, Heene DL. Tissue factor pathway inhibitor: proposed heparin recognition region. Blood Coagul Fibrinolysis. 1995;6(Suppl 1):S50–6. [PubMed] [Google Scholar]
- 30.Sandset PM, Abildgaard U, Larsen ML. Heparin induces release of extrinsic coagulation pathway inhibitor (EPI). Thromb Res. 1988;50:803–13. doi: 10.1016/0049-3848(88)90340-4. [DOI] [PubMed] [Google Scholar]
- 31.Cunningham AC, Hasty KA, Enghild JJ, Mast AE. Structural and functional characterization of tissue factor pathway inhibitor following degradation by matrix metalloproteinase-8. Biochem J. 2002;367:451–8. doi: 10.1042/BJ20020696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanse SM, Declerck PJ, Ruf W, Broze G, Etscheid M. Factor VII-activating protease promotes the proteolysis and inhibition of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2012;32:427–33. doi: 10.1161/ATVBAHA.111.238394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broze GJ., Jr Tissue factor pathway inhibitor and the current concept of blood coagulation. Blood Coagul Fibrinolysis. 1995;6(Suppl 1):S7–13. doi: 10.1097/00001721-199506001-00002. [DOI] [PubMed] [Google Scholar]
- 34.Broze GJ, Jr, Lange GW, Duffin KL, MacPhail L. Heterogeneity of plasma tissue factor pathway inhibitor. Blood Coagul Fibrinolysis. 1994;5:551–9. [PubMed] [Google Scholar]
- 35.Mast AE, Acharya N, Malecha MJ, Hall CL, Dietzen DJ. Characterization of the association of tissue factor pathway inhibitor with human placenta. Arterioscler Thromb Vasc Biol. 2002;22:2099–104. doi: 10.1161/01.atv.0000042456.84190.f0. [DOI] [PubMed] [Google Scholar]
- 36.Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, Beale R, Svoboda P, Laterre PF, Simon S, Light B, Spapen H, Stone J, Seibert A, Peckelsen C, De DC, Postier R, Pettila V, Artigas A, Percell SR, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–47. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 37.de Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, Kesecioglu J, Levi M, van Deventer SJ, van der Poll T. Tissue factor pathway inhibitor dose-dependently inhibits coagulation activation without influencing the fibrinolytic and cytokine response during human endotoxemia. Blood. 2000;95:1124–9. [PubMed] [Google Scholar]