

Abstract
Epidemiological studies have consistently supported the notion that environmental and/or dietary factors play a central role in the aetiology of cancers of the breast and prostate. However, for more than five decades investigators have failed to identify a single cause-and-effect factor, which could be implicated; identification of a causative entity would allow the implementation of an intervention strategy in at-risk populations. This suggests a more complex pathoaetiology for these cancer sites, compared to others. When one examines the increases or decreases in incidence of specific cancers amongst migrant populations, it is notable that disease arising in colon or stomach requires one or at most two generations to exhibit a change in incidence to match that of high-incidence regions, whereas for breast or prostate cancer, at least three generations are required. This generational threshold could suggest a requirement for nonmutation-driven epigenetic alterations in the F0/F1 generations (parental/offspring adopting a more westernized lifestyle), which then predisposes the inherited genome of subsequent generations to mutagenic/genotoxic alterations leading to the development of sporadic cancer in these target sites. As such, individual susceptibility to carcinogen insult would not be based per se on polymorphisms in activating/detoxifying/repair enzymes, but on elevated accessibility of crucial target genes (e.g., oncogenes, tumour suppressor genes) or hotspots therein to mutation events. This could be termed a genomic susceptibility organizational structure (SOS). Several exposures including alcohol and heavy metals are epigens (i.e., modifiers of the epigenome), whereas others are mutagenic/genotoxic, for example, heterocyclic aromatic amines; humans are continuously and variously exposed to mixtures of these agents. Within such a transgenerational multistage model of cancer development, determining the interaction between epigenetic modification to generate a genomic SOS and genotoxic insult will facilitate a new level of understanding in the aetiology of cancer.
1. Introduction
Epidemiological studies clearly implicate environmental and/or lifestyle factors in the aetiology of cancers arising in hormone-responsive tissues, such as those from the breast or prostate [1]. This is based on the observations that incidence of these cancers is high in regions such as Northern/Western Europe and the USA, whereas recorded levels in other areas including China and India are traditionally some 10-fold lower [2] (Figure 1(a)). However, when populations migrate from these areas of low risk to high-risk regions, subsequent generations exhibit a disease incidence more in keeping with that of the host population [3, 4] (Figure 1(b)). Even amongst families identified with highly penetrant predisposing mutations in genes such as BRCA1/2 and resident in low-risk areas, there appears to be a lower incidence compared to similar familial lineages resident in a westernized environment [5]. These observations begin to lay the basis of a complex and maybe transgenerational model of cancer induction in some hormone-responsive tissues.
Figure 1.
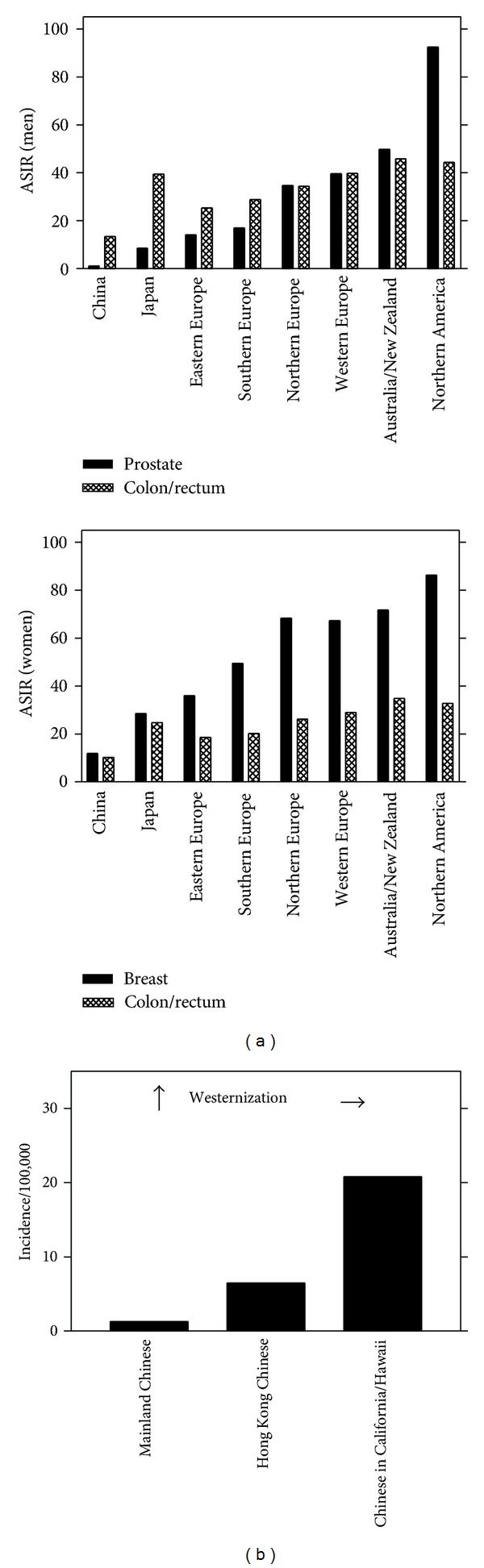
Incidence by region of breast, prostate, and colorectal cancers, and increase in prostate cancer in Chinese migrants. (a) Age-standardized incidence rate (ASIR) as estimated by Parkin et al. (1999) [2]. (b) Increasing incidence of prostate amongst Chinese migrants as estimated from Muir et al. (1991) [3].
Within migrant populations, the cancer-incidence profile changes [4], but not at the same rate for all tissue sites. The incidence of colorectal cancer rises and that of stomach cancer falls in migrant populations from Far East areas more quickly and within two generations compared to the three generations required to observe similar increases in breast and prostate cancer [1]. This suggests an additional requirement to the simple initiation-promotion model of cancer development [6, 7]. The notion of a transgenerational requirement in cancer induction is not new: albeit intrauterine exposure occurred, diethylstilbestrol (DES) gave rise to marked increases in the unusual entities of adenosis and clear-cell adenocarcinoma of the genital tract in young female daughters of mothers exposed to this agent [8]; whether there were consequences in male offspring remains to be ascertained. More recently, models such as the Agouti mouse have shown that transgenerational influences can result in offspring predisposed to a pathological state such as obesity [9], in itself a grave predisposing factor for chronic morbidities.
With more populations globally adopting a westernized lifestyle (that which could be associated with living in Northern/Western Europe and USA), there is the real possibility for a sudden surge in cancers of the breast, colon, prostate, and uterus in areas that hitherto would not have seen large rates of incidence of these conditions; in many of these regions, the question will be whether there will be a healthcare infrastructure capable of coping with markedly increased numbers of cases and, capable of providing appropriate treatment and after-patient care [10]. As far back as the 1960's, there was recognition of worldwide region-specific differences in breast cancer incidence and, correlations between calorie, protein or fat consumption, and risk were noted [11]. Even then, in addition to environmental carcinogen exposures, a hormone-mediated difference in susceptibility to breast cancer among US-resident women of different ethnic backgrounds was observed. Breast adipose tissue may act as a reservoir for lipophilic genotoxic carcinogens [12, 13], but still there is likely a requirement for a hormone-driven event [11] that develops or evolves with a phenotypic change associated with lifestyle.
Diet predisposes children from ethnically diverse populations resident in the USA to chronic conditions such as cancer later in life; this same early-life influence is noted in the UK [14, 15]. The question then is whether due to differing environmental and/or lifestyle changes, a phenotypic change occurs in migrants to Western regions (e.g., Northern/Western Europe and USA) and their children (i.e., the F0/F1 generations), and this then not so much enhances vulnerability as a mutation might, but creates accessibility to critical target sites (e.g., tumour suppressor genes (TSGs) and oncogenes) in the genome of subsequent generations (Table 1). As a consequence the F2/F3/and so forth generations inherit a structurally vulnerable genome; this may be to allow increased growth and physicality associated with Western lifestyle, but it could also open up the potential for genomic accessibility with consequent cycles of chemical-DNA adduct formation and potentially faulty repair. This parental priming of the genome template could be termed a genomic susceptibility organizational structure (SOS); within this model, later generations do not inherit predisposing mutations but a chromatin organization that allows critical genes to be better targeted by DNA-damaging agents of environmental and/or dietary origin.
Table 1.
How a genomic susceptibility organizational structure (SOS) may influence progeny responses to environmental exposures.
| Generation | Evolution of genomic SOS | |
|---|---|---|
| Chromatin modification | Cancer risk | |
| F0 | − | Low |
| F1 | + | Low |
| F2 | ++ | Emerging |
| F3 | +++ | High |
Translation of epigenetic alterations amongst migrant populations from low-risk cancer (breast and prostate) areas to high-risk regions. Parental generation has not acquired a genomic SOS, but by F1 and subsequent generations this organizational structure has emerged via environmental and/or lifestyle changes. This exposes the F2/F3 genomes to DNA damage insult.
−: not present; +: evolving; ++: evolved; +++: highly evolved.
Such developmental plasticity could be associated with epigenetic modifications. Environmental and/or lifestyle factors alter the epigenome of the F0/F1 generation amongst a migrant population to a high-risk region; these inherited epigenetic alterations would then predispose the genome of subsequent generations. This hypothesised model would explain why epidemiological studies to date have failed to identify causative factors for breast or prostate cancers although migration, hormone factors, calorie/protein/fat consumption, and exposure to carcinogens have been variously implicated. This review sets out to delineate a potential role for transgenerational epigenetic influences in the aetiology of breast and prostate cancer, cancers arising from these sites being more complex than more directly targeted tissues such as the lung, colon or stomach. It will also highlight that conventional epidemiological studies that typically set out to establish cause (or susceptible genotype) and effect, will be severely limited in their scope under this paradigm. However, it also shows why with ever increasingly large studies more robust associations with environmental causes are noted.
2. Epigenetic Alterations Underpinning Disease
Epigenetic alterations include modifications in DNA methylation, posttranslational influences on histone morphology/code (i.e., proteins forming the nucleosome core of chromatin and pivotal in structural packaging of DNA), nucleosome positioning and, the profile of microRNAs (miRNAs) and noncoding RNAs [16–18]. The most-studied alteration to date is methylation at the 5′ position of cytosines mediated by DNA methyltransferases (DNMT), predominantly in cytosine-guanine dinucleotide (CpG) sites; this generates 5-methylcytosine (5-meC), and if it occurs at sites that occupy the promoter regions of genes, this hypermethylation is correlated with transcriptional silencing.
Methylation can result in complete transcriptional repression of TSGs such as p16 [19]; if detected early enough, the specificity of this methylation pattern could be predictive for cancer types occurring at a particular or different sites [20, 21]. This has led to the development of quantitative methylation-specific PCR analyses to determine the methylation status of a panel of candidate TSGs in order to determine if one can triage for certain cancers [22]. Such methylation patterns may throw up new differentially expressed gene candidates in cancer, some of which may even be free of mutations [23]. These observations herald the possibility of epigenetic blood-based biomarkers linked to the underlying mechanism of a specific cancer pathogenesis [24]; it opens up possibilities of novel early screening tools.
Although methylation status is currently the most-studied epigenetic marker, there is increasing recognition that other modifications such as those of the histone code can modify the chromatin organization in such a way as to influence in a dominant fashion regional mutation-rate [25]; this could be a pivotal observation to be translated to the notion of the evolution of a genomic SOS in humans. Histone deacetylases are now recognized to play a role in the control of the DNA damage responses at several levels [26]. What is compelling is that these structural chromatin alterations can be inherited [27]; this remodelled genome must influence the offspring's susceptibility to damage events following carcinogen exposures in their lifetime, that is, a genomic SOS. Metabolic signals play a critical role in determining chromatin structure [28]; thus one can surmise that given a markedly different lifestyle, the same genotype could generate a markedly different phenotype through the evolution of differing profiles of epigenetic marking of the genome. When migrants arrive in a new region, an initially slowly-adopted diet and/or lifestyle will then impact on the organizational structure and relative compaction of their offspring's chromatin (Figure 2). Migrants from the Far East to Northern/Western Europe may see their children/grandchildren grow more than those with similar genotypes in previous generations; this may be looked upon as a beneficial health outcome, but does this have an impact on the accessibility of their genome to genotoxic exposures of dietary and/or environmental origin? Studies of evolution have suggested that epigenetic drivers have underpinned dramatic changes in developmental biology, an example being that of cognitive function, to a far greater extent than genetic variability [29]. Persistent physical exercise will modify the epigenome, which will alter phenotype [30]; likewise, diet and/or lifestyle will undoubtedly play a significant role in this process too.
Figure 2.
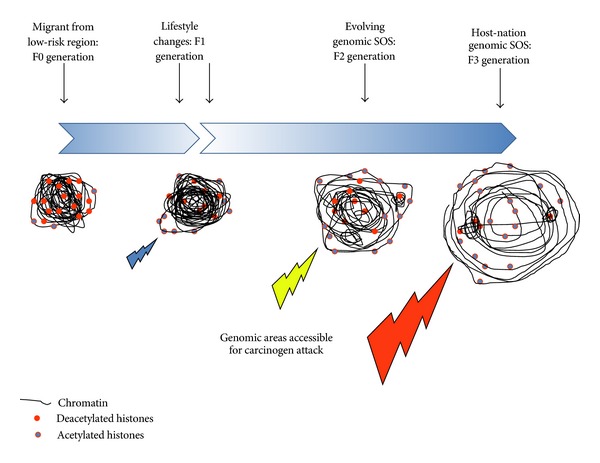
Transgenerational dependency of the genomic susceptibility organizational structure (SOS). Migrants (F0 generation) from a low-risk region for breast or prostate cancer exhibit a chromatin conformation that makes target genes often targeted for damage by environmental and/or dietary constituents less accessible. With lifestyle changes, a more accessible genome evolves in the F1 generation, as evidenced by a less compact chromatin and more acetylated histones; this creates more target sites for attack by carcinogens. Through F2, the genomic SOS further evolves, creating even more accessible sites for carcinogen attack. By F3, an evolution to more closely match that of the host nation genomic SOS has ocurred, with a maximum number of target sites for carcinogen attack. The break in the temporal arrow highlights a change of environment and/or lifestyle that leads to the generation of the genomic SOS; the increase in shade indicates its evolution.
Of additional recent interest is the role noncoding RNAs play in the pathogenesis of cancer. Although their biology and function still remain mostly obscure, they are believed to have core functions in a wide range of cellular processes, through interaction with key component proteins in the gene regulatory system. Alterations of their cell- or tissue-specific expression and/or their primary or secondary structures are thought to promote cell proliferation, invasion, and metastasis [31]. miRNAs comprise a family of small, endogenous, noncoding functional RNA molecules that have emerged as key posttranscriptional regulators of gene expression; they may again be useful biomarkers for early detection of disease-related molecular and genetic changes [32]. Noncoding miRNAs can contribute to cancer development and progression, and are differentially expressed in normal tissues compared to cancers; their expression signature may define cancer gene targets [33] or be diagnostic of the cancer type. However, not dissimilar to histone modifications, the expression profile of noncoding miRNAs constitutes an epigenetic alteration that seems to regulate male gamete production [34], CNS functions [35], cell cycle and proliferation [36], and erythrocyte development during haematopoiesis in vertebrates [37], amongst a plethora of other biological functions and mechanisms. In a mouse model, paternal exposure to the procarcinogen benzo[a]pyrene (B[a]P) affected the expression of several miRNAs, and the target genes for some of the dysregulated miRNAs were enriched in many different pathways that are likely to be relevant for the developing embryo [38]. The notion of an expression control by a posttranslational genomic signature generated by a previous generation's environmental influence, but governing the offspring's susceptibility, will pose extreme challenges for future epidemiological studies. This does not represent a simple cause-and-effect model within the individual but one modulated by ancestral environmental influences.
2.1. Lifestyle Factors That Modify the Epigenome
Early-life (pre- and/or postnatal) influences on the epigenome appear to modify risk to later-life susceptibility to chronic diseases, including cardiovascular disease, diabetes, and cancer [39]. In a number of animal models, the maternal nutritional environment appears to play a pivotal role in the long-term wellbeing of the offspring [40]; we are only now beginning to investigate how this might influence the epidemiology of chronic age-related diseases such as cancer in humans. Cancer is a heterogeneous disease, which we understand now is not only caused by genetic alterations (e.g., mutations) but also by an altered gene expression profile (i.e., epigenetic alteration) [41]. Given that we have known for years about a constitutive role for extrahepatic bioactivating enzymes capable of activating procarcinogens [42, 43], differences in expression between different regions or cell populations within the same tissue [44], and their environmentally-/lifestyle-mediated modifiable nature [45], the notion that epigenetic influences may also modify their role in cancer aetiology is not unsurprising. Differences in environmental influences mean that older monozygous twins who may have grown up in differing environments (e.g., urban versus rural) exhibit significant discordance in epigenomic markers such as genomic distribution of 5-meC DNA and histone acetylation compared to those examined in the early years of life [46]. Epigenetic changes in DNA methylation patterns at CpG sites (which could be known as epimutations) may give rise to transgenerational effects [47], although understanding these phenomena in real-world situations remains a major challenge [48]. However, the premise of this review is that a transgenerational step is required towards acquiring the elevated risk of breast or prostate cancer observed in North/Western Europe and the USA. This differs from a more cause-and-effect model seen with lung cancer induced by tobacco smoke, stomach cancer induced by nitrosamines [49], or hepatocellular carcinoma induced by aflatoxin B1 [50].
2.2. Could Epigens Modify Early-Life Risk?
Ever since valproic acid was identified as a specific histone deacetylase (HDAC) inhibitor [51], it has also been speculated that new intervention or therapeutic strategies exploiting agents that also modify transcriptional regulation in the pathogenesis of cancer via the generation of epigenetic alterations could be developed. Although this may convey a novel therapeutic strategy in the presence of disease, it also raises the possibility that continuous low-level exposures to epimutagenic agents might enhance susceptibility to genotoxic mechanisms [52]. A whole range of chemical contaminants found in the human diet and/or environment may modify the epigenomic patterns of cytosine methylation and/or histone acetylation [53]. Examples of this are the heavy metals including arsenic, nickel, chromium, and cadmium, which increase cancer incidence; these are weak mutagens but appear to be potent modifiers of the epigenome [54]. By examining morphological changes in heterochromatin and DNA methylation at gene loci in an in vitro early embryo (mouse) model, it was shown that selected environmental contaminants, including diethyl phosphate, mercury, cotinine, selenium, and octachlorodipropyl ether induced at low concentrations (i.e., ppb), marked and sometimes irreversible epigenetic alterations [55]; given the potential for real-world exposures to such exogenous agents, one might surmise that this could impact human embryological development. Of course, humans are not exposed to single test agents but are exposed continuously and variously throughout life to a changing cocktail of different contaminants [56]. Modelling the potential effects of such mixtures is a challenge, as different test agents might be similarly acting or independently acting [57, 58]; as such, they could act in combination in additive, synergistic, or inhibitory mechanisms. In populations with high exposures to persistent organic pollutants (POPs), such as Greenlandic Inuit, global methylation levels were inversely associated with blood plasma levels for several POPs [59]. Such observations make it highly likely that epigens (i.e., modifiers of the epigenome) found routinely in the human diet and/or environment could modify the human epigenome, which could add another level of complexity in the exposure-causation model of classical epidemiological studies [60]. If these heritable genomic alterations are a consequence of parental or early-life exposures, they may if they occur greatly enhance subsequent susceptibility to later-life factors in the aetiology of chronic disease.
3. Chemicals That Modify the Epigenome
There is an increasing suggestion in the academic literature that exposure to environmental contaminants may play an aetiological role in a range of disease-predisposing conditions, including obesity. Although high-density calorie diet and lack of physical activity might be the primary causes of obesity, endocrine disruptors acting as obesogens could initiate or exacerbate this morbidity [61, 62]. In addition to endocrine disruptors, there is a growing body of evidence that heavy metals, including nickel, lead, cadmium, arsenic and others, asbestos, and alcohol intake all act to variously modify the potential for epigenetic alterations [63]. Air pollution constituents, especially particulate matter (PM), appear to alter the profile of miRNAs [18]. PM is known to alter epigenetic markers (e.g., DNA methylation and histone modifications), which may contribute to air-pollution-mediated health consequences including an elevated risk for cardiovascular diseases or events; identifying individual epigenetic loci associated with dysregulated gene expression following exposure could generate novel intervention strategies mitigating the development of such adverse outcomes [64]. Ionizing radiation and nanomaterials are also thought to induce epigenetic alterations. In addition, perfluorinated compounds are of significant concern as they bioaccumulate with suggestions that in utero human exposure is associated with global hypomethylation of the genome; recently, perfluorooctanoic acid-mediated toxicity was associated with aberrant methylation of glutathione-S-transferase Pi [65], a carcinogen-detoxifying enzyme.
Chemical pollutants, dietary components, temperature changes, and other external stresses can indeed have long-lasting effects on development, metabolism, and health, sometimes even in generations subsequent to the exposed individual [66]. A growing body of epidemiological evidence demonstrates associations between parental usage (especially occupational) of pesticides, particularly insecticides, giving rise to acute lymphocytic leukaemia and brain tumours in offspring [67]. Accumulating evidence suggests that environmental and occupational exposures to natural substances, as well as man-made chemical and physical agents, play an aetiological role in human cancer; carcinogenesis may be induced by either genotoxic or nongenotoxic carcinogens (e.g., arsenic, 1,3-butadiene) that also cause prominent epigenetic changes [68]. Cadmium is a toxic, nonessential transition metal and contributes a health risk to humans, including strong associations between its exposure and various cancers or cardiovascular diseases. This agent has been shown to induce various epigenetic changes in plant and mammalian cells in vitro and in vivo, and this is likely the primary mechanism via which it mediates its toxicity [69].
The importance of early-life changes towards future susceptibility to chronic age-related diseases is gaining increasing recognition. Of major concern in this regard are observations that common environmental contaminants such a bisphenol A and phthalates can variously be hypomethylating and alter miRNA expression levels or DNMT activities [70]; the fact that such agents appear to induce low-dose effects postmaternal exposure in the genital tract of female offspring of mice [71] suggests a phenotype change associated with an epigenetic alteration that has later-life consequences. In fact, in the area of environmental epigenetics, such agents as well as other endocrine disruptors including organochlorines, are likely to play a pivotal role [72]. This could be an important link with cancers arising from hormone-responsive tissues, including the breast and prostate. Disruptors of hormonal status via epimutagenic processes are yet to be understood in terms of their long-term health consequences.
The interplay between genotoxicity and epigenetic alterations remains to be elucidated; for instance, acetylation of histones occurs during the process of DNA damage induction [73]. As direct-acting DNA-damaging agents are traditionally known as genotoxins, agents that induce aberrant epigenetic alterations may be known as epimutagens. In general, cancer is typified by global genomic hypomethylation and site-specific hypermethylation [74], especially at TSGs: the former being associated with an overactive genome and proliferation, and the latter with inactivation of genes such as TP53 that might sit at the crossroads between induction of aberrant proliferation and apoptosis. Time of exposure during life, dose, gender, and organ specificity all need to be considered in the development of epigenetic endpoints as biomarkers for exposure to epimutagenic toxicants [75]. How at different stages of life in a particular target organ there is induction of irreversible changes to the genetic material (i.e., DNA mutations) against a backdrop of putatively reversible changes to the epigenetic landscape (i.e., changes in the DNA methylation and chromatin modification state) remains to be understood [76]. Does the latter modify accessibility of the genome in a fashion that predisposes it to genotoxic insult? This may underpin the interplay between genotoxic and epigenetic mechanisms in the aetiology of cancer.
4. Epidemiology and the Epigenome
Linking exposures and their possible effects to environmental health is a major inter-disciplinary challenge [77]. Inter-individual variability increases with age and is probably mediated by an environmental modification of the epigenome; this could explain why genetically identical twins might age differently [78]. In fact, the ageing process has recently been observed to be associated with an increasing level of global genomic hypomethylation [79]. In lung cancer tissues, several gene loci were observed to be either significantly hypermethylated or hypomethylated [80]. Ideally, epidemiological studies require early biomarkers of risk so that susceptible individuals and/or populations can be identified. In theory, this would facilitate intervention studies within which damaging exposures would be reduced or removed [81]. Because epigenetic alterations are modifiable, this has enormous implications for cancer prevention and treatment [82].
In an era of epidemiology that has been largely based on attempting to correlate gene-environment interactions to identify environmental causative factors imposed on a profile of genetic susceptibility [83], determining parental or ancestral lifestyle and/or environmental influences on transgenerational susceptibility to chronic diseases such as cancer will be a major challenge. Populations that migrate from regions of low disease incidence to high-risk areas do not just face changes in environment (which may in fact become more contaminant-free and regulated in the case of westernized regions), but primarily, and especially in subsequent generations, profound cultural and lifestyle changes. This has resulted amongst Asians migrating to the UK in a higher prevalence of Type II diabetes compared to the multigenerational resident population [84], obesity and coronary heart disease [85]. Within a laboratory model such as the Agouti mouse, one may generate interesting mechanistic paradigms of transgenerational effects. However, given that human lifestyle has changed so dramatically in the last few generations, especially in Northern/Western Europe and USA, factors that were either potentially deleterious (e.g., smoking) or even beneficial (e.g., high calorie storage; [86]) may manifest an increased susceptibility or protection in a subsequent generation. Understanding ancestral influences on current risk stratification in today's global population is going to be a major challenge in coming years.
4.1. Genotoxins/Mutagens versus Epimutagens in Cancer Aetiology
The traditional model of cancer causation is based on an initiating exposure (independent of age) by a genotoxic/mutagenic agent followed by a multistage process of promotion, which may take >20 yrs in humans [87, 88]; invasive disease characterized by gross genomic instability then progresses beyond this. In the absence of the inheritance of highly-penetrant mutant alleles (e.g., BRCA1/2), this model has laid the basis for our understanding of the aetiology of sporadic cancers. The need for an initiating exposure was first proposed by Roger Case in his studies of bladder cancer incidence in dye industry workers [89, 90], and subsequent studies of the role of cigarette smoking in lung cancer [91, 92] have been pivotal in the acceptance of this theory. Molecular archaeology studies clearly demonstrate that a specific environmental/dietary chemical exposure (e.g., aflatoxin B1) can induce a particular mutation in localized “hotspots” of critical genes (e.g., the tumour suppressor TP53) in hepatocellular carcinoma [93, 94].
One can now point to several types and classes of physical and chemical environmental and dietary carcinogens, which will with or without a bioactivation step (the latter required in the case of inert chemicals) induce a genotoxic event, ultimately giving rise to mutations in target cells. These include polycyclic aromatic hydrocarbons (PAHs) such as B[a]P [95], heterocyclic aromatic amines (HAAs) such as 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) [96], physical agents such as ultraviolet (UV) or ionizing radiation [97], and materials including asbestos fibres [98, 99]. With indirect or inert chemical carcinogens, a bioactivation step is required (mediated primarily by cytochrome P450 mixed function oxidases (CYPs), but also by N-acetyltransferases (NATs) or sulfotransferases) to generate electrophilic intermediates that then give rise to covalent DNA adducts at nucleophilic sites on DNA bases, for example, C8 position of deoxyguanosine [100, 101]. The generation of base substitution or frameshift mutations then occurs via the inaccurate repair of the adducted template, which has been associated with the replication complex performing a mutagenic bypass of the lesion by a slippage mechanism [102, 103]. The question is how does the epigen determine the magnitude of the mutagen or genotoxin effect?
Chromatin accessibility plays a role in not only regulating cell-type specific gene expression [104], but also DNA repair [105]; if epigenetic mechanisms determine the accessibility of pivotal genes to electrophilic attack giving rise to DNA adducts and to subsequent faulty repair mechanisms, this might be a plausible link between the genotoxic insult and the epigenomic modification governing individual susceptibility to cancer causation. The transgenerational mechanism is that the epigenomic modification was inherited. As such, the offspring does not have an inherently susceptible genome in terms of polymorphic differences in a panel of susceptibility genes (e.g., bioactivating, DNA repair, or detoxification), but the structural dimensions of the chromatin facilitate its vulnerability to exogenously- and/or endogenously-derived insults. Thus the parental or ancestral environment and/or lifestyle generated the evolution of a genomic SOS, that is heritable, and predetermines from early life the offspring's susceptibility to genotoxic carcinogens. Within the accessible genome, there is a greatly elevated chance of the initiation-promotion multistage process of carcinogenesis occurring.
4.2. Factoring Transgenerational Epigenetic Modifications into Future Epidemiological Studies
Before embarking on an epidemiological study, sample size calculations are important to provide evidence that the proposed study is capable of detecting real associations between study factors [106]. Traditionally, to know what size of relative risk may be confidently detected with the projected size of the cohort and length of followup, one often determines the adequacy of cohort size at the planning stage of a study [107]. Disease epidemiology aiming at identification of carcinogens and quantification of associated risks has always had an apparent low resolving power, with detectable incidence or mortality increments often being orders of magnitude smaller than levels which would be of public concern. Other drawbacks of disease epidemiology are the long latency times in development of chronic diseases, difficulties in reliably tracking large population cohorts and the influence of confounders [108]. Biomarkers such as gene polymorphisms or chemical-DNA adducts were through the 1990's employed as measures to improve cause specificity [108–110]. In the main, these have proved limited in their ability to generate robust risk associations. With the increasing development of systems biology approaches that generate large and complex datasets, deriving significant associations with chronic conditions such as cancer will become more challenging [111]. For instance, in genome-wide association studies (GWAS) that aim to identify genetic variants related to diseases by examining the associations between phenotypes and hundreds of thousands of geno-typed markers, new theoretical frameworks are required [112].
Chemical-DNA adducts do occur in target tissues such as the prostate, although their levels may not correlate with expression levels of bioactivating enzymes [113]. To estimate sample size using a simple cause-and-effect model is difficult. The majority of studies have failed to provide robust associations with common complex diseases; that said increasing sample size dramatically may be capable of achieving this, and these observations have given stimulus to the implementation of biobank studies worldwide [114]. However, incorporation of a transgenerational component into epidemiology studies could require the incorporation of an uncertainty factor in their design. This could also explain why exceedingly large studies are required to robustly isolate cause and effect against the major confounder of inherited chromatin organization determined by ancestral, environmental, and/or lifestyle factors.
5. Nurturing the Epigenome
The complex interplay between nature (could be understood as genotype) and nurture (understood as lifestyle and/or environment) has been considered to hamper efforts to specify quantitatively the relative contribution of either to disease causation [115]. Questions traditionally addressed would be whether particular polymorphisms in key genes (e.g., NAT2) could be associated with a disease endpoint (e.g., bladder cancer) in Chinese subjects exposed to an agent (e.g., benzidine); here, there appears to be a protective effect of the slow acetylator genotype [116]. Although individual combinations of polymorphic traits may modify susceptibility to exposures [117], in the complex disease scenario many studies have failed to show robust risk associations with genotype [118–120]. Spontaneous deamination of 5-meC to thymidine (which is not excised or repaired) could be a significant source of signature mutations [121] in particular cancers. These C→T transitions at CpG sites appear to be prevalent mutations in TP53 in human colon cancer [122]. In the context of this hypothesis, the question might be whether the definition of an epimutagen could be expanded not just to include altering the methylome, histone code, or profile of noncoding RNAs, but also the altering of the accessibility of important regions of the chromatin to genotoxic insult.
As lifetime exposures to environmental carcinogens might be expected to increase risk, especially in genotypes perceived as vulnerable, for example, glutathione-S-transferase null [123], other lifestyle factors such as consumption of fruit and vegetables appear to be protective against risk of disease [124, 125] and even initial DNA damage [126]. However, with the emergence of the epigenomics field the role of modifiable factors takes on another dimension. One could surmise under the model hypothesized here that environmental and/or lifestyle factors typically associated with a Western region might induce the evolution of a genomic SOS. However, are there risk factors such as tobacco smoke or infection that potentiate or accelerate this and thus greatly increase the accessibility of chromatin in future generations to genotoxic insult? Conversely, are there protective factors such as healthy intakes of fresh fruit and vegetables that counteract these effects and retard the evolution of the genomic SOS? Within epidemiology, deciphering the role of transgenerational influences mediated by epigenetic marking of the chromatin may require a detailed human epigenome project [127–129]. This could allow a stratification of predisposing early-life risk based on the structural organization of the inherited genome and the expression pattern therein. Given the even more individualized nature of the epigenome compared to the genome (it being the genome modified by environment), the complexity of this task could take several years to unravel [130].
Within this context it will be interesting to determine whether the epigenome of an individual is relatively similar across all tissues or whether there are between-organ and within-organ differences in the epigenomic profile. For years there has been evidence of a field effect in cancer in which the surrounding tissue might provide the microenvironment allowing the cancerous growth. This has been associated with nuclear changes (possibly epigenetic) and downstream epigenetic-associated events including changes in gene and protein expression, DNA damage, and angiogenesis [131]. Thus in prostate, the androgen receptor promotes growth of cancer initiating cells via autonomous signalling pathways, whereas there is a lack and no apparent need for androgen receptor signalling in surrounding stroma [132]. The question would be whether there are differing epigenomic profiles even within the same tissue, some which may generate a field change necessary to nurture the promotion and progression of disease, and others necessary for the clonal expansion of an initiated cell(s). Additionally, there is emerging interest in exosomes, which are extracellular subnanosized vesicles that are believed to contain defined patterns of mRNA, miRNA, noncoding RNA, and occasionally genomic DNA [133]. This may be a relatively unexplored mechanism of intercellular communication in which transferred genetic information may induce transient or persistent phenotypic changes in recipient cells. Exosomes play a fundamental biological role in the regulation of normal physiological as well as aberrant pathological processes, via altered gene regulatory networks and/or via epigenetic programming [134]; how they may modulate the microenvironmental epigenome remains to be ascertained.
In the past, correlations of environmental exposure and cytogenetic biomarkers have been very complex [135] because of the interaction of independent factors (e.g., genotype versus exposure). Often within each factor, there will be a requirement to stratify for variables such as age or gender. Ancestral influences on the epigenome could be yet another such variable. How such a variable could be readily factored into large-scale epidemiological studies is uncertain. In general one relies on biological material that is readily available, including blood (whole serum or plasma), buccal mucosa, or urine. Circulating free DNA in plasma has often been used [136]. Whether epigenetic alterations in surrogate tissues such as peripheral blood lymphocytes correlate with exposures (ancestral versus present) and can reflect disease risk or development in a target tissue remains to be determined.
5.1. Consequences for Breast and Prostate Cancer Incidence in Traditionally Low-Incidence Regions
As more regions become increasingly westernized in their lifestyle, based on the genomic SOS model, one could surmise that the environmental and/or dietary influences are already occurring in the present generation in traditionally low-risk regions of China, India, or Japan; this will predispose the next generations to higher incidences of breast and prostate cancer. Figure 1(a) shows that incidence of colorectal cancer in Japan is now not dissimilar to levels observed in Northern/Western Europe or the USA, the former being traditionally a low-risk region for this disease. However, levels of breast or prostate cancer still remain relatively low in this region; that said, are epigenetic modifications already occurring that in the next decades will predispose this population to markedly higher levels of this disease? This transgenerational switch in chromatin accessibility could be a dichotomous marker to the offspring's commitment to an elevated risk. The development of robust and high-throughput sensor platforms examining the in situ methylation status or genomic SOS may be far off [137].
6. Conclusions
Despite an overwhelming amount of epidemiological evidence pointing to a role for environment and/or lifestyle in the aetiology of breast and prostate cancer, the failure to identify causative factors has led some to question this in favour of endogenous entities, for example, reactive oxygen species and hormones [138]. Chronic inflammation has been highlighted as a pivotal endogenous cancer-predisposing factor [139]. However, how ancestral exposures may influence the accessibility and consequent susceptibility of an individual's genome remains to be understood. This transgenerational influence may not be applicable in all cancers (e.g., lung, colorectal, or stomach) in which there might be more direct applications of the causative agent(s) (e.g., tobacco smoke constituents, HAAs, or nitrosamines) to the target site. As a consequence, such cancers exhibit a faster rate of increasing or decreasing incidence in migrant populations from low-/high-risk regions [4].
The additional generation gap between migration from a low-risk region and attaining an incidence of breast or prostate cancer similar to the host populations points to a transgenerational step. The hypothesis laid out herein is that in the F0/F1 generations there occurs an evolution towards a genomic SOS, a model within which the chromatin becomes more accessible for genotoxic insult (Figure 2). This organizational structure of the chromatin is inherited and could be considered an epimutagenic event lending increased susceptibility to the organism and preceding mutagenic events incurred via environment and/or lifestyle factors by the F2/F3 generations. Such a model could partially explain why traditional epidemiological studies have in the main failed to draw firm cause-and-effect conclusions. A combination of novel sensor technologies and epigenetic biomarkers will be needed to order to integrate ancestral epigenetic influences at the organism, organ, and within-organ levels into future studies designed to give insights into the aetiology of these cancers. If ancestral influences such as environment and/or lifestyle markedly influence susceptibility to chronic diseases such as sporadic breast or prostate cancer, this will have enormous social as well as biological implications.
Acknowledgment
F. L. Martin would like to thank Rosemere Cancer Foundation for generously funding his research over the last number of years.
Abbreviations
- PhIP:
2-Amino-1-methyl-6-phenylimidazo [4,5-b]pyridine
- B[a]P:
Benzo[a]pyrene
- CYPs:
Cytochrome P450 mixed function oxidases
- CpG:
Cytosine-guanine dinucleotide
- DES:
Diethylstilbestrol
- DNMT:
DNA methyltransferases
- GWAS:
Genome-wide association studies
- HAAs:
Heterocyclic aromatic hydrocarbons
- HDAC:
Histone deacetylase
- miRNAs:
MicroRNAs
- 5-meC:
5-methylcytosine
- NAT:
N-Acetyltransferase
- PAHs:
Polycyclic aromatic hydrocarbons
- POPs:
Persistent organic pollutants
- SOS:
Susceptibility organizational structure
- TSGs:
Tumour suppressor genes
- UV:
Ultraviolet.
References
- 1.Grover PL, Martin FL. The initiation of breast and prostate cancer. Carcinogenesis. 2002;23(7):1095–1102. doi: 10.1093/carcin/23.7.1095. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of 25 major cancers in 1990. International Journal of Cancer. 80(6):827–841. doi: 10.1002/(sici)1097-0215(19990315)80:6<827::aid-ijc6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 3.Muir CS, Nectoux J, Staszewski J. The epidemiology of prostatic cancer. Geographical distribution and time-trends. Acta Oncologica. 1991;30(2):133–140. doi: 10.3109/02841869109092336. [DOI] [PubMed] [Google Scholar]
- 4.Peto J. Cancer epidemiology in the last century and the next decade. Nature. 2001;411(6835):390–395. doi: 10.1038/35077256. [DOI] [PubMed] [Google Scholar]
- 5.Saito M, Matsuzaki M, Sakuma T, et al. Clinicopathological study of non-palpable familial breast cancer detected by screening mammography and diagnosed as DCIS. doi: 10.1007/s12282-012-0389-3. Breast Cancer. In press. [DOI] [PubMed] [Google Scholar]
- 6.Stenback F, Peto R, Shubik P. Initiation and promotion at different ages and doses in 2200 mice. III. Linear extrapolation from high doses may underestimate low-dose tumour risks. British Journal of Cancer. 1981;44(1):24–34. doi: 10.1038/bjc.1981.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy AL, Fialkow PJ. Influence of dose of initiator on two-stage skin carcinogenesis in BALB/c mice with cellular moscaicism. Carcinogenesis. 1988;9(5):751–754. doi: 10.1093/carcin/9.5.751. [DOI] [PubMed] [Google Scholar]
- 8.Herbst AL, Scully RE, Robboy SJ. The significance of adenosis and clear cell adenocarcinoma of the genital tract in young females. Journal of Reproductive Medicine for the Obstetrician and Gynecologist. 1975;15(1):5–11. [PubMed] [Google Scholar]
- 9.Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects A vy mouse offspring from obesity by modifying the fetal epigenome. Environmental Health Perspectives. 2006;114(4):567–572. doi: 10.1289/ehp.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ur Rehman M, Buttar QM, Irfan-ul-Haq Khawaja M, Rizwan-ul-Haq Khawaja M. An impending cancer crisis in developing countries: are we ready for the challenge? Asian Pacific Journal of Cancer Prevention. 2009;10(4):719–720. [PubMed] [Google Scholar]
- 11.Carroll KK, Gammal EB, Plunkett ER. Dietary fat and mammary cancer. Canadian Medical Association journal. 1968;98(12):590–594. [PMC free article] [PubMed] [Google Scholar]
- 12.Martin FL, Carmichael PL, Crofton-Sleigh C, Venitt S, Phillips DH, Grover PL. Genotoxicity of human mammary lipid. Cancer Research. 1996;56(23):5342–5346. [PubMed] [Google Scholar]
- 13.Martin FL, Venitt S, Carmichael PL, et al. DNA damage in breast epithelial cells: detection by the single-cell gel (comet) assay and induction by human mammary lipid extracts. Carcinogenesis. 1997;18(12):2299–2305. doi: 10.1093/carcin/18.12.2299. [DOI] [PubMed] [Google Scholar]
- 14.Bronner YL. Nutritional status outcomes for children: ethnic, cultural, and environmental contexts. Journal of the American Dietetic Association. 1996;96(9):891–903. doi: 10.1016/S0002-8223(96)00242-8. [DOI] [PubMed] [Google Scholar]
- 15.Maringe C, Mangtani P, Rachet B, Leon DA, Coleman MP, dos Santos Silva I. Cancer incidence in South Asian migrants to England, 1986–2004: unraveling ethnic from socioeconomic differentials. doi: 10.1002/ijc.27826. International Journal of Cancer. In press. [DOI] [PubMed] [Google Scholar]
- 16.Martin FL. Epigenomics and disease, 10th anniversary winter meeting of the UK Molecular Epidemiology Group (MEG), the Royal Statistical Society, London, UK, 8th December 2006. Mutagenesis. 2007;22(6):425–427. [Google Scholar]
- 17.Lao VV, Grady WM. Epigenetics and colorectal cancer. Nature Reviews Gastroenterology and Hepatology. 2011;8(12):686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. International Journal of Epidemiology. 2012;41(1):79–105. doi: 10.1093/ije/dyr154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merlo A, Herman JG, Mao L, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature Medicine. 1995;1(7):686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 20.Reed AL, Califano J, Cairns P, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Research. 1996;56(16):3630–3633. [PubMed] [Google Scholar]
- 21.Herman JG, Civin CI, Issa JPJ, Collector MI, Sharkis SJ, Baylin SB. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Research. 1997;57(5):837–841. [PubMed] [Google Scholar]
- 22.Snellenberg S, Strooper LM, Hesselink AT, et al. Development of a multiplex methylation-specific PCR as candidate triage test for women with an HPV-positive cervical scrape. BMC Cancer. 2012;12(1, article 551) doi: 10.1186/1471-2407-12-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JW, Kim ST, Turner AR, et al. Identification of new differentially methylated genes that have potential functional consequences in prostate cancer. PloS One. 2012;7(10) doi: 10.1371/journal.pone.0048455.e48455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heyn H, Carmona FJ, Gomez A, et al. DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as novel epigenetic biomarker. Carcinogenesis. 2013;34(1):102–108. doi: 10.1093/carcin/bgs321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuster-Böckler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488(7411):504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 26.Botrugno OA, Robert T, Vanoli F, Foiani M, Minucci S. Molecular pathways: old drugs define new pathways: non-histone acetylation at the crossroads of the DNA damage response and autophagy. Clinical Cancer Research. 2012;18(9):2436–2442. doi: 10.1158/1078-0432.CCR-11-0767. [DOI] [PubMed] [Google Scholar]
- 27.Zeybel M, Hardy T, Wong YK, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nature Medicine. 2012;18(9):1369–1377. doi: 10.1038/nm.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metabolism. 2012;16(1):9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keverne EB. Epigenetics and brain evolution. Epigenomics. 2011;3(2):183–191. doi: 10.2217/epi.11.10. [DOI] [PubMed] [Google Scholar]
- 30.Sanchis-Gomar F, Garcia-Gimenez JL, Perez-Quilis C, Gomez-Cabrera MC, Pallardo FV, Lippi G. Physical exercise as an epigenetic modulator: eustress, the “positive stress” as an effector of gene expression. The Journal of Strength & Conditioning Research. 2012;26(12):3469–3472. doi: 10.1519/JSC.0b013e31825bb594. [DOI] [PubMed] [Google Scholar]
- 31.Maruyama R, Suzuki H. Long noncoding RNA involvement in cancer. BMB Reports. 2012;45(11):604–611. doi: 10.5483/BMBRep.2012.45.11.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerjee A, Luettich K. MicroRNAs as potential biomarkers of smoking-related diseases. Biomarkers in Medicine. 2012;6(5):671–684. doi: 10.2217/bmm.12.50. [DOI] [PubMed] [Google Scholar]
- 33.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meikar O, Da Ros M, Kotaja N. Epigenetic regulation of male germ cell differentiation. Subcellular Biochemistry. 2012;61:119–138. doi: 10.1007/978-94-007-4525-4_6. [DOI] [PubMed] [Google Scholar]
- 35.Malan-Müller S, Hemmings SM, Seedat S. Big effects of small RNAs: a review of microRNAs in anxiety. doi: 10.1007/s12035-012-8374-6. Molecular Neurobiology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballarino M, Jobert L, Dembélé D, de la Grange P, Auboeuf D, Tora L. TAF15 is important for cellular proliferation and regulates the expression of a subset of cell cycle genes through miRNAs. doi: 10.1038/onc.2012.490. Oncogene. In press. [DOI] [PubMed] [Google Scholar]
- 37.Listowski MA, Heger E, Bogusławska DM, et al. microRNAs: fine tuning of erythropoiesis. Cellular and Molecular Biology Letters. 2013;18(1):34–46. doi: 10.2478/s11658-012-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brevik A, Lindeman B, Brunborg G, Duale N. Paternal benzo[a]pyrene exposure modulates microRNA expression patterns in the developing mouse embryo. International Journal of Cell Biology. 2012;2012:11 pages. doi: 10.1155/2012/407431.407431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lillycrop KA, Burdge GC. Epigenetic mechanisms linking early nutrition to long term health. Best Practice & Research Clinical Endocrinology & Metabolism. 2012;26(5):667–676. doi: 10.1016/j.beem.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Lillycrop KA. Effect of maternal diet on the epigenome: implications for human metabolic disease. Proceedings of the Nutrition Society. 2011;70(1):64–72. doi: 10.1017/S0029665110004027. [DOI] [PubMed] [Google Scholar]
- 41.Lechner M, Boshoff C, Beck S. Cancer Epigenome. Advances in Genetics. 2010;70:247–276. doi: 10.1016/B978-0-12-380866-0.60009-5. [DOI] [PubMed] [Google Scholar]
- 42.Williams JA, Martin FL, Muir GH, Hewer A, Grover PL, Phillips DH. Metabolic activation of carcinogens and expression of various cytochromes P450 in human prostate tissue. Carcinogenesis. 2000;21(9):1683–1689. doi: 10.1093/carcin/21.9.1683. [DOI] [PubMed] [Google Scholar]
- 43.Martin FL, Patel II, Sozeri O, et al. Constitutive expression of bioactivating enzymes in normal human prostate suggests a capability to activate pro-carcinogens to DNA-damaging metabolites. Prostate. 2010;70(14):1586–1599. doi: 10.1002/pros.21194. [DOI] [PubMed] [Google Scholar]
- 44.Ragavan N, Hewitt R, Cooper LJ, et al. CYP1B1 expression in prostate is higher in the peripheral than in the transition zone. Cancer Letters. 2004;215(1):69–78. doi: 10.1016/j.canlet.2004.06.051. [DOI] [PubMed] [Google Scholar]
- 45.Abass K, Lämsâ V, Reponen P, et al. Characterization of human cytochrome P450 induction by pesticides. Toxicology. 2012;294(1):17–26. doi: 10.1016/j.tox.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 46.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szarc vel Szic K, Ndlovu MN, Haegeman G, Vanden Berghe W. Nature or nurture: let food be your epigenetic medicine in chronic inflammatory disorders. Biochemical Pharmacology. 2010;80(12):1816–1832. doi: 10.1016/j.bcp.2010.07.029. [DOI] [PubMed] [Google Scholar]
- 48.Anway MD, Cupp AS, Uzumcu N, Skinner MK. Toxicology: epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308(5727):1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartsch H, Montesano R. Relevance of nitrosamines to human cancer. Carcinogenesis. 1984;5(11):1381–1393. doi: 10.1093/carcin/5.11.1381. [DOI] [PubMed] [Google Scholar]
- 50.Gouas DA, Villar S, Ortiz-Cuaran S, et al. TP53 R249S mutation, genetic variations in HBX and risk of hepatocellular carcinoma in The Gambia. Carcinogenesis. 2012;33(6):1219–1224. doi: 10.1093/carcin/bgs135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Göttlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. The EMBO Journal. 2002;20(24):6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmad T, Shekh K, Khan S, et al. Pretreatment of valproic acid, a histone deacetylase inhibitor enhances the sensitivity of peripheral blood micronucleus assay in rodents. doi: 10.1016/j.mrgentox.2012.10.009. Mutation Research. In press. [DOI] [PubMed] [Google Scholar]
- 53.Vo AT, Millis RM. Epigenetics and breast cancers. Obstetrics and Gynecology International. 2012;2012:10 pages. doi: 10.1155/2012/602720.602720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez-Zamudio R, Ha HC. Environmental epigenetics in metal exposure. Epigenetics. 2011;6(7):820–827. doi: 10.4161/epi.6.7.16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arai Y, Ohgane J, Yagi S, et al. Epigenetic assessment of environmental chemicals detected in maternal peripheral and cord blood samples. Journal of Reproduction and Development. 2011;57(4):507–517. doi: 10.1262/jrd.11-034a. [DOI] [PubMed] [Google Scholar]
- 56.Martin FL. Complex mixtures that may contain mutagenic and/or genotoxic components: a need to assess in vivo target-site effect(s) associated with in vitro-positive(s) Chemosphere. 2007;69(6):841–848. doi: 10.1016/j.chemosphere.2007.05.066. [DOI] [PubMed] [Google Scholar]
- 57.Llabjani V, Trevisan J, Jones KC, Shore RF, Martin FL. Binary mixture effects by PBDE congeners (47, 153, 183, or 209) and PCB congeners (126 or 153) in MCF-7 cells: biochemical alterations assessed by IR spectroscopy and multivariate analysis. Environmental Science and Technology. 2010;44(10):3992–3998. doi: 10.1021/es100206f. [DOI] [PubMed] [Google Scholar]
- 58.Lister LJ, Svendsen C, Wright J, Hooper HL, Spurgeon DJ. Modelling the joint effects of a metal and a pesticide on reproduction and toxicokinetics in Lumbricid earthworms. Environment International. 2011;37(4):663–670. doi: 10.1016/j.envint.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 59.Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic inuit. Environmental Health Perspectives. 2008;116(11):1547–1552. doi: 10.1289/ehp.11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuasa Y. Epigenetics in molecular epidemiology of cancer: a new scope. Advances in Genetics. 2010;71:212–235. doi: 10.1016/B978-0-12-380864-6.00007-9. [DOI] [PubMed] [Google Scholar]
- 61.Janesick A, Blumberg B. Obesogens, stem cells and the developmental programming of obesity. International Journal of Andrology. 2012;35(3):437–448. doi: 10.1111/j.1365-2605.2012.01247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karoutsou E, Polymeris A. Environmental endocrine disruptors and obesity. Endocrine Regulations. 2012;46(1):37–46. doi: 10.4149/endo_2012_01_37. [DOI] [PubMed] [Google Scholar]
- 63.Christensen BC, Marsit CJ. Epigenomics in environmental health. Frontiers in Genetics. 2011;2, article 84 doi: 10.3389/fgene.2011.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang T, Garcia JG, Zhang W. Epigenetic regulation in particulate matter-mediated cardiopulmonary toxicities: a systems biology perspective. Current Pharmacogenomics and Personalized Medicine. 2012;10(4):314–321. doi: 10.2174/187569212803901792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tian M, Peng S, Martin FL, et al. Perfluorooctanoic acid induces gene promoter hypermethylation of glutathione-S-transferase Pi in human liver L02 cells. Toxicology. 2012;296:48–55. doi: 10.1016/j.tox.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 66.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nature Reviews Genetics. 2012;13(2):97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 67.Roberts JR, Karr CJ, Council on Environmental Health Pesticide exposure in children. Pediatrics. 2012;130(6):e1765–e1788. doi: 10.1542/peds.2012-2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pogribny IP, Rusyn I. Environmental toxicants, epigenetics, and cancer. Advances in Experimental Medicine and Biology. 2013;754:215–232. doi: 10.1007/978-1-4419-9967-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang B, Li Y, Shao C, Tan Y, Cai L. Cadmium and its epigenetic effects. Current Medicinal Chemistry. 2012;19(16):2611–2620. doi: 10.2174/092986712800492913. [DOI] [PubMed] [Google Scholar]
- 70.Singh S, Li SS. Epigenetic effects of environmental chemicals bisphenol A and phthalates. International Journal of Molecular Sciences. 2012;13(8):10143–10153. doi: 10.3390/ijms130810143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Markey CM, Wadia PR, Rubin BS, Sonnenschein C, Soto AM. Long-term effects of fetal exposure to low doses of the xenoestrogen bisphenol-A in the female mouse genital tract. Biology of Reproduction. 2005;72(6):1344–1351. doi: 10.1095/biolreprod.104.036301. [DOI] [PubMed] [Google Scholar]
- 72.Fleisch AF, Wright RO, Baccarelli AA. Environmental epigenetics: a role in endocrine disease. Journal of Molecular Endocrinology. 2012;49(2):R61–R67. doi: 10.1530/JME-12-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vempati RK. DNA damage in the presence of chemical genotoxic agents induce acetylation of H3K56 and H4K16 but not H3K9 in mammalian cells. Molecular Biology Reports. 2012;39(1):303–308. doi: 10.1007/s11033-011-0739-9. [DOI] [PubMed] [Google Scholar]
- 74.Esteller M. Relevance of DNA methylation in the management of cancer. The Lancet Oncology. 2003;4(6):351–358. doi: 10.1016/s1470-2045(03)01115-x. [DOI] [PubMed] [Google Scholar]
- 75.Kim M, Bae M, Na H, Yang M. Environmental toxicants-induced epigenetic alterations and their reversers. Journal of Environmental Science and Health, Part C. 2012;30(4):323–367. doi: 10.1080/10590501.2012.731959. [DOI] [PubMed] [Google Scholar]
- 76.Talikka M, Sierro N, Ivanov NV, et al. Genomic impact of cigarette smoke, with application to three smoking-related diseases. Critical Reviews in Toxicology. 2012;42(10):877–889. doi: 10.3109/10408444.2012.725244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mattingly CJ, McKone TE, Callahan MA, Blake JA, Hubal EA. Providing the missing link: the exposure science ontology ExO. Environmental Science & Technology. 2012;46(6):3046–3053. doi: 10.1021/es2033857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steves CJ, Spector TD, Jackson SH. Ageing, genes, environment and epigenetics: what twin studies tell us now, and in the future. Age and Ageing. 2012;41(5):581–586. doi: 10.1093/ageing/afs097. [DOI] [PubMed] [Google Scholar]
- 79.Heyn H, Li N, Ferreira HJ, et al. Distinct DNA methylomes of newborns and centenarians. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(26):10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nelson HH, Marsit CJ, Christensen BC, et al. Key epigenetic changes associated with lung cancer development: results from dense methylation array profiling. Epigenetics. 2012;7(6):559–566. doi: 10.4161/epi.20219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Turner PC, Collinson AC, Cheung YB, et al. Aflatoxin exposure in utero causes growth faltering in Gambian infants. International Journal of Epidemiology. 2007;36(5):1119–1125. doi: 10.1093/ije/dym122. [DOI] [PubMed] [Google Scholar]
- 82.Verma M. Epigenetic biomarkers in cancer epidemiology. Methods in Molecular Biology. 2012;863:467–480. doi: 10.1007/978-1-61779-612-8_28. [DOI] [PubMed] [Google Scholar]
- 83.Vineis P, Kriebel D. Causal models in epidemiology: past inheritance and genetic future. Environmental Health. 2006;5, article 21 doi: 10.1186/1476-069X-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Simmons D, Williams DRR, Powell MJ. The Coventry Diabetes Study: prevalence of diabetes and impaired glucose tolerance in Europids and Asians. Quarterly Journal of Medicine. 1991;81(296):1021–1030. doi: 10.1093/qjmed/81.3.1021. [DOI] [PubMed] [Google Scholar]
- 85.Dhawan J, Bray CL, Warburton R, Ghambhir DS, Morris J. Insulin resistance, high prevalence of diabetes, and cardiovascular risk in immigrant Asians. British Heart Journal. 1994;72(5):413–421. doi: 10.1136/hrt.72.5.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carey MC, Paigen B. Epidemiology of the American Indians’ burden and its likely genetic origins. Hepatology. 2002;36(4, part 1):781–791. doi: 10.1053/jhep.2002.36545. [DOI] [PubMed] [Google Scholar]
- 87.Pitot HC. The stability of events in the natural history of neoplasia. American Journal of Pathology. 1977;89(3):703–716. [PMC free article] [PubMed] [Google Scholar]
- 88.Chouroulinkov I, Gentil A, Tierney B. Biological activities of dihydrodiols derived from two polycyclic hydrocarbons in rodent test systems. British Journal of Cancer. 1979;39(4):376–382. doi: 10.1038/bjc.1979.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.CASE RA. Incidence of death from tumours of the urinary bladder. British Journal of Preventive & Social Medicine. 1953;7(1):14–19. doi: 10.1136/jech.7.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.CASE RA. The expected frequency of bladder tumour in works populations. British Journal of iIndustrial Medicine. 1953;10(2):114–120. doi: 10.1136/oem.10.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doll R, Peto R. Mortality in relation to smoking: 20 years’ observations on male British doctors. British Medical Journal. 1976;2(6051):1525–1536. doi: 10.1136/bmj.2.6051.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Doll R, Peto R. Cigarette smoking and bronchial carcinoma: dose and time relationships among regular smokers and lifelong non-smokers. Journal of Epidemiology and Community Health. 1978;32(4):303–313. doi: 10.1136/jech.32.4.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350(6317):427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 94.Hollstein MC, Wild CP, Bleicher F, et al. p53 Mutations and aflatoxin B1 exposure in hepatocellular carcinoma patients from Thailand. International Journal of Cancer. 1993;53(1):51–55. doi: 10.1002/ijc.2910530111. [DOI] [PubMed] [Google Scholar]
- 95.Phillips DH. Fifty years of benzo(a)pyrene. Nature. 1983;303(5917):468–472. doi: 10.1038/303468a0. [DOI] [PubMed] [Google Scholar]
- 96.Pfau W, Martin FL, Cole KJ, et al. Heterocyclic aromatic amines induce DNA strand breaks and cell transformation. Carcinogenesis. 1999;20(4):545–551. doi: 10.1093/carcin/20.4.545. [DOI] [PubMed] [Google Scholar]
- 97.Yamada M, Kodama K, Fujita S, et al. Prevalence of skin neoplasms among the atomic bomb survivors. Radiation Research. 1996;146(2):223–226. [PubMed] [Google Scholar]
- 98.Doll R. Mortality from lung cancer in asbestos workers. British Journal of Industrial Medicine. 1955;12(2):81–86. doi: 10.1136/oem.12.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rushton L, Hutchings SJ, Fortunato L, et al. Occupational cancer burden in Great Britain. British Journal of Cancer. 2012;107(supplement 1):3–7. doi: 10.1038/bjc.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Philips DH. Understanding the genotoxicity of tamoxifen? Carcinogenesis. 2001;22(6):839–849. doi: 10.1093/carcin/22.6.839. [DOI] [PubMed] [Google Scholar]
- 101.Bendaly J, Metry KJ, Doll MA, et al. Role of human CYP1A1 and NAT2 in 2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine-induced mutagenicity and DNA adducts. Xenobiotica. 2009;39(5):399–406. doi: 10.1080/00498250902748953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maenhaut-Michel G, Janel-Bintz R, Samuel N, Fuchs RPP. Adducts formed by the food mutagen 2-amino-3-methylimidazo (4, 5-f) quinoline induce frameshift mutations at hot spots through an SOS-independent pathway. Molecular and General Genetics. 1997;253(5):634–641. doi: 10.1007/s004380050366. [DOI] [PubMed] [Google Scholar]
- 103.Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, Ling H. A structural gap in Dpo4 supports mutagenic bypass of a major benzo [a]pyrene dG adduct in DNA through template misalignment. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(38):14905–14910. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saeed S, Logie C, Francoijs KJ, et al. Chromatin accessibility, p300, and histone acetylation define PML-RARa and AML1-ETO binding sites in acute myeloid leukemia. Blood. 2012;120(15):3058–3068. doi: 10.1182/blood-2011-10-386086. [DOI] [PubMed] [Google Scholar]
- 105.Nalabothula N, Carrier F. Cancer cells’ epigenetic composition and predisposition to histone deacetylase inhibitor sensitization. Epigenomics. 2011;3(2):145–155. doi: 10.2217/epi.11.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Merrifield A, Smith W. Sample size calculations for the design of health studies: a review of key concepts for non-statisticians. New South Wales Public Health Bulletin. 2012;23:142–147. doi: 10.1071/NB11017. [DOI] [PubMed] [Google Scholar]
- 107.Armstrong B. A simple estimator of minimum detectable relative risk, sample size, or power in cohort studies. American Journal of Epidemiology. 1987;126(2):356–358. doi: 10.1093/aje/126.2.356. [DOI] [PubMed] [Google Scholar]
- 108.Ehrenberg L, Törnqvist M. Use of biomarkers in epidemiology: quantitative aspects. Toxicology Letters. 1992;64:485–492. doi: 10.1016/0378-4274(92)90223-7. [DOI] [PubMed] [Google Scholar]
- 109.Vineis P. The use of biomarkers in epidemiology: the example of bladder cancer. Toxicology Letters. 1992;64-65:463–467. doi: 10.1016/0378-4274(92)90220-e. [DOI] [PubMed] [Google Scholar]
- 110.Perera FP, Mooney LA, Stampfer M, et al. Associations between carcinogen-DNA damage, glutathione S-transferase genotypes, and risk of lung cancer in the prospective Physicians’ Health Cohort Study. Carcinogenesis. 2002;23(10):1641–1646. doi: 10.1093/carcin/23.10.1641. [DOI] [PubMed] [Google Scholar]
- 111.Fu WJ, Stromberg AJ, Viele K, Carroll RJ, Wu G. Statistics and bioinformatics in nutritional sciences: analysis of complex data in the era of systems biology. Journal of Nutritional Biochemistry. 2010;21(7):561–572. doi: 10.1016/j.jnutbio.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu Z, Zhao H. Statistical power of model selection strategies for genome-wide association studies. PLoS Genetics. 2009;5(7) doi: 10.1371/journal.pgen.1000582.e1000582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.John K, Ragavan N, Pratt MM, et al. Quantification of phase I/II metabolizing enzyme gene expression and polycyclic aromatic hydrocarbon-DNA adduct levels in human prostate. Prostate. 2009;69(5):505–519. doi: 10.1002/pros.20898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Burton PR, Hansell AL, Fortier I, et al. Size matters: just how big is BIG?: quantifying realistic sample size requirements for human genome epidemiology. International Journal of Epidemiology. 2009;38(1):263–273. doi: 10.1093/ije/dyn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Doll R. Nature and nurture: possibilities for cancer control. Carcinogenesis. 1996;17(2):177–184. doi: 10.1093/carcin/17.2.177. [DOI] [PubMed] [Google Scholar]
- 116.Carreón T, Ruder AM, Schulte PA, et al. NAT2 slow acetylation and bladder cancer in workers exposed to benzidine. International Journal of Cancer. 2006;118(1):161–168. doi: 10.1002/ijc.21308. [DOI] [PubMed] [Google Scholar]
- 117.Miller MC, Mohrenweiser HW, Bell DA. Genetic variability in susceptibility and response to toxicants. Toxicology Letters. 2001;120(1–3):269–280. doi: 10.1016/s0378-4274(01)00279-x. [DOI] [PubMed] [Google Scholar]
- 118.Ingelman-Sundberg M. Polymorphism of cytochrome P450 and xenobiotic toxicity. Toxicology. 2002;181-182:447–452. doi: 10.1016/s0300-483x(02)00492-4. [DOI] [PubMed] [Google Scholar]
- 119.Kidd LR, Hein DW, Woodson K, et al. Lack of association of the N-acetyltransferase NAT1*10 allele with prostate cancer incidence, grade, or stage among smokers in Finland. Biochemical Genetics. 2011;49(1-2):73–82. doi: 10.1007/s10528-010-9386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lin Y, Yagyu K, Egawa N, et al. An overview of genetic polymorphisms and pancreatic cancer risk in molecular epidemiologic studies. Journal of Epidemiology. 2011;21(1):2–12. doi: 10.2188/jea.JE20100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287(5782):560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 122.Schmutte C, Yang AS, Nguyen TT, Beart RW, Jones PA. Mechanisms for the involvement of DNA methylation in colon carcinogenesis. Cancer Research. 1996;56(10):2375–2381. [PubMed] [Google Scholar]
- 123.Steck SE, Gaudet MM, Britton JA, et al. Interactions among GSTM1, GSTT1 and GSTP1 polymorphisms, cruciferous vegetable intake and breast cancer risk. Carcinogenesis. 2007;28(9):1954–1959. doi: 10.1093/carcin/bgm141. [DOI] [PubMed] [Google Scholar]
- 124.Gonzalez CA, Riboli E, Overvad K, et al. Diet and cancer prevention: contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. European Journal of Cancer. 2010;46(14):2555–2562. doi: 10.1016/j.ejca.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 125.Masala G, Assedi M, Bendinelli B, et al. Fruit and vegetables consumption and breast cancer risk: the EPIC Italy study. Breast Cancer Research and Treatment. 2012;132(3):1127–1136. doi: 10.1007/s10549-011-1939-7. [DOI] [PubMed] [Google Scholar]
- 126.Palli D, Masala G, Vineis P, et al. Biomarkers of dietary intake of micronutrients modulate DNA adduct levels in healthy adults. Carcinogenesis. 2003;24(4):739–746. doi: 10.1093/carcin/bgg003. [DOI] [PubMed] [Google Scholar]
- 127.Esteller M. The necessity of a human epigenome project. Carcinogenesis. 2006;27(6):1121–1125. doi: 10.1093/carcin/bgl033. [DOI] [PubMed] [Google Scholar]
- 128.Kulis M, Esteller M. DNA methylation and cancer. Advances in Genetics. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 129.Beck S. Taking the measure of the methylome. Nature Biotechnology. 2010;28(10):1026–1028. doi: 10.1038/nbt1010-1026. [DOI] [PubMed] [Google Scholar]
- 130.Murrell A, Rakyan VK, Beck S. From genome to epigenome. Human Molecular Genetics. 2005;14(1):R3–R10. doi: 10.1093/hmg/ddi110. [DOI] [PubMed] [Google Scholar]
- 131.Nonn L, Ananthanarayanan V, Gann PH. Evidence for field cancerization of the prostate. Prostate. 2009;69(13):1470–1479. doi: 10.1002/pros.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vander Griend DJ, D’Antonio J, Gurel B, Antony L, DeMarzo AM, Isaacs JT. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate. 2010;70(1):90–99. doi: 10.1002/pros.21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tetta C, Ghigo E, Silengo L, Deregibus MC, Camussi G. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. doi: 10.1007/s12020-012-9839-0. Endocrine. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Human Molecular Genetics. 2012;21(1):R125–R134. doi: 10.1093/hmg/dds317. [DOI] [PubMed] [Google Scholar]
- 135.Gyorffy E, Anna L, Kovács K, Rudnai P, Schoket B. Correlation between biomarkers of human exposure to genotoxins with focus on carcinogen-DNA adducts. Mutagenesis. 2008;23(1):1–18. doi: 10.1093/mutage/gem043. [DOI] [PubMed] [Google Scholar]
- 136.Lo YMD. Fetal DNA in maternal plasma: progress through epigenetics. Annals of the New York Academy of Sciences. 2006;1075:74–80. doi: 10.1196/annals.1368.009. [DOI] [PubMed] [Google Scholar]
- 137.Kelly JG, Najand GM, Martin FL. Characterisation of DNA methylation status using spectroscopy (mid-IR versus Raman) with multivariate analysis. Journal of Biophotonics. 2011;4(5):345–354. doi: 10.1002/jbio.201000085. [DOI] [PubMed] [Google Scholar]
- 138.Ames BN, Gold LS. Paracelsus to parascience: the environmental cancer distraction. Mutation Research. 2000;447(1):3–13. doi: 10.1016/s0027-5107(99)00194-3. [DOI] [PubMed] [Google Scholar]
- 139.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nature Reviews Cancer. 2003;3(4):276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]