

Abstract
Purpose. Malignant rhabdoid tumor (MRT) is an uncommon tumor that rarely occurs outside of renal and central nervous system (CNS) sites. Data from the literature were compiled to determine prognostic factors, including both demographic and treatment variables of malignant rhabdoid tumor, focusing on those tumors arising in extra-renal, extra-CNS (ER/EC MRT) sites. Patients and Methods. A systematic review and meta-analysis was performed by extracting demographic, treatment, and survival follow up on 167 cases of primary ER/EC MRT identified in the literature. Results. No survival differences were observed between those treated with or without radiation, or with or without chemotherapy. A Cox regression of overall survival revealed several independent prognostic factors. Surgical excision had a 74% (P = 0.0003) improvement in survival. Actinomycin had a 73% (P = 0.093) improvement in survival. Older age was associated with improved survival. The four-year survival, by Kaplan-Meier estimates, comparing patients less than two years old versus older than two at diagnosis was 11% versus 35%, respectively (P = 0.0001, Log-Rank). Conclusion. ER/EC MRT is a rare, soft-tissue tumor with a poor prognosis most commonly occurring in children. Surgical resection, treatment with actinomycin, and older age at diagnosis are all associated with improved survival.
1. Introduction
Malignant rhabdoid tumor (MRT) was first described as a rhabdomyosarcomatous subtype of Wilms Tumor in 1978 [1] and recognized as a distinct entity in 1981 [2]. Malignant rhabdoid tumors most commonly occur in children with extrarenal variants seen in the CNS, liver, female genital tract, and soft tissues. Extrarenal malignant rhabdoid tumor is a rare tumor with a poor prognosis. The medical literature is replete with papers describing the highly lethal nature of MRT. We report on demographics and treatment variables as prognostic after compiling data from the literature on extrarenal extra-CNS MRT.
2. Methods
2.1. Eligibility and Search Strategy
A search was conducted for pure primary extrarenal extracentral nervous system malignant rhabdoid tumors. Ovid Medline was searched for articles containing “rhabdoid tumor” or listed under the MeSH “rhabdoid tumor” in English. This search was conducted between the years of 1981 and April 2006 as MRT was not recognized as a distinct entity until 1981. As this is a study of extrarenal extracentral nervous system MRT cases of paraspinal tumors were excluded. Cases described as carcinomas or other primary tumor types with “rhabdoid features” or a “rhabdoid component” were excluded. The inclusion criteria strategy was conducted with the understanding that there is not complete consensus on what constitutes a rhabdoid tumor. Our goal was to capture as many reports of malignant rhabdoid tumor as possible without including cases that could be deemed questionable.
2.2. Data Abstraction
Data from 85 reports in the literature were compiled. Seven patients were dropped from analyses as they appeared to overlap between studies. This resulted in the identification of a total of 167 patients with extrarenal extracentral nervous system MRT. Follow-up data was available for 139 patients.
A database was constructed and available information was compiled including age at diagnosis, sex, site of primary tumor, time since diagnosis at the last followup, chemotherapy, individual chemotherapy drugs, radiation treatment, and surgery (either partial or complete resection). Life table analysis was performed in order to provide Kaplan-Meier estimates of survival after stratification separately for age and sex [3]. Differences in survival among subgroups were analyzed using a Cox proportional hazards analysis on other variables. Relative risks of death, ratio of mortality rates between subgroups, were estimated from the Cox regression model with and without adjustment for other factors [4].
3. Results
Ninety-one patients were males and 76 patients were females giving a male to female ratio of 1.2. Age at diagnosis ranged from 0 weeks to 84 years old with a median age of 76 months (6 years) and a mean age of 197 months (16 years). As shown in Figure 1 the distribution of age at diagnosis was skewed towards younger patients. The mean time from diagnosis to the last followup was 19.3 months with a range of 0.25–192 months in the 139 patients where it was known. Overall survival at 4 years for the 139 patients with available follow-up data was 0.245 ± 0.083 (95% confidence interval). Of the 75 males with available follow-up data, overall survival at 4 years was 0.196 ± 0.112 (95% confidence interval). Of the 62 females with followup, overall survival at 4 years was 0.304 ± 0.128 (95% confidence interval). Survival between males and females was not statistically different (P = 0.36 Wilcoxon, P = 0.26 Log-rank). Refs for table from MRT search on March 13, 2006 [5–85].
Figure 1.
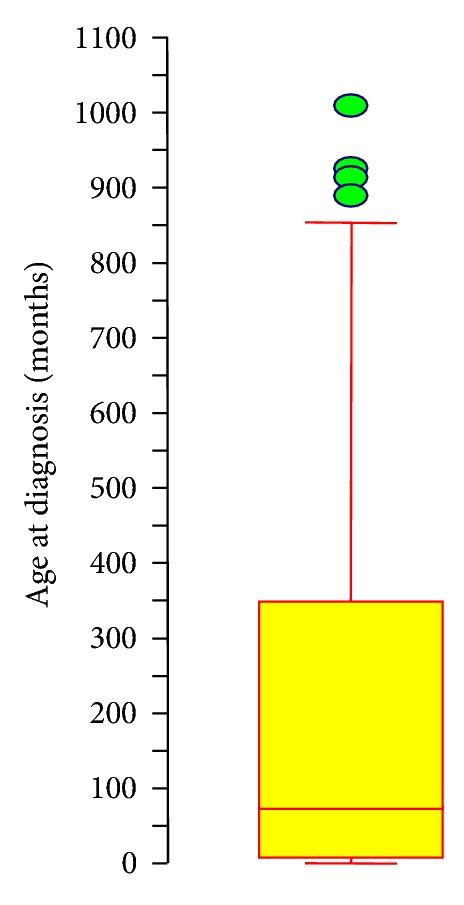
Age Boxplot. Boxplot showing median age (solid line within box) with borders of box representing 25th and 75th percentiles with 95% confidence intervals. Circles indicate individual outliers, the oldest of which is 1008 months at diagnosis.
3.1. Analysis of Age at Diagnosis and Survival
Patients were stratified into 5 age classes based on age at diagnosis: Class 1 (0–5 months), Class 2 (6–11 months), Class 3 (13–23 months), Class 4 (24–119 months or 2–10 years), and Class 5 (>120 months or 10 years) (Figure 2). These age classes were chosen to coincide with a recent report on renal malignant rhabdoid tumor and illustrate differences in survival based on age [86]. The age classes under 2 years (Classes 1, 2, and 3) were indistinguishable. The 2–10-year-old group (Class 4) had the best prognosis and was significantly different from the combined Classes 1, 2, and 3 (P = 0.0001 Wilcoxon). The 2–10-year-old group (Class 4) appeared to have a better prognosis than the >10-year-old group (Class 5), but this was not significant (P = 0.18 Wilcoxon). The >10-year-old group was different from the 0–2-year-old group (P < 0.0001 Wilcoxon). Based on the above results, the ages at diagnosis were lumped into two groups: less than 2 years old and greater than or equal to 2 years. Overall survival in these two groups was significantly different (P < 0.0001 Wilcoxon) (Figure 3), with survival statistics as seen in Table 1.
Figure 2.
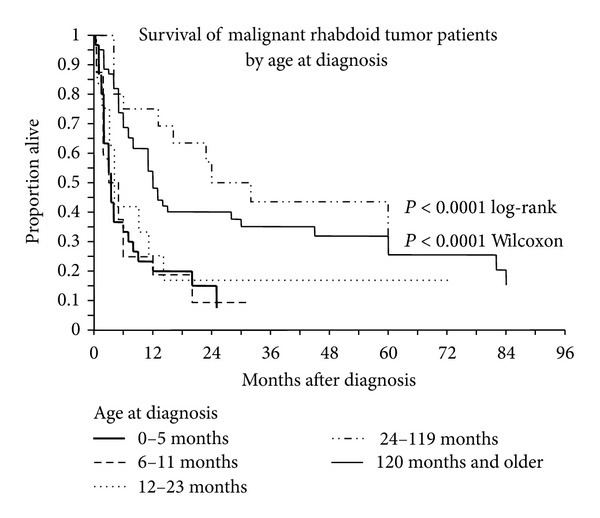
Analysis of age at diagnosis and survival. Kaplan-Meier survival curves of patients diagnosed with extrarenal extra-CNS malignant rhabdoid tumor. Age classes under 2 years old were indistinguishable. Age class 2–10 years had the best prognosis and were significantly different than those age classes under 2, but were not different from the >10-year-old age group. The >10 age group was significantly different from the <2-year-old group.
Figure 3.
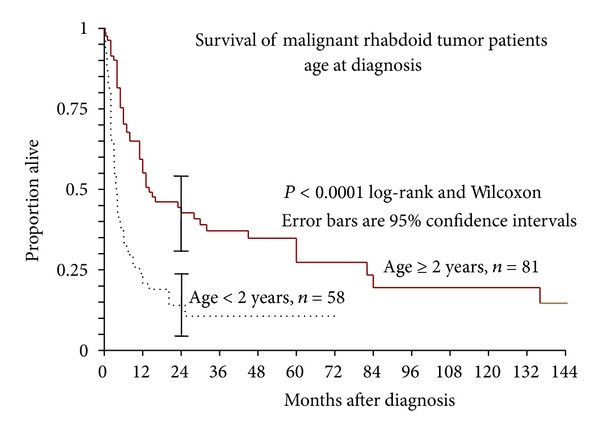
Kaplan-Meier survival of patients diagnosed with extrarenal extra-CNS malignant rhabdoid tumor with four-year survival comparing patients less than two years old versus older than two at diagnosis was 11% versus 35%, respectively (P = 0.0001).
Table 1.
Kaplan-Meier survival estimates showing the proportion of patients surviving at given time from diagnosis.
| Age at diagnosis | <2 years | ≥2 years |
|---|---|---|
| N | 58 | 81 |
| Deaths | 50 | 53 |
| 2-Year K-M Estimate | 0.1422 | 0.4773 |
| 95% CI. | 0.0476 to 02369 | 0.3121 to 0.5424 |
| 5-Year K-M Estimate | 0.1067 | 0.2737 |
| 95% CI. | 0.0135 to 0.1999 | 0.1548 to 0.3925 |
Of 50 deaths in the <2 group, only one occurred at more than two years after diagnosis (25 months). Of 53 deaths in the older group, 8 occurred after two years, with the last at 11.3 years.
3.2. Analysis of Treatment and Survival
Sixty-nine patients with available followup were reported as undergoing surgical resection (partial or complete) while 18 patients did not undergo surgery (Figure 4). Most of the articles did not specify whether surgical resections were partial or complete. Therefore, we compared patients who had not had surgical resection to patients who had any type of surgical resection (partial or complete). Surgical resection was a highly significant factor reducing the risk of death by 74% (P = 0.0003; hazard ratio 0.26, 95% C.I. 0.12 to 0.55). Most of the patients that did not have surgery were reported as having an unresectable tumor.
Figure 4.
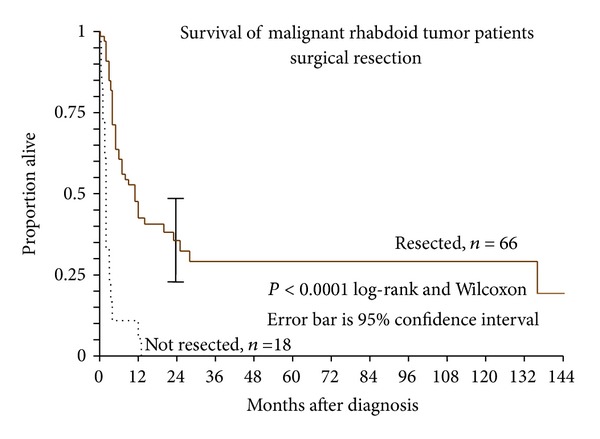
Kaplan-Meier survival of malignant rhabdoid tumor patients with and without surgical resection.
Resected patients were significantly older at diagnosis than those who went unresected (P = 0.0027 by Mann-Whitney “U” test). Median age at diagnosis for resected patients was 168 months (14.0 years); for nonresected patients this figure was 5 months (0.42 years). While this marked age difference could be due to differences in resectability of a tumor in an older child versus a neonate, unfortunately, the lack of data regarding actual resectability, as opposed to whether or not a surgical resection was done, precluded a more in-depth analysis of this potential issue. Despite their association, age at diagnosis and resection separately had a significant impact on overall survival (Table 2).
Table 2.
Results of Cox regression of overall survival on six cofactors.
| Cofactor | P value | Hazard Ratio |
|---|---|---|
| Resection (Y/N) | 0.0003 | 0.26 |
| Actinomycin (Y/N) | 0.0093 | 0.28 |
| Age ≥ 2 years (Y/N) | 0.0140 | 0.43 |
| Male (Y/N) | 0.46 | 1.26 |
| Radiation (Y/N) | 0.94 | 0.97 |
| Chemotherapy (Y/N) | 0.99 | 1.01 |
Ninety-six patients with available followup received some form of chemotherapy while 15 did not. Eighty-nine of the patients receiving chemotherapy had a multidrug regimen, but the regimens were not consistent. There was no statistically significant survival benefit with or without chemotherapy as a whole (P = 0.75). However, inclusion of actinomycin in a multidrug regimen was associated with a 73% reduced risk of death (P = 0.0093, hazard ratio 0.28, Table 2), which held true for all age groups. Seventeen patients with available followup received actinomycin while 77 patients did not (Figure 5). A univariate analysis suggested that the regimens containing actinomycin had a longer survival and this was independently significant upon Cox regression. While this does not demonstrate a superior efficacy of actinomycin in this tumor, it suggests that actinomycin should be included in multidrug chemoregimens for this tumor.
Figure 5.
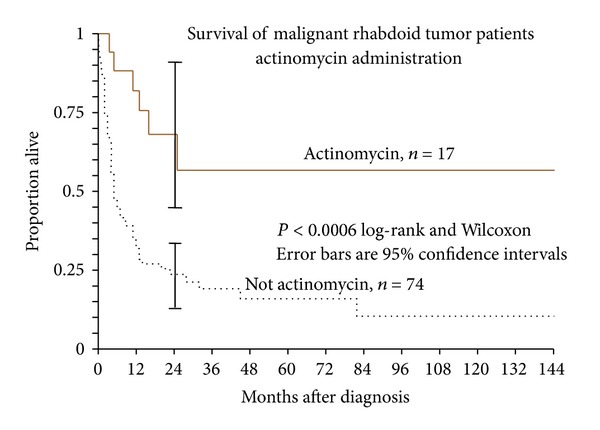
Kaplan-Meier survival estimates of patients treated with and without actinomycin. Actinomycin is associated with a 74% reduced risk of death (P = 0.0059) which held true for all age groups.
Fifty-nine patients with available followup were reported as receiving doxorubicin as part of their multidrug regimen, while 35 patients were reported as not receiving doxorubicin. Inclusion of doxorubicin in a multidrug regimen did not have a significant impact on survival (P = 0.84). Seventeen patients were reported as receiving cisplatin while 78 patients were reported as not having received cisplatin. Inclusion of cisplatin in a multidrug regimen was associated with a 111% increased risk of death (P = 0.0466). The increase in risk associated with cisplatin was analyzed further by looking at associations among cofactors. Age at diagnosis and cisplatin had a weak association, (P = 0.07 by Mann-Whitney “U” test). Median age of diagnosis for patients taking cisplatin was 12 months (1.0 year), while for those not taking cisplatin the median age at diagnosis was 81.6 months (6.8 years). Resection and cisplatin also had a weak association (2 × 2 table, Fisher's exact probability, P = 0.067) with cisplatin associated with the unresected patients. From the associations between age at diagnosis, resection, and cisplatin, it appears that younger patients were receiving cisplatin, but that their disease severity rendered it ineffective.
Forty-five patients were reported to have received radiation therapy while 66 were reported as not having received RT. There was no statistically significant effect of treatment with or without RT (P = 0.6705). Table 2 displays a Cox regression of six of the cofactors discussed.
3.3. Analysis of Tumors of Soft Tissues
In this analysis tumors of the ovary (1 case), bladder (2), colon (2), small intestine (2), esophagus (1), liver (16), prostate (3), lungs (2), thymus (1), thyroid (1), stomach (1), uterus (5), heart (1), and lacrimal gland (1) were excluded. Tumors of the soft tissue showed a weak trend toward improved survival with a decreased risk of death of 40% (P = 0.11; hazard ratio 0.605, 95% C.I. 0.321 to 1.14). There was no difference between the “soft tissue” group and excluded group in regards to the report of a surgical resection (P = 0.27). The analysis of the soft tissue subgroup showed the same trends in regards to age at diagnosis as the ER/EC MRT group as a whole.
Further analysis of the “soft tissue” group showed 7 sites with more than 3 reported cases: chest wall (11 cases), inguinal (5), neck (15), orbit (4), pelvis (8), shoulder (8), and vulva (7). Chi-squared and Cox regression analysis showed no evidence that site in these subgroups had any effect on rate of resection or survival.
Finally, we examined subgroups of cases that included 15 in the neck, 32 in the extremities (20 proximal and 12 distal), and 67 involving the trunk (one of them also in the neck and one also in the thigh). Statistic chi-squared showed no difference in rates of resection between these sites. A Cox regression of these subgroups showed no difference in survival.
4. Discussion
Reports in the literature have shown poor long-term survival rates in patients with extrarenal extra-CNS MRT. Particularly troubling are the dismal survival rates for the very young. Our reported overall survival for patients diagnosed at less than two years of age of 11% is close to the 9.1% survival for fetal and neonate ER/EC MRT reported by Isaacs [87]. Tomlinson et al. demonstrated a strong correlation of increasing survival with increasing age at diagnosis for patients with renal malignant rhabdoid tumors which is similar to what we have found for ER/EC MRT [86]. Similar to our study, a German review of 70 pediatric cases of malignant rhabdoid tumor of varying locations showed no improvement in outcome with radiotherapy [88].
Absence of INI1 is valuable in confirming the diagnosis of renal or extrarenal MRT versus other tumors with focal rhabdoid appearance [89]. INI1, associated with chromatin remodeling and expressed in all tissues, is a product of the hSNF5/INI1 tumor suppressor gene which has been demonstrated to be frequently mutated or deleted in MRTs [90, 91]. Cytogenetic study of malignant rhabdoid tumors (renal and extrarenal) has shown a region of common deletion at 22q11. Analysis of chromosome 22q has been used as an aid to the diagnosis of rhabdoid tumors [24]. A putative tumor suppressor gene locus, hSNF5/INI1, has been identified. Mitotic recombination, nondisjunction, duplication, or deletion in the proximal part of chromosome 22q appears to be associated with hSNF5/INI1 inactivation [92]. Reexpression of the hSNF5 gene in MRT cell lines induces G1 cell cycle arrest and activation of senescence-associated proteins [93–96].
Genetic alterations in chromatin remodeling complexes appear to be responsible for the formation of cytoplasmic perinuclear inclusion bodies seen in MRT. The cytokeratin (CK) 8 gene analyzed from human MRT showed missense mutations in genes whose products are involved in lateral protofilament-protofilament interactions and phosphorylation sites important to filament organization [97]. Alterations to regions involved in microfilament conformational change appear to interfere with chromatin remodeling in MRT.
Current understanding of the genetic similarities observed in rhabdoid tumors would support establishing common treatment protocols; however, the relative rarity of the disease has made this difficult. Resections to the extent possible, chemotherapy, and radiation therapy are often employed together in the treatment of this disease. Chemotherapy regimes for treatment of MRT have included various combinations of cisplatinum, cyclophosphamide, adriamycin, and VP-16 [20, 66, 98]. There are reports of using the German Society of Pediatric Oncology Protocol (HIT, procarbazine, ifosfamide, VP-16, methotrexate, cytosine-arabinoside, and cisplatin) [98]. With no well-established adjunct treatment protocols our data suggests that an aggressive surgical excision is indicated in cases of operable MRT. In addition, our retrospective review and analysis of the literature would support the consideration of a chemotherapy regime that includes treatment with actinomycin.
Acknowledgments
The authors would like to thank Paul Lender, Department of Orthopaedics, University of Minnesota. All authors have read and approved the paper.
References
- 1.Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumor. Results from the first national Wilms’ tumor study. Cancer. 1978;41(5):1937–1948. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 2.Haas JE, Palmer NF, Weinberg AG, Beckwith JB. Ultrastructure of malignant rhabdoid tumor of the kidney. A distinctive renal tumor of children. Human Pathology. 1981;12(7):646–657. doi: 10.1016/s0046-8177(81)80050-0. [DOI] [PubMed] [Google Scholar]
- 3.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association. 1958;53(282):456–481. [Google Scholar]
- 4.Cox DR. Regression models and life-tables. Journal of the Royal Statistical Society Series B. 1972;34(2):187–220. [Google Scholar]
- 5.Hsueh S, Chang TC. Malignant rhabdoid tumor of the uterine corpus. Gynecologic Oncology. 1996;61(1):142–146. doi: 10.1006/gyno.1996.0113. [DOI] [PubMed] [Google Scholar]
- 6.Igarashi T, Sasano H, Konno R, et al. Malignant rhabdoid tumor of the vulva: case report with cytological, immunohistochemical, ultrastructural and DNA ploidy studies and a review of the literature. Pathology International. 1998;48(11):887–891. doi: 10.1111/j.1440-1827.1998.tb03856.x. [DOI] [PubMed] [Google Scholar]
- 7.Muramatsu M, Kotake S, Yoshikawa K, Sasamoto Y, Matsuda H, Yamawaki S. The development of malignant rhabdoid tumor in a patient with Behcet’s disease treated with ciclosporin. Graefe’s Archive for Clinical and Experimental Ophthalmology. 1998;236(10):798–799. doi: 10.1007/s004170050162. [DOI] [PubMed] [Google Scholar]
- 8.Donner LR, Rao A, Truss LM, Dobin SM. Translocation (8;13)(q24.2;q33) in a malignant rhabdoid tumor of the liver. Cancer Genetics and Cytogenetics. 2000;116(2):153–157. doi: 10.1016/s0165-4608(99)00131-4. [DOI] [PubMed] [Google Scholar]
- 9.Alfonso I, Papazian O, Prieto G, Alfonso DT, Melnick SJ. Neoplasm as a cause of brachial plexus palsy in neonates. Pediatric Neurology. 2000;22(4):309–311. doi: 10.1016/s0887-8994(99)00144-7. [DOI] [PubMed] [Google Scholar]
- 10.Ravindra KV, Cullinane C, Lewis IJ, Squire BR, Stringer MD. Long-term survival after spontaneous rupture of a malignant rhabdoid tumor of the liver. Journal of Pediatric Surgery. 2002;37(10):1488–1490. doi: 10.1053/jpsu.2002.35427. [DOI] [PubMed] [Google Scholar]
- 11.Yaris N, Cobanoglu U, Dilber E, Ahmetolu A, Saruhan H, Ökten A. Malignant rhabdoid tumor of adrenal gland. Medical and Pediatric Oncology. 2002;39(2):128–131. doi: 10.1002/mpo.10059. [DOI] [PubMed] [Google Scholar]
- 12.Salamanca J, Rodríguez-Peralto JL, Azorín D, Ballestín C, de Agustín P. Paratesticular congenital malignant rhabdoid tumor diagnosed by fine-needle aspiration cytology. A case report. Diagnostic Cytopathology. 2004;30(1):46–50. doi: 10.1002/dc.10410. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Jung SM, Chao TC. Malignant rhabdoid tumor of the small intestine in an adult: a case report with immunohistochemical and ultrastructural findings. Digestive Diseases and Sciences. 1998;43(5):975–979. doi: 10.1023/a:1018866314989. [DOI] [PubMed] [Google Scholar]
- 14.Lee JR, Chamberlain CR, Gerrity RG, McKee EM, Gadacz TR, Rao RN. Malignant rhabdoid tumor of the duodenum. Annals of Diagnostic Pathology. 1998;2(1):25–30. doi: 10.1016/s1092-9134(98)80032-4. [DOI] [PubMed] [Google Scholar]
- 15.García-Bustínduy M, Ávarez-Arguelles H, Guimeriá F, et al. Malignant rhabdoid tumor beside benign skin mesenchymal neoplasm with myofibromatous features. Journal of Cutaneous Pathology. 1999;26(10):509–515. doi: 10.1111/j.1600-0560.1999.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 16.Brand A, Covert A. Malignant rhabdoid tumor of the vulva: case report and review of the literature with emphasis on clinical management and outcome. Gynecologic Oncology. 2001;80(1):99–103. doi: 10.1006/gyno.2000.6032. [DOI] [PubMed] [Google Scholar]
- 17.Staehelin F, Bissig H, Hösli I, et al. Inv(11)(p13p15) and Myf-3(MyoD1) in a malignant extrarenal rhabdoid tumor of a premature newborn. Pediatric Research. 2000;48(4):463–467. doi: 10.1203/00006450-200010000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Sajedi M, Wolff JEA, Egeler RM, et al. Congenital extrarenal non-central nervous system malignant rhabdoid tumor. Journal of Pediatric Hematology/Oncology. 2002;24(4):316–320. doi: 10.1097/00043426-200205000-00020. [DOI] [PubMed] [Google Scholar]
- 19.Katzenstein HM, Kletzel M, Reynolds M, Superina R, Gonzales-Crussi F. Metastatic malignant rhabdoid tumor of the liver treated with tandem high-dose therapy and autologous peripheral blood stem cell rescue. Medical and Pediatric Oncology. 2003;40(3):199–201. doi: 10.1002/mpo.10149. [DOI] [PubMed] [Google Scholar]
- 20.Yuri T, Danbara N, Shikata N, et al. Malignant rhabdoid tumor of the liver: case report and literature review. Pathology International. 2004;54(8):623–629. doi: 10.1111/j.1440-1827.2004.01672.x. [DOI] [PubMed] [Google Scholar]
- 21.Morgan MB, Stevens L, Patterson J, Tannenbaum M. Cutaneous epithelioid malignant nerve sheath tumor with rhabdoid features: a histologic, immunohistochemical, and ultrastructural study of three cases. Journal of Cutaneous Pathology. 2000;27(10):529–534. doi: 10.1034/j.1600-0560.2000.027010529.x. [DOI] [PubMed] [Google Scholar]
- 22.Drut R, Drut RM. Renal and extrarenal congenital rhabdoid tumor:diagnosis by fine-needle aspiration biopsy and FISH. Diagnostic Cytopathology. 2002;27(1):32–34. doi: 10.1002/dc.10040. [DOI] [PubMed] [Google Scholar]
- 23.Gottlieb C, Nijhawan N, Chorneyko K, O’Grady KF, Harvey JT. Congenital orbital and disseminated extrarenal malignant rhabdoid tumor. Ophthalmic Plastic and Reconstructive Surgery. 2005;21(1):76–79. doi: 10.1097/01.iop.0000150353.90937.b4. [DOI] [PubMed] [Google Scholar]
- 24.Simons J, Teshima I, Zielenska M, et al. Analysis of chromosome 22q as an aid to the diagnosis of rhabdoid tumor: a case report. American Journal of Surgical Pathology. 1999;23(8):982–988. doi: 10.1097/00000478-199908000-00018. [DOI] [PubMed] [Google Scholar]
- 25.White FV, Dehner LP, Belchis DA, et al. Congenital disseminated malignant rhabdoid tumor: a distinct clinicopathologic entity demonstrating abnormalities of chromosome 22q11. American Journal of Surgical Pathology. 1999;23(3):249–256. doi: 10.1097/00000478-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Gfinduz K, Shields JA, Eagle RC, Jr., Shields CL, de Potter P, Klombers L. Malignant rhabdoid tumor of the orbit. Archives of Ophthalmology. 1998;116(2):243–246. doi: 10.1001/archopht.116.2.243. [DOI] [PubMed] [Google Scholar]
- 27.Johnson LN, Sexton FM, Goldberg SH. Poorly differentiated primary orbital sarcoma (presumed malignant rhabdoid tumor): radiologic and histopathologic correlation. Archives of Ophthalmology. 1991;109(9):1275–1278. doi: 10.1001/archopht.1991.01080090101031. [DOI] [PubMed] [Google Scholar]
- 28.Frierson HF, Jr., Mills SE, Innes DJ., Jr. Malignant rhabdoid tumor of the pelvis. Cancer. 1985;55(9):1963–1967. doi: 10.1002/1097-0142(19850501)55:9<1963::aid-cncr2820550922>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 29.Uchida H, Yokoyama S, Nakayama I, Zeze K. An autopsy case of malignant rhabdoid tumor arising from soft parts in the left inguinal region. Acta Pathologica Japonica. 1988;38(8):1087–1096. doi: 10.1111/j.1440-1827.1988.tb02381.x. [DOI] [PubMed] [Google Scholar]
- 30.Kent AL, Mahoney DH, Jr., Gresik MV, Steuber CP, Fernbach DJ. Malignant rhabdoid tumor of the extremity. Cancer. 1987;60(5):1056–1059. doi: 10.1002/1097-0142(19870901)60:5<1056::aid-cncr2820600521>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 31.Small EJ, Gordon GJ, Dahms BB. Malignant rhabdoid tumor of the heart in an infant. Cancer. 1985;55(12):2850–2853. doi: 10.1002/1097-0142(19850615)55:12<2850::aid-cncr2820551224>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 32.Tsokos M, Kouraklis G, Chandra RS, Bhagavan BS, Triche TJ. Malignant rhabdoid tumor of the kidney and soft tissues. Evidence for a diverse morphological and immunocytochemical phenotype. Archives of Pathology and Laboratory Medicine. 1989;113(2):115–120. [PubMed] [Google Scholar]
- 33.Tsujimura T, Wada A, Kawano K, Iwasa A, Mizutani S. A case of malignant rhabdoid tumor arising from soft parts in the prepubic region. Acta Pathologica Japonica. 1989;39(10):677–682. doi: 10.1111/j.1440-1827.1989.tb02416.x. [DOI] [PubMed] [Google Scholar]
- 34.Perez-Atayde AR, Newbury R, Fletcher JA, Barnhill R, Gellis S. Congenital “neurovascular hamartoma” of the skin: a possible marker of malignant rhabdoid tumor. American Journal of Surgical Pathology. 1994;18(10):1030–1038. doi: 10.1097/00000478-199410000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Perrone T, Swanson PE, Twiggs L, Ulbright TM, Dehner LP. Malignant rhabdoid tumor of the vulva: is distinction from epithelioid sarcoma possible? A pathologic and immunohistochemical study. American Journal of Surgical Pathology. 1989;13(10):848–858. [PubMed] [Google Scholar]
- 36.Koibuchi Y, Lino Y, Joshita T, et al. Malignant rhabdoid tumor of the breast: a case report. Japanese Journal of Clinical Oncology. 1995;25(6):273–277. [PubMed] [Google Scholar]
- 37.Dabbs DJ, Park HK. Malignant rhabdoid skin tumor: an uncommon primary skin neoplasm. Ultrastructural and immunohistochemical analysis. Journal of Cutaneous Pathology. 1988;15(2):109–115. doi: 10.1111/j.1600-0560.1988.tb00529.x. [DOI] [PubMed] [Google Scholar]
- 38.Weyman C, Dolson L, Kedar A. Secretion of vasointestinal peptide by a primary liver tumor with rhabdoid features. Journal of Surgical Oncology. 1993;54(4):267–270. doi: 10.1002/jso.2930540417. [DOI] [PubMed] [Google Scholar]
- 39.Lee TL, Lee SH, Lin RY, Chuang SM, Leu SH. Malignant rhabdoid tumor arising from soft parts of the right thigh with unusual neurologic manifestation: report of a case. Journal of the Formosan Medical Association. 1992;91(9):907–911. [PubMed] [Google Scholar]
- 40.Cho KR, Rosenshein NB, Epstein JI. Malignant rhabdoid tumor of the uterus. International Journal of Gynecological Pathology. 1989;8(4):381–387. doi: 10.1097/00004347-198912000-00010. [DOI] [PubMed] [Google Scholar]
- 41.Ekfors TO, Ahon HJ, Kekomaki M. Malignant rhabdoid tumor of the prostatic region. Immunohistological and ultrastructural evidence for epithelial origin. Virchows Archiv—A Pathological Anatomy and Histopathology. 1985;406(3):381–388. doi: 10.1007/BF00704307. [DOI] [PubMed] [Google Scholar]
- 42.Okada A, Takehara H, Masamune K, et al. A newborn case of extrarenal malignant rhabdoid tumor. Tokushima Journal of Experimental Medicine. 1993;40(1-2):109–112. [PubMed] [Google Scholar]
- 43.Lupi G, Jin R, Clemente C. Malignant rhabdoid tumor of the vulva: a case report and review of the literature. Tumori. 1996;82(1):93–95. doi: 10.1177/030089169608200120. [DOI] [PubMed] [Google Scholar]
- 44.Kaiserling E, Ruck P, Handgretinger R, Leipoldt M, Hipfel R. Immunohistochemical and cytogenetic findings in malignant rhabdoid tumor. General and Diagnostic Pathology. 1995;141(5-6):327–337. [PubMed] [Google Scholar]
- 45.Sert MB, Onsrud M, Perrone T, Abbas F, Currie JL. Malignant rhabdoid tumor of the vulva. Case report. European Journal of Gynaecological Oncology. 1999;20(4):258–261. [PubMed] [Google Scholar]
- 46.Haidopoulos D, Elsheikh A, Vlahos G, et al. Malignant rhabdoid tumor of the clitoris in an elderly patient: report of a case. European Journal of Gynaecological Oncology. 2002;23(5):447–449. [PubMed] [Google Scholar]
- 47.Marcus VA, Viloria J, Owen D, Tsao MS. Malignant rhabdoid tumor of the colon: report of a case with molecular analysis. Diseases of the Colon and Rectum. 1996;39(11):1322–1326. doi: 10.1007/BF02055131. [DOI] [PubMed] [Google Scholar]
- 48.Pettinato G, Manivel JC, d’Amore ES, Petrella G. Extrarenal primitive malignant tumor with rhabdoid features: fine-needle aspiration cytology, immunocytochemistry, and electron microscopy of a case. Diagnostic Cytopathology. 1991;7(2):178–183. doi: 10.1002/dc.2840070215. [DOI] [PubMed] [Google Scholar]
- 49.Akhtar M, Kfoury H, Haider A, Sackey K, Ali MA. Fine-needle aspiration biopsy diagnosis of extrarenal malignant rhabdoid tumor. Diagnostic Cytopathology. 1994;11(3):271–276. doi: 10.1002/dc.2840110315. [DOI] [PubMed] [Google Scholar]
- 50.Parham DM, Peiper SC, Robicheaux G, Ribeiro RC, Douglass EC. Malignant rhabdoid tumor of the liver. Evidence for epithelial differentiation. Archives of Pathology and Laboratory Medicine. 1988;112(1):61–64. [PubMed] [Google Scholar]
- 51.Hsueh C, Kuo TT. Congenital malignant rhabdoid tumor presenting as a cutaneous nodule: report of 2 cases with review of the literature. Archives of Pathology and Laboratory Medicine. 1998;122(12):1099–1102. [PubMed] [Google Scholar]
- 52.Pogačnik A, Zidar N. Malignant rhabdoid tumor of the liver diagnosed by fine needle aspiration cytology: a case report. Acta Cytologica. 1997;41(2):539–543. doi: 10.1159/000332553. [DOI] [PubMed] [Google Scholar]
- 53.Tsuneyoshi M, Daimaru Y, Hashimoto H, Enjoji M. Malignant soft tissue neoplasms with the histologic features of renal rhabdoid tumors: an ultrastructural and immunohistochemical study. Human Pathology. 1985;16(12):1235–1242. doi: 10.1016/s0046-8177(85)80036-8. [DOI] [PubMed] [Google Scholar]
- 54.Niffenegger JH, Jakobiec FA, Shore JW, Albert DM. Adult extrarenal rhabdoid tumor of the lacrimal gland. Ophthalmology. 1992;99(4):567–574. doi: 10.1016/s0161-6420(92)31948-7. [DOI] [PubMed] [Google Scholar]
- 55.Albregts AE, Hebert AA, Aboul-Nasr RA, Raney RB. Malignant rhabdoid tumor presenting as a hemangioma. Pediatric Dermatology. 1996;13(6):468–471. doi: 10.1111/j.1525-1470.1996.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt D, Leuschner I, Harms D, Sprenger E, Schafer HJ. Malignant rhabdoid tumor. A morphological and flow cytometric study. Pathology Research and Practice. 1989;184(2):202–210. doi: 10.1016/S0344-0338(89)80121-9. [DOI] [PubMed] [Google Scholar]
- 57.Rootman J, Damji KF, Dimmick JE. Malignant rhabdoid tumor of the orbit. Ophthalmology. 1989;96(11):1650–1654. doi: 10.1016/s0161-6420(89)32666-2. [DOI] [PubMed] [Google Scholar]
- 58.Ohgaki M, Higuchi A, Chou H, et al. An extrarenal malignant rhabdoid tumor suspected to originate from the mesentery in an adult: report of a case. Surgery Today. 2003;33(7):556–559. doi: 10.1007/s10595-002-2518-8. [DOI] [PubMed] [Google Scholar]
- 59.Nakamura I, Nakano K, Nakayama K, et al. Malignant rhabdoid tumor of the colon: report of a case. Surgery Today. 1999;29(10):1083–1087. doi: 10.1007/s005950050649. [DOI] [PubMed] [Google Scholar]
- 60.Fabre A, Eyden B, Ali HH. Soft-tissue extrarenal rhabdoid tumor with a unique long-term survival. Ultrastructural Pathology. 2004;28(1):49–52. [PubMed] [Google Scholar]
- 61.Parham DM, Weeks DA, Beckwith JB. The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohistochemistry or electron microscopy. American Journal of Surgical Pathology. 18(10):1010–1029. doi: 10.1097/00000478-199410000-00005. [DOI] [PubMed] [Google Scholar]
- 62.Gururangan S, Bowman LC, Parham DM, et al. Primary extracranial rhabdoid tumors: clinicopathologic features and response to ifosfamide. Cancer. 1993;71(8):2653–2659. doi: 10.1002/1097-0142(19930415)71:8<2653::aid-cncr2820710834>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 63.Kodet R, Newton WA, Jr., Sachs N, et al. Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the intergroup rhabdomyosarcoma study. Human Pathology. 1991;22(7):674–684. doi: 10.1016/0046-8177(91)90289-2. [DOI] [PubMed] [Google Scholar]
- 64.Sotelo-Avila C, Gonzalez-Crussi F, deMello D, et al. Renal and extrarenal rhabdoid tumors in children: a clinicopathologic study of 14 patients. Seminars in Diagnostic Pathology. 1986;3(2):151–163. [PubMed] [Google Scholar]
- 65.Fanburg-Smith JC, Hengge M, Hengge UR, Smith JSC, Jr., Miettinen M. Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immunohistochemical study of 18 cases. Annals of Diagnostic Pathology. 1998;2(6):351–362. doi: 10.1016/s1092-9134(98)80038-5. [DOI] [PubMed] [Google Scholar]
- 66.Hunt SJ, Anderson WD. Malignant rhabdoid tumor of the liver. A distinct clinicopathologic entity. American Journal of Clinical Pathology. 1990;94(5):645–648. doi: 10.1093/ajcp/94.5.645. [DOI] [PubMed] [Google Scholar]
- 67.Kelly DM, Jones M, Humphreys S, Howard ER. Spontaneous rupture of a malignant rhabdoid tumour of the liver. Pediatric Surgery International. 1998;14(1-2):111–112. doi: 10.1007/s003830050453. [DOI] [PubMed] [Google Scholar]
- 68.Rosty C, Peter M, Zucman J, Validire P, Delattre O, Aurias A. Cytogenetic and molecular analysis of a t(1,22)(p36,q11. 2) in a rhabdoid tumor with a putative homozygous deletion of chromosome 22. Genes Chromosomes Cancer. 1998;21(2):82–89. [PubMed] [Google Scholar]
- 69.Comtesse PPFGP, Simons A, Siepman A, et al. Isochromosome (12p) and peritriploidy in a highly malignant extrarenal rhabdoid tumor. Cancer Genetics and Cytogenetics. 1999;109(2):175–177. doi: 10.1016/s0165-4608(98)00164-2. [DOI] [PubMed] [Google Scholar]
- 70.Duvdevani M, Nass D, Neumann Y, Leibovitch I, Ramon J, Mor Y. Pure rhabdoid tumor of the bladder. Journal of Urology. 2001;166(6, article 2337) [PubMed] [Google Scholar]
- 71.Knapik J, Yachnis AT, Ripley D, et al. Aggressive uterine sarcoma with rhabdoid features: diagnosis by peritoneal fluid cytology and absence of INI1 gene mutation. Human Pathology. 2001;32(8):884–886. doi: 10.1053/hupa.2001.26476. [DOI] [PubMed] [Google Scholar]
- 72.Fowler JC, Ehrlich P, Wenger S, Ducatman BS. Pathologic quiz case: a 10-year-old girl with a pelvic mass. Archives of Pathology and Laboratory Medicine. 2003;127(5):633–635. doi: 10.5858/2003-127-0633-PQCAYO. [DOI] [PubMed] [Google Scholar]
- 73.Chang JH, Dikranian AH, Johnston WH, Storch SK, Hurwitz RS. Malignant extrarenal rhabdoid tumor of the bladder: 9-year survival after chemotherapy and partial cystectomy. Journal of Urology. 2004;171(2 I):820–821. doi: 10.1097/01.ju.0000107014.40195.cf. [DOI] [PubMed] [Google Scholar]
- 74.Perlman EJ, Ali SZ, Robinson R, Lindato R, Griffin CA. Infantile extrarenal rhabdoid tumor. Pediatric and Developmental Pathology. 1998;1(2):149–152. doi: 10.1007/s100249900019. [DOI] [PubMed] [Google Scholar]
- 75.Figarola MS, Khader SM. Pediatric case of the day. Radiographics. 1999;19(6):1693–1695. doi: 10.1148/radiographics.19.6.g99no141693. [DOI] [PubMed] [Google Scholar]
- 76.Castellino SM, Powers R, Kalwinsky D, DeVoe M. Abdominal rhabdoid tumor presenting as fetal hydrops: a case report. Journal of Pediatric Hematology/Oncology. 2001;23(4):258–259. doi: 10.1097/00043426-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 77.Helmke L, Engler S, Mattke A, Henne-Bruns D. Extrarenal malignant rhabdoid tumors in childhood. Medical and Pediatric Oncology. 2001;36(2):317–319. doi: 10.1002/1096-911X(20010201)36:2<317::AID-MPO1073>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 78.Tsuda H, Maed K, Hashiguchi Y, et al. Malignant rhabdoid tumour of the uterine cervix. British Journal of Obstetrics and Gynaecology. 2001;108(1):120–123. doi: 10.1111/j.1471-0528.2001.00019.x. [DOI] [PubMed] [Google Scholar]
- 79.Sangueza OP, Meshul CK, Sangueza P, Mendoza R. Rhabdoid tumor of the skin. International Journal of Dermatology. 1992;31(7):484–487. doi: 10.1111/j.1365-4362.1992.tb02695.x. [DOI] [PubMed] [Google Scholar]
- 80.Leath CA, III, Huh WK, Conner M, Barnes MN., III Primary extrarenal rhabdoid tumor of the ovary: a case report. Journal of Reproductive Medicine for the Obstetrician and Gynecologist. 2003;48(4):283–286. [PubMed] [Google Scholar]
- 81.Sinha Roy S, Mukherji SK, Castillo M, O’Connell T. MRI of congenital rhabdoid tumor of the neck: case report. Neuroradiology. 1996;38(4):373–374. doi: 10.1007/BF00596592. [DOI] [PubMed] [Google Scholar]
- 82.Rubenchik I, Dardick I, Auger M. Cytopathology and ultrastructure of primary rhabdoid tumor of lung. Ultrastructural Pathology. 1996;20(4):355–360. doi: 10.3109/01913129609016337. [DOI] [PubMed] [Google Scholar]
- 83.Mazzocchi M, Chiummariello S, Bistoni G, Marchetti F, Alfano C. Extrarenal malignant rhabdoid tumour of the heel—a case report. Anticancer Research. 2005;25(6 C):4573–4576. [PubMed] [Google Scholar]
- 84.Boscaino A, Donofrio V, Tornillo L, Staibano S, de Rosa G. Primary rhabdoid tumour of the skin in a 14-month-old child. Dermatology. 1994;188(4):322–325. doi: 10.1159/000247176. [DOI] [PubMed] [Google Scholar]
- 85.Read HS, Webb JN, Macintyre IMC. Malignant rhabdoid tumour of stomach. Histopathology. 1996;29(5):474–477. [PubMed] [Google Scholar]
- 86.Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the national Wilms’ tumor study: age at diagnosis as a prognostic factor. Journal of Clinical Oncology. 2005;23(30):7641–7645. doi: 10.1200/JCO.2004.00.8110. [DOI] [PubMed] [Google Scholar]
- 87.Isaacs H., Jr. Fetal and neonatal rhabdoid tumor. Journal of Pediatric Surgery. 2010;45(3):619–626. doi: 10.1016/j.jpedsurg.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 88.Reinhard H, Reinert J, Beier R, et al. Rhabdoid tumors in children: prognostic factors in 70 patients diagnosed in Germany. Oncology Reports. 2008;19(3):819–823. [PubMed] [Google Scholar]
- 89.Hoot AC, Russo P, Judkins AR, Perlman EJ, Biegel JA. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. American Journal of Surgical Pathology. 2004;28(11):1485–1491. doi: 10.1097/01.pas.0000141390.14548.34. [DOI] [PubMed] [Google Scholar]
- 90.Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clinical Cancer Research. 2002;8(11):3461–3467. [PubMed] [Google Scholar]
- 91.Roberts CWM, Orkin SH. The SWI/SNF complex—chromatin and cancer. Nature Reviews Cancer. 2004;4(2):133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 92.Rousseau-Merck MF, Versteege I, Legrand I, et al. hSNF5/INI1 inactivation is mainly associated with homozygous deletions and mitotic recombinations in rhabdoid tumors. Cancer Research. 1999;59(13):3152–3156. [PubMed] [Google Scholar]
- 93.Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. 2002;21(34):5193–5203. doi: 10.1038/sj.onc.1205706. [DOI] [PubMed] [Google Scholar]
- 94.Reincke BS, Rosson GB, Oswald BW, Wright CF. INI1 expression induces cell cycle arrest and markers of senescence in malignant rhabdoid tumor cells. Journal of Cellular Physiology. 2003;194(3):303–313. doi: 10.1002/jcp.10201. [DOI] [PubMed] [Google Scholar]
- 95.Versteege I, Medjkane S, Rouillard D, Delattre O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene. 2002;21(42):6403–6412. doi: 10.1038/sj.onc.1205841. [DOI] [PubMed] [Google Scholar]
- 96.Vries RGJ, Bezrookove V, Zuijderduijn LMP, et al. Cancer-associated mutations in chromatin remodeler hSNF5 promote chromosomal instability by compromising the mitotic checkpoint. Genes and Development. 2005;19(6):665–670. doi: 10.1101/gad.335805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shiratsuchi H, Saito T, Sakamoto A, et al. Mutation analysis of human cytokeratin 8 gene in malignant rhabdoid tumor: a possible association with intracytoplasmic inclusion body formation. Modern Pathology. 2002;15(2):146–153. doi: 10.1038/modpathol.3880506. [DOI] [PubMed] [Google Scholar]
- 98.Behring B, Brück W, Goebel HH, et al. Immunohistochemistry of primary central nervous system malignant rhabdoid tumors: report of five cases and review of the literature. Acta Neuropathologica. 1996;91(6):578–586. doi: 10.1007/s004010050470. [DOI] [PubMed] [Google Scholar]