

Abstract
Acute pancreatitis (AP) is a common clinical condition with an incidence of about 300 or more patients per million annually. About 10%-15% of patients will develop severe acute pancreatitis (SAP) and of those, 10%-30% may die due to SAP-associated complications. Despite the improvements done in the diagnosis and management of AP, the mortality rate has not significantly declined during the last decades. Toll-like receptors (TLRs) are pattern-recognition receptors that seem to play a major role in the development of numerous diseases, which make these molecules attractive as potential therapeutic targets. TLRs are involved in the development of the systemic inflammatory response syndrome, a potentially lethal complication in SAP. In the present review, we explore the current knowledge about the role of different TLRs that have been described associated with AP. The main candidate for targeting seems to be TLR4, which recognizes numerous damage-associated molecular patterns related to AP. TLR2 has also been linked with AP, but there are only limited studies that exclusively studied its role in AP. There is also data suggesting that TLR9 may play a role in AP.
Keywords: Acute pancreatitis, Severe acute pancreatitis, Pathophysiological mechanism, Toll-like receptors, Intervention
INTRODUCTION
A central characteristic of the innate immune system is its capability to identify constitutive and conserved products of microbial metabolism. Several metabolic pathways are unique for invading microorganisms and absent in host cells. Being essential for survival, they are even highly conserved among a given class of microorganisms. A classical example is lipopolysaccharide (LPS), a molecule made by Gram-negative bacteria, but not by eukaryotic cells.
Molecules found in microorganisms, but not in host cells, can serve as molecular signatures that can be recognised by the innate immune system, starting an immunological response against the invasion and if necessary, assisting in the activation of the adaptive immune system[1]. As these molecules are conserved molecular patterns, they are called pathogen-associated molecular patterns (PAMPs) and interact with pattern-recognition receptors (PRRs), which are receptors of the innate immune system.
PAMPs are marvellous targets for innate immune recognition, as they are only produced by microbes, are invariant between microorganisms of a given class and are essential for microbial survival. Therefore, PRRs representes a vital component of the immune system, which probably developed early during evolution, as PRRs are found in all mammals, invertebrates and even in plants. PRRs are expressed on the cell surface or in intracellular compartments, but they can be secreted into the blood stream and tissue fluids as well[2]. However, the role of PRRs in immune recognition is not solely associated to PAMPs, since PRRs in addition can recognize alarmins[3]. Alarmins are endogenous molecules released into the extracellular compartment by activated or necrotic cells in response to stress or tissue damage[4]. Even extracellular matrix molecules are alarmins when up-regulated upon injury or degraded following tissue damage[5]. Alarmins and PAMPs constitute damage-associated molecular patterns (DAMPs), which are the main targets of PRRs[3,4].
The relative recent discovery of a main PRR family, toll-like receptors (TLRs), has vastly increased our comprehension of the physiological and pathophysiological role of the innate immune system. While mice have twelve TLRs (TLR1 to TLR9 and TLR11 to TLR13), humans express only ten functional TLRs (TLR1 to TLR10) and ligands have been identified for all human TLRs, except for TLR10[3]. A selection of PAMPs and alarmins recognized by human TLRs are shown in Tables 1 and 2.
Table 1.
Human Toll-like receptors: Localization, known pathogen-associated molecular patterns and their producing microorganisms
| Localization | PAMP | Origin of PAMP | |
| TLR1 | Plasma membrane | Soluble factors | N. meningitidis |
| Triacyl lipopeptides | Bacteria, mycobacteria | ||
| TLR2 | Plasma membrane | Glycoinositolphospholipids | Trypanosoma cruzi |
| Glycolipids | Treponema maltophilum | ||
| Haemagglutinin | Virus | ||
| Lipoarabinomannan | Mycobacteria | ||
| Lipoprotein/lipopeptides | Various pathogens | ||
| Lipoteichoic acid | Gram-positive bacteria | ||
| Peptidoglycan | Gram-positive bacteria | ||
| Phenol-soluble modulin | S. epidermidis | ||
| Porins | Neisseria | ||
| Zymosan | Fungi | ||
| TLR3 | Endosome | Double-stranded RNA | Virus |
| TLR4 | Plasma membrane | Envelope protein | Mouse-mammary tumour virus |
| Fusion protein | Respiratory syncytial virus | ||
| Heat-shock protein 60 | Chlamydia pneumoniae | ||
| Lipopolysaccharide | Gram-negative bacteria | ||
| Taxol | Plants | ||
| TLR5 | Plasma membrane | Flagellin | Bacteria |
| TLR6 | Plasma membrane | Diacyl lipopeptides | Mycoplasma |
| Lipoteichoic acid | Gram-positive bacteria | ||
| Zymosan | Fungi | ||
| TLR7 | Endosome | Single-stranded RNA | Virus |
| TLR8 | Endosome | Single-stranded RNA | Virus |
| TLR9 | Endosome | DNA (CpG) | Bacteria, virus |
| Haemozoin | Plasmodium spp. | ||
| Rhodnius spp. | |||
| Schistosoma spp. | |||
| TLR10 | Endosome | Not determined | Not determined |
Modified after Akira et al[7]. TLR: Toll-like receptor; PAMP: Pathogen-associated molecular patterns.
Table 2.
Human toll-like receptors and a selection of known alarmins
| Proteins, peptides | Fatty acids, lipoproteins | Proteoglycans, glycosaminoglycans | |
| TLR1 | β-defensin-3 | ||
| TLR2 | Antiphospholipid antibodies β-defensin-3 | Serum amyloid A | Biglycan |
| Eosinophil-derived neurotoxin | Hyaluronic acid fragments | ||
| Heat-shock protein 60, 70, Gp96 | Versiglycan | ||
| High-mobility group protein B1 (HMGB1) | |||
| HMGB1-nucleosome complexes | |||
| Surfactant protein A, D | |||
| TLR4 | Antiphospholipid antibodies | Oxidised low-density protein | Biglycan |
| β-defensin-2 | Saturated fatty acids | Heparan sulphate fragment | |
| Fibrinogen | Serum amyloid A | Hyaluronic acid fragment | |
| Fibronectin (extra domain-A) | |||
| High-mobility group protein B1 | |||
| Heat-shock protein 60, 70, 72, 22, Gp96 | |||
| Lactoferrin MRP8, MRP14 | |||
| Neutrophil elastase | |||
| Surfactant protein A, D | |||
| Tenascin-C (fibrinogen-like globe) |
Modified after Paccinini et al[5]. TLR: Toll-like receptor.
TLRs are type I transmembrane glycoproteins composed of an extracellular N-terminal that contains leucine-rich repeats, a single transmembrane domain and an intracellular C-terminal tail known as the Toll/IL-1 receptor (TIR)[6]. When TLRs form heterodimers or homodimers, an activating signal is started and TIR-TIR dimers are involved in the recruitment of five known signalling adaptor molecules: Myeloid differentiation primary response protein 88 (MyD88), TIR domain-containing adaptor protein (TIRAP), TIRAP inducing interferon β (TRIF), TRIF-related adaptor molecule (TRAM) and sterile-alpha and Armadillo containing motif protein[7-9].
Once the adaptor molecules are recruited, TLRs can activate two major intracellular signalling pathways. All TLRs except TLR3 can activate a MyD88-dependent pathway, in which IL-1R-associated kinases (IRAK), TNF receptor-associated factor 6 (TRAF-6) and mitogen-activated kinases are involved[5]. This pathway results in the transcription of pro-inflammatory genes through the activation of nuclear factor κβ (NFκB) and/or the activation of activating protein 1[9]. An alternative, non MyD88-dependent pathway can be activated by TLR3 and TLR4. In the TRIF pathway, the activation of interferon-regulated factors (IRF) via TRIF leads to the synthesis of interferon (IFN)[5].
As TLRs recognize several PAMPs and DAMPs, their involvement in the pathophysiology of several diseases has become a major research field[8,10]. Moreover, recent studies have reported the importance of NFκB pathways in the development of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS)[11,12]. However, NFκB pathways are not restricted to TLRs, which converts the elucidation of the role of TLRs in SIRS and MODS into a tremendous challenging task.
SIRS is a nonspecific condition that can be caused by infection, ischemia, trauma, and inflammation, or by the combination of several insults. Considered by many as a self-defense mechanism, SIRS results in a complex inflammatory cascade that involve humoral and cellular responses, complement, and cytokines cascades. Severe complications depend on the underlying etiology and the magnitude of the inflammatory response, and may include e.g., single or multiple organ failure[13].
Acute pancreatitis (AP) is one common cause of SIRS and MODS. AP occurs with an incidence of about 300 or more patients per million annually[14,15], reported to have increased during the last decades[16,17]. Since the principal risk factors for AP are gallstones and excessive alcohol intake[18,19], plausible the augmented incidence reflecting the overweight epidemic observed in the Western World[20-22].
Most cases of AP are mild and self-limiting, with only brief need of clinical support[14,15]. However, about 10%-15% of patients will develop severe acute pancreatitis (SAP) and of those, 10%-30% may die due to SAP-associated complications (Table 3)[23]. Complications may be local or extra-pancreatic and in up to one third, the pancreatic injury leads to pancreatic necrosis, acute fluid collections and pseudocyst formation. SIRS may develop early during the course of SAP and cause e.g., adult respiratory dysfunction syndrome, acute renal and liver failure[24]. SIRS in association with SAP may also result in MODS, which carries a 40% mortality rate[15]. A model for the course of AP is shown (Figure 1).
Table 3.
Complications of severe acute pancreatitis
| Pancreatic | Systemic |
| Abscess | Acute kidney failure |
| Fat necrosis | Acute liver failure |
| Hemorrhages | Adult respiratory distress syndrome |
| Infected necrosis | Disseminated intravascular coagulation |
| Pseudocyst formation | Encephalopathy |
| Sterile necrosis | Gut ischemia |
| Hypocalcemia | |
| Paralytic ileus | |
| Shock |
Modified after Baddeley et al[24].
Figure 1.
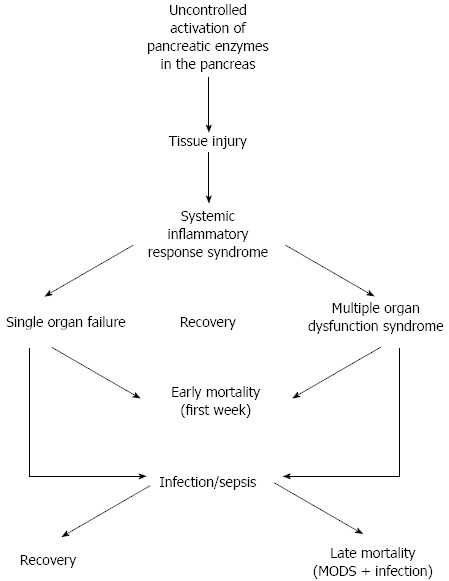
Systemic inflammatory response syndrome in acute pancreatitis.
Despite the improvements done in the diagnosis and management of SAP, the rate of mortality has only marginally declined during the last decades[24]. This is of particular concern, considering that AP patient numbers has been persistently increasing. In order to decrease the mortality rates, it is imperative to find new therapeutic strategies that target the underlying pathophysiological mechanism of SAP.
TLRs seem to play a major role in the development of numerous diseases[5,10], which make these molecules attractive as potential therapeutic targets. Since SIRS is a potentially lethal complication in AP, and TLRs are involved in the development of SIRS[25]; it is plausible that TLRs play a major role in severe acute pancreatitis.
In the present paper, we aim to explore the current knowledge about the role of different TLRs that have been reported associated with AP. In addition, future therapeutic strategies will be discussed.
TLR2-FAR AWAY FROM THE ANSWER
TLR2 is expressed on the plasma membrane of a large diversity of cells, including monocytes and macrophages, dendritic cells, polymorphonuclear leukocytes, B cells, T cells and microglia. This PRR recognises a wide range of DAMPs (Tables 1 and 2) and forms heterodimers with TLR1, TLR6 or TLR10[10]. Usually associated to the innate immune response against Gram-positive bacteria (several of its ligands origin in these microorganisms), TLR2 signals through a MyD88-dependent pathway (Figure 2A). CD14 is a protein found either in soluble form or anchored into the plasma membrane by a glycosylphosphatidylinositol tail. It has been reported that CD14 is required for the activation of TLR2-TLR6 dimers, since it facilitates the transport of specific ligands, such as peptidoglycan or lipoteichoic acid. Additionally, CD36, a membrane protein found in lipids rafts, seems to be important in the activation of TLR2-TLR6 pathways. However, it is unknown how CD36 enhances the formation of TL2-TLR6 dimers. CD44 is another membrane protein that has been associated to TLR2 and appears to enhance TLR2-mediated pro-inflammatory response[26]. However, the mechanisms are still unknown. There are also reports suggesting that CD44 may interact with TLR2, down-regulating TLR2-mediated inflammation[27,28].
Figure 2.
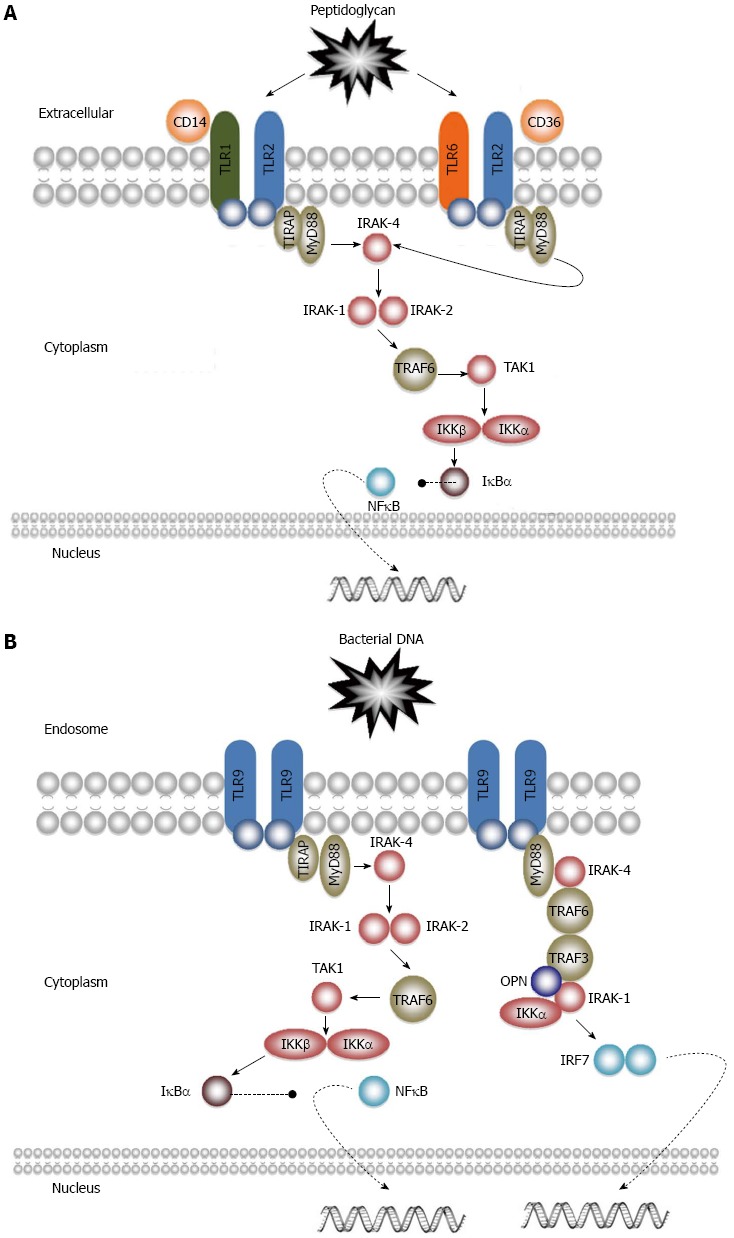
Toll-like receptor 2 and receptor 9 signalling pathways. A: Receptor 2; B: Receptor 9.
Upon dimerization with TLR1, TLR6 or TLR10, TIR-TIR dimers recruit TIRAP, which is needed for the further recruitment of MyD88. MyD88 interacts with IRAK-4, which phosphorylates IRAK-1 and IRAK-2. IRAK-1 and IRAK-2 then activate TRAF-6, which in turn activates TGF-β activated kinase-1 (TAK-1). TAK-1 phosphorylates I-κ-β kinase β (IKKβ), which together with IKKα phosphorylate I-κ-β kinase α (IKKα). Upon phosphorylation IκBα becomes inactive, allowing the nuclear translocation of NFκB, with subsequent production of several cytokines and molecules involved in immune responses (Table 4)[29].
Table 4.
A selection of genes regulated by nuclear factor κB
| Acute phase proteins | C-reactive protein |
| Complement factor Bf | |
| Complement factor C3 | |
| TNFβ | |
| Adhesion molecules | E-selectin |
| Intracellular cell adhesion molecule 1 | |
| Vascular cell adhesion molecule 1 | |
| Chemokines | IL8 |
| Monocyte chemoattractant protein-1 | |
| Chemokine C-C motif ligand 5 (also known as RANTES) | |
| Cytokines | IL2, 6, 12 |
| ILβ | |
| IL1β | |
| TNFβ | |
| TNFα | |
| Growth factors | Granulocyte colony stimulating factor |
| Granulocyte-macrophage colony stimulating factor | |
| Macrophage colony stimulating factor |
Modified after Christman et al[11]. TLR: Toll-like receptor; TNF: Tumour necrosis factor; IL: Interleukin.
Besides its role in infectious diseases, TLR2 has also been reported involved in several non-infectious disorders, including atherosclerosis, asthma, renal disease, systemic lupus erythematosus and even sporadic colorectal cancer[30-34]. Though, the role of TLR2 in AP has only been evaluated in a limited number of studies.
Pancreatic damage
The TLR2mRNA expression was found to be increased in the pancreas in cerulein-induced AP in rats[35]. Furthermore, TLR2 levels in the gland were also increased, as well as TNF-α, intracellular adhesion molecule 1 and IL-6. TLR2mRNA overexpression seemed to be caused by peroxisome proliferator-activated receptor-α (PPAR-α), since WY14643, a synthetic PPAR-α antagonist, decreased TLR2 levels in treated animals. Even if the observed TLR2mRNA overexpression was associated with PPAR-α, it is uncertain how important TLR2 was in the development of inflammation and tissue damage. Possibly, hyaluronic acid fragments found in the injured pancreas could be a major source of TLR2 upregulation[36]. Small molecular weight hyaluronic acid fragments seem to need TLR2 in order to stimulate the pro-inflammatory state in mouse macrophages[37]. Heat shock proteins may leak into the extracellular compartment after necrotic cell death in AP, interact with CD14/TLR2 and induce the production of inflammatory cytokines (especially TNF-α)[38].
Lung injury
Acute lung injury (ALI) is possibly the most serious complication associated to SAP, since it accounts for most deaths in untreated patients and in hospitalised patients that die during the first week after the onset of AP[39].
In 2005, Wu et al[40] described that TLR2mRNA overexpression in the lungs in a sodium taurocholate-based AP rat model was coupled to elevated TNF-α levels and lower nitric oxide (NO) levels, when compared to controls. Moreover, L-Arg administration decreased TLR2mRNA expression and pulmonary TNF-α levels. Since L-Arg stimulates the production of NO, it is difficult to evaluate if the anti-inflammatory response was caused by NO itself or by a L-Arg-mediated TLR2 down-regulation. Chloroquine showed similar effects (both TLR2mRNA and TNF-α were decreased) but the lung injury was not significantly ameliorated. Chloroquine is a well-known anti-malaria drug that prevents against endosomal acidification[41] and is used to study the role of intracellular TLRs[42].
The authors concluded that SAP-associated ALI was coupled to higher TLR2 levels in the lungs and that reduced NO levels were in part responsible for the grade of inflammation, since NO has known anti-inflammatory properties. However, how NO levels are associated with TLR2 and its potential therapeutic use in AP, is still unknown.
Matsumura et al[43] reported that the expression of TLR2mRNA decreased in pulmonary macrophages in deoxycholate-induced AP in rats. Macrophages were obtained through bronchoalveolar lavage fluid (6 h after induction) and the cells were exposed to lipoteichoic acid. No change in TLR2mRNA was initially observed, but after six hours TLR2mRNA expression significantly decreased in macrophages. Additionally, the production of TNF-α after lipoteichoic acid stimulation was reduced. An important observation made was the increment of bacterial translocation (cultured from mesenteric lymph nodes) 18 h after AP-induction, indicating that impaired TLR2mRNA expression in pulmonary macrophages could partially be responsible for the development of ALI in SAP complicated by sepsis. It is widely accepted that TLR2-/- mice are vulnerable to infections[44,45]. Pulmonary macrophages have been pointed out as major players in ALI in SAP, as monocyte chemoattractant protein-1 and TNF-α are released by these cells[46]. Hence, diminished TLR2mRNA expression in pulmonary macrophages under AP is doubtfully important for the development of ALI when TLR2-related PAMPs are absent.
Liver involvement
Liver failure is a major complication in AP. Besides, there is increasing data suggesting that activated Kupffer cells mediates the inflammatory response and pulmonary damage seen in SAP[47].
Xiong et al[48] showed that hepatic TLR2mRNA expression was increased in AP-induced mice. The incremented TL2mRNA expression was observed 3 h after the induction, peaking at 12 h. Likewise, severe damage in the liver and SIRS ensued in the animals. However, in order to strengthen the immunological reaction, LPS was injected as well, leading to a TLR4mRNA expression increment in the liver. Thus, it is difficult to establish if SAP-related SIRS is associated with an increase in TLR2 or TLR4. Possibly, a combined increment of both TLRs account for the observed severity. Zhang et al[49] obtained similar results in a taurocholate-based rat model. Besides, decreased hepatic TLR2mRNA expression, chloroquine or L-Arg administration attenuated the liver injury and decreased TNF-α levels. Increased pro-inflammatory cytokines and chemokines are released following overexpression of TLR2mRNA in the liver of untreated animals. As previously shown in the lungs[40], NO levels also decreased in the liver. This is of special interest since reduced NO levels correlated with diminished secretion of anti-inflammatory mediators.
Even if several studies associate TLR2 to AP, it is still debated if TLR2 plays a pathophysiological role. Awla et al[50] reported that the progression of SAP was not significantly different in TLR2-/- mice when compared with wild-type mice. No significant differences were observed concerning pancreatic tissue damage, chemokine formation and neutrophil recruitment in taurocholate-induced AP.
TLR2 and AP in humans
In humans, microsatellite polymorphism in intron 2 in the human TLR2 gene was associated with an increased risk for AP in Japan[51]. For this experiment, DNA was harvested from 202 patients of which 80 were diagnosed with SAP. When compared to healthy Japanese controls, AP patients showed significantly increased polymorphism rates in TLR2 genes. Since the same polymorphism has been associated with susceptibility to colorectal cancer, tuberculosis, rheumatoid arthritis and sarcoidosis[34,52-54]; it is conceivable that TLR2 signalling is altered if intron 2 is changed. However, it remains to be elucidated if the relationship between changed TLR2 genes and the risk for AP is just limited to Japanese subjects, or if the results can be extrapolated to other populations. Other studies have shown that gene-related vulnerability for AP in a country could not be demonstrated in other populations[55,56].
A very interesting observation was made by Szabo et al[57]. Monocytes were harvested from healthy volunteers before alcohol consumption and 24 h thereafter. The cells showed a decreased pro-inflammatory profile. Furthermore, in vitro acute alcohol stimulation of monocytes activated the pro-inflammatory state and decreased IL-production when TLR2 and TLR4 ligands were added. However, if only TLR2 ligands were added, no significant changes were observed. Therefore, acute alcohol stimulation appears to inhibit TLR2 expression in monocytes, worsening by this way the innate immune response against microorganisms that produce TLR2 specific ligands. Acute alcohol stimulation seems to be a double edged sword, since it could induce both anti and pro-inflammatory responses depending on which TLRs are involved.
In summary, TLR2 seems to be up regulated in the pancreas, lungs and liver in experimental AP animals. Moreover, TLR2-deficiency or inhibition (chloroquine or L-Arg) seems to ameliorate ALI in SAP.
How the increased TLR2 levels in vital organs are associated with SAP is still unknown. It is plausible that the overexpression of TLR2 in the pancreas, lungs and liver, amplifies the inflammatory response observed in SIRS when DAMPs released in SAP are recognised.
Despite an association found in Japan, the relationship between modified human TLR2 genes and AP has not been reported in other populations. Even if TLR2 seems to be relevant for the development of ALI in SAP, further research has to be done before this important PRR can be considered as a potential therapeutic target in AP.
TLR4-THE MAIN CANDIDATE?
TLR4 was the first TLR identified and is widely expressed on the plasma membrane of various immune cells, including macrophages and dendritic cells[58]. TLR4 recognises several DAMPs, including LPS, fibrinogen, and various heat shock proteins (Tables 1 and 2). Upon activation, TLR4 forms homodimers or heterodimers with TLR6. The TLR4 recognition of many DAMPs demands several accessory molecules. As for TLR2, CD14 is needed for binding LPS to TLR4 dimers. Another vital protein for LPS recognition is myeloid differentiation protein-2[3].
In the same matter as other TLRs, except TLR3, TLR4 signals through a MyD88-dependent pathway, leading to the activation of NFκB (see previous section). Besides, TLR4 can signal via a TRIF-dependent pathway. Once activated, TIR-TIR dimers recruit TRIF and TRAM. TRIF then activates I-κ-β kinase epsilon (IKK epsilon), which binds to TANK-binding kinase 1 (TBK1). Finally, IKK epsilon-TBK1 phosphorylates and activates IRF3, culminating in the transcription of IFN-α and IFN-β (Figure 3)[26].
Figure 3.
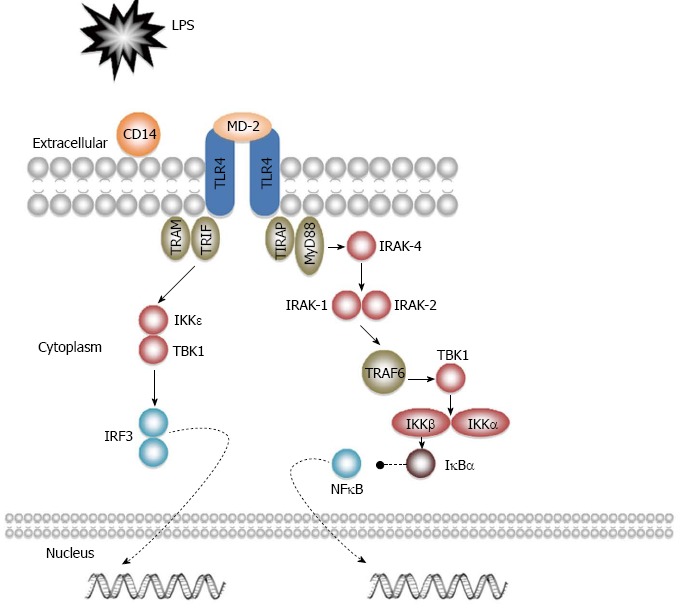
Toll-like receptor 4 signalling pathways.
TLR4 was initially mainly studied because of its involvement in Gram-negative bacterial infection, but has in recent years also been associated with an increasing number of diseases. Many reports suggest its involvement in atherosclerosis, liver disease, obesity, cardiac disease, and renal disease, among others[59-63].
Bacteria are important microorganisms in AP. In acute haemorrhagic-necrotizing pancreatitis, intestinal bacterial translocation into the injured/necrotic pancreas or the systemic circulation, may lead to SIRS, ALI and MODS[16-19]. As TLR4 is related to LPS and a wide range of alarmins released during inflammation and tissue damage, its role in AP appears to be highly possible.
Pancreatic damage
As for TLR2, overexpressed TLR4mRNA was also found in the pancreas in rats with cerulein-induced AP[35]. Li et al[64,65] reported (in the same animal model) that in both healthy and AP-induced animals, TLR4 was expressed mainly in the epithelium of the pancreatic duct and the pancreatic microcirculation. Even if some staining was observed in endocrine islets, no TLR4 protein was detected in the acinar cells. Likewise, TLR4 mRNA overexpression was observed, peaking after 1 h and returning to base levels after 4 h. In taurocholate-induced AP in TLR4-/- mice[50], serum amylase and myeloperoxidase levels in the pancreas decreased as compared to wild-type induced mice. Additionally, acinar cell necrosis, oedema, and haemorrhage significantly reduced. Interestingly, pancreas and serum levels of CXCL2, a chemokine, released by monocytes and macrophages, recruiting neutrophils, were strongly diminished.
Opposite results were presented by Ding et al[66] using the same AP model. No significant changes in serum amylase levels and pancreatic histological scores were observed between TLR4-/- and the controls. Curiously, TLR4-/- mice showed decreased levels of pro-apoptotic proteins in the pancreas 2 h after induction, but not after 4 h. TLR4 can increase the apoptotic rate in different cells[67]. As some studies suggest that apoptosis is favourable in AP[68,69], it is feasible that TLR4 is involved both in the beginning and in the resolution of AP.
Wang et al[70] showed that the administration of anti-CD14 antibody prior to cerulein-induction and LPS challenge in mice, reduced the severity of pancreatic injury, decreased pancreatic myeloperoxidase activity and down-regulated the secretion of pro-inflammatory cytokines.
TLR4 signalling pathways have been investigated in AP. Ding et al[71] showed in TLR4-/- mice that IRAK-4 could play a role in AP, independently of TLR4. In this experiment, lacking TLR4-mediated response did not result in increased IRAK-4 levels as was expected. This suggest that TLRs other than TLR4 may also play a role in AP, as the activation of TLR2 and TLR9 also results in decreased IRAK-4 in macrophages[72]. The hypothesis is supported by a report in which TRAF6 mediated inflammation in AP in TLR4-/- mice[73]. The author concluded that TLR4 may not be exclusively required for initiating AP, but its signal pathway may be of importance.
However, the association between alarmins released and TLR4-mediated inflammation in AP appears to be robust. In mononuclear inflammatory cells, the enzyme pancreatic elastase seems to activate NFκB, inducing TNF-α secretion[74,75]. Hietaranta et al[76] found that when human myeloid cells were exposed to porcine elastase, increased expression of NFκB and AP1 was achieved. The effects of elastase seem to be mediated by TLR4, since the blocking of TLR4 with a specific neutralizing antibody strongly prevented the expression of pro-inflammatory factors in elastase-exposed cells. Premature elastase activation has been reported in AP, implying its role in the disease[77]. Hence, TLR4-mediated inflammation may be important in AP. Blocking elastase might attenuate the severity of AP.
The degradation of matrix components appears to be connected with the pathophysiology of AP. Reports indicate that low molecular weight polysaccharides from degraded hyaluronan activates dendritic cells through TLR4[78]. Besides, dendritic cells might protect the pancreas against cell stress in AP[79]. Blycans and oligosaccharides of hyaluronan seem to signal via TLR4 and induce the production and secretion of great quantities of TNF-α and macrophage inflammatory protein-2 in macrophages[80]. Elastase and trypsin, both involved in the initiation of AP, cleaves heparin sulphate (HS) from cell surfaces and extra cellular matrix in vitro[81]. This, probably may in turn lead to free endogenous HS in damaged tissues in AP. Johnson et al[25] demonstrated that the rapid degradation of HS in mice causes a TLR4-mediated SIRS-like reaction in mice. When soluble HS was injected intraperitoneally, almost all wild type, but no TLR4-deficient mice, perish. This reaction was HS specific since molecules structurally similar to HS did not stimulate TLR4 and HS was not contaminated with LPS. Moreover, wild type, but not TLR4-deficient, mice had increased TNF-α serum levels 1 h after administration. Continuing in this line, Axelsson et al[82] confirmed that HS is involved in the initiation of AP in the rat.
Akbarshahi et al[83] later showed that the HS-induced TLR4-dependent immune response in the murine pancreas is IRF3-mediated. When TLR4-/- or MyD88-/- mice were challenged with HS or LPS, myeloperoxidase activity was annulled in the pancreas. The same pattern with HS was observed in IRF3-/- mice. However, LPS administration initiated a strong immune response in these animals. This outcome could explain preceding reports about discrepancies between TLR4-deficiency and decreased TRAF6 and IRAK-4 levels in AP[71,73]. Additionally, in this experiment, pre-treatment with the TLR4 antagonist eritoran inhibited the otherwise occurring increase in myeloperoxidase production in HS-treated wild type mouse pancreas. Eritoran is a synthetic Lipid A analogue that bind to TLR4/myeloid differentiation protein-2, thereby competing with Lipid A, resulting in inhibited LPS-mediated immune response[84].
Possibly, the inhibition of TLR4, elastase or HS, alone or in combination, could ameliorate pancreatic damage in AP, thus considerably reducing the risk for developing SAP.
Lung injury
As the role of TLR4 in AP has been investigated previously together with TLR2, similar results have been reported (see TLR2 section). Briefly, TLR4mRNA expression was increased in the lungs of AP-induced rats; leading to increased TNF-α levels combined with decreased NO levels. Chloroquine and L-Arg decreased TLR4mRNA expression and attenuated the lung injury[40]. Moreover, pulmonary macrophages decreased the TLR4mRNA expression, which caused decreased LPS-induced TNF-α levels and predisposition for intestinal bacterial translocation[41].
Pastor et al[85] concluded that TLR4 may not play a role in AP-associated ALI, but it may participate in pulmonary injury mediated by endotoxemia. In a cerulein-induced pancreatitis model in TLR4-/- mice there were no significant changes in serum amylase levels, pancreatic myeloperoxidase activity, and pancreatic oedema and acinar necrosis when compared to wild type mice. Additionally, cerulein-related pulmonary damage did not decrease in TLR4-/-. Although, when cerulein was combined with LPS, a significant decrease in pulmonary damage was reported. Thus, TLR4 would only be of importance in AP-related ALI when the disease is worsened by sepsis.
Sharif et al[86] obtained pole opposed results. TLR4-/- mice showed decreased serum amylase activity and pancreatic damage (oedema, myeloperoxidase activity, necrosis). Importantly, myeloperoxidase activity in the lungs also decreased, indicating reduced neutrophil sequestration. Furthermore, when AP was induced in CD14-/- mice, pancreatic and pulmonary damage were reduced as previously observed in TLR4-/- animals. Since TLR4/CD14 are very important for LPS-mediated TLR4 activation, TLR4 appears to be involved in the development of ALI in AP, even when endotoxemia is absent. Still, the subject is controversial, since ALI can ensure LPS-mediated TLR4/CD11b activation in CD14-/- mice[87].
Matsuda et al[88] proposed that the TLR4-associated inflammatory response in AP complicated by endotoxemia is mediated by macrophage migration inhibitory protein (MIF). Increased MIF expression in the lungs was observed after cerulein/LPS administration in mice. MIF levels were coupled to ALI and increased TLR4 expression in the lungs. Moreover, AP-induced MIF-/- mice showed lower TLR4 expression than in wild type mice; and anti-MIF antibody administration greatly suppressed the pulmonary expression of TLR4. It is important to stress that MIF can mediate inflammation independently of TLR4. For instance, the induction of cytosolic phospholipase A2, an enzyme that has been linked to ALI, may be MIF-mediated[89,90].
The role of TLR4 in AP-associated ALI is debated. While it is generally accepted that TLR4 plays a role in the course of ALI when endotoxemia ensues in AP; most reports are divided between those that suggests TLR4 as an important player (even in the absence of endotoxemia) and those that minimize its role. Fortunately, this discrepancy may lead to new experiments that focuses not only in LPS but also in the wide range of alarmins related to AP, ALI and TLR4. For instance, neuropeptide substance P, which has pro-inflammatory properties that increase vascular permeability and is correlated to AP and ALI[91]; has also been associated to the up regulation of TLR4 in AP[92]. Moreover, extracellular heat shock protein 70 can induce SIRS-like immune responses in cerulein-challenged mice[93]. The action of this protein appears to be mediated by TLR4, as TLR4-/- mice did not showed the same outcome.
Liver, kidneys and intestine
Similarly to TLR2, TLR4mRNA expression is increased in the liver during AP-associated SIRS[48,49]. Briefly, after AP-induction, higher levels of liver enzymes and TNF-α were observed. Hepatic NO levels decreased. All these changes appeared to be related to the TLR4mRNA overexpression in the liver. The administration of chloroquine and L-Arg decreased TLR4mRNA expression, thus reducing the liver damage.
Peng et al[94] showed that the deletion of TLR4 attenuated liver injury in AP. AP was induced in mice by choline-deficient ethionine diet. Besides the expected augmentation of TLR4 mRNA in mice liver, an increment in protein kinase C-zeta (PKCζ) was detected. However, induced TLR4-/- mice showed less apoptosis in hepatic cells and the hepatic PKCζmRNA expression was clearly reduced. PKCζ activates NFκB, which is essential for the production of cytokines in Kupffer cells[95]. Consequently, TLR4-deficiency protects the liver in AP through down-regulation of PKCζ, resulting in less inflammation and hepatic cell apoptosis.
Sawa et al[96] showed that in SAP, TLR4 expression is not only increased in the liver, but also in the kidneys and small intestine. Closed duodenal loop operation was performed in mice. The increased expression of TLR4 occurred 4 h after the ligation and returned to baseline after 12 h. Curiously, TLR4-deficient mice showed the same tendency and histological analyses of the liver and kidney did not show any differences between the different groups. However, apoptosis was seen in the liver and kidney of ligated TLR4-deficient, but not in wild type, mice. Additionally, Gram-negative bacterial translocation into the pancreas occurred in higher rates in TLR4-deficient animals. The translocation ensued 12 h after induction, but not before. Even Gram-positive bacterial translocation was registered, but the difference between the groups was not significant. Bacterial translocation highly correlated to fluctuations in TLR4 expression. According to the authors, as MODS develops in the early or the late phase of SAP, the early inhibition and late stimulation of TLR4 could result in a milder clinical course, since it could prevent MODS and bacterial translocation. Yet, results from another experiment in mice challenges this hypothesis[97]. van Westerloo et al showed that the immune response against Escherichia coli in AP-induced TLR4-/- mice was not different from that observed in controls. Nevertheless, there is a chance that mice developed the compensatory anti-inflammatory response syndrome (CARS), which is generally (but not exclusively) related to SIRS and sepsis[98]. CARS is a systemic deactivation of the immune system, that can ensue after SIRS and sepsis in order to restore homeostasis[99]. Thus, CARS could explain why LPS-mediated TLR4-activation was not observed in TLR4+/+ mice after bacterial challenge in AP.
TLR4 and AP in humans
Despite controversy, there is an increasing amount of data suggesting that TLR4 plays a role in experimental AP in rodents. However, in humans, the role of TLR4 in AP patients has been investigated mainly indirectly via DNA analysis or immune cells taken from blood.
In a Chinese study 310 patients were diagnosed with AP according to the Atlanta severity classification[100,101]. Pancreatic necrosis was recognized in 115 patients and in 37 of those, TLR4 mutation (896G allele) was identified. When compared to AP patients without TLR4 mutation (896A allele), patients with mutated TLR4 had an increased morbidity following Gram-negative infection. When compared to healthy volunteers (n = 80), TLR4 mutation frequency was significantly higher in patients with pancreatic necrotic infection. Thus, according to this study, TLR4 Asp299Gly polymorphism appears to be associated with an increased risk for pancreatic necrotic infection in AP. However, this study may have limited value as all patients were recruited from one hospital located in southwestern China, thus questioning its representative value for the whole Chinese population. In a later study, Zhang et al[102] could not find any significant difference in the occurrence of Asp299Gly polymorphism between 238 Chinese AP patients and 121 healthy volunteers.
DNA samples were harvested from 92 AP patients in a Hungarian medical centre[103]. Not only TLR4 Asp299Gly polymorphism was considered, but also as Thr399Ile polymorphism was taken into account. No significant differences were detected between AP (n = 42) and SAP patients (n = 50) in any polymorphism analysed. Likewise, no differences were found between the 92 patients and healthy controls (n = 200). Perhaps, the strongest data against the involvement of TLR4 mutations in AP comes from an intercontinental study reported by Guenther et al[104]. No significant differences could be found between 521 AP patients (343 from Germany and 178 from United States) and healthy controls (128 Germans and. 265 Americans, respectively) in the incidence of Asp299Gly and/or Thr399Ile polymorphism. Thus, current knowledge indicates that the investigated TLR4 polymorphisms are not important for the development and clinical course of AP, even if these mutation have been linked to impairment of LPS-induced TLR4-mediated immune response in humans[105].
In vitro acute alcohol stimulation seems to inhibit TLR4 expression in human monocytes[57]. When LPS was added, a decreased secretion of pro-inflammatory cytokines was observed. Intriguingly, the effect was abolished if LPS and TLR2 ligands were added at the same time to the cells. Li et al[106] showed that the expression of TLR4 in human peripheral blood mononuclear cells in mild AP, raises in the beginning of the disease to return to baseline levels after a week, when the patients recovered. As TNF-α and IL-6 showed the same pattern, the authors concluded that TLR4 might play an important role in the pathogenesis of AP.
In summary, there is strong data indicating that TLR4 plays an important role in experimental AP in rodents. Nevertheless, contrary results have been published. While most studies emphasize the role of TLR4 in the pancreas in AP, its role in AP-associated ALI is controversial. There is some evidence linking TLR4 levels and tissue damage in the liver in AP. Likewise, the general opinion is that TLR4 is very important in the development of endotoxemia in AP.
In humans, DNA mutations in TLR4 genes do not seem to be of importance for the development or the clinical course of AP, but TLR4 expression changes in monocytes and macrophages may be of significance. Unfortunately, the latter is only based on a very limited number of studies. There are some promising reports about the use of herbs from traditional Chinese medicine in AP that may act through TLR4 inhibition[107-109]. Salvia miltiorrhizae (also known as danshen) appears to inhibit the binding of LPS to TLR4 in the rat liver in SAP, which may reduce bacterial translocation and liver injury. Moreover, treatment with emodin and/or baicalin reduced serum levels of amylase, IL-6 and TNF-α in AP-induced rats. Pancreatic damage and ALI were also ameliorated. The effect of emodin and baicalin appears to be mediated by TLR4, since decreased TLR4mRNA expression and protein levels were found in the pancreas and lungs in treated animals. Its implication in experimental AP in rodents makes TLR4 a potentially very promising therapeutic target in AP, its utility though to be demonstrated in humans in future studies.
TLR9-UNEXPLORED IMPLICATIONS
As for TLR2 and TLR4, TLR9 is expressed intracellularly (endosomes) in several immune cells, including B cells and dendritic cells[110]. TLR9 recognises unmethylated CpG dinucleotides found in bacteria and virus[111,112]. CpG motifs are rare in vertebrate DNA (less 1% in human genome), but they are common in bacterial DNA. Besides, if present in vertebrate DNA, CpG motifs use to be methylated. CpG DNA directly stimulates B cells, macrophages and dendritic cells to secrete cytokines[113]. TLR9 can also recognize haemozoin, which is a disposal product formed from the digestion of erythrocytes by some parasites (e.g., Plasmodium spp.)[114].
Upon activation, TLR9 signals through a MyD88-dependent pathway, leading to the production of pro-inflammatory cytokines (see previous sections). This pathway is mainly observed in macrophages, B cells, conventional dendritic cells and plasmacytoid dendritic cells. However, in plasmacytoid dendritic cells, TLR9 can induce the production of type I IFN through a different pathway. After the recruitment of Myd88 and IRAK-4, these interact with TRAF6, TRAF3, IRAK-1, IKKα and osteopontin. IRAK-1 and IKKα phosphorylate and activate IRF7, culminating in the production of type I IFN (Figure 2B)[26]. Apart from its role in bacterial, viral or malaria infection; TLR9 has been associated with SLE and cancer[115,116].
TLR9 and AP
To date, there are only two publications in which the role of TLR9 in AP has been investigated. Zeng et al[117] reported that TLR9 was expressed in rat pancreas in cerulein-induced AP. TLR9 staining was detected both in the pancreas from AP-induced rats, as in controls. The epithelium of the pancreatic duct and pancreatic microcirculation were the main sites for TLR9 staining in AP-induced animals. Controls showed staining in the vasculature, but not in the pancreatic duct. Moreover, no staining was detected in pancreatic acinar cells in either group. Interestingly, a similar pattern of staining has been reported for TLR4 in AP[64]. TLR9mRNA expression was increased after 30 min, peaked at 1 h, and remained high for the first eight hours after cerulein challenge.
In a more extensive study, Hoque et al[118] found that TLR9 is involved in pancreatic acinar cell death in AP-induced mice. After cerulein-administration, TLR9-/- mice had less oedema, leukocyte infiltration and IL-1βmRNA expression in the pancreas. Even if several cell types express TLR9 in the pancreas, bone marrow-derived CD45+ cells (mainly macrophages) had the highest expression level. Furthermore, the action of a TLR9 antagonist (IRS954) was evaluated. TLR9+/+ animals had IRS954 administered 1 h before inducing AP by cerulein. Pancreatic oedema, leukocyte infiltration and pancreatic cell apoptosis were ameliorated. Moreover, pancreatic pro-IL-1β elevation was also reduced, as serum amylase levels. These results were observed at 1 h after AP-induction.
The authors speculated if IRS954 might have a therapeutic value. Interestingly, the administration of IRS954 after cerulein challenge reduced pancreatic oedema, leukocyte infiltration and pancreatic cell apoptosis. Similar results were observed when another model for AP was used. Pre-treatment with IRS954 at 1 h after AP induction through taurolithocholic acid 3-sulphate challenge in TLR9+/+, reduced serum amylase elevation, pancreatic necrosis and inflammatory cell infiltration in mice lungs. Additionally, in vitro experiments were performed. When isolated peritoneal macrophages were exposed to pancreatic homogenate and DNA, significant NFκB activation was observed. Interestingly, pre-treatment with IRS954 reduced NFκB activation to baseline levels.
IRS954 appears to be a good candidate for further investigation. Like TLR2 and TLR4, TLR9 may be important when AP is aggravated by sepsis. Reports indicate that during sepsis, TLR9 is expressed both in murine and human adrenal glands and its stimulation leads to corticosterone release and inflammatory response[119]. Macrophages and NK-cells in the liver mediates liver toxicity and express high levels of TLR9 in murine peritonitis[120]. Chloroquine inhibits TLR9 and may prevent sepsis-induced acute kidney injury in mice[121]. Additionally, TLR9-/- mice have reduced mortality in polymicrobial sepsis[122].
Still, the number of studies is very limited. Hence, it is impossible to conclude if TLR9 plays an important role in AP with a clinical course complicated with our without sepsis.
Closing remarks
An increasing number of publications suggest that TLRs are involved in the pathophysiology of several important medical conditions. In acute pancreatitis, TLRs appear to play a role, but a general consensus has not been achieved, since contradictory results have been presented.
The main candidate for targeting seems to be TLR4, which recognizes numerous DAMPs associated to AP[123,124]. TLR2 has also been linked to AP, but there are few studies that exclusively has studied its role in AP. There is data suggesting that TLR9 also can play a role in AP. The associations found between TLRs and AP, as possible novel types of therapy are presented (Tables 5 and 6). There is also some evidence indicating that TLR4 and TLR3 may be involved in chronic pancreatitis[125,126]. This is of particular importance since chronic pancreatitis may develop following repeated AP episodes, as chronic pancreatitis is to some extent a risk factor for pancreatic cancer[127].
Table 5.
Toll-like receptors and acute pancreatitis
| Observed change | Tissue/cell | Ref. | |
| TLR2 | Increased mRNA expression | Pancreas | [35] |
| Lungs | [40] | ||
| Liver | [48,49] | ||
| Increased protein levels | Pancreas | [35] | |
| Liver | [48] | ||
| Decreased mRNA expression | Pulmonary macrophages | [43] | |
| TLR4 | Increased mRNA expression | Pancreas | [35,64,73,92] |
| Lungs | [40,88] | ||
| Liver | [48,94,96] | ||
| Kidney | [96] | ||
| Increased protein levels | Small intestine | [96] | |
| Pancreas | [35,92] | ||
| Lungs | [88] | ||
| Liver | [48,49] | ||
| Intestine | [65] | ||
| Blood monocytes | [106] | ||
| Decreased mRNA expression | Pulmonary macrophages | [43] | |
| TLR9 | Increased mRNA expression | Pancreas | [117,118] |
| Increased protein levels | Pancreas | [118] |
TLR: Toll-like receptor.
Table 6.
Toll-like receptor intervention in acute pancreatitis
| Substance | Ref. | |
| TLR2 | Chloroquine | [40,49] |
| L-Arg | [40,49] | |
| WY14643 | [35] | |
| TLR4 | Anti-MIF antibody | [88] |
| Baicalin | [107] | |
| Chloroquine | [40,49] | |
| Emodin | [107] | |
| Eritoran | [83] | |
| L-Arg | [40,49] | |
| Salvia miltiorrhizae | [108,109] | |
| TLR9 | IRS954 | [118] |
TLR: Toll-like receptor; MIF: Migration inhibitory protein.
CONCLUSION
To our knowledge, this is the first time the current understanding of the role of TLRs in acute pancreatitis is summarized. Of course, further research and elucidation of involved mechanisms is warranted, hopefully stimulated by the present review giving an update and state-of-the-art concerning the role of TLRs in acute pancreatitis and its potential future clinical implications.
Footnotes
P- Reviewers Coelho AMM, Hac SAY, Maria D, Blanco-Guillermo I S- Editor Huang XZ L- Editor A E- Editor Zhang DN
References
- 1.Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 1:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 3.Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol. 2012;12:168–179. doi: 10.1038/nri3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010:672395. doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hedayat M, Netea MG, Rezaei N. Targeting of Toll-like receptors: a decade of progress in combating infectious diseases. Lancet Infect Dis. 2011;11:702–712. doi: 10.1016/S1473-3099(11)70099-8. [DOI] [PubMed] [Google Scholar]
- 7.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 8.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 9.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 10.O'Neill LA, Bryant CE, Doyle SL. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev. 2009;61:177–197. doi: 10.1124/pr.109.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christman JW, Lancaster LH, Blackwell TS. Nuclear factor kappa B: a pivotal role in the systemic inflammatory response syndrome and new target for therapy. Intensive Care Med. 1998;24:1131–1138. doi: 10.1007/s001340050735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abraham E. Nuclear factor-kappaB and its role in sepsis-associated organ failure. J Infect Dis. 2003;187 Suppl 2:S364–S369. doi: 10.1086/374750. [DOI] [PubMed] [Google Scholar]
- 13.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 14.Appelros S, Borgström A. Incidence, aetiology and mortality rate of acute pancreatitis over 10 years in a defined urban population in Sweden. Br J Surg. 1999;86:465–470. doi: 10.1046/j.1365-2168.1999.01049.x. [DOI] [PubMed] [Google Scholar]
- 15.Andersson R, Andersson B, Haraldsen P, Drewsen G, Eckerwall G. Incidence, management and recurrence rate of acute pancreatitis. Scand J Gastroenterol. 2004;39:891–894. doi: 10.1080/00365520410007061. [DOI] [PubMed] [Google Scholar]
- 16.Kingsnorth A, O'Reilly D. Acute pancreatitis. BMJ. 2006;332:1072–1076. doi: 10.1136/bmj.332.7549.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldacre MJ, Roberts SE. Hospital admission for acute pancreatitis in an English population, 1963-98: database study of incidence and mortality. BMJ. 2004;328:1466–1469. doi: 10.1136/bmj.328.7454.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med. 2006;354:2142–2150. doi: 10.1056/NEJMcp054958. [DOI] [PubMed] [Google Scholar]
- 19.Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet. 2008;371:143–152. doi: 10.1016/S0140-6736(08)60107-5. [DOI] [PubMed] [Google Scholar]
- 20.Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet. 2011;377:557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 22.Margetts B. WHO global strategy on diet, physical activity and health.Editorial. Public Health Nutr. 2004;7:361–363. doi: 10.1079/PHN2004622. [DOI] [PubMed] [Google Scholar]
- 23.McKay CJ, Imrie CW. The continuing challenge of early mortality in acute pancreatitis. Br J Surg. 2004;91:1243–1244. doi: 10.1002/bjs.4750. [DOI] [PubMed] [Google Scholar]
- 24.Baddeley R, Skipworth J, Pereira S. Acute Pancreatitis. Med. 2011;39(2):108–115. [Google Scholar]
- 25.Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor 4. J Immunol. 2004;172:20–24. doi: 10.4049/jimmunol.172.1.20. [DOI] [PubMed] [Google Scholar]
- 26.Abe T, Fukuhara T, Wen X, Ninomiya A, Moriishi K, Maehara Y, Takeuchi O, Kawai T, Akira S, Matsuura Y. CD44 participates in IP-10 induction in cells in which hepatitis C virus RNA is replicating, through an interaction with Toll-like receptor 2 and hyaluronan. J Virol. 2012;86:6159–6170. doi: 10.1128/JVI.06872-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawana H, Karaki H, Higashi M, Miyazaki M, Hilberg F, Kitagawa M, Harigaya K. CD44 suppresses TLR-mediated inflammation. J Immunol. 2008;180:4235–4245. doi: 10.4049/jimmunol.180.6.4235. [DOI] [PubMed] [Google Scholar]
- 28.van der Windt GJ, van 't Veer C, Florquin S, van der Poll T. CD44 deficiency is associated with enhanced Escherichia coli-induced proinflammatory cytokine and chemokine release by peritoneal macrophages. Infect Immun. 2010;78:115–124. doi: 10.1128/IAI.00949-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Satoh M, Ishikawa Y, Minami Y, Takahashi Y, Nakamura M. Role of Toll like receptor signaling pathway in ischemic coronary artery disease. Front Biosci. 2008;13:6708–6715. doi: 10.2741/3183. [DOI] [PubMed] [Google Scholar]
- 31.Eder W, Klimecki W, Yu L, von Mutius E, Riedler J, Braun-Fahrländer C, Nowak D, Martinez FD. Toll-like receptor 2 as a major gene for asthma in children of European farmers. J Allergy Clin Immunol. 2004;113:482–488. doi: 10.1016/j.jaci.2003.12.374. [DOI] [PubMed] [Google Scholar]
- 32.Anders HJ, Banas B, Schlöndorff D. Signaling danger: toll-like receptors and their potential roles in kidney disease. J Am Soc Nephrol. 2004;15:854–867. doi: 10.1097/01.asn.0000121781.89599.16. [DOI] [PubMed] [Google Scholar]
- 33.Urbonaviciute V, Fürnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boraska Jelavić T, Barisić M, Drmic Hofman I, Boraska V, Vrdoljak E, Peruzović M, Hozo I, Puljiz Z, Terzić J. Microsatelite GT polymorphism in the toll-like receptor 2 is associated with colorectal cancer. Clin Genet. 2006;70:156–160. doi: 10.1111/j.1399-0004.2006.00651.x. [DOI] [PubMed] [Google Scholar]
- 35.Ding JL, Zhou ZG, Zhou XY, Zhou B, Wang L, Wang R, Zhan L, Sun XF, Li Y. Attenuation of Acute Pancreatitis by Peroxisome Proliferator-Activated Receptor-α in Rats: The Effect on Toll-Like Receptor Signaling Pathways. Pancreas. 2013;42:114–122. doi: 10.1097/MPA.0b013e3182550cc4. [DOI] [PubMed] [Google Scholar]
- 36.Lorne E, Dupont H, Abraham E. Toll-like receptors 2 and 4: initiators of non-septic inflammation in critical care medicine? Intensive Care Med. 2010;36:1826–1835. doi: 10.1007/s00134-010-1983-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 38.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 39.Zhou MT, Chen CS, Chen BC, Zhang QY, Andersson R. Acute lung injury and ARDS in acute pancreatitis: mechanisms and potential intervention. World J Gastroenterol. 2010;16:2094–2099. doi: 10.3748/wjg.v16.i17.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu HS, Zhang L, Chen Y, Guo XJ, Wang L, Xu JB, Wang CY, Zhang JH. Effect of nitric oxide on toll-like receptor 2 and 4 gene expression in rats with acute lung injury complicated by acute hemorrhage necrotizing pancreatitis. Hepatobiliary Pancreat Dis Int. 2005;4:609–613. [PubMed] [Google Scholar]
- 41.Steinman RM, Mellman IS, Muller WA, Cohn ZA. Endocytosis and the recycling of plasma membrane. J Cell Biol. 1983;96:1–27. doi: 10.1083/jcb.96.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hart OM, Athie-Morales V, O'Connor GM, Gardiner CM. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol. 2005;175:1636–1642. doi: 10.4049/jimmunol.175.3.1636. [DOI] [PubMed] [Google Scholar]
- 43.Matsumura N, Takeyama Y, Ueda T, Yasuda T, Shinzeki M, Sawa H, Nakajima T, Kuroda Y. Decreased expression of Toll-like receptor 2 and 4 on macrophages in experimental severe acute pancreatitis. Kobe J Med Sci. 2007;53:219–227. [PubMed] [Google Scholar]
- 44.Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- 45.Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, Kirschning CJ. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. 2003;170:438–444. doi: 10.4049/jimmunol.170.1.438. [DOI] [PubMed] [Google Scholar]
- 46.Gea-Sorlí S, Guillamat R, Serrano-Mollar A, Closa D. Activation of lung macrophage subpopulations in experimental acute pancreatitis. J Pathol. 2011;223:417–424. doi: 10.1002/path.2814. [DOI] [PubMed] [Google Scholar]
- 47.Folch-Puy E. Importance of the liver in systemic complications associated with acute pancreatitis: the role of Kupffer cells. J Pathol. 2007;211:383–388. doi: 10.1002/path.2123. [DOI] [PubMed] [Google Scholar]
- 48.Xiong J, Zhu ZH, Liu JS, Wang Y, Wu HS. The expression of toll-like receptor 2, 4 of livers in mice with systemic inflammatory response syndrome. Hepatobiliary Pancreat Dis Int. 2006;5:143–146. [PubMed] [Google Scholar]
- 49.Zhang L, Wu HS, Chen Y, Guo XJ, Wang L, Wang CY, Zhang JH, Tian Y. Role of nitric oxide in Toll-like receptor 2 and 4 mRNA expression in liver of acute hemorrhagic necrotizing pancreatitis rats. World J Gastroenterol. 2006;12:485–488. doi: 10.3748/wjg.v12.i3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Awla D, Abdulla A, Regnér S, Thorlacius H. TLR4 but not TLR2 regulates inflammation and tissue damage in acute pancreatitis induced by retrograde infusion of taurocholate. Inflamm Res. 2011;60:1093–1098. doi: 10.1007/s00011-011-0370-1. [DOI] [PubMed] [Google Scholar]
- 51.Takagi Y, Masamune A, Kume K, Satoh A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Microsatellite polymorphism in intron 2 of human Toll-like receptor 2 gene is associated with susceptibility to acute pancreatitis in Japan. Hum Immunol. 2009;70:200–204. doi: 10.1016/j.humimm.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Yim JJ, Lee HW, Lee HS, Kim YW, Han SK, Shim YS, Holland SM. The association between microsatellite polymorphisms in intron II of the human Toll-like receptor 2 gene and tuberculosis among Koreans. Genes Immun. 2006;7:150–155. doi: 10.1038/sj.gene.6364274. [DOI] [PubMed] [Google Scholar]
- 53.Lee EY, Yim JJ, Lee HS, Lee YJ, Lee EB, Song YW. Dinucleotide repeat polymorphism in intron II of human Toll-like receptor 2 gene and susceptibility to rheumatoid arthritis. Int J Immunogenet. 2006;33:211–215. doi: 10.1111/j.1744-313X.2006.00599.x. [DOI] [PubMed] [Google Scholar]
- 54.Veltkamp M, Wijnen PA, van Moorsel CH, Rijkers GT, Ruven HJ, Heron M, Bekers O, Claessen AM, Drent M, van den Bosch JM, et al. Linkage between Toll-like receptor (TLR) 2 promotor and intron polymorphisms: functional effects and relevance to sarcoidosis. Clin Exp Immunol. 2007;149:453–462. doi: 10.1111/j.1365-2249.2007.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahman SH, Ibrahim K, Larvin M, Kingsnorth A, McMahon MJ. Association of antioxidant enzyme gene polymorphisms and glutathione status with severe acute pancreatitis. Gastroenterology. 2004;126:1312–1322. doi: 10.1053/j.gastro.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Bhat YM, Papachristou GI, Park JS, Lamb J, Slivka A, Whitcomb DC. Functional polymorphisms of the GSTT-1 gene do not predict the severity of acute pancreatitis in the United States. Pancreatology. 2007;7:180–186. doi: 10.1159/000104243. [DOI] [PubMed] [Google Scholar]
- 57.Szabo G, Mandrekar P, Oak S, Mayerle J. Effect of ethanol on inflammatory responses. Implications for pancreatitis. Pancreatology. 2007;7:115–123. doi: 10.1159/000104236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 59.Ding Y, Subramanian S, Montes VN, Goodspeed L, Wang S, Han C, Teresa AS, Kim J, O'Brien KD, Chait A. Toll-like receptor 4 deficiency decreases atherosclerosis but does not protect against inflammation in obese low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:1596–1604. doi: 10.1161/ATVBAHA.112.249847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sahin H, Trautwein C, Wasmuth HE. TLR4 stresses the liver. Gut. 2012;61:1241–1242. doi: 10.1136/gutjnl-2012-302188. [DOI] [PubMed] [Google Scholar]
- 61.Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Brüning JC. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10:249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kashiwagi M, Imanishi T, Ozaki Y, Satogami K, Masuno T, Wada T, Nakatani Y, Ishibashi K, Komukai K, Tanimoto T, et al. Differential expression of Toll-like receptor 4 and human monocyte subsets in acute myocardial infarction. Atherosclerosis. 2012;221:249–253. doi: 10.1016/j.atherosclerosis.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 63.Lepenies J, Eardley KS, Kienitz T, Hewison M, Ihl T, Stewart PM, Cockwell P, Quinkler M. Renal TLR4 mRNA expression correlates with inflammatory marker MCP-1 and profibrotic molecule TGF-β₁ in patients with chronic kidney disease. Nephron Clin Pract. 2011;119:c97–c104. doi: 10.1159/000324765. [DOI] [PubMed] [Google Scholar]
- 64.Li Y, Zhou ZG, Xia QJ, Zhang J, Li HG, Cao GQ, Wang R, Lu YL, Hu TZ. Toll-like receptor 4 detected in exocrine pancreas and the change of expression in cerulein-induced pancreatitis. Pancreas. 2005;30:375–381. doi: 10.1097/01.mpa.0000160959.21580.41. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Zhou ZG, Zhang J, Chen YD, Li HG, Gao HK, Wang R, Hu TZ. Microcirculatory detection of Toll-like receptor 4 in rat pancreas and intestine. Clin Hemorheol Microcirc. 2006;34:213–219. [PubMed] [Google Scholar]
- 66.Ding SQ, Li Y, Zhou ZG, Wang C, Zhan L, Zhou B. Toll-like receptor 4-mediated apoptosis of pancreatic cells in cerulein-induced acute pancreatitis in mice. Hepatobiliary Pancreat Dis Int. 2010;9:645–650. [PubMed] [Google Scholar]
- 67.Salaun B, Romero P, Lebecque S. Toll-like receptors' two-edged sword: when immunity meets apoptosis. Eur J Immunol. 2007;37:3311–3318. doi: 10.1002/eji.200737744. [DOI] [PubMed] [Google Scholar]
- 68.Bhatia M. Apoptosis of pancreatic acinar cells in acute pancreatitis: is it good or bad? J Cell Mol Med. 2004;8:402–409. doi: 10.1111/j.1582-4934.2004.tb00330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao Y, Adhikari S, Clément MV, Wallig M, Bhatia M. Induction of apoptosis by crambene protects mice against acute pancreatitis via anti-inflammatory pathways. Am J Pathol. 2007;170:1521–1534. doi: 10.2353/ajpath.2007.061149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, Wu L, Wu K, Zhang R, Dong Y. Roles of endotoxin-related signaling molecules in the progression of acute necrotizing pancreatitis in mice. Pancreas. 2005;31:251–257. doi: 10.1097/01.mpa.0000175179.62916.17. [DOI] [PubMed] [Google Scholar]
- 71.Ding JL, Li Y, Zhou XY, Wang L, Zhou B, Wang R, Liu HX, Zhou ZG. Potential role of the TLR4/IRAK-4 signaling pathway in the pathophysiology of acute pancreatitis in mice. Inflamm Res. 2009;58:783–790. doi: 10.1007/s00011-009-0048-0. [DOI] [PubMed] [Google Scholar]
- 72.Hatao F, Muroi M, Hiki N, Ogawa T, Mimura Y, Kaminishi M, Tanamoto K. Prolonged Toll-like receptor stimulation leads to down-regulation of IRAK-4 protein. J Leukoc Biol. 2004;76:904–908. doi: 10.1189/jlb.0504277. [DOI] [PubMed] [Google Scholar]
- 73.Zhou XY, Zhou ZG, Ding JL, Wang L, Wang R, Zhou B, Gu J, Sun XF, Li Y. TRAF6 as the key adaptor of TLR4 signaling pathway is involved in acute pancreatitis. Pancreas. 2010;39:359–366. doi: 10.1097/MPA.0b013e3181bb9073. [DOI] [PubMed] [Google Scholar]
- 74.Jaffray C, Yang J, Carter G, Mendez C, Norman J. Pancreatic elastase activates pulmonary nuclear factor kappa B and inhibitory kappa B, mimicking pancreatitis-associated adult respiratory distress syndrome. Surgery. 2000;128:225–231. doi: 10.1067/msy.2000.107419. [DOI] [PubMed] [Google Scholar]
- 75.Jaffray C, Mendez C, Denham W, Carter G, Norman J. Specific pancreatic enzymes activate macrophages to produce tumor necrosis factor-alpha: role of nuclear factor kappa B and inhibitory kappa B proteins. J Gastrointest Surg. 2000;4:370–377. doi: 10.1016/s1091-255x(00)80015-3. [DOI] [PubMed] [Google Scholar]
- 76.Hietaranta A, Mustonen H, Puolakkainen P, Haapiainen R, Kemppainen E. Proinflammatory effects of pancreatic elastase are mediated through TLR4 and NF-kappaB. Biochem Biophys Res Commun. 2004;323:192–196. doi: 10.1016/j.bbrc.2004.08.077. [DOI] [PubMed] [Google Scholar]
- 77.Lüthen R, Niederau C, Grendell JH. Intrapancreatic zymogen activation and levels of ATP and glutathione during caerulein pancreatitis in rats. Am J Physiol. 1995;268:G592–G604. doi: 10.1152/ajpgi.1995.268.4.G592. [DOI] [PubMed] [Google Scholar]
- 78.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bedrosian AS, Nguyen AH, Hackman M, Connolly MK, Malhotra A, Ibrahim J, Cieza-Rubio NE, Henning JR, Barilla R, Rehman A, et al. Dendritic cells promote pancreatic viability in mice with acute pancreatitis. Gastroenterology. 2011;141:1915–26.e1-14. doi: 10.1053/j.gastro.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Götte M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–2233. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klebanoff SJ, Kinsella MG, Wight TN. Degradation of endothelial cell matrix heparan sulfate proteoglycan by elastase and the myeloperoxidase-H2O2-chloride system. Am J Pathol. 1993;143:907–917. [PMC free article] [PubMed] [Google Scholar]
- 82.Axelsson J, Norrman G, Malmström A, Weström B, Andersson R. Initiation of acute pancreatitis by heparan sulphate in the rat. Scand J Gastroenterol. 2008;43:480–489. doi: 10.1080/00365520701733814. [DOI] [PubMed] [Google Scholar]
- 83.Akbarshahi H, Axelsson JB, Said K, Malmström A, Fischer H, Andersson R. TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J Transl Med. 2011;9:219. doi: 10.1186/1479-5876-9-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yeh YC, Ko WJ, Chan KC, Fan SZ, Tsai JC, Cheng YJ, Sun WZ. Effects of eritoran tetrasodium, a toll-like receptor 4 antagonist, on intestinal microcirculation in endotoxemic rats. Shock. 2012;37:556–561. doi: 10.1097/SHK.0b013e31824e20ef. [DOI] [PubMed] [Google Scholar]
- 85.Pastor CM, Pugin J, Kwak B, Chanson M, Mach F, Hadengue A, Frossard JL. Role of Toll-like receptor 4 on pancreatic and pulmonary injury in a mice model of acute pancreatitis associated with endotoxemia. Crit Care Med. 2004;32:1759–1763. doi: 10.1097/01.ccm.0000133020.47243.8e. [DOI] [PubMed] [Google Scholar]
- 86.Sharif R, Dawra R, Wasiluk K, Phillips P, Dudeja V, Kurt-Jones E, Finberg R, Saluja A. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut. 2009;58:813–819. doi: 10.1136/gut.2008.170423. [DOI] [PubMed] [Google Scholar]
- 87.Jeyaseelan S, Chu HW, Young SK, Freeman MW, Worthen GS. Distinct roles of pattern recognition receptors CD14 and Toll-like receptor 4 in acute lung injury. Infect Immun. 2005;73:1754–1763. doi: 10.1128/IAI.73.3.1754-1763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matsuda N, Nishihira J, Takahashi Y, Kemmotsu O, Hattori Y. Role of macrophage migration inhibitory factor in acute lung injury in mice with acute pancreatitis complicated by endotoxemia. Am J Respir Cell Mol Biol. 2006;35:198–205. doi: 10.1165/rcmb.2005-0272OC. [DOI] [PubMed] [Google Scholar]
- 89.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 90.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1:42–46. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 91.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 92.Tamizhselvi R, Shrivastava P, Koh YH, Zhang H, Bhatia M. Preprotachykinin-A gene deletion regulates hydrogen sulfide-induced toll-like receptor 4 signaling pathway in cerulein-treated pancreatic acinar cells. Pancreas. 2011;40:444–452. doi: 10.1097/MPA.0b013e31820720e6. [DOI] [PubMed] [Google Scholar]
- 93.Song JM, Liu HX, Li Y, Zeng YJ, Zhou ZG, Liu HY, Xu B, Wang L, Zhou B, Wang R. Extracellular heat-shock protein 70 aggravates cerulein-induced pancreatitis through toll-like receptor-4 in mice. Chin Med J (Engl) 2008;121:1420–1425. [PubMed] [Google Scholar]
- 94.Peng Y, Sigua CA, Rideout D, Murr MM. Deletion of toll-like receptor-4 downregulates protein kinase C-zeta and attenuates liver injury in experimental pancreatitis. Surgery. 2008;143:679–685. doi: 10.1016/j.surg.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 95.Peng Y, Sigua CA, Gallagher SF, Murr MM. Protein kinase C-zeta is critical in pancreatitis-induced apoptosis of Kupffer cells. J Gastrointest Surg. 2007;11:1253–1261. doi: 10.1007/s11605-007-0193-0. [DOI] [PubMed] [Google Scholar]
- 96.Sawa H, Ueda T, Takeyama Y, Yasuda T, Shinzeki M, Nakajima T, Kuroda Y. Role of toll-like receptor 4 in the pathophysiology of severe acute pancreatitis in mice. Surg Today. 2007;37:867–873. doi: 10.1007/s00595-007-3520-x. [DOI] [PubMed] [Google Scholar]
- 97.van Westerloo DJ, Weijer S, Bruno MJ, de Vos AF, Van't Veer C, van der Poll T. Toll-like receptor 4 deficiency and acute pancreatitis act similarly in reducing host defense during murine Escherichia coli peritonitis. Crit Care Med. 2005;33:1036–1043. doi: 10.1097/01.ccm.0000162684.11375.85. [DOI] [PubMed] [Google Scholar]
- 98.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 99.Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. 2008;29:617–625, viii. doi: 10.1016/j.ccm.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gao HK, Zhou ZG, Li Y, Chen YQ. Toll-like receptor 4 Asp299Gly polymorphism is associated with an increased risk of pancreatic necrotic infection in acute pancreatitis: a study in the Chinese population. Pancreas. 2007;34:295–298. doi: 10.1097/mpa.0b013e318032674a. [DOI] [PubMed] [Google Scholar]
- 101.Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, Ga, September 11 through 13, 1992. Arch Surg. 1993;128:586–590. doi: 10.1001/archsurg.1993.01420170122019. [DOI] [PubMed] [Google Scholar]
- 102.Zhang D, Zheng H, Zhou Y, Yu B, Li J. TLR and MBL gene polymorphisms in severe acute pancreatitis. Mol Diagn Ther. 2008;12:45–50. doi: 10.1007/BF03256267. [DOI] [PubMed] [Google Scholar]
- 103.Hofner P, Balog A, Gyulai Z, Farkas G, Rakonczay Z, Takács T, Mándi Y. Polymorphism in the IL-8 gene, but not in the TLR4 gene, increases the severity of acute pancreatitis. Pancreatology. 2006;6:542–548. doi: 10.1159/000097363. [DOI] [PubMed] [Google Scholar]
- 104.Guenther A, Aghdassi A, Muddana V, Rau B, Schulz HU, Mayerle J, Kraft M, Whitcomb DC, Lerch MM, Weiss FU. Toll-like receptor 4 polymorphisms in German and US patients are not associated with occurrence or severity of acute pancreatitis. Gut. 2010;59:1154–1155. doi: 10.1136/gut.2009.192492. [DOI] [PubMed] [Google Scholar]
- 105.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 106.Li HG, Zhou ZG, Li Y, Zheng XL, Lei S, Zhu L, Wang Y. Alterations of Toll-like receptor 4 expression on peripheral blood monocytes during the early stage of human acute pancreatitis. Dig Dis Sci. 2007;52:1973–1978. doi: 10.1007/s10620-006-9211-4. [DOI] [PubMed] [Google Scholar]
- 107.Li Z, Xia X, Zhang S, Zhang A, Bo W, Zhou R. Up-regulation of Toll-like receptor 4 was suppressed by emodin and baicalin in the setting of acute pancreatitis. Biomed Pharmacother. 2009;63:120–128. doi: 10.1016/j.biopha.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 108.Zhang X, Liu D, Wu D, Zhu C, Ye J, Wang K, Peng L, Zhuo G. Effect of salvia miltiorrhizae on the expressions of TLR4 protein in the liver of rats with SAP or OJ. Inflammation. 2009;32:151–162. doi: 10.1007/s10753-009-9114-6. [DOI] [PubMed] [Google Scholar]
- 109.Xiping Z, Dijiong W, Jianfeng L, Qihui C, Jing Y, Penghui J, Meijuan Y, Ninni Z. Effects of Salvia miltiorrhizae on ICAM-1, TLR4, NF-kappaB and Bax proteins expression in multiple organs of rats with severe acute pancreatitis or obstructive jaundice. Inflammation. 2009;32:218–232. doi: 10.1007/s10753-009-9124-4. [DOI] [PubMed] [Google Scholar]
- 110.Wagner H. The immunobiology of the TLR9 subfamily. Trends Immunol. 2004;25:381–386. doi: 10.1016/j.it.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 111.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 112.Zauner L, Nadal D. Understanding TLR9 action in Epstein-Barr virus infection. Front Biosci. 2012;17:1219–1231. doi: 10.2741/3982. [DOI] [PubMed] [Google Scholar]
- 113.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 114.Gowda NM, Wu X, Gowda DC. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J Immunol. 2012;188:5073–5085. doi: 10.4049/jimmunol.1102143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ghaly NR, Kotb NA, Nagy HM, Rageh ES. Toll-like receptor 9 in systemic lupus erythematosus, impact on glucocorticoid treatment. J Dermatolog Treat. 2012;2012:Epub ahead of print. doi: 10.3109/09546634.2012.697110. [DOI] [PubMed] [Google Scholar]
- 116.Noack J, Jordi M, Zauner L, Alessi D, Burch A, Tinguely M, Hersberger M, Bernasconi M, Nadal D. TLR9 agonists induced cell death in Burkitt's lymphoma cells is variable and influenced by TLR9 polymorphism. Cell Death Dis. 2012;3:e323. doi: 10.1038/cddis.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zeng YJ, Song JM, Li Y, Wang R, Zhou B, Zhou ZG, Liu HY, Xu B. Toll-like receptor 9 is expressed in rat pancreas and is involved in cerulein-induced pancreatitis. Pancreas. 2008;36:212–214. doi: 10.1097/01.MPA.0000311840.58054.35. [DOI] [PubMed] [Google Scholar]
- 118.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011;141:358–369. doi: 10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tran N, Koch A, Berkels R, Boehm O, Zacharowski PA, Baumgarten G, Knuefermann P, Schott M, Kanczkowski W, Bornstein SR, et al. Toll-like receptor 9 expression in murine and human adrenal glands and possible implications during inflammation. J Clin Endocrinol Metab. 2007;92:2773–2783. doi: 10.1210/jc.2006-2697. [DOI] [PubMed] [Google Scholar]
- 120.Tsujimoto H, Ono S, Matsumoto A, Kawabata T, Kinoshita M, Majima T, Hiraki S, Seki S, Moldawer LL, Mochizuki H. A critical role of CpG motifs in a murine peritonitis model by their binding to highly expressed toll-like receptor-9 on liver NKT cells. J Hepatol. 2006;45:836–843. doi: 10.1016/j.jhep.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 121.Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, Hu X, Zhou H, Doi K, Childs R, et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol. 2008;294:F1050–F1058. doi: 10.1152/ajprenal.00461.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Plitas G, Burt BM, Nguyen HM, Bamboat ZM, DeMatteo RP. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J Exp Med. 2008;205:1277–1283. doi: 10.1084/jem.20080162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang X, Zhu C, Wu D, Jiang X. Possible role of toll-like receptor 4 in acute pancreatitis. Pancreas. 2010;39:819–824. doi: 10.1097/MPA.0b013e3181ca065c. [DOI] [PubMed] [Google Scholar]
- 124.Hoque R, Malik AF, Gorelick F, Mehal WZ. Sterile inflammatory response in acute pancreatitis. Pancreas. 2012;41:353–357. doi: 10.1097/MPA.0b013e3182321500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vonlaufen A, Phillips PA, Xu Z, Zhang X, Yang L, Pirola RC, Wilson JS, Apte MV. Withdrawal of alcohol promotes regression while continued alcohol intake promotes persistence of LPS-induced pancreatic injury in alcohol-fed rats. Gut. 2011;60:238–246. doi: 10.1136/gut.2010.211250. [DOI] [PubMed] [Google Scholar]
- 126.Soga Y, Komori H, Miyazaki T, Arita N, Terada M, Kamada K, Tanaka Y, Fujino T, Hiasa Y, Matsuura B, et al. Toll-like receptor 3 signaling induces chronic pancreatitis through the Fas/Fas ligand-mediated cytotoxicity. Tohoku J Exp Med. 2009;217:175–184. doi: 10.1620/tjem.217.175. [DOI] [PubMed] [Google Scholar]
- 127.Dítě P, Hermanová M, Trna J, Novotný I, Růžička M, Liberda M, Bártková A. The role of chronic inflammation: chronic pancreatitis as a risk factor of pancreatic cancer. Dig Dis. 2012;30:277–283. doi: 10.1159/000336991. [DOI] [PubMed] [Google Scholar]