

Abstract
As the field of biotechnology has advanced, oral protein delivery has also made significant progress. Oral delivery is the most common method of drug administration with high levels of patient acceptance. Despite the preference of oral delivery, administration of therapeutic proteins has been extremely difficult. Increasing the bioavailability of oral protein drugs to the therapeutically acceptable level is still a challenging goal. Poor membrane permeability, high molecular weight, and enzymatic degradation of protein drugs have remained unsolved issues. Among diverse strategies, nanotechnology has provided a glimpse of hope in oral delivery of protein drugs. Nanoparticles have advantages, such as small size, high surface area, and modification using functional groups for high capacity or selectivity. Nanoparticles with peptidic ligands are especially worthy of notice because they can be used for specific targeting in the gastrointestinal (GI) tract. This article reviews the transport mechanism of the GI tract, barriers to protein absorption, current status and limitations of nanotechnology for oral protein delivery system.
Keywords: Oral protein delivery, Nanoparticles, Peptidic ligands, GI tract, Protein absorption
1. Introduction
Oral administration is most preferred because of the various advantages over other routes of drug delivery. The advantages include patient convenience and compliance, which increase the therapeutic efficacy of the drug. Oral formulations are also cheaper to produce because they do not need to be manufactured under sterile conditions [1]. Oral delivery of protein has become a pressing goal in recent years due to the increased availability of novel therapeutics through the advent of recombinant DNA technology. One of the holy grails of oral drug delivery is to deliver proteins, such as insulin, with the efficacy similar to the parenteral formulations [2].
The increasing importance of proteins can be attributed to three main developments. First, improved analytical methods have promoted discovery of numerous hormones and peptides that have found applications as biopharmaceuticals. Second, molecular biology and genetic engineering have enabled large-scale production of polypeptides previously available only in small quantities. Lastly, there is a better understanding of the role of regulatory proteins in the pathophysiology of human diseases [3, 4]. Consequently, pharmaceutical companies around the world have developed protein oral delivery technologies for producing therapeutically active ingredients in commercial scales, as listed in Table 1 [5]. Proteins have become the drugs of choice for treatment of numerous diseases as a result of their exquisite selectivity and their ability to provide effective and potent action [6]. Protein drug development, however, continues to be a formulation challenge to pharmaceutical scientists. Many protein drugs are currently used as parenteral formulations because of their poor oral bioavailability. This is due to several unfavorable physicochemical properties, such as large molecular size [7], susceptibility to enzymatic degradation, poor stability in the gastric low pH environment [8], poor penetration of the intestinal membrane, short plasma half-life, immunogenicity, and the tendency to undergo aggregation, adsorption, and denaturation [9, 10]. Enzymatic degradation and poor penetration of the intestinal membrane induce low oral bioavailability of biological molecules. The challenge here is to improve the oral bioavailability from less than 1% to at least 30–50% [11, 12]. These problems also remain unsolved. Unfavorable physicochemical properties of proteins present monumental challenges to pharmaceutical formulation scientists.
Table 1.
Protein oral delivery technologies under development by companies [5].
| Company | Product | Systems | Characteristics and advantages | Products currently available or under development |
|---|---|---|---|---|
| Emisphere | Eligen® | Carrier molecules | Facilitates the absorption of small molecules without altering chemical form, biological integrity or pharmacological properties Passive transcellular transport enables drug molecules of all sizes to cross the cell membrane |
Calcitonin, GPL-1, PYY, insulin, growth hormone, parathyroid hormone, heparin |
| Altus | CLEC® | Protein crystallization | Catalysts containing the enzyme alcohol dehydrogenase (ADH) Protein stabilization against proteolysis and self-digestion |
Calcitonin, other polypeptides, lipases, esterases, and proteases |
| Generex | Oral-Lyn™ | Spray device and aerosol particles | Penetrate the buccal epithelium Treatment of Type 1 & 2 diabetes |
Insulin, Macrotonin |
| NOBEX/Biocon | HIM2 | Amphiphilic oligomers | Resist enzyme digestion and increase membrane permeation | Insulin, enkephalin, calcitonin, parathyroid hormone |
| Apollo Life Sciences | Oradel™ | Nanoparticles | Protection of the drug payload from digestive enzymes and transport of protein-based drugs and antibodies across the intestinal wall transporting both small and large molecules (up to 150 kDa in size) for protein-based drugs and antibodies | Insulin and oral delivery of anti-inflammatory proteins(TNF blocker) |
| Autoimmune Incorporated/Eli-Lilly | AI-401 | Oral formulation | Protect proteins from enzyme digestion. Oral tolerance therapy Treatment of Type 1 diabetes but also for prevention of progression |
Insulin |
| Provalis PLC | Macrulin™ | Lipid-based water-in-oil microemulsion | Protect proteins from proteolysis or acidic degradation, and enhance the protein absorption in GIT treatment of Type 2 diabetes | Insulin, salmon calcitonin |
| Endorex | Orasome™ | Polymerized liposomes | Protect proteins from the stomach and upper GIT | Insulin and growth hormone, vaccines |
Designing and formulating a protein drug for delivery through the gastrointestinal (GI) tract requires innovative and practical strategies. Various strategies currently under investigation include chemical modification, formulation vehicles, protease inhibitors, absorption enhancers and muco-adhesive polymers. Among them, nanoparticles as a carrier or a device have become the focus of attention in this field recently. The nanoparticles possess certain advantages such as greater stability during storage, stability in vivo after administration and ease of scale-up without an aseptic process for oral administration [13]. The major goals in using nanoparticles as a drug delivery system are to control particle size, surface properties and release of active pharmaceutical ingredients for achieving the site-specific action of the drug at the therapeutically optimal rate and dose regimen. Especially, nanoparticles with peptidic ligands as formulation hold out considerable promise for the future because all benefits collectively can make a significant synergistic effect. The following sections briefly review the transport mechanisms, barriers to absorption for oral protein delivery, targeted nanoparticles for protein oral delivery by using peptidic ligands.
2. Transport mechanisms in the GI tract
There are four distinct mechanisms for molecules to cross the cell membrane: via paracellular, transcellular, carrier-mediated, and receptor-mediated transport (Fig. 1). Absorption through each pathway is dependent on different physical characteristics, such as molecular weight, hydrophobicity, ionization constants, and pH stability of absorbing molecules as well as biological barriers that restrict protein absorption from the GI tract. Thus, an understanding of biomolecules and these distinct mechanisms are important in designing delivery systems for oral protein drugs.
Figure 1.
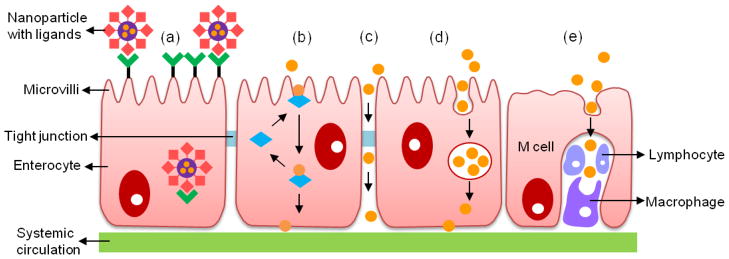
Schematic representation of the transport mechanisms: (a) receptor-mediated transport; (b) carrier-mediated transport; (c) paracellular transport; (d) transcellular transport; and (e) M cell mediated transport (i.e., phagocytosis by M cells).
2.1. Paracellular transport
Paracellular transport is the pathway of substances across an epithelium by passing through the intercellular spaces in between epithelial cells. Paracellular transport is passive and results from diffusion. This transport is under the control of tight junctions. A tight junction constitutes the major rate limiting barrier towards the paracellular transport for permeation of ions and larger substances [14]. The dimension of the paracellular space is on the order of 10 Å. The average size of aqueous pores created by epithelial tight junctions is approximately 7–9 Å for the jejunum, 3–4 Å for the ileum, and 8–9 Å for the colon in the human intestine [15]. This data suggests that solutes with a molecular radius exceeding 15 Å (approximately 3.5 kDa) cannot be transported via this route [16]. Furthermore, tight junctions comprise only about 0.01% of the total absorption surface area of the intestine [17]. Consequently, one would conclude that protein delivery across mucosal epithelia using paracellular transport is severely restricted. However, paracellular transport varies enormously among epithelia in terms of electrical resistance and shows small differences in ionic selectivity. The paracellular transport complements the transcellular mechanism by defining the degree and selectivity of reverse leak for ions and solutes, making an important tissue-specific contribution to overall transport [18, 19]. The tight junction shares biophysical properties with conventional ion channels, including size and charge selectivity, dependency of permeability on the ion concentration, competition between permeant molecules, anomalous mole-fraction effects, and sensitivity to pH [20]. The paracellular pathway is not largely determined by the hydrogen bonding capacity and lipophilicity.
2.2. Transcellular transport
Transcellular transport occurs through the intestinal epithelial cells by transcytosis, a particular process by which particles are taken up by cells. A typical example is the movement of glucose from the intestinal lumen to extracellular fluid by epithelial cells. This starts with an endocytic process that takes place at the cell apical membrane. Then, particles are transported through the cells and released at the basolateral pole [21]. The basolateral membrane is thinner and more permeable than the apical membrane because the protein-to-lipid ratio is very low in the basolateral membrane. Transport of particles by the transcellular transport depends on several factors: (i) various physicochemical properties of particles, such as size, lipophilicity, hydrogen bond potential, charge, surface hydrophobicity or the presence of a ligand at the particle surface; (ii) the physiology of the GI tract; and (iii) the animal model used to study the uptake [22, 23].
Enterocytes and M cells are the primary intestinal cells for transport. Enterocytes represent the majority of cells lining the gastrointestinal tract and M cells are mainly located within the epithelium of Peyer’s patches and represent a very small proportion of the intestinal epithelium (5% of the human follicle-associated epithelium (FAE), i.e., about 1% of the total intestinal surface) [24]. M cells in the follicle-associated epithelium (FAE) of Peyer’s patches are specialized for an antigen. M cells deliver proteins and peptides from the lumen to the underlying lymphoid tissues for the induction of immune responses. However, M cells are also exploited by a range of pathogens as a route for host invasion [25]. Furthermore, M cells represent a potential portal for oral delivery of proteins and peptides due to their high endocytosis ability. M cells possess a high transcytotic capacity and transport a wide variety of materials, including nanoparticles [26, 27]. M cells take up macromolecules, particles and microorganisms by adsorptive endocytosis via clathrin-coated pits and vesicles, fluid phase endocytosis and phagocytosis [28]. Although there has been some controversy in the literature on the extent of particle absorption, there is evidence that particle translocation can occur across enterocytes in the villipart of the intestine [29, 30]. However, the number of particles translocated through these routes is mostly very low because of the low endocytic activity of the enterocytes. It has been generally observed that the bulk of particle translocation mainly occurs in FAE [29, 31, 32]. As a result, many researchers have studied with great interest the Peyer’s patches and M cells which have adapted to absorb a large range of materials. Nevertheless, this route is limited to the transport of relatively low-molecular-weight lipophilic drugs. Furthermore, studies in humans have demonstrated that absorption by the transcellular route decreases significantly in the colon, whereas no such gradient exists for the paracellular route [33].
2.3. Carrier-mediated transport
Drugs are transferred across the cell membrane or entire cell and then released from the basal surface of the enterocyte into circulation [34]. The process is suitable and utilized by small hydrophilic molecules [35]. Active absorption requires energy-dependent uptake of specific molecules by carriers. The carriers recognize target molecules through membrane receptors and transport them across the membranes into the GI epithelium, even against the concentration gradient and in trace quantities. For example, small di/tripeptides (including β-lactam antibiotics and angiotensin-converting enzyme (ACE) inhibitors), monosaccharides, and amino acids are transported transcellularly by a carrier-mediated transportprocess [36]. Shah and Shen investigated the carrier-mediated transport of insulin across Caco-2 cell monolayers. They observed that transport of the conjugated insulin was mediated via the transferrin receptor and not through the insulin receptor. The authors found that insulin-transferrin (In-Tf) transport across the Caco-2 cell monolayers increased by 5- to 15-fold compared to free insulin [37].
2.4. Receptor-mediated transport
In receptor-mediated transport, protein drugs act either as a receptor specific ligand for surface-attached receptors or as a receptor for surface-attached ligands [38]. Receptor-mediated transport has also been exploited to increase the oral bioavailability of protein drugs by modification such as receptor specific ligands with peptide and protein drugs. This transportation entails cell invagination, which leads to formation of a vesicle. This transportation, in general, is known as endocytosis and comprises phagocytosis, pinocytosis, receptor-mediated endocytosis (clathrin-mediated), and potocytosis (nonclathrin-mediated) [39]. The first step in this process includes binding of the ligand to a specific cell-surface receptor, receptor clustering and internalization through coated vesicles into endosomal acidic compartments. The subsequent pathway is strongly dependent on the type of the receptor/ligand pair; the low endosomal pH may or may not trigger dissociation of the receptor and ligand, and sorting processes may lead to degradative lysosomal compartments. After protein drugs are transported to the GI tract, they take access to the systemic circulation via two separate and functionally distinct absorption pathways: portal blood and the intestinal lymphatics. The physicochemical and metabolic features of the protein drug and the characteristics of the formulation largely control the relative proportion of protein drug absorbed via these two pathways. Portal blood represents the major pathway for the majority of orally administered protein drugs. During this process, hydrophilic ligands are carried to the liver via the hepatic portal vein, and then by the hepatic artery gain access to the systemic circulation, for subsequent delivery to their sites of action. On the other hand, highly lipophilic ligands (log P >5) that cross the same epithelial barrier are transported to the intestinal lymphatics, which directly deliver them to the vena cava, thereby bypassing the hepatic first-pass metabolism [40].
3. Barriers to Protein Absorption
3.1. Gastrointestinal barriers
An understanding of the GI tract and of drug target sites offers an opportunity for targeted oral delivery of proteins [41]. The GI tract has various proteolytic enzymes such as trypsin, chymotrypsin, and elastase which are endopeptidases. Carboxypeptidase A and aminopeptidase are exopeptidases which are also involved as proteolytic enzymes [42]. Table 2 shows various proteases along with their sites of action [41, 43]. Endopeptidases hydrolyze the bond internal to the terminal bonds of the peptide chain, while exopeptidases hydrolyze the bond linking the NH2–terminal or the COOH-terminal amino acid to the peptide chain. Enzymatic degradation can occur at the lumen, brush border, the cytosol of the enterocytes, and even in the lysosomes and other cell organelles [44].
Table 2.
A list of various proteases along with their sites of action [41].
| Types | Enzymes | Major Site of Action |
|---|---|---|
| Gastric proteases | Pepsins (aspartic proteases) | Broad activity, hydrolyzes many peptide bond peptides |
| Brush border proteases | Aminopeptidase A | Aminopeptidases are N-terminopeptidases, degrading mostly 3–10 amino acid residue-dipeptides and amino acids |
| Aminopeptidase N | ||
| Aminooligopeptidase | ||
| Dipeptidylaminopeptidase IV | ||
| Carboxypeptidase | ||
| Cystosolic proteases | Di- and tripeptidase | 2–3 aminopeptide amino acids |
| Intestinal pancreatic proteases | Trypsin (endopeptidase) | Peptide bonds of basic amino acids/peptides |
| α-chymotrypsin (endopeptidase) | Peptide bonds of hydrophobic amino acids/peptides | |
| Elastase (endopeptidase) | Peptide bonds of smaller and nonaromatic amino acids/peptides | |
| Carboxypeptidases (exopeptidase) | A: C-terminal amino acid | |
| B: C-terminal basic amino acid | ||
| Brush border proteases | Aminopeptidase A | Aminopeptidases are N-terminopeptidases, degrading mostly 3–10 amino acid residue-dipeptides and amino acids |
| Aminopeptidase N | ||
| Aminooligopeptidase | ||
| Dipeptidylaminopeptidase IV | ||
| Carboxypeptidase |
The stomach produces gastric juice having hydrochloric acid (HCl), potassium chloride (KCl) and sodium chloride (NaCl). The acidic environment with a pH range of 1.5 to 3.5 induces proteolysis of proteins and peptides into constituent aminoacids, dipeptides, and tripeptides for absorption. Pepsin is the first in a series of enzymes that digest protein. In the stomach, protein chains bind in the deep active site groove of pepsin and are broken into smaller pieces. Pepsin acts within the stomach so its optimum pH is around 2, an acidic pH. When the enzyme passes into the duodenum it meets a higher pH and its enzyme activity ends. Rapid pH changes also affect the degradation of ingested proteins and peptides. The pH is increased from 2 to about 6 when proteins move from the stomach to the duodenum. This wide pH range covers the isoelectric points of many peptides and proteins to precipitate them. These precipitated proteins do not rapidly redissolve upon pH change [45–47].
The small intestine is generally the major place for absorption of food. But the enzymatic activity of proteases is also higher than in any other segment of the GI tract. The main parts of the small intestine are the duodenum, jejunum, and ileum. The brush border is the name for the microvilli-covered surface of the epithelial cells found in the small intestine. The brush border membrane contains sucrase and more than a dozen of peptidases. They together have a broad specificity and can degrade both proteins and peptides [42]. Brush border enzyme activity is generally greater in the duodenum and the jejunum than in the ileum. The duodenum has pancreatic proteases consisting of endopeptidases and exopeptidases. They can create severe conditions for ingested peptides and proteins. In the duodenum, pancreatic secretions increase the pH of the enteric juice for the action of other digestive enzymes, for example, trypsin. Apart from the areas these peptidases reach, there are areas in the jejunum and ileum where the aminopeptidases activity is about 20–30% of the aminopeptidases activity in other neighboring areas. Such areas are known as Peyer’s patches and are a potential targeting site for the delivery of proteins and peptides [36, 48].
3.2. Mucosal barrier
Mucus plays an important role in determining the absorption and bioavailability of orally administered drugs. The mucosal barrier consists of three protective components. These provide additional resistance for the mucosal surface of the stomach. The first is a compact epithelial cell lining which is bound by tight junctions that repel harsh fluids that may injure the mucosal lining. The second is a special mucus blanket. The mucus blanket is derived from mucus secreted by surface epithelial cells and mucosal neck cells. This insoluble mucus forms a protective gel-like coating over the entire surface of the gastric mucosa. The third consists of bicarbonate ions. The bicarbonate ions are secreted by the surface epithelial cells [49, 50]. Glycocalyx is one of the main mucosal barrier components against delivery proteins via the oral route. The glycocalyx, which is atop the epithelial cells, is a fuzzy and fibrous coat that is weakly acidic and consists of sulfated mucopolysaccharides. Goblet cells secrete mucus, which lines the top of the glycocalyx [51]. The mucus consists of mucin glycoproteins, enzymes, electrolytes and water [52]. The cohesive and adhesive nature of the mucus layer is due to the presence of mucin glycoprotein [53, 54]. These kinds of complex surfaces are protected by highly viscoelastic layers. In contrast to proteases, the mucin lining presents a physical barrier rather than a chemical one. It is reported that the mucin layer is thickest in the stomach and colon, whereas in the small intestine the thickness varies depending on the extent of digestive activity [55].
The mucus and glycocalyx layers are the first and foremost barriers to peptides and proteins, which must first diffuse through these layers to reach the cellular membrane. Due to the viscosity and the interactive nature of these layers, they offer a certain level of resistance to the protein drug diffusion. Electrostatic adhesive interactions with mucin fibers and particle aggregation affect the nanoparticle transport rate. Negatively charged carboxylate- and sulfate-modified particles showed a higher transport rate than near neutral or positively charged amine modified particles. The amine nanoparticle transport is severely limited, likely by particle aggregation and electrostatic adhesive interaction with mucin fibers [56]. The interaction of particle and mucus made by electrostatic/ionic interactions, van der Waals interactions, hydrophobic forces, and hydrogen bonding influence the nanoparticle retention at the mucosal surface [57]. After diffusing through the mucus and glycocalyx, the protein drug reaches the epithelial surface [58]. The protein drugs must adhere to the mucus and must cross the mucus layer. However, drugs delivered to mucosal surfaces are usually efficiently removed by mucus clearance mechanisms [59]. Mucus continuously traps and removes pathogens and foreign particles in order to protect the epithelial surface. For this reason, low tissue permeability is currently one of the biggest hurdles to orally administrated drugs. Nanoparticles as drug carriers are a good alternative to diffuse into the mucus layer and avoid elimination by mucilliary clearance. However, there is a size limit to cross the intestinal mucosal barrier because the mesh-pore spacing of the mucus is 50–1800 nm [60]. Many researchers have reported that the transport of nanoparticles at various mucosal sites is highly dependent on its size. The pore network accommodates the movement of a number of particles as long as hydrophobic and electrostatic mucoadhesive forces can be minimized. Many studies have shown that nanoparticles under 200 nm size effectively diffuse through the mucus [61]. Aoki et al. investigated the contribution of the mucus/glycocalyx layers in rat small intestine as a diffusional or enzymatic barrier to the absorption of insulin by in vitro studies [62]. Their studies also suggest the possibility of mucus/glycocalyx layers acting as an enzymatic but not a diffusional barrier, irrespective of the intestinal region. Morishita et al. reported using an in situ absorption study with different intestinal segment loops to increase the insulin absorption from the ileum, the distal part of the small intestine [63]. Lai et al. focused their research on mucoadhesive nanoparticles. Strong interactions with mucus could increase retention at the mucosal surface. These interactions are driven by hydrogen bonding, van der Waals interactions, polymer chain interpenetration, hydrophobic forces, and electrostatic/ionic interactions [64].
4. Strategies for oral protein delivery
4.1. Nanotechnology and protein delivery
Advances in biotechnology have resulted in discovery of a large number of therapeutic and antigenic proteins. Currently, more than 100 peptide and protein drug products are under clinical investigation and about 30 compounds have received FDA approval [65]. Each year new therapeutic proteins are introduced into the market. Many researchers have studied to find more suitable oral protein delivery systems. Important efforts have already been focused on the design of carriers for transport of proteins across mucosal and intestinal barriers. Nanotechnology has shown a potential for delivery of proteins [66–68]. Table 3 lists potential applications of nanotechnology for oral delivery and targeting of therapeutic and diagnostic agents [69].
Table 3.
Potential applications for nanotechnologies in drug delivery [69].
| Material/technique | Characteristics | Medical applications |
|---|---|---|
| Ligands attached to nanoparticles | Surface modification with functional groups High degree of engineering precision Control the size of the nanoparticles |
Labeling, tracing and imaging Sensing and detection Recognition and attachment to damaged or diseased tissue followed by release of therapeutic compound |
| Quantum dots | Emit different wavelengths over abroad range of the light spectrum from visible to infrared, depending on their size and chemical composition Influence the fluorescence properties of the particles |
Fluorescent probes Detection and targeting |
| Nanocapsules | Consists of a shell and a space Can be made in specific sizes, shapes, and in reasonable quantities Control the release of substances or protect them from the environment Higher safety and efficacy Evasion of the host immune system and delivery of therapeutic agent to target sites |
Slowly release loading drugs Lipid nanocapsules as nanocarriers e.g. Buckyball-based treatment for AIDS |
| Nanoporous materials | Ability of nanopores of certain sizes to let some substances pass and others not, or to force molecules | Nanoporous membranes for molecules like DNA and RNA Can be coupled to sensors or used for drug-delivering implants |
| Polymers | Allow for judicious selection for targeting and delivery Can be used to improve the function of the nanoparticle High degree of engineering precision |
Drug carrying devices or implants Combining multi-modal therapy and imaging |
| Sorting biomolecules and precise sorting | Nanopores capable of rapid and precise sorting | Gene analysis and sequencing |
The US National Nanotechnology Initiative (NNI, http://www.nano.gov), launched in October 2000, provides a federal vision for nanotechnology-based investments through the coordination of 16 US departments and independent agencies. The potential research and development targets by 2015 for the NNI are shown in Table 4. They include no suffering and death from treated cancers, advanced materials and manufacturing, pharmaceutical synthesis and delivery, converging nanoscale technologies, and life-cycle biocompatible/sustainable development. The targets are really ambitious and it may take much beyond 2015 to achieve them.
Table 4.
Targets for the US National Nanotechnology Initiative.
| Research and development targets related to drug delivery/diagnosis
|
| Advanced materials and manufacturing: one-half from molecular level |
| Converging technologies from nanoscale |
| Life-cycle biocompatible/sustainable development |
| No suffering and death from cancer when treated |
| Pharmaceuticals synthesis and delivery: one-half on nanoscale level
|
| Research and development targets not directly related drug delivery/diagnosis
|
| Control of nanoparticles in air, soils, and waters |
| Education: nanoscale instead of microscale based |
| Nanoscale visualization and simulation of three-dimensional domains |
| New catalysts for chemical manufacturing |
| Transistor beyond/integrated CMOS < 10 nm |
The major goals in designing nanoparticles as a delivery system are to control particle size, surface properties [70], and release kinetics of pharmacologically active ingredients in order to achieve the site-specific action of the drug at the therapeutically optimal rate and dose regimen [71]. The advantages of using nanoparticles as a drug delivery system are listed in Table 5 [72]. The efficiency of drug delivery is directly related to particle size because particle size can enhance bioavailability and enable more precise targeting to the level of direct intracellular delivery [73]. Some investigators have observed that the number of nanoparticles which cross the intestinal epithelium is greater than the number of microspheres, and that not only the M cells but also the normal enterocytes are involved in the transport [74–76]. Nanoparticles allow penetration of cell membranes, binding, and encapsulation of the protein drugs inside a matrix and protect them against enzymatic and hydrolytic degradation [72]. There are many different techniques for making the nanoparticle with various biomaterials of polymers, lipids, and lectins. Emulsion polymerization, interfacial polymerization, emulsification evaporation, solvent displacement, salting out, emulsification diffusion, and desolvation are popular methods for making nanoparticles [77, 78]. The solvent displacement and salting out are more useful because they provide less stress to protein drugs. Chemical structures and surface characteristics have a significant influence on the physicochemical properties of nanoparticles and their behavior when they are exposed to physiological media [76].
Table 5.
Advantages of using nanoparticles as a drug delivery system [72].
|
4.2. Targeted nanoparticles with peptidic ligands
As stated earlier, there are a number of limitations to the oral delivery of proteins despite the progress of the knowledge in this field. The barriers to protein bioavailability after oral administration are intestinal membrane permeability, molecule size, intestinal and hepatic metabolism, and lastly solubility. Therefore, its administration has been restricted to the invasive route [79, 80]. The dosage form must initially stabilize the drug for oral delivery, making it easy to take orally [81]. It must then protect the drug from the extreme acidity and action of pepsin in the stomach. In the intestine, the drug must be protected from the many enzymes that are present in the intestinal lumen. In addition, the formulation must facilitate both aqueous solubility at neutral pH and lipid layer penetration for protein molecules to cross the intestinal membrane and then the basal membrane for entry into the blood stream. To ensure enteric protection and to improve bioavailability of proteins, diverse formulations have been developed, taking into account these restrictive parameters.
Modifying nanoparticles by coupling a targeting molecule at their surface could represent a more efficient way to enhance oral uptake of nanoparticles. Optimum contact between a carrier and a target biological surface is necessary to increase drug absorption. The nanoparticles should be able to make a strong interaction with the epithelial surface [82]. Extensive efforts have been devoted to achieving the so-called ‘active targeting’ of nanoparticles in order to deliver drugs to the right targets, based on molecular recognition processes such as ligand-receptor or antigen-antibody interactions. Targeting with small ligands appears more likely to succeed since they are easier to handle and manufacture. Furthermore, it could be advantageous when the active targeting ligands are used in combination with the long-circulating nanoparticles to maximize the likelihood of success in the active targeting of nanoparticles. In addition, ligands conjugated to the surface of engineered nanoparticles can influence the mode of cellular internalization. Ligands such as folic acid, albumin, and cholesterol have been shown to facilitate uptake through caveolin-mediated endocytosis, whereas ligands for glycol receptors promote clathrin-mediated endocytosis [83]. Ligands play important roles in dictating nanoparticle size, shape, and interparticle spacing, and also in determining the properties of the interface between the ligands and the nanoparticle surface as well as the interface between the nanoparticle and its environment. Indeed, particles have been decorated by adsorption or covalent attachment of peptidic ligands interacting with surface receptors to target the epithelium, with an expectation that such interactions will lead to a greater uptake and delivery of nanoparticles [84–86]. Ligands specific for certain cell types can be incorporated into drug delivery platforms to localize delivery to specific cells and tissues, thereby reducing required dosage and minimizing side effects. Huang et al. demonstrated the effects of goblet cell-targeting nanoparticles on the oral absorption of insulin in vitro, ex vivo and in vivo, and identified the targeting mechanism as well as the influence of mucus. The nanoparticles were modified with a CSKSSDYQC (CSK) targeting peptide. The CSK peptide modified NPs facilitated the uptake in the villi. In transport studies across a Caco-2/HT29-MTX co-culture cell monolayer, the CSK peptide modification also showed enhanced transport ability, even if the targeting recognition was partially affected by mucus. CSK modified NPs produced a better effect with a 1.5 fold higher relative bioavailability compared to unmodified ones [86]. Angelo et al. have fused TNF with the ACDCRGDCFCG peptide, a ligand of αV integrins by recombinant DNA technology. Subnanogram doses of this conjugate with melphalan were sufficient to induce antitumor effects in tumor-bearing mice, the ACGDRGDCFCG-mouse, TNF conjugate bound TNF receptors and trigger death signals. The Figure 2 shows that RGD-mTNF, although less active than NGR-mTNF on a molar basis, is capable of inducing antitumor effects in the pictogram range. This result indicates that a peptidic ligand improves antitumor activity of the drug while reducing side effects [87].
Figure 2.

The effect of ACGDRGDCFCG-murine tumor necrosis factor-conjugate (RGD-mTNF) on tumor growth and body weight of animal-bearing RMA tumors. 5 mice/group were treated intraperitoneal at day 10 with melphalan alone or in combination with RGD-mTNF or CNGRCG-mTNF conjugate (NGR-mTNF) at the indicated doses (A). Loss of animal weight at day 1 and day 4 after treatment (B) [87].
Table 6 shows various protein/peptidic ligands, functional activities, and characteristics. Figure 3 shows targeted nanoparticles with peptidic ligands.
Table 6.
Proteins/targeting ligands and functional activity [88].
| Protein/ligand | Functional activity and characteristics | References |
|---|---|---|
| Transferrin | Iron uptake occurs via the internalization of iron-loaded transferring mediated by the interaction with the Transferrin receptor Widely applied as a targeting ligand in the active targeting of anticancer agents, proteins and genes to primary proliferating cells via transferrin receptors |
[89–93] |
| Insulin | A hormone that regulates blood glucose levels, A small protein |
[94] |
| Elastin | A cross-linked protein in the extracellular matrix that provides elasticity for many tissues | [95] |
| Albumin | The major serum protein, binds a wide variety of lipophilic compounds including steroids | [96] |
| RGD peptide | Increases cell spreading, differentiation, and enhances DNA synthesis The RGD sequence can bind to multiple integrin species and also minimizes the risk of immune reactivity or pathogen transfer, particularly when xenograftor cadaveric protein sources are utilized |
[97–101] |
| Lectin (WGA) | Binds to the Caco-2 cell surface and human enterocytes | [84, 102–108] |
Figure 3.

Schematic illustration of targeted nanoparticles with peptidic ligands.
A peptide as a ligand has many advantages. Peptides can be synthesized by chemical methods on a large scale. These advantages and anticipated improvements in conjugation techniques can bring about a great improvement of their application in diagnosis and therapy. To screen peptide libraries produced by either chemical synthesis [109] or phage display [110–113] is a main method to select useful peptide ligands. The peptide library is widely applicable to both in vitro and in vivo studies. Moreover, the peptide library can be used to identify peptides even though receptors are unknown. Various peptide ligands have been found out for various types of receptors or cells, such as integrin receptors [114, 115], cardiomyocytes [116], thrombin receptors[117], tumor cells [116, 118–120], intestinal tissue, M cells, and pancreatic β cells [121]. In vitro and in vivo target specific ligand modified nanoparticle applications have been conducted by a large number of research groups to identify peptidic ligand administration routes including oral drug administration for targeting various organs and tissues. The sequence of peptidic ligands has been typically screened by analysis of comparative superiority in transcytosis efficacy across target cell layer in vitro and in vivo among a huge number of candidates [112]. Receptor-mediated endocytosis is a process that transports peptidic ligands into a target cell. The specificity results from a receptor-ligand interaction. The receptors on the plasma membrane of the target tissue specifically bind to peptidic lignads on the outside of the cell. The peptidic ligands and their receptors accumulate in coated pits and are internalized as receptor-ligand complexes. After internalized, endocytosed peptidic ligands are delivered into endosome. And then, endocytosed peptidic ligands are transported into lysosomes where hydrolytic digestion starts. The subsequent pathways are strongly dependent on the type of receptor/ligand pair [89]. Cho et al. reported to develop an efficient oral vaccine carrier which specifically targets the follicle-associated epithelium region of Peyer’s patch. M cell-homing peptide ligand was selected by the phase display technique. The CKSTHPLSC (CKS9) peptide sequence was immobilized chitosan nanoparticles (CKS9-CNs). The target specificity was evaluated by an in vitro transcytosis assay and in vivo assays. In vivo localization of CKS9 was compared with CSK9 in rat small intestinal tissues. Their tissue specific localization was monitored under fluorescence-microscopy (Fig. 4). The CKS9-CNs were spread more effectively across the M cell model and accumulated more specifically into Peyer’s patch regions in comparison with CNs [112].
Figure 4.

In vivo localization of CKS9 compared with CSK9 in rat small intestinal tissues. Chemically synthesized CKS9 or CSK9 was injected into closed ileal loops and their tissue specific localization was monitored under fluorescence-microscopy; (a and b) in vivo localization of CSK9 peptides in Peyer’s patches and Non- Peyer’s patches. (c and d) in vivo localization of CKS9 peptides in Peyer’s patches and Non- Peyer’s patches. Green and red fluorescent signals in each panel indicate the location of the peptides and mucus layer in rat small intestinal tissues (closed ileal loops), respectively. Scale bars indicate 50 mm in Peyer’s patches and 20 mm in Non- Peyer’s patches, respectively [112].
Schneider et al. studied improving the transport of vaccine-loaded nanoparticles. The phage display screening was used to identify peptides targeting human M cells. Phage libraries have been used to select a peptide utilizing an in vitro model of the human follicle-associated epithelium (FAE) containing both Caco-2 and M cells. Clones were sequenced after five rounds of selection in the human FAE in vitro model. Three identical clones (CTGKSC, PAVLG and LRVG) appeared at high frequency after selection on both mono-and co-cultures (Table 7). The presence of LRVG and CTGKSC peptides modified nanoparticles significantly increased their transport across the cell layer by 8 and 4 times, respectively, when compared to non-modified nanoparticles. The transport of PAVLG-modified nanoparticles, on the other hand, was the same as that of non-modified nanoparticles. Two peptides could be used significantly to enhance the transport of vaccine-loaded nanoparticles across the intestinal mucosal barrier [113].
Table 7.
Phage selection. Clones were sequenced after five rounds of selection in the human FAE in vitro model. T5C were clones selected for their ability to induce phage transcytosis on co-cultures and T5M on monocultures. Numbers 1 and 2 represent both sequencing in which 12 and 50 clones were analyzed respectively. Bold type represents identical clones recovered at high frequency after selection on mono- and co-cultures.
| Clone name | Sequence/peptide | Frequency |
|---|---|---|
| T5C1-2/3/5/11-T5C2-1/4 | L-R-V-G-stop | 6 |
| T5C1-1 - T5C2-3/5/8/10/12/14/17/19/22/24/26/28/38/40/42/45/47 | C-T-G-K-S-c-stop | 25 |
| T5C1-4 | C-stop | 1 |
| T5C1-6 | C-E-G-P-L-K-P-stop | 1 |
| T5C1-7 | C-G-G-X-D-N-S-C | |
| T5C1-8 - T5C2-2/13/20/30/41 | S-stop | 6 |
| T5C1-9 | C-A-P-I-L-F-P-R-C | 1 |
| T5C1-10 - T5C2-7/29/34/36/37 | P-A-stop | 6 |
| T5C1-12 - T5C2-9 | C-L-E-S-K-K-K-T-C | 2 |
| T5C2-21/27 | P-A-V-L-G | 2 |
| T5C2-6 | C-R-M-K | 1 |
| T5C2-18 | C-E-K-R | 1 |
| T5C2-25 | C-I-G-K-R-D-A-K-H | 1 |
| T5C2-32 | C-R-R-stop | 1 |
| T5C2-33 | C-K-S-G-G-T-S-A-C | 1 |
| T5C2-35 | C-R-S-G-T-S-R-S-C | 1 |
| T5C2-46 | C-R-D-stop | 1 |
| T5M1-2/4/8 - T5M2-9/11/18/23 | P-A-V-L-G | 7 |
| T5M1-1 - T5M2-1/3/7/20/24/26/28/30/35/38/45/48/49 | C-T-G-K-S-C | 25 |
| T5M1-3 - T5M2-4 | S-A-stop | 2 |
| T5M1-5 - T5M2-16/21 | P-A-stop | 2 |
| T5M1-6 | C-I-E-V-P-C | 1 |
| T5M1-7 | C-G-E-K-K-M-R-C | 1 |
| T5M1-9 | C-G-K-S-T-K-N-W | 1 |
| T5M1-10 - T5M2-5/8/10/17/22 | S-stop | 6 |
| T5M1-11 | P-A-R-L-A-R-L | 1 |
| T5M2-2/13/14/19/25 | L-R-V-G | 5 |
| T5M2-36/46/47 | C-P-F-D-S-stop | 3 |
| T5M2-6/12 | C-K-stop | 2 |
| T5M2-29 | L-V-G-G-H-C-G-E-C | 1 |
| T5M2-15 | C-Q-E-A-T-N-R-K-C | 1 |
| T5M2-37 | C-T-G-K-R | 1 |
Different types of targeting molecules have been tested but the most studied has been the lectin family. Lectins are proteins that bind to highly-specific carbohydrate moieties of the glycocalyx of the intestinal enterocytes and the mucus layer [122]. They are involved in many cell recognition and adhesion processes. Their conjugation to polymeric nanoparticles significantly increases their transport across the intestinal mucosa by efficiently increasing interactions with the mucus [27, 123, 124] and/or the surface of the epithelial cells [125] and by promoting particle translocation [123, 126]. The association of lectins with nanoparticles can be achieved by adsorption or covalent coupling, with a definite preference for a covalent linkage, if conjugation does not affect lectin activity and specificity. As an alternative to injection, oral administration of lectin conjugated nanoparticles loaded with insulin, enhanced the intestinal absorption of insulin enough to drop the glucose level in blood [127]. Even if insulin is a hydrophilic peptide, it can be incorporated with high efficiency (about 98%) nanoparticles showing good physical stability and sustained drug release behavior [128]. YaShu et al. demonstrated that the highest amount of lectin conjugated nanoparticles was detected in the small intestine, suggesting an increase of intestinal bioadhesion and endocytosis. This result represented an increase of almost 1.4–3.1 fold across the intestine compared to <4.9% for the uptake of unconjugated nanoparticles.
Peptidic ligands like the well-known arginine-glycine-aspartic acid (RGD) and cell penetrating peptides have also been covalently linked on polymers before the formation of nanoparticles [129]. The RGD or CPP target β1 integrins localized at the apical pole of M cells [130]. Covalent binding on PEG chains favors RGD presentation and then targeting [131]. Ligands can also be non-covalently attached to PEG chains. The presence of RGD peptides on the surface has recently been shown to induce a 50-fold increase in transport across the human intestine epithelial cells compared to blank PS particles [132]. The RGD motif has previously been demonstrated to promote cell attachment to hydrophobic substrates [133, 134]. Recently, Gref et al. have grafted biotin molecules on PEG chains and exploited the strongest biological, non-covalent interactions. The non-covalent coupling method is an attractive method for modifying the surface of a nanoparticle such as a PCL-PEG-avidin-biotin ligand. A biotin–lectin ligand was incubated with nanoparticles in the presence of avidin. This process led to the formation of a nanoparticle–biotin–avidin/biotin–lectin complex. The main advantage of this technique lies in the variety of biotinylated ligands that could be grafted at the nanoparticle surface. Thus, the surface properties of the nanoparticles can be modified either by improving non-specific interactions with the cell apical surface or by grafting specific ligand targeting epithelial intestinal cells [102].
Conclusions
Poor intestinal absorption of the protein drugs is due to their unfavorable physicochemical properties, such as high molecular weight and susceptibility to enzymatic hydrolysis. In addition, there are several biological barriers to intestinal absorption of protein drugs in the GI tract. The low bioavailability of protein drugs remain to be an important issue requiring active research. The nanoparticles with peptidic ligands have many advantages to solve the limitations mentioned above. They can be used to target the epithelium, with an expectation that such interactions will lead to a greater uptake and delivery of the drug. Much research, however, is yet to be done to determine the exact mechanism of the nanoparticulate uptake and subsequent clearance, associated potential for in vivo nano-toxicology, tissue specific targeting, and modulation of GI transit. To translate the potential into real products, practical formulations need to be developed. The nanoparticles with peptidic ligands would be a promising candidate for oral protein delivery. The potential of nanoparticles in drug delivery has remained. Challenges to developing protein formulations for oral delivery are still significant, and the quest to overcome the problem is ongoing.
Acknowledgments
This study was supported in part by NIH through grants CA129287 and GM095879, and the Showalter Research Trust Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Salama NN, Eddington ND, Fasano A. Tight junction modulation and its relationship to drug delivery. Adv Drug Deliv Rev. 2006;58:15–28. doi: 10.1016/j.addr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman A, Ziv E. Pharmacokinetic considerations of new insulin formulations and routes of administration. Clin Pharmacokinet. 1997;33:285–301. doi: 10.2165/00003088-199733040-00004. [DOI] [PubMed] [Google Scholar]
- 3.Adessi C, Soto C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr Med Chem. 2002;9:963–978. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 4.Adessi C, Soto C. Strategies to Improve Stability and Bioavailability of Peptide Drugs Frontiers in Medicinal Chemistry. 2004;1:513–527. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 5.Park K, Kwon IC, Park K. Oral protein delivery: Current status and future prospect. React Funct Polym. 2011;71:280–287. [Google Scholar]
- 6.Hussain N, Jaitley V, Florence AT. Recent advances in the understanding of uptake of microparticulates across the gastrointestinal lymphatics. Adv Drug Deliv Rev. 2001;50:107–142. doi: 10.1016/s0169-409x(01)00152-1. [DOI] [PubMed] [Google Scholar]
- 7.Donovan MD, Flynn GL, Amidon GL. Absorption of Polyethylene Glycols 600 Through 2000: The Molecular Weight Dependence of Gastrointestinal and Nasal Absorption. Pharm Res. 1990;7:863–868. doi: 10.1023/a:1015921101465. [DOI] [PubMed] [Google Scholar]
- 8.Ikesue K, Kopečkovà P, Kopeček J. Degradation of proteins by guinea pig intestinal enzymes. Int J Pharm. 1993;95:171–179. [Google Scholar]
- 9.Saffran M, Kumar G, Savariar C, Burnham J, Williams F, Neckers D. A new approach to the oral administration of insulin and other peptide drugs. Science. 1986;233:1081–1084. doi: 10.1126/science.3526553. [DOI] [PubMed] [Google Scholar]
- 10.Fix JA. Oral controlled release technology for peptides: status and future prospects. Pharm Res. 1996;13:1760–1764. doi: 10.1023/a:1016008419367. [DOI] [PubMed] [Google Scholar]
- 11.Lee HJ. Protein drug oral delivery: the recent progress. Arch Pharm Res. 2002;25:572–584. doi: 10.1007/BF02976925. [DOI] [PubMed] [Google Scholar]
- 12.Lee VH, DKS, MGG, WR Oral route of protein and peptide drug delivery. Peptide and protein drug delivery. 1991:691–738. [Google Scholar]
- 13.Kreuter J. Nanoparticulate systems in drug delivery and targeting. J Drug Target. 1995;3:171–173. doi: 10.3109/10611869509015940. [DOI] [PubMed] [Google Scholar]
- 14.Madara JL. Regulation of the movement of solutes across tight junctions. Annu Rev Physiol. 1998;60:143–159. doi: 10.1146/annurev.physiol.60.1.143. [DOI] [PubMed] [Google Scholar]
- 15.Tomita M, Shiga M, Hayashi M, Awazu S. Enhancement of colonic drug absorption by the paracellular permeation route. Pharm Res. 1988;5:341–346. doi: 10.1023/a:1015999309353. [DOI] [PubMed] [Google Scholar]
- 16.Rubas W, Cromwell ME, Shahrokh Z, Villagran J, Nguyen TN, Wellton M, Nguyen TH, Mrsny RJ. Flux measurements across Caco-2 monolayers may predict transport in human large intestinal tissue. J Pharm Sci. 1996;85:165–169. doi: 10.1021/js950267+. [DOI] [PubMed] [Google Scholar]
- 17.Pappenheimer JR. Physiological regulation of transepithelial impedance in the intestinal mucosa of rats and hamsters. J Membr Biol. 1987;100:137–148. doi: 10.1007/BF02209146. [DOI] [PubMed] [Google Scholar]
- 18.Barry PH. Ionic permeation mechanisms in epithelia: biionic potentials, dilution potentials, conductances, and streaming potentials. Methods Enzymol. 1989;171:678–715. doi: 10.1016/s0076-6879(89)71038-7. [DOI] [PubMed] [Google Scholar]
- 19.Powell DW. Barrier function of epithelia. Am J Physiol. 1981;241:G275–288. doi: 10.1152/ajpgi.1981.241.4.G275. [DOI] [PubMed] [Google Scholar]
- 20.Tang VW, Goodenough DA. Paracellular Ion Channel at the Tight Junction. Biophys J. 2003;84:1660–1673. doi: 10.1016/S0006-3495(03)74975-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shakweh M, Ponchel G, Fattal E. Particle uptake by Peyer’s patches: a pathway for drug and vaccine delivery. Expert Opin Drug Deliv. 2004;1:141–163. doi: 10.1517/17425247.1.1.141. [DOI] [PubMed] [Google Scholar]
- 22.Florence AT. Issues in oral nanoparticle drug carrier uptake and targeting. J Drug Target. 2004;12:65–70. doi: 10.1080/10611860410001693706. [DOI] [PubMed] [Google Scholar]
- 23.Burton PS, Conradi RA, Hilgers AR. (B) Mechanisms of peptide and protein absorption: (2) Transcellular mechanism of peptide and protein absorption: passive aspects. Adv Drug Deliv Rev. 1991;7:365–385. [Google Scholar]
- 24.Giannasca PJ, Giannasca KT, Leichtner AM, Neutra MR. Human intestinal M cells display the sialyl Lewis A antigen. Infect Immun. 1999;67:946–953. doi: 10.1128/iai.67.2.946-953.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gebert A, Rothkötter H-J, Pabst R. M Cells in Peyer’s Patches of the Intestine. In: Kwang WJ, editor. Int Rev Cytol. Academic Press; 1996. pp. 91–159. [DOI] [PubMed] [Google Scholar]
- 26.Frey A, Neutra MR. Targeting of mucosal vaccines to Peyer’s patch M cells. Behring Inst Mitt. 1997;98:376–389. [PubMed] [Google Scholar]
- 27.Clark MA, Hirst BH, Jepson MA. Lectin-mediated mucosal delivery of drugs and microparticles. Adv Drug Deliv Rev. 2000;43:207–223. doi: 10.1016/s0169-409x(00)00070-3. [DOI] [PubMed] [Google Scholar]
- 28.Buda A, Sands C, Jepson MA. Use of fluorescence imaging to investigate the structure and function of intestinal M cells. Adv Drug Deliv Rev. 2005;57:123–134. doi: 10.1016/j.addr.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Jani PU, Florence AT, McCarthy DE. Further histological evidence of the gastrointestinal absorption of polystyrene nanospheres in the rat. Int J Pharm. 1992;84:245–252. [Google Scholar]
- 30.Kataoka K, Tabata J, Yamamoto M, Toyota T. The association of gap junctions with large particles in the crypt epithelium of the rat small intestine. Arch Histol Cytol. 1989;52:81–86. doi: 10.1679/aohc.52.81. [DOI] [PubMed] [Google Scholar]
- 31.Lavelle EC, Sharif S, Thomas NW, Holland J, Davis SS. The importance of gastrointestinal uptake of particles in the design of oral delivery systems. Adv Drug Deliv Rev. 1995;18:5–22. [Google Scholar]
- 32.O’Hagan DT. Intestinal translocation of particulates — implications for drug and antigen delivery. Adv Drug Deliv Rev. 1990;5:265–285. [Google Scholar]
- 33.Hebden JM, Wilson CG, Spiller RC, Gilchrist PJ, Blackshaw E, Frier ME, Perkins AC. Regional differences in quinine absorption from the undisturbed human colon assessed using a timed release delivery system. Pharm Res. 1999;16:1087–1092. doi: 10.1023/a:1018948102778. [DOI] [PubMed] [Google Scholar]
- 34.Russell-Jones GJ. Carrier-mediated transport, oral drug delivery. In: Mathiowitz E, editor. Encyclopedia of controlled drug delivery. Vol. 1. New York, NY: John Wiley & Sons; 1999. pp. 173–184. [Google Scholar]
- 35.Barthe L, Woodley J, Houin G. Gastrointestinal absorption of drugs: methods and studies. Fundam Clin Pharmacol. 1999;13:154–168. doi: 10.1111/j.1472-8206.1999.tb00334.x. [DOI] [PubMed] [Google Scholar]
- 36.Bai JPF, Amidon GL. Structural Specificity of Mucosal-Cell Transport and Metabolism of Peptide Drugs: Implication for Oral Peptide Drug Delivery. Pharm Res. 1992;9:969–978. doi: 10.1023/a:1015885823793. [DOI] [PubMed] [Google Scholar]
- 37.Shah D, Shen WC. Transcellular delivery of an insulin-transferrin conjugate in enterocyte-like Caco-2 cells. J Pharm Sci. 1996;85:1306–1311. doi: 10.1021/js9601400. [DOI] [PubMed] [Google Scholar]
- 38.Russell-Jones GJ. The potential use of receptor-mediated endocytosis for oral drug delivery. Adv Drug Deliv Rev. 1996;20:83–97. doi: 10.1016/s0169-409x(00)00127-7. [DOI] [PubMed] [Google Scholar]
- 39.Swaan PW. Recent advances in intestinal macromolecular drug delivery via receptor-mediated transport pathways. Pharm Res. 1998;15:826–834. doi: 10.1023/a:1011908128045. [DOI] [PubMed] [Google Scholar]
- 40.Charman WN, Porter CJH. Lipophilic prodrugs designed for intestinal lymphatic transport. Adv Drug Deliv Rev. 1996;19:149–169. [Google Scholar]
- 41.Wang W. Oral protein drug delivery. J Drug Target. 1996;4:195–232. doi: 10.3109/10611869608995624. [DOI] [PubMed] [Google Scholar]
- 42.Woodley JF. Enzymatic barriers for GI peptide and protein delivery. Crit Rev Ther Drug Carrier Syst. 1994;11:61–95. [PubMed] [Google Scholar]
- 43.Patel G, Misra A. 10 - Oral Delivery of Proteins and Peptides: Concepts and Applications. In: Ambikanandan M, editor. Challenges in Delivery of Therapeutic Genomics and Proteomics. Elsevier; London: 2011. pp. 481–529. [Google Scholar]
- 44.Langguth P, Bohner V, Heizmann J, Merkle HP, Wolffram S, Amidon GL, Yamashita S. The challenge of proteolytic enzymes in intestinal peptide delivery. J Control Release. 1997;46:39–57. [Google Scholar]
- 45.Mrsny R. Challenges for the oral delivery of proteins and peptides: theoretical and practical approaches to their delivery. Greenwood, SC: Capsugel Symposia Series; 1991. p. 452. [Google Scholar]
- 46.Ganong WF. Regulation of gastrointestinal function. In: Ganong WF, editor. Review of medical physiology. 12. Lange Medical Publications; 1983. pp. 394–420. [Google Scholar]
- 47.Mrsny RJ. Capsugel Library Symposia series. 1991. Challenges for the oral delivery of proteins and peptides: theoretical and practical approaches to their delivery; pp. 45–52. [Google Scholar]
- 48.Hayakawa E, Lee VH. Aminopeptidase activity in the jejunal and ileal Peyer’s patches of the albino rabbit. Pharm Res. 1992;9:535–540. doi: 10.1023/a:1015800615674. [DOI] [PubMed] [Google Scholar]
- 49.Skillman JJ, Gould SA, Chung RS, Silen W. The gastric mucosal barrier: clinical and experimental studies in critically ill and normal man, and in the rabbit. Ann Surg. 1970;172:564–584. doi: 10.1097/00000658-197010000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meyer RA, McGinley D, Posalaky Z. The gastric mucosal barrier: structure of intercellular junctions in the dog. J Ultrastruct Res. 1984;86:192–201. doi: 10.1016/s0022-5320(84)80058-1. [DOI] [PubMed] [Google Scholar]
- 51.MacAdam A. The effect of gastro-intestinal mucus on drug absorption. Adv Drug Deliv Rev. 1993;11:201–220. [Google Scholar]
- 52.Phelps CF. Biosynthesis of mucus glycoprotein. Br Med Bull. 1978;34:43–48. doi: 10.1093/oxfordjournals.bmb.a071456. [DOI] [PubMed] [Google Scholar]
- 53.Allen A, Garner A. Mucus and bicarbonate secretion in the stomach and their possible role in mucosal protection. Gut. 1980;21:249–262. doi: 10.1136/gut.21.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kompella UB, Lee VH. Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Adv Drug Deliv Rev. 2001;46:211–245. doi: 10.1016/s0169-409x(00)00137-x. [DOI] [PubMed] [Google Scholar]
- 55.Kerss S, Allen A, Garner A. A simple method for measuring thickness of the mucus gel layer adherent to rat, frog and human gastric mucosa: influence of feeding, prostaglandin, N-acetylcysteine and other agents. Clin Sci. 1982;63:187–195. doi: 10.1042/cs0630187. [DOI] [PubMed] [Google Scholar]
- 56.Crater JS, Carrier RL. Barrier properties of gastrointestinal mucus to nanoparticle transport. Macromol Biosci. 2010;10:1473–1483. doi: 10.1002/mabi.201000137. [DOI] [PubMed] [Google Scholar]
- 57.Lai SK, Wang Y-Y, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee VHL, Yamamoto A. Penetration and enzymatic barriers to peptide and protein absorption. Adv Drug Deliv Rev. 1989;4:171–207. [Google Scholar]
- 59.Tang BC, Dawson M, Lai SK, Wang YY, Suk JS, Yang M, Zeitlin P, Boyle MP, Fu J, Hanes J. Biodegradable polymer nanoparticles that rapidly penetrate the human mucus barrier. Proc Natl Acad Sci U S A. 2009;106:19268–19273. doi: 10.1073/pnas.0905998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lai SK, Wang YY, Hida K, Cone R, Hanes J. Nanoparticles reveal that human cervicovaginal mucus is riddled with pores larger than viruses. Proc Natl Acad Sci U S A. 2010;7:59603. doi: 10.1073/pnas.0911748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Primard C, Rochereau N, Luciani E, Genin C, Delair T, Paul S, Verrier B. Traffic of poly(lactic acid) nanoparticulate vaccine vehicle from intestinal mucus to sub-epithelial immune competent cells. Biomaterials. 2010;31:6060–6068. doi: 10.1016/j.biomaterials.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 62.Aoki Y, Morishita M, Asai K, Akikusa B, Hosoda S, Takayama K. Region-dependent role of the mucous/glycocalyx layers in insulin permeation across rat small intestinal membrane. Pharm Res. 2005;22:1854–1862. doi: 10.1007/s11095-005-6137-z. [DOI] [PubMed] [Google Scholar]
- 63.Morishita M, Morishita I, Takayama K, Machida Y, Nagai T. Site-dependent effect of aprotinin, sodium caprate, Na2EDTA and sodium glycocholate on intestinal absorption of insulin. Biol Pharm Bull. 1993;16:68–72. doi: 10.1248/bpb.16.68. [DOI] [PubMed] [Google Scholar]
- 64.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maria Burke SW. Biopharmaceuticals: a new era of discovery in the biotechnology revolution. PJB Publications; 2001. [Google Scholar]
- 66.Almeida AJ, Alpar HO, Brown MR. Immune response to nasal delivery of antigenically intact tetanus toxoid associated with poly(L-lactic acid) microspheres in rats, rabbits and guinea-pigs. J Pharm Pharmacol. 1993;45:198–203. doi: 10.1111/j.2042-7158.1993.tb05532.x. [DOI] [PubMed] [Google Scholar]
- 67.Maloy KJ, Donachie AM, O’Hagan DT, Mowat AM. Induction of mucosal and systemic immune responses by immunization with ovalbumin entrapped in poly(lactide-co- glycolide) microparticles. Immunology. 1994;81:661–667. [PMC free article] [PubMed] [Google Scholar]
- 68.Carino GP, Jacob JS, Mathiowitz E. Nanosphere based oral insulin delivery. J Control Release. 2000;65:261–269. doi: 10.1016/s0168-3659(99)00247-3. [DOI] [PubMed] [Google Scholar]
- 69.Emerich DF, Thanos CG. The pinpoint promise of nanoparticle-based drug delivery and molecular diagnosis. Biomol Eng. 2006;23:171–184. doi: 10.1016/j.bioeng.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 70.Jahanshahi M, Zhang Z, Lyddiatt A. Subtractive chromatography for purification and recovery of nano-bioproducts. IEE Proc Nanobiotechnol. 2005;152:121–126. doi: 10.1049/ip-nbt:20045004. [DOI] [PubMed] [Google Scholar]
- 71.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70:1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 72.Mohanraj V, Chen Y. Nanoparticles – A Review. Trop J Pharm Res. 2006;5:561–573. [Google Scholar]
- 73.Galindo-Rodriguez SA, Allemann E, Fessi H, Doelker E. Polymeric nanoparticles for oral delivery of drugs and vaccines: a critical evaluation of in vivo studies. Crit Rev Ther Drug Carrier Syst. 2005;22:419–464. doi: 10.1615/critrevtherdrugcarriersyst.v22.i5.10. [DOI] [PubMed] [Google Scholar]
- 74.Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13:1838–1845. doi: 10.1023/a:1016085108889. [DOI] [PubMed] [Google Scholar]
- 75.McClean S, Prosser E, Meehan E, O’Malley D, Clarke N, Ramtoola Z, Brayden D. Binding and uptake of biodegradable poly-dl-lactide micro- and nanoparticles in intestinal epithelia. Eur J Pharm Sci. 1998;6:153–163. doi: 10.1016/s0928-0987(97)10007-0. [DOI] [PubMed] [Google Scholar]
- 76.Jung T, Kamm W, Breitenbach A, Kaiserling E, Xiao JX, Kissel T. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur J Pharm Biopharm. 2000;50:147–160. doi: 10.1016/s0939-6411(00)00084-9. [DOI] [PubMed] [Google Scholar]
- 77.Allemann E, Gurny R, Doelker E. Drug-loaded nanoparticles - Preparation methods and drug targeting issues. Eur J Pharm Biopharm. 1993;39:173–191. [Google Scholar]
- 78.Quintanar-Guerrero D, Allemann E, Fessi H, Doelker E. Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm. 1998;24:1113–1128. doi: 10.3109/03639049809108571. [DOI] [PubMed] [Google Scholar]
- 79.Landry DVBFB, Spenlehauer G, Veillard M, Kreuter J. Influence of the coating agents on the degradation of poly(D,Llactic acid) nanoparticles in model digestive fluids. STP pharma : sciences techniques et pratiques pharmaceutiques. 1996;6:195–202. [Google Scholar]
- 80.Le Ray AM, Vert M, Gautier JC, Benoît JP. Fate of [14C]poly(DL-lactide-co-glycolide) nanoparticles after intravenous and oral administration to mice. Int J Pharm. 1994;106:201–211. [Google Scholar]
- 81.Sayani AP, Chien YW. Systemic delivery of peptides and proteins across absorptive mucosae. Crit Rev Ther Drug Carrier Syst. 1996;13:85–184. [PubMed] [Google Scholar]
- 82.Ezpeleta I, Arangoa MA, Irache JM, Stainmesse S, Chabenat C, Popineau Y, Orecchioni A-M. Preparation of Ulex europaeus lectin-gliadin nanoparticle conjugates and their interaction with gastrointestinal mucus. Int J Pharm. 1999;191:25–32. doi: 10.1016/s0378-5173(99)00232-x. [DOI] [PubMed] [Google Scholar]
- 83.Oldenborg PA, Gresham HD, Chen Y, Izui S, Lindberg FP. Lethal autoimmune hemolytic anemia in CD47-deficient nonobese diabetic (NOD) mice. Blood. 2002;99:3500–3504. doi: 10.1182/blood.v99.10.3500. [DOI] [PubMed] [Google Scholar]
- 84.Hussain N. Ligand-mediated tissue specific drug delivery. Adv Drug Deliv Rev. 2000;43:95–100. doi: 10.1016/s0169-409x(00)00066-1. [DOI] [PubMed] [Google Scholar]
- 85.Lee GY, Kim J-H, Oh GT, Lee B-H, Kwon IC, Kim I-S. Molecular targeting of atherosclerotic plaques by a stabilin-2-specific peptide ligand. J Control Release. 2011;155:211–217. doi: 10.1016/j.jconrel.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 86.Jin Y, Song Y, Zhu X, Zhou D, Chen C, Zhang Z, Huang Y. Goblet cell-targeting nanoparticles for oral insulin delivery and the influence of mucus on insulin transport. Biomaterials. 2012;33:1573–1582. doi: 10.1016/j.biomaterials.2011.10.075. [DOI] [PubMed] [Google Scholar]
- 87.Curnis F, Gasparri A, Sacchi A, Longhi R, Corti A. Coupling tumor necrosis factor-alpha with αV integrin ligands improves its antineoplastic activity. Cancer Res. 2004;64:565–571. doi: 10.1158/0008-5472.can-03-1753. [DOI] [PubMed] [Google Scholar]
- 88.Gupta AK, Gupta M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials. 2005;26:3995–4021. doi: 10.1016/j.biomaterials.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 89.Wagner E, Curiel D, Cotten M. Delivery of drugs, proteins and genes into cells using transferrin as a ligand for receptor-mediated endocytosis. Adv Drug Deliv Rev. 1994;14:113–135. [Google Scholar]
- 90.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 91.Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 92.Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54:561–587. doi: 10.1124/pr.54.4.561. [DOI] [PubMed] [Google Scholar]
- 93.Berry CC, Charles S, Wells S, Dalby MJ, Curtis ASG. The influence of transferrin stabilised magnetic nanoparticles on human dermal fibroblasts in culture. Int J Pharm. 2004;269:211–225. doi: 10.1016/j.ijpharm.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 94.Gupta AK, Berry CC, Gupta M, Curtis ASG. Receptor-mediated targeting of magnetic nanoparticles using insulin as a surface ligand to prevent endocytosis. IEEE Trans NanoBioscience. 2003;2:255–261. doi: 10.1109/tnb.2003.820279. [DOI] [PubMed] [Google Scholar]
- 95.Debelle L, Tamburro AM. Elastin: molecular description and function. Int J Biochem Cell Biol. 1999;31:261–272. doi: 10.1016/s1357-2725(98)00098-3. [DOI] [PubMed] [Google Scholar]
- 96.Baker ME. Albumin’s role in steroid hormone action and the origins of vertebrates: is albumin an essential protein? FEBS lett. 1998;439:9–12. doi: 10.1016/s0014-5793(98)01346-5. [DOI] [PubMed] [Google Scholar]
- 97.Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Bi. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 98.Andersson M, Fromell K, Gullberg E, Artursson P, Caldwell KD. Characterization of surface-modified nanoparticles for in vivo biointeraction. A sedimentation field flow fractionation study. Anal Chem. 2005;77:5488–5493. doi: 10.1021/ac050631h. [DOI] [PubMed] [Google Scholar]
- 99.Hersel U, Dahmen C, Kessler H. RGD modified polymers: biomaterials for stimulated cell adhesion and beyond. Biomaterials. 2003;24:4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 100.Bellis SL. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials. 2011;32:4205–4210. doi: 10.1016/j.biomaterials.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Montet X, Funovics M, Montet-Abou K, Weissleder R, Josephson L. Multivalent effects of RGD peptides obtained by nanoparticle display. J Med Chem. 2006;49:6087–6093. doi: 10.1021/jm060515m. [DOI] [PubMed] [Google Scholar]
- 102.Gref R, Couvreur P, Barratt G, Mysiakine E. Surface-engineered nanoparticles for multiple ligand coupling. Biomaterials. 2003;24:4529–4537. doi: 10.1016/s0142-9612(03)00348-x. [DOI] [PubMed] [Google Scholar]
- 103.Weissenböck A, Wirth M, Gabor F. WGA-grafted PLGA-nanospheres: preparation and association with Caco-2 single cells. J Control Release. 2004;99:383–392. doi: 10.1016/j.jconrel.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 104.Mo Y, Lim L-Y. Paclitaxel-loaded PLGA nanoparticles: Potentiation of anticancer activity by surface conjugation with wheat germ agglutinin. J Control Release. 2005;108:244–262. doi: 10.1016/j.jconrel.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 105.Mo Y, Lim LY. Preparation and in vitro anticancer activity of wheat germ agglutinin (WGA)-conjugated PLGA nanoparticles loaded with paclitaxel and isopropyl myristate. J Control Release. 2005;107:30–42. doi: 10.1016/j.jconrel.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 106.Gao X, Wang T, Wu B, Chen J, Chen J, Yue Y, Dai N, Chen H, Jiang X. Quantum dots for tracking cellular transport of lectin-functionalized nanoparticles. BBBC. 2008;377:35–40. doi: 10.1016/j.bbrc.2008.09.077. [DOI] [PubMed] [Google Scholar]
- 107.Jeong YI, Seo SJ, Park IK, Lee HC, Kang IC, Akaike T, Cho CS. Cellular recognition of paclitaxel-loaded polymeric nanoparticles composed of poly(gamma-benzyl L-glutamate) and poly(ethylene glycol) diblock copolymer endcapped with galactose moiety. Int J Pharm. 2005;296:151–161. doi: 10.1016/j.ijpharm.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 108.Yin Y, Chen D, Qiao M, Wei X, Hu H. Lectin-conjugated PLGA nanoparticles loaded with thymopentin: Ex vivo bioadhesion and in vivo biodistribution. J Control Release. 2007;123:27–38. doi: 10.1016/j.jconrel.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 109.Marasco D, Perretta G, Sabatella M, Ruvo M. Past and future perspectives of synthetic peptide libraries. Curr Protein Pept Sci. 2008;9:447–467. doi: 10.2174/138920308785915209. [DOI] [PubMed] [Google Scholar]
- 110.Smith GP, Petrenko VA. Phage Display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 111.Koivunen E, Arap W, Rajotte D, Lahdenranta J, Pasqualini R. Identification of receptor ligands with phage display peptide libraries. J Nucl Med. 1999;40:883–888. [PubMed] [Google Scholar]
- 112.Yoo M-K, Kang S-K, Choi J-H, Park I-K, Na H-S, Lee H-C, Kim E-B, Lee N-K, Nah J-W, Choi Y-J, Cho C-S. Targeted delivery of chitosan nanoparticles to Peyer’s patch using M cell-homing peptide selected by phage display technique. Biomaterials. 2010;31:7738–7747. doi: 10.1016/j.biomaterials.2010.06.059. [DOI] [PubMed] [Google Scholar]
- 113.Fievez V, Plapied L, Plaideau C, Legendre D, des Rieux A, Pourcelle V, Freichels H, Jerome C, Marchand J, Preat V, Schneider YJ. In vitro identification of targeting ligands of human M cells by phage display. Int J Pharm. 2010;394:35–42. doi: 10.1016/j.ijpharm.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 114.Koivunen E, Wang B, Ruoslahti E. Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnol. 1995;13:265–270. doi: 10.1038/nbt0395-265. [DOI] [PubMed] [Google Scholar]
- 115.Pasqualini R, Koivunen E, Ruoslahti E. Alpha v integrins as receptors for tumor targeting by circulating ligands. Nat Biotechnol. 1997;15:542–546. doi: 10.1038/nbt0697-542. [DOI] [PubMed] [Google Scholar]
- 116.McGuire MJ, Samli KN, Johnston SA, Brown KC. In vitro Selection of a Peptide with High Selectivity for Cardiomyocytes in vivo. J Mol Biol. 2004;342:171–182. doi: 10.1016/j.jmb.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 117.Doorbar J, Winter G. Isolation of a Peptide Antagonist to the Thrombin Receptor using Phage Display. J Mol Biol. 1994;244:361–369. doi: 10.1006/jmbi.1994.1736. [DOI] [PubMed] [Google Scholar]
- 118.Oyama T, Rombel IT, Samli KN, Zhou X, Brown KC. Isolation of multiple cell-binding ligands from different phage displayed-peptide libraries. Biosens Bioelectron. 2006;21:1867–1875. doi: 10.1016/j.bios.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 119.Demirgoz D, Garg A, Kokkoli E. PR_b-targeted PEGylated liposomes for prostate cancer therapy. Langmuir. 2008;24:13518–13524. doi: 10.1021/la801961r. [DOI] [PubMed] [Google Scholar]
- 120.Mamaeva V, Rosenholm JM, Bate-Eya LT, Bergman L, Peuhu E, Duchanoy A, Fortelius LE, Landor S, Toivola DM, Linden M, Sahlgren C. Mesoporous silica nanoparticles as drug delivery systems for targeted inhibition of Notch signaling in cancer. Molecular Therapy. 2011;19:1538–1546. doi: 10.1038/mt.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Samli KN, McGuire MJ, Newgard CB, Johnston SA, Brown KC. Peptide-mediated targeting of the islets of Langerhans. Diabetes. 2005;54:2013–2108. doi: 10.2337/diabetes.54.7.2103. [DOI] [PubMed] [Google Scholar]
- 122.Goldstein IJ, Hughes RC, Monsigny M, Osawa T, Sharon N. What should be called a lectin? Nature. 1980;285:66–66. [Google Scholar]
- 123.Irache JM, Durrer C, Duchene D, Ponchel G. Preparation and characterization of lectin-latex conjugates for specific bioadhesion. Biomaterials. 1994;15:899–904. doi: 10.1016/0142-9612(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 124.Irache JM, Durrer C, Duchene D, Ponchel G. Bioadhesion of lectin-latex conjugates to rat intestinal mucosa. Pharm Res. 1996;13:1716–1719. doi: 10.1023/a:1016405126656. [DOI] [PubMed] [Google Scholar]
- 125.Lehr CM, Bouwstra JA, Kok W, Noach AB, de Boer AG, Junginger HE. Bioadhesion by means of specific binding of tomato lectin. Pharm Res. 1992;9:547–553. doi: 10.1023/a:1015804816582. [DOI] [PubMed] [Google Scholar]
- 126.Russell-Jones GJ, Veitch H, Arthur L. Lectin-mediated transport of nanoparticles across Caco-2 and OK cells. Int J Pharm. 1999;190:165–174. doi: 10.1016/s0378-5173(99)00254-9. [DOI] [PubMed] [Google Scholar]
- 127.Zhang N, Ping Q, Huang G, Xu W, Cheng Y, Han X. Lectin-modified solid lipid nanoparticles as carriers for oral administration of insulin. Int J Pharm. 2006;327:153–159. doi: 10.1016/j.ijpharm.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 128.Liu J, Gong T, Wang C, Zhong Z, Zhang Z. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: Preparation and characterization. Int J Pharm. 2007;340:153–162. doi: 10.1016/j.ijpharm.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 129.Arosio D, Manzoni L, Araldi EM, Scolastico C. Cyclic RGD functionalized gold nanoparticles for tumor targeting. Bioconjugate Chem. 2011;22:664–672. doi: 10.1021/bc100448r. [DOI] [PubMed] [Google Scholar]
- 130.Fievez V, Plapied L, des Rieux A, Pourcelle V, Freichels H, Wascotte V, Vanderhaeghen M-L, Jerôme C, Vanderplasschen A, Marchand-Brynaert J, Schneider Y-J, Préat V. Targeting nanoparticles to M cells with non-peptidic ligands for oral vaccination. Eur J Pharm Biopharm. 2009;73:16–24. doi: 10.1016/j.ejpb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 131.Kim WJ, Yockman JW, Lee M, Jeong JH, Kim YH, Kim SW. Soluble Flt-1 gene delivery using PEI-g-PEG-RGD conjugate for anti-angiogenesis. J Control Release. 2005;106:224–234. doi: 10.1016/j.jconrel.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 132.Gullberg E. Particle Transcytosis Across the Human Intestinal Epithelium : Model Development and Target Identification for Improved Drug Delivery. Acta Universitatis Upsaliensis. 2005 [Google Scholar]
- 133.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 134.Neff JA, Caldwell KD, Tresco PA. A novel method for surface modification to promote cell attachment to hydrophobic substrates. J Biomed Mater Res. 1998;40:511–519. doi: 10.1002/(sici)1097-4636(19980615)40:4<511::aid-jbm1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]