

Abstract
In severely injured and hypoperfused trauma patients, endogenous acute coagulopathy (EAC) is associated with an increased morbidity and mortality. Recent human data correlate this coagulopathy with activation of the protein C pathway. To examine the mechanistic role of protein C in the development of EAC, we used a mouse model of trauma and hemorrhagic shock, characterized by the combination of tissue injury and severe metabolic acidosis. Mice were subjected to one of four treatment groups: 1) C, control; 2) T, trauma (laparotomy); 3) H, hemorrhage (MAP, 35 mmHg × 60 min); 4) TH, trauma + hemorrhage. After 60 min, blood was drawn for analysis. Compared with C mice, the TH mice had a significantly elevated activated partial thromboplastin time (23.3 vs. 34.5 s) and significantly increased levels of activated protein C (aPC; 2.30 vs. 13.58 ng/mL). In contrast, T and H mice did not develop an elevated activated partial thromboplastin time or increased aPC. Selective inhibition of the anticoagulant property of aPC prevented the coagulopathy seen in response to trauma/hemorrhage (23.5 vs. 38.6 s [inhibitory vs. control monoclonal antibody]) with no impact on survival during the shock period. However, complete blockade of both the anticoagulant and cytoprotective functions of aPC caused 100% mortality within 45 min of shock, with histopathology evidence of pulmonary thrombosis and perivascular hemorrhage. These results indicate that our unique mouse model of T/H shock mimics our previous observations in trauma patients and demonstrates that EAC is mediated by the activation of the protein C pathway. In addition, the cytoprotective effect of protein C activation seems to be necessary for survival of the initial shock injury.
Keywords: Trauma, shock, hemorrhage, hypoperfusion, coagulation, survival
INTRODUCTION
Trauma remains the leading cause of death and disability in adults, eclipsing ischemic heart disease, cerebrovascular disease and human immunodeficiency virus/AIDS (1). Worldwide, one in seven deaths is due to injury, and this is expected to rise to one in five in the next 15 years, despite continuing advances in resuscitation, trauma surgery, and critical care (2). Hemorrhage is responsible for 40% of early trauma deaths, and efforts to control hemorrhage and restore circulatory homeostasis form the core of the therapeutic approach to traumatic injuries (3).
Perturbations in blood coagulation are common after major trauma and are associated with poor outcomes (1, 4). Classically, coagulopathy associated with trauma is thought to be due to the consumption of coagulation factors, acidosis, dilution from intravenous blood and fluid therapy, and hypothermia (5). This coagulopathy can be described as systemic acquired coagulopathy (SAC) (6). The abnormalities associated with SAC have been partially characterized in animal models and extensive clinical human research (7–9). In addition, there is extensive literature exploring the ideal resuscitative protocol and treatment of SAC (5, 7, 10).
More recently, it has been recognized that some trauma patients present with an early coagulopathy that is physiologically and mechanistically distinct from SAC. Two recent studies have identified an acute traumatic coagulopathy, present on arrival in the emergency department, in 25% of patients with major trauma (11, 12). This posttraumatic endogenous acute coagulopathy (EAC) is associated with higher transfusion requirements, a greater incidence of multiple organ dysfunction syndrome, longer intensive care unit and hospital stays, and a 4-fold increased risk of mortality compared with those with normal coagulation (11, 12). In examining the mechanism for this EAC, we reported that the combination of traumatic injury and hypoperfusion (shock) resulted in a coagulopathy that was associated with a reduction in protein C (PC) levels (13).
Protein C is a plasma serine protease that is activated through a thrombin-dependent reaction also involving thrombomodulin and the endothelial protein C receptor (14). Once activated, aPC exerts its anticoagulant effects by irreversibly inactivating factors Va and VIIIa (14). In addition, aPC has anticoagulant activity through its derepression of fibrinolysis by directly inhibiting plasminogen activator inhibitor 1 (15). Activated protein C (aPC) also acts via the cell surface receptor, protease-activated receptor 1 (PAR-1) to produce several cytoprotective effects (16). These effects include anti-inflammatory properties, antiapoptotic activity, and protection of endothelial barrier function (17–19).
Several mouse models have been used to study the effects of trauma and hemorrhagic shock on the following: survival, organ perfusion, immune response, inflammation, injury to lung and liver, and the impact of sex on the response to trauma and hemorrhage (20–25). In addition, although other animal models have examined the coagulopathy associated with the classic mechanisms of hypothermia, dilution, factor consumption, and acidosis, no published models have been adapted to study EAC associated with trauma (7–9, 26). Therefore, the first aim of the present study was to optimize a preexisting mouse model of trauma and hemorrhagic shock (20) to develop the first animal model of EAC that is seen after tissue injury and hypoperfusion in trauma patients. Second, we set out to use this model to examine the hypothesis drawn from our correlative human data that activation of PC is the primary mechanism responsible for the development of posttraumatic EAC. Finally, using our model, we also questioned whether this activation of PC could be a physiologic response to severe trauma/hemorrhage necessary for survival of the initial shock injury.
MATERIALS AND METHODS
Mouse model of trauma-hemorrhage and acute traumatic coagulopathy
Protocol was developed, based in principle, on the mouse model of traumatic shock extensively published by several investigators (20, 22, 27). The experiments were conducted in accordance with National Institutes of Health (Bethesda, Md) guidelines and approved by the University of California, San Francisco Institutional Animal Care and Use Committee.
Male C57BL/6J mice 8 to 10 weeks old (Jackson Laboratory, West, Sacramento, Calif) were used in all experiments. Animals were allowed water and food ad libitum and given at least 24 h to acclimate to the housing facility. The mice were anesthetized with isoflurane 1.2% in air:O2 at 1:0.5 L/min. The animals were then secured with plastic tape in a supine position on a firm plastic board. A lubricated rectal temperature probe was inserted and continuously monitored. A heat lamp was used to maintain core body temperature between 36 and 37°C throughout the experiment. As a model of soft tissue trauma, a sterile 2-cm midline laparotomy was performed. After inspection of underlying organs to verify absence of damage from the laparotomy, the wound was closed with a single layer of sterile wound clips (Reflex 7-mm wound clips; Braintree Scientific, Braintree, Mass). Then, the left femoral artery and right femoral vein were cannulated with PE-10 tubing preflushed with isotonic sodium chloride solution. After the laparotomy and vessel cannulations, all incisions were bathed with lidocaine 1% for analgesia, and the isoflurane was discontinued to allow emergence from general anesthesia.
MAP was monitored by attaching the left femoral arterial catheter to a pressure transducer (TSD104A; Biopac Systems, Goleta, Calif) and amplifier (MP1004-CE; Biopac Systems). The transducer output was analyzed using AcqKnowledge Software (Biopac Systems) with the arterial waveform continuously displayed. Upon emergence from anesthesia, baseline MAP greater than 90 mmHg was confirmed before initiation of the shock period. Shock was induced by withdrawing blood into a 1-mL syringe containing 3.2% sodium citrate via the left femoral arterial catheter 10 min after discontinuation of anesthesia at a rate of 0.2 mL/min. Arterial pressure was measured every minute during hemorrhage until the target MAP (35 mmHg) was achieved. This target MAP (35 ± 5 mmHg) was maintained over the course of the 60-min shock period by repeatedly removing additional aliquots of blood. Temperature, MAP, and respiratory rate were recorded every 5 min. Tachypnea seen in the trauma + hemorrhage (TH) and hemorrhage alone (H) groups at the end of the shock period was used as a reassuring surrogate marker for metabolic acidosis and systemic hypoperfusion, although no interventions were protocolized to target a specific respiratory rate (Fig. 1). Control (C), trauma (T), and hemorrhagic shock (H) mice underwent the same anesthetic, catheter placement, and 60-min period of board stress. Hemorrhagic mice underwent hemorrhagic shock only (no laparotomy), the T mice underwent laparotomy only (no hemorrhagic shock), and the C mice did not receive either the laparotomy or the hemorrhagic shock. Throughout the shock period, isotonic sodium chloride solution was used to gently flush the catheters to maintain catheter patency. Total volume of isotonic sodium chloride solution (crystalloid) administered to the animals was minimal and is presented in the Table 1.
Fig. 1. Hemodynamic and respiratory responses to hemorrhagic shock.
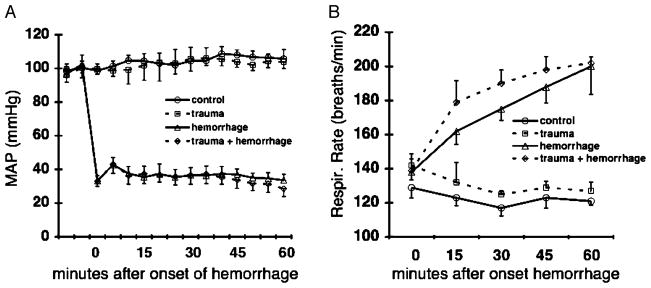
A–B, Changes in MAP (A) and respiratory rate before and during hemorrhagic shock (B). Control mice underwent catheter placement only. Trauma mice received 2-cm midline laparotomy and closure, followed by catheter placement. Hemorrhage mice underwent catheter placement, followed by hemorrhagic shock (blood withdrawal(s) necessary to maintain MAP = 35 ± 5 mmHg for 60 min). Trauma + hemorrhage mice received 2-cm midline laparotomy and closure, catheter placement, and hemorrhagic shock. Data are expressed as mean ± SD (n = 10 per group).
Table 1.
Characteristics of the model and experimental groups
| Control | Trauma | Hemorrhage | Trauma + hemorrhage | TH mAb 1761 | TH mAb 1591 | TH mAb 1609 | |
|---|---|---|---|---|---|---|---|
| Weight, g | 25.1 ± 1.1 | 24.7 ± 1.7 | 23.7 ± 1.7 | 24.2 ± 2.8 | 25.3 ± 1.6 | 24.9 ± 1.2 | 24.8 ± 1.3 |
| Hemorrhage, %ETBV | 0 | 0 | 40.5 ± 1.8 | 37.0 ± 3.5 | 34.2 ± 5.1 | 35.1 ± 2.5 | 30.8 ± 3.7 |
| Crystalloid, %ETBV | 4.0 ± 0.9 | 6.1 ± 1.7 | 6.0 ± 1.5 | 6.2 ± 1.9 | 7.9 ± 2.2 | 8.0 ± 1.3 | 7.0 ± 1.8 |
| Surgery time, min | 26 ± 7 | 32 ± 3 | 23 ± 5 | 33 ± 10 | 39 ± 6 | 36 ± 7 | 35 ± 4 |
| Hgb: end-shock, g/dL | 13.1 ± 0.6 | 13.5 ± 0.7 | 9.1 ± 0.5 | 10.1 ± 1.0 | 9.2 ± 1.3 | 10.1 ± 0.9 | 10.9 ± 1.5 |
| Lactate: end-shock, mM | 4.6 ± 0.7 | 6.5 ± 0.8 | 10.7 ± 3.6 | 10.7 ± 2.1 | 9.7 ± 1.8 | 10.4 ± 3.3 | 13.2 ± 4.6 |
Groups treated as described in the “Materials and Methods” section. Hemorrhage is total blood volume removed over course of 60-min shock period. Crystalloid is total crystalloid volume administered during surgery and shock period. ETBV = weight (g) × 0.077 mL/g. Hemoglobin (Hgb) and lactate values are measured at the end of 60-min shock period.
After 60 min of shock (H, TH) or board stress (C, T), mice were quickly euthanized with an overdose of isoflurane. Blood was drawn via inferior vena cava (IVC) puncture into a 1-mL syringe containing either 3.2% sodium citrate (for analysis of lactate, hemoglobin, activated partial thromboplastin time [aPTT], aPC) or heparin sulfate (for blood gas analysis and correlative lactate measurement). To preserve the aPC for later analysis, an aliquot of citrated whole blood was removed and mixed with the reversible protease inhibitor benzamidine (final concentration, 1 mM). All citrated blood samples were centrifuged at 4,000 rpm × 10 min, the supernatant removed and centrifuged at 13,000 rpm × 10 min to remove remaining cells and platelets. The final plasma supernatant was then removed and stored at −80°C until the time of analysis. After euthanasia and IVC blood draw, the lungs were removed and formalin-fixed. Organs were subsequently paraffin-embedded, sectioned, and hematoxylin and eosin stained. Blinded pathological assessment was performed by trained pathologists from the Department of Pathology at University of California at San Francisco.
Reagents
Monoclonal antibodies (mAbs) blocking specific functions of PC were generously provided by Charles Esmon, Ph.D. (Oklahoma Medical Research Foundation, Oklahoma City, Okla). Monoclonal antibody 1609 inhibits binding to cell surfaces and thus blocks both the anticoagulant and cytoprotective functions of PC. Monoclonal antibody 1591 selectively inhibits only the anticoagulant function of aPC but allows binding to cell surfaces and thereby cellular signaling (manuscript in preparation). Monoclonal antibody 1761 is an immunoglobulin G1 rat-antimouse isotype control antibody. Animals pretreated with mAbs received 10 mg/kg of mAb via the right femoral venous catheter 10 min before initiation of hemorrhagic shock. Gentle aspiration of venous blood after administration of mAb confirmed successful intravenous injection.
Assays
Activated partial thromboplastin time was performed on mouse plasma using the STACompact coagulation analyzer (Diagnostica Stago, Inc., Parsippany, NJ). Hemoglobin values were measured on whole blood samples using the B-Hemoglobin Photometer (HemoCue AB, Angelholm, Sweden).
Blood gas measurements were performed immediately on heparinized whole blood using an Osmetech OPTI CCA blood gas analyzer (OPTI Medical, Inc. Roswell, Ga). Lactate measurements were also performed immediately on all whole blood samples using the Accutrend Lactate analyzer (Roche Diagnostics, Indianapolis, Ind).
Activated protein C levels were measured in our laboratory using an assay specific for the activated form of PC developed in the laboratory of Esmon (28). Briefly, a 96-well vinyl plate was coated overnight at 4°C with monoclonal antibody AMGDPC 1587 at 5 μg/mL in plating buffer (0.02 M Tris, 0.1 M NaCl, pH 7.5). Plates were then blocked with blocking buffer (0.02 M Tris, 0.1 M NaCl, 1% bovine serum albumin, pH 7.5). Standard dilutions of mouse aPC and mouse plasma samples were loaded onto the plate and incubated at room temperature for 2 h on a 150-rpm rocker. Plates were washed with wash buffer (0.02 M Tris, 0.1 M NaCl, 0.05% Tween, pH 7.5). Spectrazyme PCA at 1 mM in plating buffer was added and incubated at 37°C for 24 h. Absorbance was measured at 405 nm with sample concentration derived from standard curve.
Statistical analysis
All data expressed as mean ± SD. Because of the sample sizes of the experimental groups, all comparison of continuous variables between groups was done using a nonparametric Kruskal-Wallis one-way ANOVA. A Mann-Whitney test was then used as a post hoc test for intergroup comparison. Survival data were analyzed using a Kaplan-Meier analysis for the curve with a log-rank test for significance. Significance is defined as a P value less than 0.05.
RESULTS
Mouse model of acute traumatic coagulopathy
Systemic physiology
As shown in Figure 1, MAP was successfully held within the goal range of 35 ± 5 mmHg for the 60-min shock period in both the H and TH groups. In contrast, the C and T groups remained normotensive throughout the 60-min period. To maintain a MAP = 35 ± 5 mmHg, animals undergoing H alone required 5.2 ± 1.1 (mean ± SD) blood withdrawals ranging in volume from 0.03 to 0.10 mL over the course of the 60-min shock period, whereas those in the TH group required 4.9 ± 0.7 withdrawals with volumes between 0.03 and 0.09 mL (P = not significant [NS]). Total hemorrhage volume, as defined as percentage of estimated total blood volume (ETBV), was not significantly different between the H and TH groups (Table 1). Interestingly, the H and TH groups each developed a progressive tachypnea over the course of the shock period, whereas the C and T groups sustained a normal respiratory rate (Fig. 1).
Hypoperfusion
The H and TH groups each developed a significant and severe metabolic acidosis after 60 min of hemorrhagic shock. These groups each manifested significantly decreased pH, decreased base excess, and increased lactate when compared with the C and T groups (Fig. 2, A–C). In this model, clinically significant base excess is defined as less than −6 mM, which corresponds to a plasma lactate value of 7.0 mM (Fig. 2D). Measurement of plasma lactate values in each individual animal allowed direct confirmation of a significant hypoperfusion injury in all experiments.
Fig. 2. Systemic markers of hypoperfusion in mice after TH shock.

A–D, Mice underwent either TH, H, T, or C (no trauma, no shock). Blood was drawn after 60 min of hemorrhagic shock via IVC puncture. Data expressed as mean ± SD (n = 5 per group). A, *P < 0.02 (C vs. H, T vs. H); **P < 0.02 (C vs. TH, T vs. TH). B, *P < 0.01 (C vs. T); **P < 0.01 (C vs. H, T vs. H); ***P < 0.01 (C vs. TH, T vs. TH). C, *P < 0.01 (C vs. H, T vs. H); **P < 0.01 (C vs. TH, T vs. TH).
Acute traumatic coagulopathy
As demonstrated in Figure 3, trauma + hemorrhagic shock is associated with increases in aPTT and aPC levels, similar to those seen in coagulopathic human trauma patients. In this model, the development of coagulopathy requires traumatic injury and a significant hypoperfusion injury because the presence of T or H was not sufficient to increase aPTT or aPC levels. Interestingly, as compared with C mice, those few TH mice with a lactate value less than 7 mM did not have elevated aPTT (23.3 vs. 25.6 s; P = NS) or aPC (2.30 vs. 1.96 ng/mL; P = NS; data not shown). Classic mechanisms of SAC were minimized throughout the experiment to isolate the mechanism(s) of EAC. Core temperature was monitored and maintained between 36 and 37°C throughout the shock period and catheter placement. In addition, dilution of coagulation factors by crystalloid administration was minimal and not significantly different between experimental groups (Table 1). Comparable relative volumes of crystalloid for a 70-kg human patient are approximately 350 mL.
Fig. 3. Trauma + hemorrhagic shock is associated with increases in aPTT (in seconds) and aPC (in nanograms per milliliter) in mice.
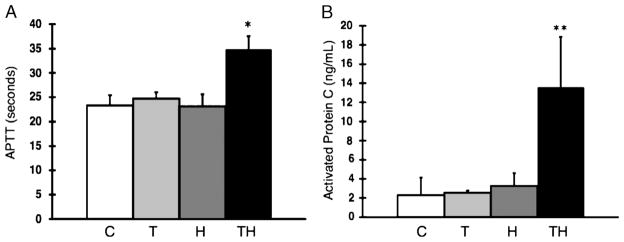
A and B, Mice underwent either TH, H, T, or C (no trauma, no shock). Activated partial thromboplastin time (in seconds) and aPC (ng/mL) levels were measured after 60 min of hemorrhagic shock. Data were expressed as mean ± SD. C (n = 6), T (n = 6), H (n = 10), TH (n = 10). A, *P = 0.02 (C vs. TH, T vs. TH, H vs. TH). B, **P < 0.02 (C vs. TH, T vs. TH, H vs. TH).
Role of aPC in acute traumatic coagulopathy and traumatic shock
Inhibition of aPC anticoagulant function prevents development of acute traumatic coagulopathy
Trauma and hemorrhagic mice pretreated with mAb 1591, which selectively inhibits the anticoagulant function of aPC, do not develop an increased aPTT after traumatic shock (Fig. 4). However, TH mice pretreated with an isotype control mAb1761 develop acute traumatic coagulopathy with an elevated aPTT (Fig. 4). Total hemorrhage volume (34.2 ± 5.1 vs. 35.1 ± 2.5 %ETBV), total crystalloid volume (7.9 ± 2.2 vs. 8.0 ± 1.3 %ETBV), and end-shock lactate (9.7 ± 1.8 vs. 10.4 ± 3.3 mM) were not significantly different between the mAb1761-pretreated and mAb1591-pretreated groups, respectively (Table 1). Pretreatment of C mice with either mAb (1761 vs. 1591) had no effect on aPTT values.
Fig. 4. Inhibition of the anticoagulant function of PC prevents the development of acute traumatic coagulopathy in mice.

Mice were pretreated with a mAb that inhibits the anticoagulant function of PC (mAb 1591) or with an isotype control mAb (mAb 1761). After 10 min, the mice then underwent TH. Activated partial thromboplastin time values were measured after 60 min of hemorrhagic shock. Data were expressed as mean ± SD. C, 1761 (n = 5); TH, 1761 (n = 9); C, 1591 (n = 5); TH, 1591 (n = 10). *P < 0.05 (C vs. TH mAb 1761). *P < 0.05 (TH [1761] vs. TH [1591]).
Complete inhibition of aPC prevents survival of traumatic shock
Trauma and hemorrhagic mice pretreated with mAb1609, which completely inhibits both the anticoagulant and cytoprotective functions of PC, suffer 100% mortality within 45 min of shock (Fig. 5). In contrast, TH mice pretreated with either mAb1761 or mAb1591 have similar survival curves with 20% mortality after 60 min of shock. All C mice (no trauma, no shock) in all treatment groups (mAbs 1761, 1591, and 1609) survived the 60-min protocol (data not shown). Histological analysis of pulmonary tissues revealed evidence of diffuse pulmonary arteriolar thrombosis as well as perivascular and alveolar hemorrhage in the TH mice pretreated with mAb 1609 (Fig. 6). Trauma and hemorrhagic mice pretreated with mAbs 1761 and 1591 did not develop any pulmonary pathology. In addition, C mice pretreated with mAbs, either 1761, 1591, or 1609, had normal pulmonary histology (not shown).
Fig. 5. Complete inhibition of PC before TH leads to 100% mortality within 45 min of shock.

Mice were pretreated with one of three monoclonal antibodies: mAb 1609 inhibits both the cytoprotective and anticoagulant functions of PC, mAb 1591 inhibits only the anticoagulant function of PC, and mAb 1761 is an isotype control monoclonal antibody. After 10 min, the mice then underwent TH. n = 14 mice per group. All C mice (no trauma, no shock) in all treatment groups (mAbs 1761, 1591, and 1609) survived (curves are not shown).
Fig. 6. Complete inhibition of PC before TH leads to pulmonary artery thrombosis, perivascular hemorrhage, and alveolar hemorrhage in mice.
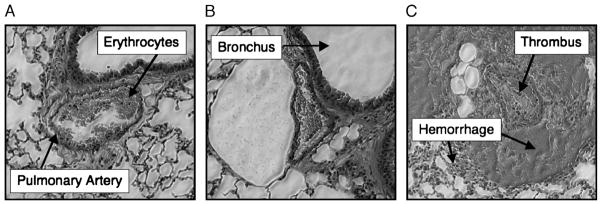
A–C, Mice were pretreated with one of three monoclonal antibodies: A, mAb 1761, an isotype control monoclonal antibody; B, mAb 1591, inhibits only the anticoagulant function of PC; C, mAb 1609, inhibits both the cytoprotective and anticoagulant functions of PC. After 10 min, the mice then underwent TH. Lungs were harvested after 60 min of hemorrhagic shock or at the time of death. Lungs were fixed, sectioned, and hematoxylin and eosin stained. Images obtained at 2003 magnification. All C mice (no trauma, no shock) in all treatment groups (mAbs 1761, 1591, and 1609) had no pulmonary pathology (sections not shown).
DISCUSSION
In this study, we present the development of the first animal model of posttraumatic EAC. Our group has previously shown that the combination of trauma and shock (tissue hypoperfusion) in human patients is associated with an activation of PC, early coagulopathy, and poor outcome (13). Although highly suggestive of an aPC mediated early traumatic coagulopathy, these human data were observational and correlative, requiring mechanistic confirmation. To provide mechanistic confirmation of our human data, we developed a translational mouse model of acute traumatic coagulopathy. Using this mouse model, we demonstrated that the combination of tissue injury and tissue hypoperfusion is required to produce an early traumatic coagulopathy. The coagulopathy seems to be mediated by aPC because blocking the anticoagulant activity of this enzyme prevents coagulopathy (Fig. 4).
Coagulopathy after traumatic injury has long been appreciated as an ominous complication and management challenge (29, 30). Our previously published human data showed a coagulopathy that was independent of traditional causes of posttraumatic SAC (hypothermia, dilution, consumption, or acidemia) due to an EAC (13). This EAC begins nearly immediately after trauma in the presence of shock and is distinct from SAC. Because we were interested in elucidating the mechanism of EAC, we attempted to minimize the impact of SAC in our model to isolate the development and mechanism(s) of EAC. As such, normothermia was maintained throughout the entire protocol, and dilution of coagulation factors with crystalloid was minimal. Although acidemia is known to contribute to coagulopathy, in our study, it was not sufficient on its own to trigger a coagulopathy because the mice subjected to H developed a significant and severe acidosis but did not become coagulopathic. As a result, hypoperfusion is clearly required in conjunction with traumatic injury to manifest EAC, whereas acidemia alone likely exacerbates and perpetuates the mechanisms and development of SAC. The posttraumatic coagulopathy is likely one of multiple phenotypes beginning with EAC and transitioning to SAC over time.
Consistent with observations in human trauma patients, our mouse model of EAC requires both tissue injury and severe hypoperfusion to manifest the coagulopathy. However, consistent with clinical observation of trauma patients, we occasionally observed heterogeneity produced by the injury/ shock protocol, with rare mice not manifesting a significant hypoperfusion injury despite the same degree of hypotension. As a result, we confirmed the degree of hypoperfusion injury in each animal by measuring plasma lactate levels to appropriately group those individuals with documented severe hypoperfusion. Interestingly, those few animals not manifesting a severe hypoperfusion injury did not activate PC and did not develop EAC in response to traumatic injury and hypotension. Conversely, all mice that developed a severe hypoperfusion injury had an increased aPC and coagulopathy.
Our human data suggest that consumption of PC through conversion to aPC is associated with posttraumatic EAC (unpublished data) (13). The present study provides the mechanistic proof that our interpretation of the human correlative data is correct. Furthermore, as the first animal model of EAC, these data demonstrate that the anticoagulant function of aPC is a primary mechanism responsible for the development of EAC. Selective inhibition of PC’s anticoagulant function in our animal model effectively prevented the development of EAC in response to injury and hypoperfusion. This result is consistent with our published clinical observations that activation of the PC pathway correlates with the development of EAC in injured and hypoperfused trauma patients.
Although PC is clearly activated in response to the combination of traumatic injury and hypoperfusion, the mechanism of this PC activation is, as yet, unknown. Perhaps, a threshold of inflammation, injury, and/or hypoxia is required to trigger the activation of PC. Human data suggest that the activation of complement subsequently triggers the activation of PC in response to trauma and hypoperfusion (31). In vitro and additional in vivo experiments are presently underway in our laboratory to further elucidate the mechanism of this PC activation after trauma and hypoperfusion.
Activated PC is a serine protease that exerts its anticoagulant effects by irreversibly inactivating factors Va and VIIIa and by derepressing fibrinolysis through its inhibitory actions on plasminogen activator inhibitor 1 (15). In addition to its anticoagulant effects, aPC proteolytically activates the cell surface receptor PAR-1 to produce several cytoprotective effects, including anti-inflammatory properties, antiapoptotic activity, and protection of endothelial barrier function (17–19). Several animal models suggest that these cytoprotective properties of aPC are involved in protective responses to ischemic injury in the brain, spinal cord, heart, kidney, liver and intestine, as well as conferring protection from injury in models of sepsis (32–38). In our study, we observed that selective blockade of PC’s anticoagulant function effectively inhibits the development of EAC without an impact on survival of shock. However, complete blockade of both the anticoagulant and cytoprotective functions of aPC result in 100% mortality within 45 min of shock. This finding suggests that activation of PC in response to traumatic injury and severe hypoperfusion is required for acute survival during shock.
Our findings suggest a crucial role for the cytoprotective effects of aPC in the response to trauma and hypoperfusion. The mechanism(s) responsible for precipitating the pulmonary thrombosis, alveolar and perivascular hemorrhage, and subsequent respiratory arrest in the setting of complete aPC blockade are unclear. The thrombotic response might be amplified by the loss of the cytoprotective effects of the aPC, thereby leading to exposure of the procoagulant membrane surfaces required for thrombosis. Perhaps, some degree of PAR-1 and/or endothelial PC receptor binding is required to maintain endothelial homeostasis. Recent data from Toltl et al. (39) implicate the cytoprotective function of aPC in the down-regulation of increased tissue factor expression in response to LPS stimulation in monocytes. As such, complete inhibition of aPC could allow an unchecked increase in tissue factor expression/activity in response to trauma and hypoperfusion, thereby creating a disrupted and procoagulant endothelium. Perhaps, the combination of anticoagulant and cytoprotective functions of aPC is required to prevent the observed mortality. Conversely, the activation of PC in response to injury and hypoperfusion might be necessary only for the cytoprotective effects, whereas the accompanying coagulopathy mediated by the anticoagulant properties of aPC is merely a matter of complete activation of a protein with multiple functions. These questions are certainly intriguing and are the focus of additional experiments in our laboratory.
Several limitations exist with this study. First, one must always accept the inherent limitation of using a mouse model to draw conclusions on human disease. Use of a larger mammalian model may be more applicable to human processes, but the ability to use genetically engineered mice in future experiments makes development of this model particularly useful. Second, whereas our present study focuses on the impact of the PC system on EAC, other anticoagulant pathways such as antithrombin III or tissue factor pathway inhibitor may also be involved. Because of our supportive human clinical data implicating PC in EAC and the extremely small volumes of plasma acquired from our mouse model, we chose to focus our study on the role of aPC in EAC. Finally, whereas our results suggest that the cytoprotective properties of aPC are necessary for survival of shock, this interpretation is limited by the absence of selective blockade of the cytoprotective functions of PC. Unfortunately, such inhibitory antibodies do not exist, and blockade of the PAR-1 receptor, although inhibiting the cytoprotective properties conferred by aPC binding, would also inhibit the binding of other PAR-1 ligands, including thrombin.
Most trauma patients die secondary to ongoing hemorrhage shortly after injury (3). Concurrent coagulopathy can exacerbate this hemorrhage and make surgical repair of damaged structures considerably more difficult. Although present treatment and resuscitative strategies focus on minimizing the development of SAC, the presence of EAC upon admission to the emergency department places trauma surgeons, anesthesiologists, and patients at an early disadvantage and demands additional treatment strategies focused on the mechanisms of EAC. As such, our study provides early evidence that selective blockade of the anticoagulant function of aPC may reduce or eliminate EAC without negatively impacting survival during traumatic injury and hypoperfusion. Specific treatment directed at EAC via blockade of aPC anticoagulant function could potentially result in less coagulopathic (medical) hemorrhage, thereby facilitating surgical control of traumatic hemorrhage and minimizing resuscitative interventions that could precipitate or exacerbate SAC. Most importantly, early control of coagulopathy and hemorrhage could result in decreased mortality secondary to hemorrhage in trauma patients. Nevertheless, additional investigation is required to examine the long-term impact of selective anticoagulant aPC blockade both during and after resuscitation.
Acknowledgments
The author Chesebro was supported by a National Institutes of Health T32 training grant. Supported in part by the National Institutes of Health (grant nos. K08 GM-085689 and RO1 GM-62188 to M.J.C. and J.F.P., respectively, and a T32 training grant awarded through the Department of Anesthesia and Perioperative Care, University of California, San Francisco, to B.B.C.) and an American Association for the Surgery of Trauma hemostasis and resuscitation scholarship (to M.J.C.).
The authors would like to acknowledge the time and energy of A. Ayala, J. Lomas-Niera, and C.S. Chung at the Shock-Trauma Research Laboratories, Rhode Island Hospital, and Brown University, Providence, RI, for teaching murine vascular surgery techniques and strategies for the management of hemorrhagic shock.
Footnotes
Institution at which work was performed: The Departments of Surgery and Anesthesia at San Francisco General Hospital, University of California San Francisco, CA.
References
- 1.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Injury Chart Book. Geneva, Switzerland: World Health Organization; 2002. [Google Scholar]
- 3.Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, Pons PT. Epidemiology of trauma deaths: a reassessment. J Trauma. 1995;38:185–193. doi: 10.1097/00005373-199502000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Gando S, Nanzaki S, Kemmotsu O. Disseminated intravascular coagulation and sustained systemic inflammatory response syndrome predict organ dysfunctions after trauma: application of clinical decision analysis. Ann Surg. 1999;229:121–127. doi: 10.1097/00000658-199901000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armand R, Hess JR. Treating coagulopathy in trauma patients. Transfus Med Rev. 2003;17:223–231. doi: 10.1016/s0887-7963(03)00022-1. [DOI] [PubMed] [Google Scholar]
- 6.Tieu BH, Holcomb JB, Schreiber MA. Coagulopathy: its pathophysiology and treatment in the injured patient. World J Surg. 2007;31:1055–1064. doi: 10.1007/s00268-006-0653-9. [DOI] [PubMed] [Google Scholar]
- 7.Cho SD, Holcomb JB, Tieu BH, Englehart MS, Morris MS, Karahan ZA, Underwood SA, Muller PJ, Prince MD, Medina L, et al. Reproducibility of an animal model simulating complex combat-related injury in a multiple-institution format. Shock. 2009;31:87–96. doi: 10.1097/SHK.0b013e3181777ffb. [DOI] [PubMed] [Google Scholar]
- 8.Fries D, Haas T, Klingler A, Streif W, Klima G, Martini J, Wagner-Berger H, Innerhofer P. Efficacy of fibrinogen and prothrombin complex concentrate used to reverse dilutional coagulopathy—a porcine model. Br J Anaesth. 2006;97:460–467. doi: 10.1093/bja/ael191. [DOI] [PubMed] [Google Scholar]
- 9.Martini WZ, Pusateri AE, Uscilowicz JM, Delgado AV, Holcomb JB. Independent contributions of hypothermia and acidosis to coagulopathy in swine. J Trauma. 1010;58:1002–1009. doi: 10.1097/01.ta.0000156246.53383.9f. [DOI] [PubMed] [Google Scholar]
- 10.Holcomb JB, Jenkins D, Rhee P, Johannigman J, Mahoney P, Mehta S, Cox ED, Gehrke MJ, Beilman GJ, Schreiber M, et al. Damage control resuscitation: directly addressing the early coagulopathy of trauma. J Trauma. 2007;62:307–310. doi: 10.1097/TA.0b013e3180324124. [DOI] [PubMed] [Google Scholar]
- 11.Brohi K, Singh J, Heron M, Coats T. Acute traumatic coagulopathy. J Trauma. 2003;54:1127–1130. doi: 10.1097/01.TA.0000069184.82147.06. [DOI] [PubMed] [Google Scholar]
- 12.MacLeod JB, Lynn M, McKenney MG, Cohn SM, Murtha M. Early coagulopathy predicts mortality in trauma. J Trauma. 2003;55:39–44. doi: 10.1097/01.TA.0000075338.21177.EF. [DOI] [PubMed] [Google Scholar]
- 13.Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. 2007;245:812–818. doi: 10.1097/01.sla.0000256862.79374.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 15.Rezaie AR. Vitronectin functions as a cofactor for rapid inhibition of activated protein C by plasminogen activator inhibitor–1. Implications for the mechanism of profibrinolytic action of activated protein C. J Biol Chem. 2001;276:15567–15570. doi: 10.1074/jbc.C100123200. [DOI] [PubMed] [Google Scholar]
- 16.Riewald M, Ruf W. Protease-activated receptor–1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J Biol Chem. 2005;280:19808–19814. doi: 10.1074/jbc.M500747200. [DOI] [PubMed] [Google Scholar]
- 17.Nick JA, Coldren CD, Geraci MW, Poch KR, Fouty BW, O’Brien J, Gruber M, Zarini S, Murphy RC, Kuhn K, et al. Recombinant human activated protein C reduces human endotoxin–induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood. 2004;104:3878–3885. doi: 10.1182/blood-2004-06-2140. [DOI] [PubMed] [Google Scholar]
- 18.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 19.Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1–phosphate receptor transactivation. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- 20.Wang P, Ba ZF, Burkhardt J, Chaudry IH. Trauma-hemorrhage and resuscitation in the mouse: effects on cardiac output and organ blood flow. Am J Physiol. 1993;264:H1166–H1173. doi: 10.1152/ajpheart.1993.264.4.H1166. [DOI] [PubMed] [Google Scholar]
- 21.Wichmann MW, Ayala A, Chaudry IH. Severe depression of host immune functions following closed-bone fracture, soft-tissue trauma, and hemorrhagic shock. Crit Care Med. 1998;26:1372–1378. doi: 10.1097/00003246-199808000-00024. [DOI] [PubMed] [Google Scholar]
- 22.Chow CC, Clermont G, Kumar R, Lagoa C, Tawadrous Z, Gallo D, Betten B, Bartels J, Constantine G, Fink MP, et al. The acute inflammatory response in diverse shock states. Shock. 2005;24:74–84. doi: 10.1097/01.shk.0000168526.97716.f3. [DOI] [PubMed] [Google Scholar]
- 23.Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock. 2003;19:358–365. doi: 10.1097/00024382-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Prince JM, Levy RM, Yang R, Mollen KP, Fink MP, Vodovotz Y, Billiar TR. Toll-like receptor–4 signaling mediates hepatic injury and systemic inflammation in hemorrhagic shock. J Am Coll Surg. 2006;202:407–417. doi: 10.1016/j.jamcollsurg.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Angele MK, Ayala A, Monfils BA, Cioffi WG, Bland KI, Chaudry IH. Testosterone and/or low estradiol: normally required but harmful immunologically for males after trauma-hemorrhage. J Trauma. 1998;44:78–85. doi: 10.1097/00005373-199801000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Clarke BJ, Sridhara S, Woskowska Z, Blajchman MA. Consumption of plasma factor VII in a rabbit model of non-overt disseminated intravascular coagulation. Thromb Res. 2002;108:329–334. doi: 10.1016/s0049-3848(03)00066-5. [DOI] [PubMed] [Google Scholar]
- 27.Xu YX, Ayala A, Chaudry IH. Prolonged immunodepression after trauma and hemorrhagic shock. J Trauma. 1998;44:335–341. doi: 10.1097/00005373-199802000-00018. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Zheng X, Gu J, Hunter J, Ferrell GL, Lupu F, Esmon NL, Esmon CT. Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost. 2005;3:1351–1359. doi: 10.1111/j.1538-7836.2005.01385.x. [DOI] [PubMed] [Google Scholar]
- 29.Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg. 1971;174:794–801. doi: 10.1097/00000658-197111000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber MA. Coagulopathy in the trauma patient. Curr Opin Crit Care. 2005;11:590–597. doi: 10.1097/01.ccx.0000186374.49320.ab. [DOI] [PubMed] [Google Scholar]
- 31.Ganter MT, Brohi K, Cohen MJ, Shaffer LA, Walsh MC, Stahl GL, Pittet JF. Role of the alternative pathway in the early complement activation following major trauma. Shock. 2007;28:29–34. doi: 10.1097/shk.0b013e3180342439. [DOI] [PubMed] [Google Scholar]
- 32.Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- 33.Hirose K, Okajima K, Taoka Y, Uchiba M, Tagami H, Nakano K, Utoh J, Okabe H, Kitamura N. Activated protein C reduces the ischemia/reperfusion-induced spinal cord injury in rats by inhibiting neutrophil activation. Ann Surg. 2000;232:272–280. doi: 10.1097/00000658-200008000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pirat B, Muderrisoglu H, Unal MT, Ozdemir H, Yildirir A, Yucel M, Turkoglu S. Recombinant human-activated protein C inhibits cardiomyocyte apoptosis in a rat model of myocardial ischemia-reperfusion. Coron Artery Dis. 2007;18:61–66. doi: 10.1097/MCA.0b013e328010a44a. [DOI] [PubMed] [Google Scholar]
- 35.Mizutani A, Okajima K, Uchiba M, Noguchi T. Activated protein C reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation. Blood. 2000;95:3781–3787. [PubMed] [Google Scholar]
- 36.Kuriyama N, Isaji S, Hamada T, Kishiwada M, Ohsawa I, Usui M, Sakurai H, Tabata M, Suzuki K, Uemoto S. Activated protein C prevents hepatic ischaemia-reperfusion injury in rats. Liver Int. 2009;29:299–307. doi: 10.1111/j.1478-3231.2008.01796.x. [DOI] [PubMed] [Google Scholar]
- 37.Teke Z, Sacar M, Yenisey C, Atalay AO, Kavak T, Erdem E. Activated protein C attenuates intestinal mucosal injury after mesenteric ischemia/reperfusion. J Surg Res. 2008;149:219–230. doi: 10.1016/j.jss.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, Castellino FJ, Mackman N, Griffin JH, Weiler H. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toltl LJ, Beaudin S, Liaw PC. Activated protein C up-regulates IL-10 and inhibits tissue factor in blood monocytes. J Immunol. 2008;181:2165–2173. doi: 10.4049/jimmunol.181.3.2165. [DOI] [PubMed] [Google Scholar]