

Abstract
We describe here the design, synthesis and thermodynamic characterization of fluorinated β-hairpin constructs. Introduction of hexafluoroleucine (Hfl) did not perturb β-hairpin formation, as judged by 1H NMR structures of four peptides determined to <1 Å backbone RMSDs, allowing direct comparison of thermodynamic stabilities of fluorinated peptides to their hydrocarbon counterparts. Judicious fluorination of peptides often results in increased thermal and chemical stability of the resultant folded structures. However, we found that when cross-strand residue partners were varied, the sidechain interaction energies followed the order Leu-Leu > Hfl-Leu > Hfl-Hfl. All peptides were more structured in 90% MeOH than in aqueous buffers. The peptides with Hfl-Leu or Hfl-Hfl cross-strand partners showed increased interaction energies in this solvent compared to water, in contrast to the insignificant effect on Leu-Leu. Our results inform the binding and assembly of peptides containing Hfl in the context of β-sheet structures and may be useful in interpreting binding of fluorinated ligands and peptides to biological targets.
Introduction
About a fifth of the drugs on the market and about a third of agrochemicals contain the element fluorine.1 Fluorine has intrigued chemists and biologists because of its unique properties and a near complete absence in soft tissue.2,3 Carbon-bound fluorine continues to resist being boxed into usual parametric molecular modeling programs. Therefore, a detailed understanding of the interactions of fluorinated molecules with biological targets remains an active area of research.
Fluorination modulates the properties of lipids4, pharmaceuticals1, and peptides5, where dramatic differences are observed depending upon the degree of fluorination. Fluorocarbons have exceedingly low polarizabilities for their size, and hence have lower interaction energies than hydrocarbons.6,7 The difference in interaction energy relative to size has been used to explain the immiscibility of hydrocarbons and fluorocarbons. However, it is not clear how this property relates to fluorinated amino acids in folded peptides, which have a lower degree of fluorination. The influence of fluorination on the transition temperature of vesicles from gel to liquid crystal phase, an indicator of increased interactions, has been investigated. In derivatives with fewer than five perfluorinated carbons the transition temperature decreases. However, with at least eight perfluorocarbons, the transition temperature increases.4,8 It has been suggested that as the length of the fluorinated chain increases, its low cohesion is offset by increased hydrophobicity. It is worthwhile to note that while perfluorination leads to molecules that are highly nonpolar, monofluorination can lead to dipolar interactions.9 Studies that directly compare the interaction of trifluoromethyl groups with both hydrocarbon and fluorinated binding partners are therefore valuable.10,11
Protein folding in water is frequently driven by the hydrophobic effect, therefore, increasing the hydrophobicity of an interior residue usually increases the stability of a folded structure.12 This has been effectively achieved by the substitution of a methyl group with a trifluoromethyl group.10,11,13–19 The hydrophobic effect is based on the principle that nonpolar surfaces cannot compete with the strong attraction of water for itself and are driven out of solvent.12 This principle applies equally to fluorinated and hydrocarbon groups. For example, the binding affinity to carbonic anhydrase was correlated to the surface area of the hydrophobic tail for cyclic, branched, and fluorinated ligands.20 When normalized for surface areas, the hydrophobicities of hydrocarbons and fluorocarbons are similar.20 This has also been observed with highly fluorinated coiled coil systems and β-peptide assemblies.21 More recent studies have examined the role of both hydrophobic surface area and inductive effects on the properties of ligands or proteins. In addition to their enhanced hydrophobic surface area, strongly electron-withdrawing fluorinated pendant groups perturb the Lewis basicity or hydrogen-bonding interactions of proximal functional groups.22,23 In previous studies, either the ligand or the protein was fluorinated, but not both. Herein, we have evaluated differences in the interactions of fluorinated and non-fluorinated compounds with both fluorinated and non-fluorinated binding partners.
Studies in our laboratory and others have focused on substituting Leu and Hfl into the core of peptides and proteins containing the coiled coil motif. However in such constructs, stabilities are intimately linked to packing, and the oligomerization state. Marsh and co-workers observed that in aqueous medium, Hfl (substituted at only one hydrophobic packing layer, therefore not “fluorous”) and Leu-substituted antiparallel 4-helix bundles form heterodimers, and suggested that fluorination does not lead to the segregation of fluorinated peptides from their hydrocarbon counterparts.24 However, we have previously demonstrated that in a different construct, Hfl-substituted coiled coils and Leu-substituted coiled coils strongly favor homodimer formation.10 In order to explore the interactions of Hfl in hydrophobic environments, Bilgiçer et al. substituted the core residues of a coiled coil motif with either Leu or Hfl, while the exterior residues were substituted with hydrophobic amino acids.13 The presence of the hydrophobic amino acids leads to partitioning of the peptides into micelles and peptides substituted with Hfl aggregated into higher order assemblies.13 These results showed that Hfl-substituted peptides oligomerize in the nonpolar context of membranes. In the coiled coil motifs described, 6–12 Hfl residues were incorporated into each peptide chain, resulting in oligomerized constructs containing 14–48 Hfl residues at the interface. Thus, these represent highly fluorinated systems.
We report here constructs with only one or two Hfl residues that allow quantitative evaluation of Hfl-Hfl and Hfl-Leu interactions. The degree of fluorination, as well as the environment, organic versus aqueous solution, influences the interactions of fluorinated amino acids. We report here a system designed to determine interaction energies of Leu and Hfl (hexafluoroleucine) side chains and dissect factors that contribute to the conformational stability of peptides.25,26 In addition, the envisaged constructs also allow evaluation of the extent to which fluorination stabilizes or destabilizes β-hairpin motifs.27–29 While much information has been gleaned from the α-helical coiled coil systems previously studied,10,11,13–19,21 we sought to incorporate fluorinated amino acids into a simpler scaffold (β-hairpins), allowing for facile synthesis and characterization. Such systems have been previously used to evaluate interaction energies and therefore provide a database for comparison. For instance, aromatic,30 π-cation,31 and hydrophobic32 interactions have been quantified using various β-turn scaffolds. Furthermore, β-hairpins can inhibit protein-protein interactions,33–36 bind specifically to ssDNA,37,38 and potentially serve as lead compounds for drug discovery. We studied cross-strand interactions in two complementary scaffolds. In one scaffold developed by Cochran, we interrogated interactions at non-hydrogen-bonded residues,39,40 and in another reported by Bartlett, we analyzed interactions between residues located at the free N and C termini.41–44 These results provide insight into how fluorine influences interactions between biological molecules – an important factor in drug design.1,45
Experimental Section
Synthesis
Synthesis of amino acids and peptides, purification procedures and characterization are included in the Supporting Information. A description of reagents, preparation of stock solutions, and buffers is also included.
Determination of Ceff for scaffold I peptides
The disulfide bonded scaffold I peptides were allowed to undergo thiol exchange in a mixture of oxidized and reduced glutathione where:
| Equation 1 |
GSH and GSSG are reduced and oxidized glutathione, respectively, and pepred and pepox are reduced and oxidized peptide, respectively. The stability of the scaffold I constructs was evaluated by comparing Ceff values for each peptide, where:39,40
| Equation 2 |
Higher Ceff values indicate that more of the peptide is in the folded form. The ratio of oxidized to reduced peptides was determined at 20°C, pH 8.1 and quantified by HPLC. We examined Ceff at 375, 37.5, and 18.75 μM peptide concentrations. In order to evaluate the constructs, Ceff at 18.75 μM peptide concentrations was converted to ΔG, where:
| Equation 3 |
The ΔG values are for the reaction shown in Equation 1. Note that the ΔG of folding cannot be directly measured, as Ceff is also a function of glutathione concentrations. Detailed experimental procedures at each concentration are provided in the Supporting Information.
Scaffold II: Determination of Fraction Folded (χβ)
In order to evaluate the stability of the peptides prepared with scaffold II, CD spectra from 320–240 nm were obtained at 15, 37.5 and 60 μM concentrations in 10 mM phosphate buffer, pH 7.0, 25 °C. Raw CD data at 282 nm were converted to molar ellipticity [θ]obs in deg·cm2/dmol using equation 4:42
| Equation 4 |
Where the CD signal at 282 nm was attributed exclusively to the Ati residue (1,2-dihydro-3(6H)-pyridinone) (Figure 1). Ati may be referred to by its single-letter amino acid code “@” (Figure 1). Molar ellipticities were converted to fraction β-turn (χβ) using the following equation:
| Equation 5 |
Figure 1.

Structures of Scaffold I and II peptides. (A) Variants LL1 through AL1 were prepared to examine scaffold I. (B) Molecular structures of Hfl and Ati (or single-letter code “@”). (C) Variants LL2 through GG2 were prepared to examine scaffold II. Substitutions were made at positions X1 and X2 (in red). For scaffold II, additional control folded and unfolded peptides were prepared with the sequences Val-Ati-Thr and Cyc = Val-DPro-Ala-Val-Ati-Val-DPro-Ala-Val-Val (cyclic), respectively.
The molar ellipticity for the unfolded state, [θ]0, was obtained from the linear tripeptide “V@T” (−2.62 × 104 deg·cm2/dmol, see Supporting Information). The molar ellipticity for the fully-folded state, [θ]100, was obtained from the cyclic control peptide (Cyc, Fig. 1) (−19.96 × 104 deg·cm2/dmol, see Supporting Information). The CD spectra showed no change in molar ellipticity over the concentration range studied (15 to 60 μM) suggesting that the construct is monomeric in the range studied. We believe this species to be a monomer, based on literature precedence, and our NMR studies on four of the peptides in 30% MeOH at 1 mM peptide concentrations. In order to improve the statistical significance of the data, χβ was measured five times for each peptide at 15 μM peptide concentrations. The free energies of folding (ΔG°XY, Figure 3A) were obtained using the following equation:
| Equation 6 |
Figure 3.
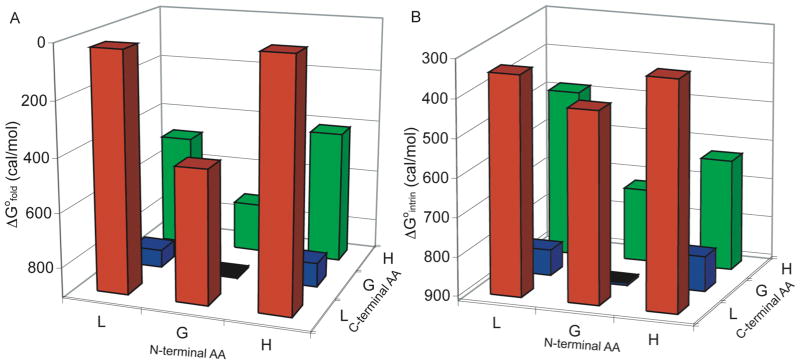
(A) ΔG°XY for Scaffold II Peptides. (B) ΔG°intrin for Scaffold II Peptides. Conditions: 15 μM peptide, 10 mM phosphate buffer, pH 7.0, 25 °C. The baseline is set at 912 cal/mol, corresponding to the stability of GG2. Intrinsic stabilities were calculated using Equation 7.
Similar procedures were used to determine ΔG°XY in 90% MeOH and 60% trifluoroethanol (TFE). Detailed experimental procedures are provided in the Supporting Information.
Scaffold II: Temperature-Dependent CD Measurements
Variable temperature data in aqueous solutions were collected at 5 μM concentrations from 5 to 95 °C and monitored at 282 nm. Variable temperature data in mixed organic/aqueous solutions were collected from 5 to 60 °C. Additional experimental details, denaturation curves, and a description of the thermodynamic analysis performed are provided in the Supporting Information.
Scaffold II: Determination of Peptide Structure by NMR
Detailed experimental procedures and calculations are provided in the Supporting Information. Briefly, 2D NMR experiments on scaffold II peptides were performed in 30% CD3OD at pH 7.0 on a Bruker AMX-500 spectrophotometer. Two-dimensional ROESY spectra were collected for each peptide with 100 ms mixing times. Additional ROESY, NOESY, COSY, and TOCSY spectra were collected as described in the Supporting Information. Spectral assignments were made as described in the Supporting Information. Restraints and additional parameter files were developed and input into CNSsolve v1.146 for structural refinement, as described in the Supporting Information.
Results
Construct Design
We surveyed the scaffolds previously used to study interaction energies, and chose scaffolds I and II that were synthetically tractable.39,41–44,47,48 Cochran and co-workers identified a relatively short and stable β-turn construct47 (scaffold I; Figure 1A). The hairpin turn is promoted by a Gly-Asn sequence, and the N and C-terminal Cys residues form a disulfide bond, stabilizing the hairpin. They determined the hairpin stability of a series of variants by measuring the extent of disulfide bond formation (Ceff) between the terminal cysteine residues in a redox-controlled environment.40 It was reasoned that hairpin formation would position the terminal residues to be proximal promoting the formation of a disulfide bond. The Ceff values were correlated with Hα chemical shift values for several residues, establishing that increased Ceff corresponds with increased β-hairpin stability. Cochran and co-workers systematically substituted the amino acids at interacting residues 3 and 8 (X1 and X2; Figure 1).40 The backbone atoms of these residues are not engaged in H-bonding, so any changes in interaction energies are a direct result of sidechain interactions. In general, Cochran and co-workers observed that increasing the hydrophobicity of the X1 and X2 residues increased the stability of the turn.40 We anticipated that substitution of Leu with Hfl at one or both positions would increase the β-turn propensity of the sequence due to the enhanced hydrophobicity of Hfl relative to Leu. However, it was unclear if Leu-Leu, Hfl-Leu, or Hfl-Hfl pairings would contribute to the stability of the hairpin in a synergistic manner. A series of variants with Leu, Hfl, and Ala substitutions (Figure 1A) were synthesized to evaluate these interactions. Leu and Hfl were substituted in all possible combinations, while Ala was substituted only at the X1 position. Peptide AL1 has alanine at X1 and leucine at X2, where the remainder of the sequence is defined by scaffold I. Peptides LL1, LH1, HL1, HH1, and AH1 are named so that H in the peptide identifier stands for hexafluoroleucine, while L and A stand for leucine and alanine (Figure 1A).
Bartlett and co-workers have developed a different construct (Scaffold II) to quantify interactions between side chains in β-hairpins.41–44 In this construct, the turn is promoted by a DPro-Ala sequence. Introduction of an Ati residue restricts the flexibility of the backbone, leading to β-sheet stabilization (Figure 1B). Although Ati is able to engage in cross-strand H-bonds of the usual register, it lacks the ability to H-bond on the external face of the hairpin, mitigating oligomerization of the motif. Furthermore, UV absorption allows for detection of the folded state by monitoring of the CD signal at 282 nm.41,42 This provides a technical advantage, since natural β-turn peptides are generally only modestly stable in aqueous solution. A series of variants was prepared to study cross-strand interactions at the terminal positions (X1 and X2).41 Hydrophobic residues were consistently preferred, and favorable side chain-side chain interactions were observed for hydrophobic residues. We envisioned that this system would reveal differences between Leu-Leu, Hfl-Leu, and Hfl-Hfl interactions. Furthermore, this system is amenable to thermal analysis, allowing determination of thermodynamic parameters that dictate folding. Constructs with Leu, Hfl, and Gly substitutions in all possible combinations were synthesized (Figure 1C). The peptide GL2 was composed of Gly at X1 and Leu at X2, where the remainder of the sequence was defined by scaffold II. The peptides LL2, LH2, HL2, HH2, HG2, LG2, GH2, and GG2 were named according to the convention described previously (Figure 1C).
Determination of Peptide Stabilities and Concentration-Dependent Effects
For scaffold I peptides, Ceff was measured at 18.75, 37.5, and 375 μM peptide concentrations. At 375 μM peptide concentrations, a slightly higher Ceff was observed for each peptide. Minor differences in Ceff at 375 μM concentrations were expected, since a different concentration of the glutathione stock solutions was required for these experiments (see Supporting Information). Ceff for peptides at 375 μM peptide concentrations for the non-fluorinated peptides was found to be in agreement with previous reports.39,40 HL1 showed anomalously large Ceff and variability at 375 μM, but behaved consistently at lower concentrations (Figure 2A).
Figure 2.
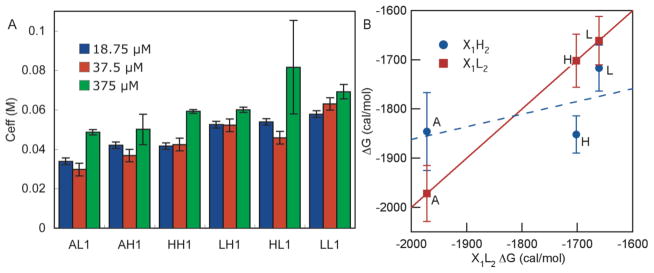
(A) Ceff values at 18.75, 37.5, and 375 μM Peptide Concentrations. See Supporting Information for experimental conditions. At 18.75 and 37.5 μM, error bars represent the 95% confidence interval from multiple comparison tests, where the data was collected from three independent experiments for a total of six HPLC traces. The error bars for experiments performed at 375 μM represent the standard deviations of three independent experiments, where the average of five HPLC traces were used for each experiment. Uniformity of the data at 18.75 μM and 37.5 μM was determined using Bartlett’s test (p = 0.05). (B) ΔG for glutathione-peptide disulfide exchange at 18.75 μM peptide concentrations, scaffold I. Peptides with Hfl at X2 (blue circles, X1-Hfl2 series) are plotted against the corresponding peptide that contains Leu at X2 (red squares, X1-Leu2 series). The identity of the amino acid at the X1 position is indicated next to each point. The X1-Leu2 series is plotted against itself (red squares) for reference. Error bars are the standard deviations.
For scaffold II peptides, thermodynamic parameters were extracted assuming a two-state model where the peptide is monomeric, and either fully folded (state 1) or unfolded (state 2). This model assumes that the unfolded state does not contribute to the CD signal that we are monitoring for the folded peptide. This model further assumes that, if present, partially folded states are at insignificant concentrations, or do not significantly affect the CD signal. These assumptions are consistent with the observation of cooperative stabilizing effects upon simultaneous mutation of the terminal or interior residues.41 A two-state model is also consistent with 1-dimensional 1H NMR spectra of LL2 and LH2 in buffer, that showed two resonances for each amide proton in aqueous solution at ambient temperatures, and the 19F NMR spectrum of LH2, which showed two resonances for each trifluoromethyl group at ambient temperature (see Supplementary Information). In aqueous solution, the observation of two signals for each amide is probably due to cis/trans isomerization of 3-Val-4-D-Pro amide bond.).49 The cis conformation of this bond provides the appropriate geometry for the turn sequence while the trans amide bond does not.49 Thus, the observed cis/trans isomerization of this bond suggests that a proportion of the unfolded peptide was in the extended random coil conformation, whereas the folded peptide can only be formed upon isomerization.49 In 30% MeOH at 283 K, one set of resonances was observed, consistent with a peptide in the favored cis conformation of the 3-Val-4-D-Pro amide bond.
In addition, for scaffold II peptides, no concentration dependence of the molar ellipticities was observed from 15 -60 μM concentrations in buffer. All NOESY crosspeaks in 30% methanol (1 mM peptide) could be accounted for, assuming a monomeric species. Together the data suggest a single monomeric species in solution. Therefore, ΔG°XY was calculated from χβ, as determined by CD, according to equations 5 and 6.
Determination of β-sheet propensities for Hfl
β-sheet propensities are determined from the relative stabilities of constructs in which the side chains of interest do not interact with other side chains. Minor and Kim50 found that β-sheet propensities for natural amino acids vary by ~2 kcal/mol in a model based on the streptococcal protein G β1 domain (β1-domain). They also demonstrated that β-sheet propensity increases in the series Ala, Val, Ile, 50 and propensities follow the same order for scaffold I40 and scaffold II41 peptides. These previously established correlations between β-sheet propensity and hydrophobicity suggest our model systems are appropriate. It is worthwhile to note that context plays a significant role in β-sheet propensities51, and indeed we observe that context influences the β-sheet propensities of Leu and Hfl. For scaffold I peptides, we found that AH1 was more stable than AL1 by 130 cal/mol at 18.75 μM peptide concentration, suggesting that Hfl had only a slightly higher β-sheet propensity than Leu (Figure 2). Examination of the stabilities (ΔGXY) of the Gly derivatives for scaffold II peptides (Figure 3A, peptides GL2, LG2, GH2, and HG2) showed that Leu and Hfl have similar β-sheet propensities, although β-sheet propensity of Hfl was higher than Leu at the C-terminus, but lower than Leu at the N-terminus. In this case, the overall stability of scaffold II was more sequence-dependent, varying from 841 to 413 cal/mol. The basicity of the amino group of Hfl is 100-fold lower than that of Leu, reflecting an inductive effect of the CF3 groups on the pKa52 underscoring that electrostatic interactions between the N and C termini of the folded hairpin contribute to the overall stability. Since scaffold I peptides are substituted at non-hydrogen bonding positions, they should be less sensitive to electronic effects. Further, it is possible that diagonal interactions between the sidechains at positions 1 (Leu or Hfl) and 6 (Val) are more favorable for Leu than for Hfl in scaffold II. Work by Gellman and co-workers has demonstrated that these diagonal interactions contribute to the overall stability of β-hairpins, and that this pairing is directional, based on the overall twist of the hairpin sequence53. Indeed, between residues 1 and 6 we observe two NOE crosspeaks for HH2 and HL2, three crosspeaks for LH2, and four crosspeaks for LL2. This suggests stronger 1–6 diagonal interactions for LH2 and LL2 than for HH2 and HL2.
Cheng and co-workers recently determined the β-sheet propensity of selected fluorinated amino acids by substitutions at residue 53 of the β1-domain and examining the stability of the prepared constructs.27 Substitutions at position 53 of β1 were more sensitive than scaffold I or scaffold II, nevertheless, the results of Cheng and coworkers are consistent with our observations. For example, substituting Ala for Leu at residue 53 of β1-domain leads to ~1 kcal/mol increase in stability, while a similar substitution on scaffold I to provided only a 270 cal/mol increase. Taken together, these results demonstrate that propensities are dependent on sequence context, and in most instances are higher for Hfl than for Leu. Raleigh and co-workers used a buried position and found that trifluorovaline provides even higher stability.27 b
Determination of Interaction Energies for Scaffold I
Smith and Regan first demonstrated that side chain interaction energies were significant in β-sheet folding in β1 model proteins.54 Using a double-mutant analysis, they observed interaction energies for aromatic and hydrophobic amino acids of ~0.2–0.6 kcal/mol.54 Their methods were used to analyze scaffold II and are described in the next section. Cochran and co-workers argued that the synergistic effects observed by Smith and Regan did not necessarily imply greater interaction energies, but were related to intrinsic properties of the individual amino acids.55 Using scaffold I and other constructs, they demonstrated that amino acid preference at one position was the same if the cross-strand position was occupied by aromatic (Tyr and Phe), hydrophobic (Val and Leu), or hydrophilic (Thr) amino acids. Moreover, a linear free-energy relationship was observed between the data sets where deviations from linearity would have indicated specific side chain-side chain interactions. We employed the analysis used by Cochran and co-workers to determine if differences in stabilities for scaffold I peptides related to intrinsic properties of the amino acids or side chain-side chain interactions.39,40 This method was particularly applicable to this scaffold, since substitutions at positions X1 and X2 are essentially equivalent.39,40 The ΔG of the X1-Hfl2 series was compared to the X1-Leu2 series in Figure 2B. A linear free energy relationship was not observed between the X1-Leu2 and X1-Hfl2 series, as indicated by the R2 value of 0.312 for a linear fit. HH1 was less stable than LL1 by 190 cal/mol. Both the HL1 and LH1 analogs displayed Ceff values similar to that of LL1 at 18.75 μM peptide concentration, indicating that the Leu-Hfl interaction or Leu-Leu interaction was more stabilizing than the Hfl-Hfl interaction.
While Hfl has a similar β-sheet propensity to Leu, it forms weaker interactions with Leu or Hfl. It is possible that the larger aliphatic sidechain of Hfl in the disulfide-bound peptide diminishes the stability of HH1. However, large aromatic side chains (in particular Trp) have been previously accommodated in this scaffold resulting in highly stable constructs.56
Determination of Intrinsic Stabilities and Interaction Energies for Scaffold II
For scaffold II peptides, both the intrinsic stabilities and the interaction energies were calculated using a double-mutant analysis method. Intrinsic stability accounts for the contributions of an individual amino acid to the stability of a construct in the absence of interactions with its cross-strand partner.41 The intrinsic stability for each peptide was calculated from the corresponding peptides that have Gly at the cross-strand position. For example, ΔG° for LH2 was calculated by summing the stability of LG2 and GH2, then subtracting the stability of GG2. The general equation is:
| Equation 7 |
As shown in Figure 3B, in the absence of any side chain-side chain interactions, HL2 has the highest intrinsic stability. This reflects the preference for Hfl at the N-terminus and Leu at the C–terminus. The interaction energy reflects the contribution of side chain interactions to the overall stability. Interaction energies between side chains of interest were obtained by subtracting the ΔG°XY from the intrinsic stability:41
| Equation 8 |
As shown in Figure 4B, the interaction energies obtained from scaffold II were consistent with those obtained from scaffold I. Hfl-Hfl side chains exhibit weak interactions, contributing only ~210 cal/mol to stability. Leu-Hfl side chains exhibited modest interactions, from 310 to 350 cal/mol. Leu-Leu interactions were about as strong as Leu-Hfl interactions, contributing 320 cal/mol of stability at 298 K. While the differences between side chain-side chain interaction energies were small, these results clearly demonstrate that Hfl interactions were not guided by the hydrophobic effect alone. If this were the case, Hfl-Hfl interactions would have been the strongest rather than the weakest.
Figure 4.
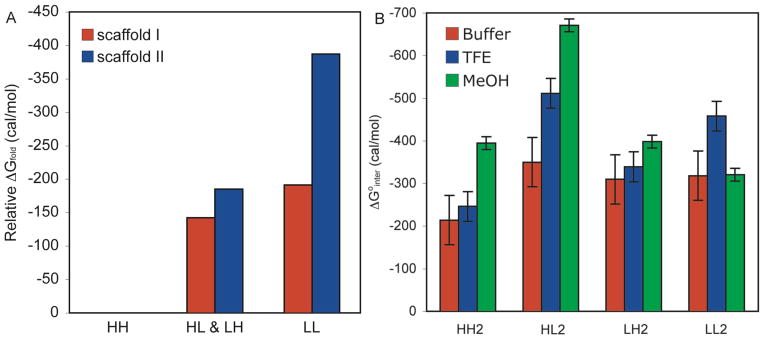
(A) Comparison of Scaffold I and Scaffold II Stabilities. The stabilities relative to HH are shown. The average stability for the peptides substituted with both Hfl and Leu is shown. For both scaffolds, peptides substituted with Hfl at both X1 and X2 were the least stable. (B) ΔG°interact for Scaffold II Peptides. Red, 15 μM peptide, 10 mM phosphate buffer, pH 7.0, 25 °C. Blue, 15 μM peptide, 40% 10 mM phosphate buffer, pH 7.0, 60% TFE, 25 °C. Green, 15 μM peptide, 10% 10 mM phosphate buffer, pH 7.0, 90% MeOH, 25 °C. Error bars are the 95% confidence intervals from five independent measurements as determined using a multiple comparison test. Interaction energies were calculated using Equation 8.
Scaffold II: Measurement of Thermodynamic Parameters in Various Media
Temperature dependence of hairpin stability has been used to extract the thermodynamic parameters ΔS, ΔH, and ΔCp of folding. Searle and co-workers have observed entropy-driven ΔG values for 16-residue hairpins, which indicated that the burial of hydrophobic surface area was energetically relevant to hairpin folding.57,58 The addition of MeOH improved the stability of the constructs, and resulted in enthalpy-driven unfolding.57 We used a similar strategy to study scaffold II peptides (see Supporting Information). With the exception of LH2, the ΔS° values were negative for these peptides, where the driving force for folding was found in ΔH° at 298 K. Given the small size of the construct (8 residues) it was not surprising that enthalpic factors (such as H-bonding) play a more significant role in folding. These data were analyzed to determine the contribution of side chain-side chain interaction energies to ΔH°interact, ΔS°interact, and ΔCp interact. Interaction entropies, enthalpies, and heat capacities were calculated from the corresponding Gly derivatives as described for ΔG°XY(inter) in equations 7 and 8. For these peptides, the magnitude of both the interaction entropy and enthalpy increased with increasing peptide stability.
For each peptide, χβ was determined in 60% TFE and 90% MeOH (see Supporting Information). All peptides displayed increasing χβ with increasing portions of organic solvents. Furthermore, temperature-dependent CD data were collected in 90% MeOH from 5–60 °C and in 60% TFE. These data were fit for the thermodynamic parameters ΔS°, ΔH°, and ΔCp and interaction entropies and enthalpies were calculated (see Supporting Information). In all cases ΔG°XY(inter) was enthalpy-driven at 298 K, and the more stable peptides had a more negative ΔH°. The interaction energies are plotted in Figure 4B. Hfl-Hfl and Hfl-Leu side chain interactions were stronger in 90% MeOH than in aqueous buffer, whereas Leu-Leu interactions were similar in 90% MeOH to those observed in water. In 60% TFE, Leu-Leu interactions were strengthened, but the effect on Hfl-Hfl and Hfl-Leu interactions was more modest than in 90% MeOH suggesting that fluorinated solvents influence intermolecular interactions in nontrivial ways.
Scaffold II: Determination of Structure by NMR
We performed NMR experiments to verify that all peptides form expected β-hairpin structures, and that the sidechains of interest interact in the folded form. We chose 30% MeOH for solution conditions as all members of the LL2, HL2, LH2, and HH2 peptide series were well folded as judged from the CD spectra and are present as monomers (as judged from the 1H and 19F NMR spectra). The 1H NMR spectra (pH 7.0, 10 °C) showed good dispersion and narrow linewidths, suggesting that the peptides are well-folded and good candidates for structure determination using NMR.
For the four residues with observable NH resonances (residues 3, 6, 7, and 8), the three-bond coupling constants, 3JHN,Hα, were between 8.4–9.5 Hz for all peptides, indicating an HN-Hα dihedral angle of approximately −139°, consistent with an antiparallel β-sheet.59 The formation of β-hairpin structures is supported by the presence of strong Hα-NH ROE cross-peaks for residues (i, i+1) and cross-strand ROE peaks between residues 2 and 7.59 All of the peptides showed cross-peaks that included sidechain protons indicating cross-strand interactions. All peptides showed crosspeaks between residues 1 and 6, as expected for a β-hairpin with a type I’ or type II’ turn 53. HH2, LH2, and HL2 showed crosspeaks between residues 2 and 7, while HL2 and LL2 showed crosspeaks between residues 1 and 8, indicating that β-hairpin structure extends to the termini of the sequence. Hα resonances of residues 1, 3, and 7 were shifted downfield from the resonances expected for a random coil (Figure 5), consistent with β-sheet formation while that of residues 4 and 8 were shifted upfield, consistent with a turn sequence at residue 4 and residue 8 being at the C-terminus.60 The observation of full intensities for NH resonances at pH 7.0 suggested that these amides were protected from the solvent and consistent with inter-strand H-bonding.
Figure 5.

Hα Chemical shift deviations from random coil values for (a) HH2, (b) HL2, (c) LH2, and (d) LL2. Chemical shift deviations are not shown for Hfl since insufficient data is available. Conditions: 30% CD3OD, 10 mM sodium phosphate, 10 °C, pH 7.0.
Approximately 73 conformational restraints were obtained for each peptide and were input into CNSsolve for structure calculation (Table 1). The resulting statistical analysis of the final structures showed total energies of about 8.8 kcal/mol and the backbone rms deviations of superimposed structures between 0.4 and 0.8 Å. An overlay of the peptide backbones and NMR ensembles for each peptide are provided in the Supporting Information. The average structures for the peptides in this series show similar conformations in solution and that the sidechains of interest interact (Figure 6). In addition, the sidechains alternated from one side of the backbone to the other, consistent with the expected β-hairpin motif. A preservation of conformation in the peptides allowed us to correlate the nature of interactions with fluorinated amino acid side chains to the observed energetic difference in side chain-side chain interactions. For each peptide, the hydrophobic sidechains of residues one and eight interacted in the ensemble of structures, though not necessarily in the averaged structure. Individual members of all of the ensembles showed significant deviations from the average structure, particularly in residues 1, 2, and 8 that may be in part due to flexibility at the termini. Part of the observed flexibility for residues 1, 2, and 4 is contributed by their lack of NH protons thus missing structural constraints available in the other residues.
Table 1.
NMR Structural Data and Refinement Statistics
| NMR Structural Data and Refinement Statistics | LL2 | HH2 | HL2 | LH2 |
|---|---|---|---|---|
| Experimental restraints | ||||
|
| ||||
| distance restraints from ROEs/NOEs | 48 | 68 | 67 | 77 |
| dihedral angle restraints | 10 | 8 | 6 | 9 |
| H-bonding restraints | 6 | 6 | 6 | 6 |
|
| ||||
| RMS deviations from experimental data | ||||
|
| ||||
| average distance restraint violation (Å) | 0.048 ± 0.013 | 0.044 ± 0.012 | 0.032 ± 0.097 | 0.076 ± 0.007 |
| distance restraint violations > 0.5 Å | 0 | 0 | 0 | 0 |
| average dihedral angle restraint violations | 0.13 ± 0.14 | 0.21 ± 0.24 | 0.086 ± 0.075 | 0.231 ± 0.086 |
| dihedral angle restraint violations > 5° | 0 ± 0.00 | 0 ± 0.00 | 0 ± 0.00 | 0 ± 0.00 |
|
| ||||
| RMS deviations from ideal stereochemistry | ||||
|
| ||||
| bonds (Å) | 0.0033 ± 0.0004 | 0.0038 ± 0.0004 | 0.0032 ± 0.0006 | 0.0051 ± 0.0051 |
| angles (deg) | 0.660 ± 0.029 | 0.771 ± 0.032 | 0.671 ± 0.028 | 0.866 ± 0.035 |
| impropers (deg) | 0.328 ± 0.057 | 0.10 ± 0.32 | 0.37 ± 0.12 | 0.770 ± 0.057 |
|
| ||||
| Ramachandran analysis of the structures* | ||||
|
| ||||
| residues in favored regions | 3 | 2 | 2 | 1 |
| residues in additionally allowed regions | 3 | 2 | 3 | 4 |
| residues in generously allowed regions | 0 | 0 | 0 | 0 |
| residues in disallowed regions | 0 | 0 | 0 | 0 |
|
| ||||
| Lennard-Jones potential energies | ||||
|
| ||||
| after annealing (kcal·mol-1) | 29.9 ± 9.7 | 11.2 ± 4.5 | 25.8 ± 4.0 | 65.6 ± 6.9 |
| ensemble average (kcal·mol-1) | 5.8 ± 6.1 | −13 ± 11 | −1.6 ± 6.7 | 43.9 ± 9.9 |
|
| ||||
| Coordinate precision (Å) | ||||
|
| ||||
| backbone | 0.82 ± 0.32 | 0.35 ± 0.6 | 0.44 ± 0.21 | 0.41 ± 0.17 |
| heavy atoms | 1.64 ± 0.39 | 1.20 ± 0.27 | 1.35 ± 0.44 | 1.06 ± 0.23 |
| sidechain precision, residues 1,8 ** | 2.5, 2.4 | 2.7, 1.1 | 2.2, 1.1 | 2.5, 1.3 |
| No. of ensemble structures of 30 where sidechains 1& 8 are very close, close*** | 5, 1 | 0, 20 | 21, 9 | 6, 6 |
Hfl, Ati, and Pro residues not included in this analysis.
The coordinate precision of the heavy atoms in sidechains 1 & 8 respectively after annealing
H-H, H-F, or F-F distances between residues 1 and 8 that are “very close”(<3.6 Å apart), and “close” (between 3.6 Å and 5.3 Å apart).
Figure 6.

NMR solution structures for scaffold II peptides. (A) Average structures for LL2 peptide, (B) HH2, (C) LH2, (D) HL2. N = blue, O = red, C = grey, F = green, H = light grey. The terminal H and F atoms on residues 1 and 8 are depicted as balls. Conditions: 30% CD3OD, 10 mM sodium phosphate, 10 °C, pH 7.0.
In order to evaluate the extent to which side chains 1 and 8 interact, the distances between Leu methyl protons and the analogous fluorines in Hfl were measured in the structural ensembles. A “very close” interaction was scored for distances of <3.6 Å, and “close” interactions were scored for distances between 3.6 and 5.3 Å. Sidechain interactions were observed in all peptides (Table 1).39 For LL2, five structures displayed very close interactions and one structure had close interactions. The interaction distance for the average was only 3.8 Å, indicating that these residues were on average proximal, but more dynamic than in other peptides. For HH2, close interactions were observed in 20 structures, but very close interactions were not observed suggesting that the larger trifluoromethyl groups tend to be further apart than methyl groups. The interactions were observed for HL2 in all 30 structures, whereas interactions were observed in only 12 structures for LH2. Interestingly, we observed that flexibility is not an indicator of overall stability. LL2 is the most flexible peptide, as judged by the coordinate precision of the backbone atoms (Table 1), yet it is more stable than the more rigid HH2. Notably, LL2 has a favorable energetic contribution from TΔS°interact while HH2 does not (see Supplemental Information), illustrating the complex interplay of entropic and enthalpic parameters in determining interaction energies and peptide stabilities.
Discussion
When considering conformational stabilities of peptides, steric clashes, backbone perturbations, inductive effects22,61, and the interaction energies of proximal sidechains all play an important role. We examined two series of peptides to demonstrate the effect of side chain interactions on conformational stability. We chose to study hairpin constructs, where sidechains are partially solvent-exposed, and thus able to accommodate larger groups. We demonstrated by NMR that the substitution of Leu with Hfl does not significantly perturb the peptide backbone and further confirmed that sidechain sterics are not an issue for scaffold II peptides. We found similar results when substitutions were performed at non-hydrogen bonded positions on a peptide (scaffold I) and the N and C termini of a peptide (scaffold II). This demonstrates that the observed differences in interaction energies are not the result of inductive effects that may perturb hydrogen bonding of backbone atoms, as these effects would be structure-specific. Therefore, our study is directly able to compare interactions of Leu and Hfl.
One might expect that the Hfl-substituted constructs would be more stable than Leu-substituted constructs, since Hfl is more hydrophobic. However, we found that Hfl-Hfl interactions were weaker than Leu-Leu or Leu-Hfl interactions. This demonstrates that the effect of substitution of hydrogen with fluorine depends upon the subtle interplay of polarizability, dipolar interactions, and hydrophobicity. It is possible that the low polarizability relative to volume of CF3 vs. CH3 influences the interaction energies of Hfl.62 We also found that the Hfl-Leu interface had an interaction energy that was similar to the Leu-Leu interface in water. It is possible that this interaction is influenced by dipolar interactions, since the C-F bond (μ = 1.85 D), and the C-H bond (μ = 0.4 D) have dipoles in opposite directions. Dipolar interactions must also be considered when examining solvent effects, as dipolar interactions are often magnified with decreasing solvent polarity.63 The differences observed between interaction energies in 90% MeOH and 60% TFE for each of the sidechain pairs serve only to highlight the complexity of these interactions.
In this construct, the side chains are not well-shielded from water in the folded state. In contrast, previous work demonstrated that fluorinated amino acid substitutions into coiled coils were generally stabilizing. An understanding of how these interactions were altered by the hydrophobicity of the medium directs us towards methods for targeting membrane-bound proteins or hydrophobic binding pockets.
This construct may also be used to probe other molecular interactions, such as interactions of Hfl with aromatic residues. Information on these interactions may have broader implications in drug design and improve our fundamental understanding of interactions involving fluorinated compounds.
Conclusions
A methyl to trifluoromethyl substitution is frequently used in pharmaceutical design, as this substitution can greatly alter the specificity and potency of a drug. For example, the changing the trifluoromethyl group on fluoxetine (Prozac) to a methyl group leads to a 26 fold decrease in specificity.64 We have evaluated the interaction energies of Hfl-Hfl, Leu-Hfl and Leu-Leu in two different scaffolds to analyze the interactions of trifluoromethyl with both hydrocarbon and fluorocarbon groups in β-hairpin motifs. Our results suggest that the binding of methyl to trifluoromethyl groups depend on more than just size and hydrophobicity and advance our understanding of interactions of fluorinated amino acids in biological contexts.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM65500 to K.K.). The ESI–MS and NMR facilities at Tufts are supported by the NSF (0320783 and 0821508). GC was supported as a GAANN fellow for part of this work (US Department of Education). We thank Vijay K. Murthy, Ragnhild Whitaker, and Yuqi Liu for helpful discussions.
Footnotes
Supporting Information Available
Synthetic methods, accompanying analytical data, thermodynamic analysis, NMR fitting procedures, CD spectra and fitting curves, and HPLC traces. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Muller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 2.Yoder NC, Yuksel D, Dafik L, Kumar K. Curr Opin Chem Biol. 2006;10:576–583. doi: 10.1016/j.cbpa.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Dafik L, d’Alarcao M, Kumar K. J Med Chem. 2010;53:4277–4284. doi: 10.1021/jm100374g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riess JG. Tetrahedron. 2002;58:4113–4131. [Google Scholar]
- 5.Meng H, Clark GA, Kumar K. In: Fluorine in Bioorganic and Medicinal Chemistry. Ojima I, Taguchi T, editors. Blackwell-Wiley Publishing; New York: 2009. [Google Scholar]
- 6.Scott RL. J Am Chem Soc. 1948;70:4090–4093. doi: 10.1021/ja01192a036. [DOI] [PubMed] [Google Scholar]
- 7.Hildebrand JH, Cochran DRF. J Am Chem Soc. 1949;71:22–25. [Google Scholar]
- 8.Santaella C, Vierling P, Riess JG, Gulikkrzywicki T, Gulik A, Monasse B. Biochim Biophys Acta-Biomembr. 1994;1190:25–39. doi: 10.1016/0005-2736(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 9.Biffinger JC, Kim HW, DiMagno SG. Chembiochem. 2004;5:622–627. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 10.Bilgicer B, Xing X, Kumar K. J Am Chem Soc. 2001;123:11815–11816. doi: 10.1021/ja016767o. [DOI] [PubMed] [Google Scholar]
- 11.Bilgicer B, Kumar K. Tetrahedron. 2002;58:4105. [Google Scholar]
- 12.Widom B, Bhimalapuram P, Koga K. Phys Chem Chem Phys. 2003;5:3085–3093. [Google Scholar]
- 13.Bilgicer B, Kumar K. Proc Nat Acad Sci. 2004;101:15324–15329. doi: 10.1073/pnas.0403314101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bilgicer B, Fichera A, Kumar K. J Am Chem Soc. 2001;123:4393–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]
- 15.Stevens MM, Flynn NT, Wang C, Tirrell DA, Langer R. Adv Mater. 2004;16:915–918. [Google Scholar]
- 16.Son S, Tanrikulu IC, Tirrell DA. Chembiochem. 2006;7:1251–1257. doi: 10.1002/cbic.200500420. [DOI] [PubMed] [Google Scholar]
- 17.Wang P, Tang Y, Tirrell DA. J Am Chem Soc. 2003;125:6900–6906. doi: 10.1021/ja0298287. [DOI] [PubMed] [Google Scholar]
- 18.Niemz A, Tirrell DA. J Am Chem Soc. 2001;123:7407–7413. doi: 10.1021/ja004351p. [DOI] [PubMed] [Google Scholar]
- 19.Tang Y, Ghirlanda G, Vaidehi N, Kua J, Mainz DT, Goddard WA, DeGrado WF, Tirrell DA. Biochemistry. 2001;40:2790–2796. doi: 10.1021/bi0022588. [DOI] [PubMed] [Google Scholar]
- 20.Gao JM, Qiao S, Whitesides GM. J Med Chem. 1995;38:2292–2301. doi: 10.1021/jm00013a005. [DOI] [PubMed] [Google Scholar]
- 21.(a) Molski M, Goodman J, Craig C, Meng H, Kumar K, Schepartz A. J Am Chem Soc. 2010;132:3658–3659. doi: 10.1021/ja910903c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Buer BC, Meagher JL, Stuckey JA, Marsh ENG. Proc Natl Acad Sci USA. 2012;109:4810–4815. doi: 10.1073/pnas.1120112109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnamurthy VM, Bohall BR, Kim CY, Moustakas DT, Christianson DW, Whitesides GM. Chem-Asian J. 2007;2:94–105. doi: 10.1002/asia.200600360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee A, Mirica KA, Whitesides GM. J Phys Chem B. 2011;115:1199–1210. doi: 10.1021/jp107765h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottler LM, de la Salud-Bea R, Marsh ENG. Biochemistry. 2008;47:4484–4490. doi: 10.1021/bi702476f. [DOI] [PubMed] [Google Scholar]
- 25.Meng H, Krishnaji ST, Beinborn M, Kumar K. J Med Chem. 2008;51:7303–7307. doi: 10.1021/jm8008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng H, Kumar K. J Am Chem Soc. 2007;129:15615–15622. doi: 10.1021/ja075373f. [DOI] [PubMed] [Google Scholar]
- 27.Chiu HP, Kokona B, Fairman R, Cheng RP. J Am Chem Soc. 2009;131:13192. doi: 10.1021/ja903631h. [DOI] [PubMed] [Google Scholar]
- 28.Senguen FT, Doran TM, Anderson EA, Nilsson BL. Mol Biosyst. 2011;7:497–510. doi: 10.1039/c0mb00210k. [DOI] [PubMed] [Google Scholar]
- 29.Senguen FT, Lee NR, Gu XF, Ryan DM, Doran TM, Anderson EA, Nilsson BL. Mol Biosyst. 2011;7:486–496. doi: 10.1039/c0mb00080a. [DOI] [PubMed] [Google Scholar]
- 30.Tatko CD, Waters ML. J Am Chem Soc. 2002;124:9372–9373. doi: 10.1021/ja0262481. [DOI] [PubMed] [Google Scholar]
- 31.Tsou LK, Tatko CD, Waters ML. J Am Chem Soc. 2002;124:14917–14921. doi: 10.1021/ja026721a. [DOI] [PubMed] [Google Scholar]
- 32.Griffiths-Jones SR, Maynard AJ, Searle MS. J Mol Biol. 1999;292:1051–1069. doi: 10.1006/jmbi.1999.3119. [DOI] [PubMed] [Google Scholar]
- 33.Gordon NC, Pan B, Hymowitz SG, Yin JP, Kelley RF, Cochran AG, Yan MH, Dixit VM, Fairbrother WJ, Starovasnik MA. Biochemistry. 2003;42:5977–5983. doi: 10.1021/bi034017g. [DOI] [PubMed] [Google Scholar]
- 34.Skelton NJ, Chen YM, Dubree N, Quan C, Jackson DY, Cochran A, Zobel K, Deshayes K, Baca M, Pisabarro MT, Lowman HB. Biochemistry. 2001;40:8487–8498. doi: 10.1021/bi0103866. [DOI] [PubMed] [Google Scholar]
- 35.Fairbrother WJ, Christinger HW, Cochran AG, Fuh C, Keenan CJ, Quan C, Shriver SK, Tom JYK, Wells JA, Cunningham BC. Biochemistry. 1998;37:17754–17764. doi: 10.1021/bi981931e. [DOI] [PubMed] [Google Scholar]
- 36.Wiesmann C, Christinger HW, Cochran AG, Cunningham BC, Fairbrother WJ, Keenan CJ, Meng G, de Vos AM. Biochemistry. 1998;37:17765–17772. doi: 10.1021/bi9819327. [DOI] [PubMed] [Google Scholar]
- 37.Butterfield SM, Sweeney MM, Waters ML. J Org Chem. 2005;70:1105–1114. doi: 10.1021/jo0491105. [DOI] [PubMed] [Google Scholar]
- 38.Butterfield SM, Cooper WJ, Waters ML. J Am Chem Soc. 2005;127:24–25. doi: 10.1021/ja045002o. [DOI] [PubMed] [Google Scholar]
- 39.Cochran AG, Tong RT, Starovasnik MA, Park EJ, McDowell RS, Theaker JE, Skelton NJ. J Am Chem Soc. 2001;123:625–632. doi: 10.1021/ja003369x. [DOI] [PubMed] [Google Scholar]
- 40.Russell SJ, Cochran AG. J Am Chem Soc. 2000;122:12600–12601. [Google Scholar]
- 41.Phillips ST, Piersanti G, Bartlett PA. Proc Natl Acad Sci USA. 2005;102:13737–13742. doi: 10.1073/pnas.0506646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips ST, Blasdel LK, Bartlett PA. J Am Chem Soc. 2005;127:4193–4198. doi: 10.1021/ja045122w. [DOI] [PubMed] [Google Scholar]
- 43.Phillips ST, Piersanti G, Ruth M, Gubernator N, van Lengerich B, Bartlett PA. Organic Letters. 2004;6:4483–4485. doi: 10.1021/ol048262j. [DOI] [PubMed] [Google Scholar]
- 44.Phillips ST, Rezac M, Abel U, Kossenjans M, Bartlett PA. J Am Chem Soc. 2002;124:58–66. doi: 10.1021/ja0168460. [DOI] [PubMed] [Google Scholar]
- 45.Hagmann WK. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 46.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 47.Kotz JD, Bond CJ, Cochran AG. Eur J Biochem. 2004;271:1623–1629. doi: 10.1111/j.1432-1033.2004.04076.x. [DOI] [PubMed] [Google Scholar]
- 48.Russell SJ, Blandl T, Skelton NJ, Cochran AG. J Am Chem Soc. 2003;125:388–395. doi: 10.1021/ja028075l. [DOI] [PubMed] [Google Scholar]
- 49.Dyson HJ, Rance M, Houghten RA, Lerner RA, Wright PE. J Mol Biol. 1988;201:161–200. doi: 10.1016/0022-2836(88)90446-9. [DOI] [PubMed] [Google Scholar]
- 50.Minor DL, Kim PS. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 51.Minor DL, Kim PS. Nature. 1994;371:264–267. doi: 10.1038/371264a0. [DOI] [PubMed] [Google Scholar]
- 52.Zhang HS, Pan J, Hogen-Esch TE. Macromolecules. 1998;31:2815–2821. [Google Scholar]
- 53.Syud FA, Stanger HE, Gellman SH. J Am Chem Soc. 2001;123:8667–8677. doi: 10.1021/ja0109803. [DOI] [PubMed] [Google Scholar]
- 54.Smith CK, Regan L. Science. 1995;270:980–982. doi: 10.1126/science.270.5238.980. [DOI] [PubMed] [Google Scholar]
- 55.Distefano MD, Zhong A, Cochran AG. J Mol Biol. 2002;322:179–188. doi: 10.1016/s0022-2836(02)00738-6. [DOI] [PubMed] [Google Scholar]
- 56.Cochran AG, Skelton NJ, Starovasnik MA. Proc Natl Acad Sci USA. 2001;98:5578–5583. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baldwin RL. Proc Natl Acad Sci USA. 1986;83:8069–8072. doi: 10.1073/pnas.83.21.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maynard AJ, Sharman GJ, Searle MS. J Am Chem Soc. 1998;120:1996–2007. [Google Scholar]
- 59.Wüthrich K. NMR of Proteins and Nucleic Acids. Wiley; New York: 1986. [Google Scholar]
- 60.Williamson MP. Biopolymers. 1990;29:1428–1431. [Google Scholar]
- 61.Gorske BC, Blackwell HE. J Am Chem Soc. 2006;128:14378–14387. doi: 10.1021/ja065248o. [DOI] [PubMed] [Google Scholar]
- 62.Dunitz JD. Chembiochem. 2004;5:614–621. doi: 10.1002/cbic.200300801. [DOI] [PubMed] [Google Scholar]
- 63.Weber G. Protein Interactions. Chapman Hall; New York: 1992. [Google Scholar]
- 64.Wong DT, Bymaster FP, Engleman EA. Life Sci. 1995;57:411–441. doi: 10.1016/0024-3205(95)00209-o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.