

Abstract
Chronic infection by either hepatitis B virus (HBV) or hepatitis C virus (HCV) share epidemiological characteristics with risks for development of severe complications such as liver cirrhosis and hepatocellular carcinoma. HBV and HCV also share a high genetic variability. Among highly variable regions, viral genes encoding surface proteins (hepatitis B surface antigen, E1/E2 HCV glycoproteins) play key roles in the stimulation of the host-related immune response and viral entry into hepatocytes. Specific segments of HBV envelope proteins (preS1, “a” determinant) are crucial in the entry process into permissive cells. HCV entry is a complex multistep process involving multiple cell cofactors (glycosaminoglycans, low density lipoprotein receptor, SR-B1, CD81, claudin-1, occludin, EGFR, EphA2) in the interaction with HCV E1/E2 envelope glycoproteins. In vitro both viruses can be controlled by antibody-mediated neutralization targeting viral envelope, also essential in preventing HBV infection in vivo as observed through successful vaccination using HBs antigen. But preventive vaccination and/or therapeutic pressure can influence HBV and HCV variability. For HBV, the patterns of antiviral drug resistance in chronic hepatitis are complex and the original pol/S gene overlap has to be taken into account. Treatment-induced HBV mutations in pol could indeed generate S mutants with subsequent modified antigenicity or increased cancer induction. Variability of HBV and HCV envelope proteins combining high exposure to selective pressures and crucial functional roles require investigation in the context of diagnostic, vaccination and treatment tools. In this editorial a synthesis is performed of HBV and HCV envelope properties at the entry step and as antigenic proteins, and the subsequent clinical impact.
Keywords: Hepatitis B, Hepatitis C, Viral envelope glycoproteins, Clinical outcome
INTRODUCTION
Approximately 400 million people are currently living with chronic infection due to hepatitis B virus (HBV) and the number is approaching 160 million for chronic hepatitis C virus (HCV)-related infection[1]. Besides sharing many epidemiological features, HBV and HCV can both provoke chronic infections after an acute phase, due to a failure of humoral and cellular immune host responses[2-5].
HBV and HCV also share some biological similarities, especially the high frequency of mutations occuring during viral replication. As a result both viruses can develop mutations leading to drug failure, especially for drugs with low potency and a low genetic barrier to resistance such as with anti-HBV lamivudine monotherapy. It is expected this will be less likely during treatment by entecavir or tenofovir which exhibit potent anti-HBV activity and high genetic barriers to resistance.
Viral dynamics are rapid with daily production of 1012-13 virions for HBV and 1012 for HCV. The half-life of free viral particles is short, less than 4 h for HBV[6,7] and 2-3 h for HCV[8]. Although both HBV and HCV can produce consistently high levels of virus in serum, they do so using viral templates with very different life-spans. The template for HBV replication, covalently closed circular DNA (cccDNA), resides in the nucleus of infected hepatocytes and is very stable in chronic infection, likely persisting for months[9], if not longer. On the other hand the intracytoplasmic minus strand RNA HCV template only persists for a few hours. Correspondingly HBV eradication is more difficult and only occurs after several years of viral suppression by therapy. Another characteristic of HBV is that its genome consists of overlapping reading frames: as a consequence, mutation rates to functional virus are lower for HBV compared to HCV which does not contain any overlapping gene. The counterpart of this molecular HBV characteristic is that changes at one position may affect the structure and function of more than one viral protein: the polymerase gene overlaps with the envelope gene, and consequently mutations have yet been published in polymerase gene under treatment pressure can produce mutations in the envelope S gene[10].
Despite the availability of an effective vaccine against HBV, new infections are still highly frequent worldwide. Hence, a careful examination of circulating viral strains must be considered in order to possibly adapt the antigenic vaccine content. To date however no effective vaccine is available against HCV. For both viruses, surface envelope glycoproteins are the external antigens first encountered by host-related immune responses during primary infection. These proteins contain both highly variable and conserved regions, the latter providing possible targets for preventive immunization and/or complementary therapeutic approaches.
The aim of this editorial is to give an overview of the current knowledge on hepatitis B and hepatitis C envelope glycoproteins, however with a particular emphasis on HBV. Structural and functional data will be described on the role of these proteins in the viral entry step into hepatocytes and in the stimulation of host-related responses and clinical impacts for HBV will also be described.
GENERAL PRESENTATION OF HBV ENVELOPE GLYCOPROTEINS AND ANIMAL/CELL CULTURES SYSTEMS SUITABLE FOR THEIR INVESTIGATION
For more than 20 years, primary human hepatocytes (PHH) from surgically excised liver specimens have been the only available in vitro HBV infection model. The transfection of hepatocyte-related cells (based on primary hepatocytes or hepatic cell lines which are easier to handle) with replication-competent HBV genomes allowed the production of secreted infectious virions. Regardless of the difficulty in maintaining these in vitro models, PHH cultures have for a long time been the only way to study viral infectivity. Due to poor efficiency of HBV infection in this model, virus amplification required the use of dimethylsulfoxide (DMSO) and of 4% polyethylene glycol during cultivation and infection, respectively. Primary hepatocyte cultures from Tupaia belangeri can also be infected with HBV, as efficiently as PHH cultures, but with fewer restrictions. Among human hepatic cell lines, the optimal system for various experiments was shown to be HepaRG cells, recently established from a liver tumor, and which become susceptible to HBV upon treatment with DMSO and hydrocortisone[11,12] (Table 1).
Table 1.
Cell culture systems and in vitro models developed to study hepatitis B virus and hepatitis C virus envelope glycoproteins
| In vitro model/cell culture system | Step of the viral cycle | Benefits and major findings | Drawbacks | |
| HBV | Primary hepatocyte cultures | Not easy to handle | ||
| PHH | Replication | No need for DMSO and hydrocortisone for PTH system | Cells cannot be propagated in vitro | |
| PTH | Addition of growth factors | |||
| Hepatic cell lines | ||||
| HepG2 | Binding and infection | Specific binding and uptake | No productive infection | |
| HepaRG | (HBV and HDV) | Cellular determinants of hepatocyte differentiation and their influence on HBV infection | Addition of DMSO and hydrocortisone | |
| HCV | Recombinant E2 glycoprotein: Truncated soluble form of recombinant E2 glycoprotein | Entry process | Identification of two major receptors CD81 and SR-BI | Different behavior from E1 E2 heterodimers |
| Interaction with heparan sulfate proteoglycans | Binding to various cell lines different from hepatocytes | |||
| VLPs: Self assembly of HCV structural proteins produced in insect or mammalian cells using a recombinant virus | Entry process | E1–E2 heterodimers at virion surface | Difference in glycosylation status | |
| Cell attachment | Difficult to prepare | |||
| Attractive vaccine candidate | Non replicative | |||
| HCVpp: Unmodified HCV envelope glycoproteins assembled onto retroviral or lentiviral core particles | Entry process | Study of infectivity and neutralization | Only the very early steps of viral cycle | |
| No association with lipoproteins | ||||
| No budding at the ER | ||||
| HCVcc: Transfection of one HCV strain sequence (JFH1) from a Japanese patient with fulminant hepatitis, in Huh 7 cell line. | Entire life cycle | Entry process +++ | Restricted to Huh-7 cell line | |
| Replication | Restricted to JFH1 non structural proteins sequence | |||
| Virus production | ||||
| Screening of antiviral molecules |
HBV: Hepatitis B virus; HCV: Hepatitis C virus; VLP: Virus-like particles; HCVpp: HCV pseudotype particles; HCVcc: Cell culture derived HCV; HDV: Hepatitis D virus; DMSO: Dimethyl sulfoxide; SR-BI: scavenger receptor class B type I; ER: Endoplasmic reticulum; PHH: Primary human hepatocytes; PTH: Primary Tupaia belangeri hepatocytes; DMSO: Dimethylsulfoxide.
Many discoveries about the viral replication cycle, persistence and clearance have been obtained using animal models such as chimpanzee, gibbon or tupaia infected with HBV[13,14]. Moreover, humanized uPA/SCID mice have been recently used to study HBV/hepatitis D virus coinfection and represent an interesting model allowing us to investigate new antiviral drugs[15]. Experimental systems have also been provided by other Hepadnaviridae such as the duck HBV transmissible to many duck species or the woodchuck hepatitis virus, restricted to the North-Eastern American woodchuck[13].
Hepadnaviruses are characterized by specific liver tropism and high species specificity, restricting in vivo infection to their natural host or closely related species and contributing to the difficulty in developing suitable models of HBV infection[11].
HBV envelope glycoproteins are key viral elements in the viral cycle and in the stimulation of host-related immunity. Within virions, HBV surface envelope antigen (HBsAg) includes three viral surface glycoproteins, named large, medium, and small proteins (LHBs, MHBs and SHBs). They exhibit various distributions either in virions or in subviral particles. Their expression is directed by one stop codon and three start codons in a single open reading frame for translation into preS1, preS2 and S proteins[16]. The preS1 protein [108 or 119 amino acids (aa) depending on the genotype] is present only in LHBs; the preS2 protein (55 aa) is present in LHBs and MHBs; and the S protein (226 aa) is shared by LHBs, MHBs and SHBs[11] (Figure 1).
Figure 1.
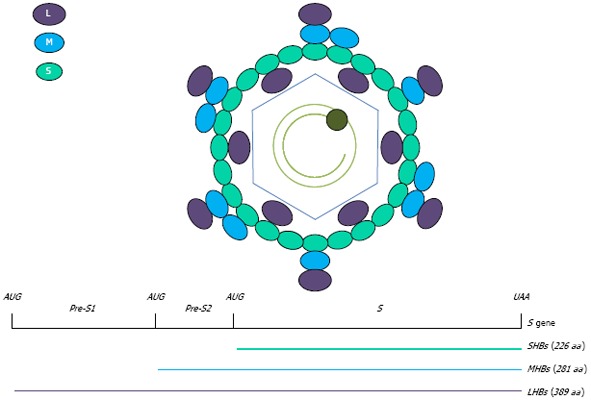
Schematic representation of hepatitis B virus envelope proteins and genome. Hepatitis B virus (HBV) surface envelope antigen includes three viral surface glycoproteins, named large, medium, and small proteins (LHBs, MHBs and SHBs). Their expression is directed by the S gene including three start codons and one stop codon in a single open reading frame.
The three HBV surface preS1, preS2 and S proteins have different functions. Within the preS1 domain, amino acids 3-77 of the Large protein are essential for infectivity as is the myristoylation of glycine at position 2. The preS2-domain is present in both M- and L-proteins but because of the cytosolic orientation of the preS1-domain in the L-protein, it is N-glycosylated only in the M-protein. Although antibodies against the N-terminal part of preS2 can inhibit HBV infection in vitro, the Medium protein of HBV is believed to play an accessory role in infectivity. While the preS-domains form linear epitopes, the S-domain forms multiprotein complexes using inter- and intramolecular disulfide bonds with the eight Cys residues within the antigenic loop. A crucial function of the S-domain is viral morphogenesis, but the first transmembrane sequence also plays a role in viral entry into hepatocytes[11]. Moreover, two different transmembrane topologies have been described for the L surface protein: L chains with an internal N-terminal preS1 part are required in virion morphogenesis, whereas the L molecules which expose their preS1 domain on the viral particle surface probably link to a putative virus receptor and could determine the species specificity and viral tropism[16]. In addition to the preS1 domain, a second infectivity determinant, located in the antigenic loop of the S domain, is also required for infectivity[17].
HBsAg plays a central role in stimulating and, because of its variability, also oppositely thwarting the host-related immune response. The main antigenic part of HBsAg, namely the “a” determinant which is the core part of the major hydrophilic region (amino acid residues 99-169 of HBsAg), is a target of neutralizing antibodies. Immunogenic T cell epitopes are also present in the HBs Ag : P1 (aa 16-33) and P4 (aa 213-226) are the dominant epitopes in vaccine immunization[18].
INTERACTION WITH PUTATIVE CELLULAR RECEPTOR(S)
HBV envelope glycoproteins are the key components contributing to the attachment to the hepatocyte plasma membrane, thus defining organ and species specificity of HBV. Viral tropism is essentially restricted to the liver[14]. Although there is considerable evidence that a HBV receptor exists on hepatocyte membranes, it still remains to be identified. Convergent data place the receptor-binding site in the preS1 segment of HBV envelope proteins that play a key role at the early entry step for further viral infectivity, as described above. The preS1 (21-47) region of the envelope protein was demonstrated to be the dominant binding site to hepatoblastoma cell line (HepG2). More recently, the region consisting of amino acids 9-18 of the preS1 domain was identified as a conserved crucial attachment site while amino acids 28-48 as an accessory binding site[19]. The receptor for the preS1 peptide seems to be a single major peptide of molecular weight 31 kD[20]. The asialoglycoprotein receptor was described as playing a role in the attachment process[21], and numerous other cellular partners have been suggested to also contribute, such as heparin or annexin V, earlier referred to as Endonexin II[22-26]. Interestingly, heparan sulfate proteoglycans emerged as potential major components in the HBV entry step[27]. However HBV entry and different steps of this process are not characterized in detail, while it is substantially known for HCV.
HOST-RELATED IMMUNE RESPONSES
During viral infection, immunological reactivity usually begins with an innate response in infected cells, where viral replication induces activation of intracellular antiviral mechanisms through transcription of interferon (IFN) inducible genes. However, in acutely HBV infected chimpanzees no specific pattern of cellular genes expression was observed during widespread expansion of HBV in the liver, revealing an absence of innate response by infected cells[28]. Interestingly, this could be thwarted by using IFN-α which has been described to inhibit HBV transcription and replication in cell culture and in humanized mice by targeting the nuclear cccDNA[29]. The adaptive immune response is responsible for viral clearance as well as HBV-related diseases pathogenesis. In patients with acute hepatitis, the T cell response is vigorous and multispecific. The CD8+ T cell response, under the control of CD4+ T cells, acts on viral clearance by triggering direct hepatocyte apoptosis and by secreting IFN-γ which inhibits HBV replication. In chronically infected patients, a weak and focused T cell response is observed (Figure 2). This seems less related to sufficient numbers of CD8+ T cells, but more to their functional alteration[30]. Human leukocyte antigen (HLA) class I and HLA class II restricted T-cell epitopes have been recently reviewed in Desmond 2008[31]. Several HBV epitopes recognized by CD4+ T cells were described in the surface protein, including S2109-134, S200-214 and S337-357, with S179-194, considered as an immunodominant HLA-DR1-restricted epitope[32].
Figure 2.
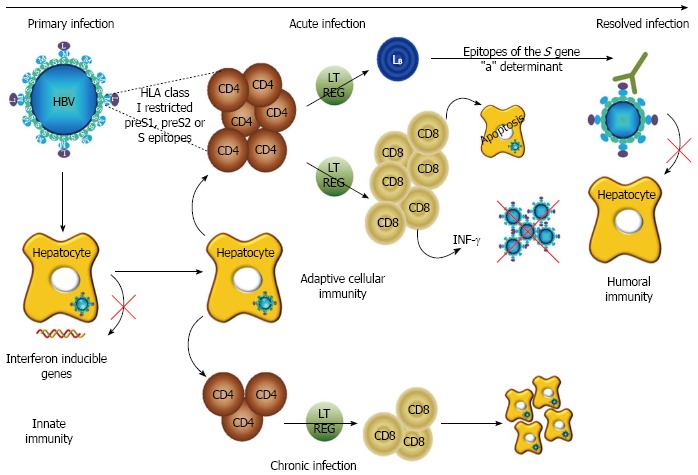
Role of the hepatitis B surface protein in the immune response to hepatitis B infection. Hepatitis B virus (HBV) primary infection does not induce activation of intracellular antiviral mechanisms through transcription of interferon inducible genes. After acute infection, the evolution to resolved hepatitis B is promoted by a vigorous and multispecific T cell response (CD4+ and CD8+ T cell) through hepatocyte apoptosis and interferon (IFN)-γ secretion, while evolution to chronicity is characterized by a weak and focused T cell response. Regulatory T cells (Treg) cells (LT REG) limit the function of effector T cells preventing excessive auto-destructive disease. A protective immunity is due in part to humoral and cellular anti-envelope response, preventing HBV attachment to hepatocytes, thus playing a critical role in prevention of infection after classical vaccination and in viral clearance in resolved hepatitis. HLA: Human leukocyte antigen.
Regulatory T (Treg) lymphocytes, known to suppress the activation, differentiation and proliferation of immune cells, could be major actors in the inadequate immune responses observed during chronic HBV infection. The Treg response protects the host by limiting liver immunopathology while at the same time favouring the virus by inhibiting a protective T-cell response. The timing of the Treg response is vital; it needs to be not too late otherwise resulting in the excessive destruction of hepatocytes by effector T-cells, and not too early which would favour the establishment of chronic infection by blocking antiviral T-cell activity[33]. As suggested by Manigold et al[33], a higher frequency of Treg cells has been described in patients with chronic infection and correlated with greater HBV DNA concentration in serum.
The humoral immune response is based on anti-envelope protective antibodies. On the one hand, antibodies complex with free particles, removing them from circulation and on the other hand they prevent HBV attachment to hepatocytes, thus playing a critical role in viral clearance. Both viral and host factors can delay or inhibit antibody-mediated neutralization of HBV. As it will be further described, viral variability contributes to the failure of the humoral response. Other factors such as HIV coinfection[34] or immunosuppression from a variety of causes can also negatively modulate humoral reactivity[35]. Additionally, the development of an immune response, usually characterized by the strong appearance of anti-HBs antibodies, may be delayed by social stress, as identified during HBV vaccination of students under exam stress[36].
GENERAL PRESENTATION OF HCV ENVELOPE GLYCOPROTEINS AND RELEVANT ANIMAL/CELL CULTURE MODELS
HCV envelope glycoprotein E1 and E2 represent crucial elements in the viral life cycle. Indeed, they are involved in virus cellular entry and are also targeted by host immune components. Thus characterization of these two proteins is essential to better understand virus-host interactions.
E1 and E2 are transmembrane glycoproteins containing an important N terminal ectodomain and a C terminal hydrophobic anchor domain. The E2 protein displays an amphipathic helix that forms a stem region, linking the ectodomain and the transmembrane region. The two glycoproteins form a non-covalent heterodimer at the viral particle surface, interacting with each other during virus entry into target cells. E1 and E2 are both stabilized by numerous disulfide bonds. Krey et al[37] described the connectivity of the E2 disulfide bonds and suggested that E2 could display a tertiary structure similar to that of class II fusion proteins, and containing three domains termed DI, DII and DIII. Their model reveals the distribution of E2 amino acids among the different domains. E1 and E2 are highly glycosylated proteins and glycosylation is needed for correct glycoprotein processing, folding and/or cellular entry.
The E2 glycoprotein contains three hypervariable regions, termed hypervariable region (HVR) 1, HVR2 and HVR3 and conformational binding sites to certain cellular co-receptors (CD81 binding site as an example). E1 and E2 glycoproteins are targets of both humoral and cellular immune responses generated during HCV infection and several specific epitopes have been described in E1 and E2.
Our knowledge of the HCV viral cycle and of the structural and functional characteristics of envelope glycoproteins has been hampered by the lack of reliable in vivo and in vitro models. Chimpanzee infection has long been the only available animal model, but with many limitations including ethical considerations, high cost, and clinical differences with human HCV infection. Other small animal models, such as humanized mice, are in development to overcome these problems.
Some in vitro models, such as soluble glycoproteins, virus like particles, and HCV pseudoparticles, have been developed that allow investigation of the viral entry process. Despite their ability to duplicate the early steps of HCV entry, none of these models support the entire HCV replication cycle or efficient production of infectious particles. In this regard an important breakthrough was achieved with the cell culture system HCVcc, which is capable of producing infectious particles. The most useful in vitro models that allow the study of HCV glycoproteins are detailed in Table 1.
INTERACTIONS OF HCV ENVELOPE GLYCOPROTEINS WITH CELLULAR RECEPTORS
The main target cells of HCV are hepatocytes. HCV enters into target cells in a complex multistep process involving the following entry host factors: tetraspanin CD81, scavenger receptor class B member I (SR-BI), Heparan Sulfate and the tight junction proteins Claudin-1 and Occludin. Other partners in HCV entry were recently characterized, such as EGFR and EphA2[38].
Highly sulfated heparan sulfate structures are cell surface factors mediating initial virus attachment prior to virus interaction with CD81 and SR-BI, which directly interact with HCV envelope glycoproteins. SR-BI may define the attachment of the virion in the form of viro-lipoparticles to the cell surface and that then favours particle interactions with CD81 and Claudin coreceptors with a role of EGFR and EphA2 activated kinases in the CD81-Claudin binary complex. CD81 and Claudin-1 are essential for the clathrin dependant particle internalization, and the engagement of CD81 promotes internalization. CD81 associates with HCV and mediates entry via Claudin-1 and occludin complexes. Claudin-1/CD81 complexes seem to localize at the basolateral surface of polarized hepatoma cells. After uncoating and internalization by clathrin-dependent endocytosis, the viral genome is released into the cytoplasm leading to its translation and replication.
INTERPLAY BETWEEN HCV ENVELOPE GLYCOPROTEINS AND THE HOST IMMUNE RESPONSE
Persistent viral infection can be explained by the fact that HCV escapes both innate and adaptative immune responses or interferes with host defense mechanisms. Despite broadly reactive neutralizing and multispecific T cell responses generated during chronic infection, in most cases viral clearance is not achieved.
HCV RNA can be detected in patient serum between one and two weeks following infection whereas the appearance of HCV specific T-lymphocytes and antibodies is delayed up to several weeks after infection. In the early phase of infection neutralizing antibodies are rapidly induced by patients who subsequently clear the virus or control viral infection whereas they are absent or of low quantity in patients who progress to chronic HCV infection[39]. Both cellular and humoral responses fail to control HCV infection during the very early phase of infection. If viral clearance is characterized by a broad vigorous and persistent T cell response, cellular immunity is weak, narrow and transient in patients chronically infected (Figure 3). Some mutations occurring in epitopes targeted by CD4+ or CD8+ T lymphocytes can show a strong inhibition of immune reactivity, such as HCV E1 226-240 and HCV E2 436-450[3]. Patients who do not clear the virus present high quantity and even cross-neutralizing antibodies during the chronic phase, but these antibodies are unable to control HCV infection.
Figure 3.
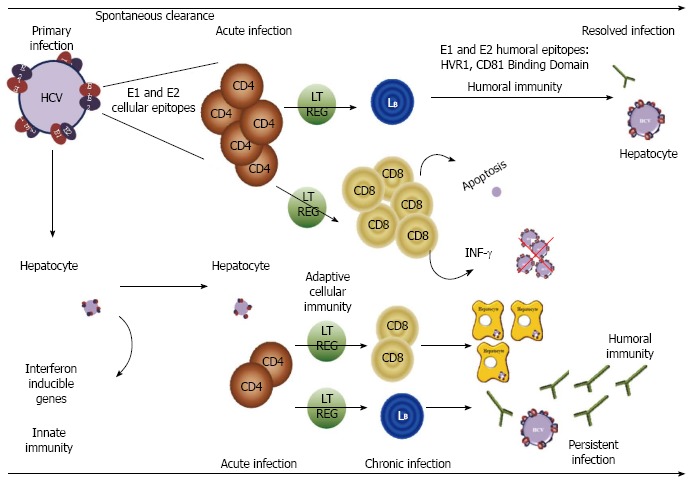
Role of hepatitis C glycoproteins in the immune response to hepatitis C infection. Following hepatitis C virus (HCV) infection, innate host response is initiated by hepatocytes producing an antiviral state by stimulating interferon inducible genes expression. In case of spontaneous clearance or resolved infection, a strong cellular response, due to CD4+ and CD8+ T lymphocytes (denoted as CD4 and CD8 in the figure), is observed in the early phase of infection. Moreover Regulatory T cells (Treg) cells (LT REG) limit the function of effector T cells. Then neutralizing antibodies are rapidly expressed. In most cases, the host adaptative immunity is unable to control HCV infection evolving to a chronic phase. The cellular response (CD4+ and CD8+ T lymphocytes and B lymphocytes, mentioned as LB in the figure) is weak and transient. The appearance of neutralizing antibodies is delayed and the humoral response not efficient. IFN: Interferon.
The failure of the humoral response is mainly due to the rapid onset of mutations in HVR1 of E2 and in other epitopes of the E1 and E2 proteins. However, HVR1, the most variable region of the HCV genome, is one of the targets of the neutralizing immune response. HVR1 may function as an “immunological decoy”, stimulating an ineffective immune response for viral clearance but inducing selection of viral variants[40].
The E2-CD81 interaction has been shown to modulate B and T cell function. During HCV infection, the E2-CD81 interaction induces hypermutation of the heavy chain immunoglobulin in B cells. Hypermutation may lower the affinity and specificity of HCV-specific antibodies, enabling HCV to escape immune surveillance. Additionally, engagement of CD81 on natural killer cells by the E2 protein has been shown to inhibit the antiviral function of NK cells early in the infection process.
GENES AND PROTEINS VARIABILITY, ESPECIALLY IN VIRAL ENVELOPE GLYCOPROTEINS
Despite the compact arrangement of its DNA genome and the overlapping open reading frames that limit genomic plasticity, HBV is evidently diverse on a global scale as indicated by the eight referenced genotypes (A-H). HCV is also characterized by high genetic variability given that six (and probably a seventh) major genotypes have been described, each including several subtypes. Intragenomic variability of HBV and HCV is further reflected in the quasispecies distribution defined by the genetic heterogeneity of the viral pool found in any infected subject at a given time point[41,42]. For both viruses, this genetic variability is a consequence firstly of the high spontaneous error rate associated with the lack of a proofreading mechanism of viral polymerases, and secondly of the structural and/or functional constraints exerted on the virus by host immunity, immunoprophylaxis or antiviral therapy pressure.
Due to the structural constraints of its genomic organization, the emergence of HBV mutants occurs more slowly than for HCV. Since the preS1 and preS2 domains overlap with the region of the polymerase gene encoding for the spacer domain, deletions in preS can occur without affecting the enzymatic properties of HBV polymerase. However, although preS1 deleted mutants are able to replicate they usually need a helper virus to infect new hepatocytes[43]. The surface S gene seems to vary less than the preS1 and preS2 regions[44]. Functional mutations that have been described in the HBV envelope genome consist of preS1 and preS2 deletions, preS2 start codon mutations, and C-terminally truncated or “a” determinant mutated S proteins. These viral variants exhibit modified replication capacities and antigenic characteristics; on the other hand they may be resistant to antiviral therapies and/or contribute to escape from host-related immune surveillance. They can also alter the sensitivity of diagnostic immunoassays. In HepG2 cells, replication of preS/S variants leads to decreased HBsAg secretion, retention of envelope proteins in the endoplasmic reticulum, less efficient virion secretion and higher amounts of cccDNA in the nucleus. In patients with HBV-related chronic liver disease, the emergence of preS/S variants appears to provoke the loss of correlation between HBV DNA replication and HBsAg synthesis/secretion[45].
Briefly for HCV, the HCV genome contains both highly conserved and highly variable regions. The most variable regions are located in NS5A and in parts of the E2 glycoprotein (HVR)[46,47]. Some mutations have been described in E2 showing an impact on HCV infectivity and/or its accessibility to antibodies or cellular response mediated by CD4+ and CD8+ T cells[3,48-51].
PREVENTIVE OPTIONS AND THE RISK OF ESCAPE MUTANTS
Vaccination using a recombinant HBV surface antigen is the most important prophylactic measure available for the prevention of new HBV infections. Immunoprophylaxis with vaccine or immunoglobulins is also recommended after exposure to blood from HBV-infected patients, in new born children from HBV-infected mothers, and to prevent recurrent HBV infection in patients receiving liver transplants for end-stage hepatitis B liver disease.
HBV surface gene variants can be selected under immune pressure after preventive immunization. This is illustrated by report of several surveys in Taiwan describing an increase in the prevalence of S gene “a” determinant mutants during a vaccination campaign[52]. In HBV DNA positive children from four sequential surveys in Taiwan, the prevalence of hepatitis B surface gene “a” determinant mutants increased from 7.8% before the vaccination program, to 19.6%, 28.1% and 23.1% at five, 10 and 15 years after the immunization period. However, lower infectivity of the G145R mutant virus, the use of a recombinant vaccine, and mutant loss with older age seem to decrease the “a” mutant prevalence in an immunized population over time[53]. In Europe and North America, mutations in the HBV S gene are mostly observed in infants born from HBV-infected mothers, in a small proportion of occult HBV infections, and in liver transplant recipients[54]. Among variants which were described, the Arg-145 and Arg-129 mutants show the lowest binding ability to monoclonal antibodies. In addition, variants epitopes in HBs antigen induced decreased or a lack of T-cell reactivity[18]. As an illustration, HBV replication was detected in vaccinated chimpanzees challenged with an HBV strain containing polymerase/envelope overlapping mutations[55].
The considerable global diversity of HCV has hampered the development of any successful vaccine. Consequently, neither vaccine, nor specific immunoglobulins are currently available for clinical practice. Current HCV vaccine research therefore follows two main pathways: (1) prophylactic; and (2) therapeutic to increase virological response to antiviral treatment and to reduce the duration of therapy. Although numerous approaches have been developed only a few of these have progressed to human trials, especially for potential therapeutic vaccines. HCV envelope proteins that induce antibodies against conserved domain involved in cell binding are considered as a promising target for vaccine research[56]. However, several mechanisms make antibody-mediated neutralization difficult: interference by interfering antibodies, glycans or lipids, the quasispecies distribution of HCV, and difficult to access cell-to-cell transfer[57].
TREATMENT AND RISK OF ESCAPE MUTANTS
The treatment of chronic hepatitis B aims to reduce liver cell inflammation, prevent the progression to cirrhosis and hepatocellular carcinoma through the suppression of viral replication, and ultimately to clear the infection. HBV treatments use the immunomodulatory agents IFN-α or its pegylated form as well as direct-acting antiviral molecules. IFN-α binding to type 1 IFN receptors activates an anti-viral state in cells by inducing stimulation of IFN genes. IFN also stimulates the cellular immune response and production of IFN-γ by CD4+ and CD8+ T cells[58]. On the other hand, nucleos(t)ide analogues block HBV polymerase functions[59]. A sustained virological response to antiviral therapy is defined by plasma HBV DNA < 2000 IU/mL six months after discontinuation of therapy, while a complete response is defined by the disappearance of both HBV DNA and HBsAg with or without appearance of anti-HBs antibodies[58]. During treatment various escape mutants can be detected. Well-characterized mutations in the reverse transcriptase (rt) gene have been described (e.g., rtM204V for lamivudine and rtN236T for adefovir). HBV resistance can develop after a single mutation with some agents or only after multiple mutations for others. Entecavir exhibits low rates of resistance, with fewer than 1% of patients on monotherapy developing resistant mutants after 5 years of treatment[59], if patients strictly respect treatment recommendations. Similarly tenofovir has a very high genetic barrier. Due to the structure of the HBV genome, mutations in the rt domains can affect the amino acid sequence of other HBV proteins, as demonstrated for the rtM204V + rtV173L + rtL180M mutations that lead to envelope changes behaving as mutants escaping vaccination in vitro[59]. In addition, studies analyzing HBV quasispecies demonstrated that resistance mutations to nucleos(t)ide analogues can cause amino acid changes within epitopes of the HBV preS/S or core antigen leading to changes in HBV immunogenicity[60].
Treatment of HCV chronic infection consists of a combination of pegylated IFN-α and ribavirin, and this combination remains the basis of gold standard treatments including the use of anti-protease molecules. The rate of sustained virological response (undetectable HCV viral load six months after the end of the therapy, detection threshold at 12-15 IU/mL) depends on the genotype of HCV with lower rates for genotype 1 and is correlated to pre-treatment HCV load and several host-related factors. Both IFN and ribavirin have broad spectrum non specific antiviral activity without direct action on viral genomes[46]. IFN produce an antiviral state by the up-regulation of IFN stimulated genes, leading to the expression of several antiviral proteins including 2’, 5’ oligoadenylate synthase, and the Mx protein. Four putative mechanisms of action have been proposed to explain the effect of ribavirin on HCV infection: modulation of the host adaptive antiviral response, HCV RNA mutagenesis, action on the intracellular GTP pool necessary for RNA synthesis and inhibition of HCV polymerase. Thus resistance to treatment is a complex mechanism involving host- and virus-related features.
Several HCV proteins (E2, core, NS5A, NS5B) are suspected to be linked with resistance mechanisms to antiviral therapy[46,61,62]. In relation to HCV envelope glycoproteins, treatment outcome can be influenced by amino acid substitutions within CD81 binding sites and HVR2 of E2[63] as well as theoretically through mutations in the PKR/eIF-2α phosphorylation homology domain of E2[61]. A lower HVR1 heterogeneity among viral variants in an individual before antiviral therapy seems to be associated with higher rates of virological response to IFN-based treatment[46,64]. HCV genetic variability in regions such as E2 HVR1 has been associated with antiviral treatment failure[46,65]. In a clinical context, Aurora et al[66] described frequent covarying amino acids positions, most often located in E1 and E2, that linked to response to treatment.
CONCLUSION
For both HBV and HCV, viral envelope glycoproteins play a key role in viral entry into hepatocytes and are exposed to host-related immune responses. Even though data that fully elucidates HBV entry into permissive cells are still lacking, specific segments of HBV envelope proteins (preS1, “a” determinant) are instrumental in the process. For HCV, numerous experimental results have recently demonstrated that viral entry is a complex multistep process involving multiple cell cofactors (glycosaminoglycans, low density lipoprotein receptor, SR-B1, CD81, claudin-1, occludin, EGFR, EphA2) in the interaction with HCV E1/E2 envelope glycoproteins. In vitro both viruses can be controlled by antibody-mediated neutralization targeting viral envelope. Neutralizing antibodies are also essential in preventing HBV infection in vivo as is well characterized through successful vaccination using HBs antigen.
Several factors, such as preventive vaccination and/or therapeutic pressure, can influence HBV and HCV variability. Moreover, for HBV, the patterns of antiviral drug resistance in chronic hepatitis are complex and the original pol/S gene overlap has to be taken into account in the analysis of public health significance of emerging genomic mutations in infected populations. Treatment-induced HBV mutations in pol could indeed generate S mutants with subsequent modified antigenicity or pathogenesis such as increased cancer induction[10].
Variability of HBV and HCV envelope proteins combining high exposure to selective pressures and crucial functional roles require investigation for the adaptation of diagnostic, vaccination and treatment tools. In this context, improved therapy for chronic HCV infection has emerged from a better understanding of the viral replication cycle, with HCV entry providing a large number of therapeutic targets[67,68]. Besides IFN-α and nucleos(t)idic analogues, complementary therapies for chronic HBV infection are desperately needed and the HBV entry process, when better characterized, can offer potential therapeutic opportunities.
Footnotes
P- Reviewers Rippe RA, Yamagiwa S S- Editor Gou SX L- Editor A E- Editor Zhang DN
References
- 1.Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect. 2011;17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 2.Thimme R, Bukh J, Spangenberg HC, Wieland S, Pemberton J, Steiger C, Govindarajan S, Purcell RH, Chisari FV. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci USA. 2002;99:15661–15668. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, McKeating JA. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132:667–678. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Qian F, Yuan Q, Li X, Wu W, Guo X, Li L. Mutations in hepatitis B virus DNA from patients with coexisting HBsAg and anti-HBs. J Clin Virol. 2011;52:198–203. doi: 10.1016/j.jcv.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 5.Baumert TF, Thimme R, von Weizsäcker F. Pathogenesis of hepatitis B virus infection. World J Gastroenterol. 2007;13:82–90. doi: 10.3748/wjg.v13.i1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray JM, Purcell RH, Wieland SF. The half-life of hepatitis B virions. Hepatology. 2006;44:1117–1121. doi: 10.1002/hep.21364. [DOI] [PubMed] [Google Scholar]
- 7.Dandri M, Murray JM, Lutgehetmann M, Volz T, Lohse AW, Petersen J. Virion half-life in chronic hepatitis B infection is strongly correlated with levels of viremia. Hepatology. 2008;48:1079–1086. doi: 10.1002/hep.22469. [DOI] [PubMed] [Google Scholar]
- 8.Neumann AU, Lam NP, Dahari H, Davidian M, Wiley TE, Mika BP, Perelson AS, Layden TJ. Differences in viral dynamics between genotypes 1 and 2 of hepatitis C virus. J Infect Dis. 2000;182:28–35. doi: 10.1086/315661. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, Miller DS, Litwin S, Furman PA, Jilbert AR, Mason WS. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75:311–322. doi: 10.1128/JVI.75.1.311-322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warner N, Locarnini S. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatology. 2008;48:88–98. doi: 10.1002/hep.22295. [DOI] [PubMed] [Google Scholar]
- 11.Glebe D, Urban S. Viral and cellular determinants involved in hepadnaviral entry. World J Gastroenterol. 2007;13:22–38. doi: 10.3748/wjg.v13.i1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci USA. 2002;99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoulim F, Berthillon P, Guerhier FL, Seigneres B, Germon S, Pichoud C, Cheng YC, Trepo C. Animal models for the study of HBV infection and the evaluation of new anti-HBV strategies. J Gastroenterol Hepatol. 2002;17 Suppl:S460–S463. doi: 10.1046/j.1440-1746.17.s4.10.x. [DOI] [PubMed] [Google Scholar]
- 14.De Meyer S, Gong ZJ, Suwandhi W, van Pelt J, Soumillion A, Yap SH. Organ and species specificity of hepatitis B virus (HBV) infection: a review of literature with a special reference to preferential attachment of HBV to human hepatocytes. J Viral Hepat. 1997;4:145–153. doi: 10.1046/j.1365-2893.1997.00126.x. [DOI] [PubMed] [Google Scholar]
- 15.Lütgehetmann M, Mancke LV, Volz T, Helbig M, Allweiss L, Bornscheuer T, Pollok JM, Lohse AW, Petersen J, Urban S, et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology. 2012;55:685–694. doi: 10.1002/hep.24758. [DOI] [PubMed] [Google Scholar]
- 16.Bruss V, Gerhardt E, Vieluf K, Wunderlich G. Functions of the large hepatitis B virus surface protein in viral particle morphogenesis. Intervirology. 1996;39:23–31. doi: 10.1159/000150471. [DOI] [PubMed] [Google Scholar]
- 17.Salisse J, Sureau C. A function essential to viral entry underlies the hepatitis B virus "a" determinant. J Virol. 2009;83:9321–9328. doi: 10.1128/JVI.00678-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauer T, Weinberger K, Jilg W. Variants of two major T cell epitopes within the hepatitis B surface antigen are not recognized by specific T helper cells of vaccinated individuals. Hepatology. 2002;35:455–465. doi: 10.1053/jhep.2002.30903. [DOI] [PubMed] [Google Scholar]
- 19.Glebe D. Attachment sites and neutralising epitopes of hepatitis B virus. Minerva Gastroenterol Dietol. 2006;52:3–21. [PubMed] [Google Scholar]
- 20.Dash S, Rao KV, Panda SK. Receptor for pre-S1(21-47) component of hepatitis B virus on the liver cell: role in virus cell interaction. J Med Virol. 1992;37:116–121. doi: 10.1002/jmv.1890370208. [DOI] [PubMed] [Google Scholar]
- 21.Stockert RJ. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol Rev. 1995;75:591–609. doi: 10.1152/physrev.1995.75.3.591. [DOI] [PubMed] [Google Scholar]
- 22.Hertogs K, Leenders WP, Depla E, De Bruin WC, Meheus L, Raymackers J, Moshage H, Yap SH. Endonexin II, present on human liver plasma membranes, is a specific binding protein of small hepatitis B virus (HBV) envelope protein. Virology. 1993;197:549–557. doi: 10.1006/viro.1993.1628. [DOI] [PubMed] [Google Scholar]
- 23.de Bruin WC, Leenders WP, Moshage H, van Haelst UJ. Species specificity for HBsAg binding protein endonexin II. J Hepatol. 1996;24:265–270. doi: 10.1016/s0168-8278(96)80003-1. [DOI] [PubMed] [Google Scholar]
- 24.De Meyer S, Gong ZJ, Hertogs K, Depla E, van Pelt JF, Roskams T, Maertens G, Yap SH. Influence of the administration of human annexin V on in vitro binding of small hepatitis B surface antigen to human and to rat hepatocytes and on in vitro hepatitis B virus infection. J Viral Hepat. 2000;7:104–114. doi: 10.1046/j.1365-2893.2000.00207.x. [DOI] [PubMed] [Google Scholar]
- 25.Yamada M, Oeda A, Jung J, Iijima M, Yoshimoto N, Niimi T, Jeong SY, Choi EK, Tanizawa K, Kuroda S. Hepatitis B virus envelope L protein-derived bio-nanocapsules: mechanisms of cellular attachment and entry into human hepatic cells. J Control Release. 2012;160:322–329. doi: 10.1016/j.jconrel.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Leistner CM, Gruen-Bernhard S, Glebe D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol. 2008;10:122–133. doi: 10.1111/j.1462-5822.2007.01023.x. [DOI] [PubMed] [Google Scholar]
- 27.Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology. 2007;46:1759–1768. doi: 10.1002/hep.21896. [DOI] [PubMed] [Google Scholar]
- 28.Chisari FV, Isogawa M, Wieland SF. Pathogenesis of hepatitis B virus infection. Pathol Biol (Paris) 2010;58:258–266. doi: 10.1016/j.patbio.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest. 2012;122:529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maini MK, Bertoletti A. How can the cellular immune response control hepatitis B virus replication? J Viral Hepat. 2000;7:321–326. doi: 10.1046/j.1365-2893.2000.00234.x. [DOI] [PubMed] [Google Scholar]
- 31.Desmond CP, Bartholomeusz A, Gaudieri S, Revill PA, Lewin SR. A systematic review of T-cell epitopes in hepatitis B virus: identification, genotypic variation and relevance to antiviral therapeutics. Antivir Ther. 2008;13:161–175. [PubMed] [Google Scholar]
- 32.Pajot A, Michel ML, Mancini-Bourgine M, Ungeheuer MN, Ojcius DM, Deng Q, Lemonnier FA, Lone YC. Identification of novel HLA-DR1-restricted epitopes from the hepatitis B virus envelope protein in mice expressing HLA-DR1 and vaccinated human subjects. Microbes Infect. 2006;8:2783–2790. doi: 10.1016/j.micinf.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 33.Manigold T, Racanelli V. T-cell regulation by CD4 regulatory T cells during hepatitis B and C virus infections: facts and controversies. Lancet Infect Dis. 2007;7:804–813. doi: 10.1016/S1473-3099(07)70289-X. [DOI] [PubMed] [Google Scholar]
- 34.van den Berg R, van Hoogstraten I, van Agtmael M. Non-responsiveness to hepatitis B vaccination in HIV seropositive patients; possible causes and solutions. AIDS Rev. 2009;11:157–164. [PubMed] [Google Scholar]
- 35.Sjogren MH. Prevention of hepatitis B in nonresponders to initial hepatitis B virus vaccination. Am J Med. 2005;118 Suppl 10A:34S–39S. doi: 10.1016/j.amjmed.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 36.Glaser R, Kiecolt-Glaser JK, Bonneau RH, Malarkey W, Kennedy S, Hughes J. Stress-induced modulation of the immune response to recombinant hepatitis B vaccine. Psychosom Med. 1992;54:22–29. doi: 10.1097/00006842-199201000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Krey T, d'Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, Petitpas I, Johansson DX, Tawar RG, Baron B, Robert B, et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010;6:e1000762. doi: 10.1371/journal.ppat.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pestka JM, Zeisel MB, Bläser E, Schürmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci USA. 2007;104:6025–6030. doi: 10.1073/pnas.0607026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bartosch B, Verney G, Dreux M, Donot P, Morice Y, Penin F, Pawlotsky JM, Lavillette D, Cosset FL. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J Virol. 2005;79:8217–8229. doi: 10.1128/JVI.79.13.8217-8229.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panjaworayan N, Brown CM. Effects of HBV Genetic Variability on RNAi Strategies. Hepat Res Treat. 2011;2011:367908. doi: 10.1155/2011/367908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simmonds P. Genetic diversity and evolution of hepatitis C virus--15 years on. J Gen Virol. 2004;85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 43.Kay A, Zoulim F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007;127:164–176. doi: 10.1016/j.virusres.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 44.Bowyer SM, Sim JG. Relationships within and between genotypes of hepatitis B virus at points across the genome: footprints of recombination in certain isolates. J Gen Virol. 2000;81:379–392. doi: 10.1099/0022-1317-81-2-379. [DOI] [PubMed] [Google Scholar]
- 45.Pollicino T, Amaddeo G, Restuccia A, Raffa G, Alibrandi A, Cutroneo G, Favaloro A, Maimone S, Squadrito G, Raimondo G. Impact of hepatitis B virus (HBV) preS/S genomic variability on HBV surface antigen and HBV DNA serum levels. Hepatology. 2012;56:434–443. doi: 10.1002/hep.25592. [DOI] [PubMed] [Google Scholar]
- 46.Wohnsland A, Hofmann WP, Sarrazin C. Viral determinants of resistance to treatment in patients with hepatitis C. Clin Microbiol Rev. 2007;20:23–38. doi: 10.1128/CMR.00010-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chayama K, Hayes CN. Hepatitis C virus: How genetic variability affects pathobiology of disease. J Gastroenterol Hepatol. 2011;26 Suppl 1:83–95. doi: 10.1111/j.1440-1746.2010.06550.x. [DOI] [PubMed] [Google Scholar]
- 48.Grove J, Nielsen S, Zhong J, Bassendine MF, Drummer HE, Balfe P, McKeating JA. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J Virol. 2008;82:12020–12029. doi: 10.1128/JVI.01569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol. 2006;80:11082–11093. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, et al. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 2012;8:e1002653. doi: 10.1371/journal.ppat.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gal-Tanamy M, Keck ZY, Yi M, McKeating JA, Patel AH, Foung SK, Lemon SM. In vitro selection of a neutralization-resistant hepatitis C virus escape mutant. Proc Natl Acad Sci USA. 2008;105:19450–19455. doi: 10.1073/pnas.0809879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang MH. Hepatitis B virus mutation in children. Indian J Pediatr. 2006;73:803–807. doi: 10.1007/BF02790390. [DOI] [PubMed] [Google Scholar]
- 53.Hsu HY, Chang MH, Ni YH, Chiang CL, Chen HL, Wu JF, Chen PJ. No increase in prevalence of hepatitis B surface antigen mutant in a population of children and adolescents who were fully covered by universal infant immunization. J Infect Dis. 2010;201:1192–1200. doi: 10.1086/651378. [DOI] [PubMed] [Google Scholar]
- 54.Tabor E. Infections by hepatitis B surface antigen gene mutants in Europe and North America. J Med Virol. 2006;78 Suppl 1:S43–S47. doi: 10.1002/jmv.20606. [DOI] [PubMed] [Google Scholar]
- 55.Kamili S, Sozzi V, Thompson G, Campbell K, Walker CM, Locarnini S, Krawczynski K. Efficacy of hepatitis B vaccine against antiviral drug-resistant hepatitis B virus mutants in the chimpanzee model. Hepatology. 2009;49:1483–1491. doi: 10.1002/hep.22796. [DOI] [PubMed] [Google Scholar]
- 56.Torresi J, Johnson D, Wedemeyer H. Progress in the development of preventive and therapeutic vaccines for hepatitis C virus. J Hepatol. 2011;54:1273–1285. doi: 10.1016/j.jhep.2010.09.040. [DOI] [PubMed] [Google Scholar]
- 57.Di Lorenzo C, Angus AG, Patel AH. Hepatitis C virus evasion mechanisms from neutralizing antibodies. Viruses. 2011;3:2280–2300. doi: 10.3390/v3112280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takkenberg RB, Weegink CJ, Zaaijer HL, Reesink HW. New developments in antiviral therapy for chronic hepatitis B. Vox Sang. 2010;98:481–494. doi: 10.1111/j.1423-0410.2009.01282.x. [DOI] [PubMed] [Google Scholar]
- 59.Bhattacharya D, Thio CL. Review of hepatitis B therapeutics. Clin Infect Dis. 2010;51:1201–1208. doi: 10.1086/656624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang YZ, Liu L, Pan MM, Wang YM, Deng GH. Evolutionary pattern of full hepatitis B virus genome during sequential nucleos(t)ide analog therapy. Antiviral Res. 2011;90:116–125. doi: 10.1016/j.antiviral.2011.03.183. [DOI] [PubMed] [Google Scholar]
- 61.Maekawa S, Enomoto N. Viral factors influencing the response to the combination therapy of peginterferon plus ribavirin in chronic hepatitis C. J Gastroenterol. 2009;44:1009–1015. doi: 10.1007/s00535-009-0126-7. [DOI] [PubMed] [Google Scholar]
- 62.Kurbanov F, Tanaka Y, Matsuura K, Sugauchi F, Elkady A, Khan A, Hasegawa I, Ohno T, Tokuda H, Mizokami M. Positive selection of core 70Q variant genotype 1b hepatitis C virus strains induced by pegylated interferon and ribavirin. J Infect Dis. 2010;201:1663–1671. doi: 10.1086/652500. [DOI] [PubMed] [Google Scholar]
- 63.Hofmann WP, Sarrazin C, Kronenberger B, Schönberger B, Bruch K, Zeuzem S. Mutations within the CD81-binding sites and hypervariable region 2 of the envelope 2 protein: correlation with treatment response in hepatitis C virus-infected patients. J Infect Dis. 2003;187:982–987. doi: 10.1086/368221. [DOI] [PubMed] [Google Scholar]
- 64.Chambers TJ, Fan X, Droll DA, Hembrador E, Slater T, Nickells MW, Dustin LB, Dibisceglie AM. Quasispecies heterogeneity within the E1/E2 region as a pretreatment variable during pegylated interferon therapy of chronic hepatitis C virus infection. J Virol. 2005;79:3071–3083. doi: 10.1128/JVI.79.5.3071-3083.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaudy C, Moreau A, Veillon P, Temoin S, Lunel F, Goudeau A. Significance of pretreatment analysis of hepatitis C virus genotype 1b hypervariable region 1 sequences to predict antiviral outcome. J Clin Microbiol. 2003;41:3615–3622. doi: 10.1128/JCM.41.8.3615-3622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aurora R, Donlin MJ, Cannon NA, Tavis JE. Genome-wide hepatitis C virus amino acid covariance networks can predict response to antiviral therapy in humans. J Clin Invest. 2009;119:225–236. doi: 10.1172/JCI37085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeisel MB, Fofana I, Fafi-Kremer S, Baumert TF. Hepatitis C virus entry into hepatocytes: molecular mechanisms and targets for antiviral therapies. J Hepatol. 2011;54:566–576. doi: 10.1016/j.jhep.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 68.Ploss A, Dubuisson J. New advances in the molecular biology of hepatitis C virus infection: towards the identification of new treatment targets. Gut. 2012;61 Suppl 1:i25–i35. doi: 10.1136/gutjnl-2012-302048. [DOI] [PubMed] [Google Scholar]