

Abstract
Combination chemoprevention for cancer was proposed a quarter of a century ago, but has not been implemented in standard medical practice owing to limited efficacy and toxicity. Recent trials have targeted inflammation and polyamine biosynthesis, both of which are increased in carcinogenesis. Preclinical studies have demonstrated that DFMO (difluoromethylornithine), an irreversible inhibitor of ODC (ornithine decarboxylase) which is the first enzyme in polyamine biosynthesis, combined with NSAIDs (non-steroidal anti-inflammatory drugs) suppresses colorectal carcinogenesis in murine models. The preclinical rationale for combination chemoprevention with DFMO and the NSAID sulindac, was strengthened by the observation that a SNP (single nucleotide polymorphism) in the ODC promoter was prognostic for adenoma recurrence in patients with prior sporadic colon polyps and predicted reduced risk of adenoma in those patients taking aspirin. Recent results from a phase III clinical trial showed a dramatic reduction in metachronous adenoma number, size and grade. Combination chemoprevention with DFMO and sulindac was not associated with any serious toxicity. A non-significant trend in subclinical ototoxicity was detected by quantitative audiology in a subset of patients identified by a genetic marker. These preclinical, translational and clinical data provide compelling evidence for the efficacy of combination chemoprevention. DFMO and sulindac is a rational strategy for the prevention of metachronous adenomas, especially in patients with significant risk for colorectal cancer. Toxicities from this combination may be limited to subsets of patients identified by either past medical history or clinical tests.
Introduction
Increased concentrations of polyamines are found in cancerous tissue [1]. Polyamines are influenced by factors such as import, export, biosynthesis and catabolism. ODC (ornithine decarboxylase) is a rationale target in several cancers, with the goal to decrease cellular polyamine pools. ODC inhibition may not be sufficient therapy in situations where polyamine synthesis is not the rate-limiting step regulating polyamine pool sizes.
The ODC gene is regulated by the Wnt signalling cascade [2]. Activated WNT signalling up-regulates MYC [3], a transcriptional activator of ODC [4]. The WNT cascade is silenced in the majority of adult intestinal tissues, but can be dysregulated through mutations. The APC (adenomatous polyposis coli) tumour suppressor gene is a component of the WNT cascade and has been identified as the germ-line mutation in FAP (familial adenomatous polyposis) [5]. Over 80% of sporadic colorectal cancers also carry mutations in APC. APC mutations and WNT signalling lead to up-regulation of MYC activity and increased expression of ODC, and increased polyamine pools. Other genetic influences of CRC (colorectal cancer) are mutant K-RAS, a proto-oncogene that also activates ODC.
Colorectal carcinogenesis is also influenced by high-fat diet and sedentary lifestyle. Long-term use of aspirin and the reduction of CRC mortality led to the COX (cyclo-oxygenase)-based hypothesis [6]. These environmental risk factors influence an individual's genetic susceptibility and produce a risk profile for cancer. A mechanism for CRC involves mutated-APC and K-RAS in combination with environmental risk factors that elevate polyamines and inflammation. A rationale strategy for cancer chemoprevention would be the combination of inhibitors of polyamine biosynthesis and inflammation.
Pre-clinical data
Cancer burden
In the United States, cancer is the leading cause of death in people under the age of 85 years [7]. There were an estimated 1437180 new cases and 565650 deaths attributed to cancer in 2008, whereas 10.8 million Americans are living with a history of cancer. Cancer therapeutics are most effective in the early stages of disease, but are less effective in treating advanced cancers. This point underscores the need for prevention, early detection and effective treatment.
The vast majority of US cancer incidence and mortality are epithelial-derived cancers of the lung, colon, breast and prostate. Epithelia provide a protective barrier while also importing nutrients and exporting waste, especially the colon. The ACS (American Cancer Society) estimated that there were 148810 new cases of colorectal cancer in 2008, with a mortality of 49960 deaths.
APC: the genetic risk factor for CRC
Carcinogenesis occurs by inactivation of a tumour suppressor gene or activation of an oncogene. Inherited loss of a single allele in the APC gene increases an individual's risk of a hereditary carcinogenesis, known as FAP [5]. FAP is characterized by an increase in adenomatous polyps of the colon with advancing age. The risk is malignant transformation of the polyps and treatment is colectomy. Greater than 80% of sporadic CRCs also have mutations in the APC gene. Shown in Figure 1 is a representation of the WNT pathway in normal tissue and in carcinogenesis.
Figure 1. The WNT pathway in normal and carcinogenic cells.

The WNT pathway in a normal, adult, colonic epithelial cell is depicted on the left-hand side of the Figure. On the basolateral portion of the cell the WNT receptor is activated and transmits the signal intracellularly to the APC, GSK-3β and β-catenin complex. GSK-3β phosphorylates β-catenin, marking it for proteosomal degradation. The WNT pathway in carcinogenesis is depicted on the right-hand side of the Figure. The WNT receptor may be activated and transmission of the signal intracellularly may occur, but inactivation of β-catenin by GSK-3β does not. Cytoplasmic accumulation of β-catenin leads to its nuclear translocation and binding with its cognate partner TCF/LEF. This heterodimerization regulates genes through transcription, notably of MYC.
The APC protein interacts with GSK-3β (glycogen synthase kinase-3β) and β-catenin. This interaction allows GSK-3β to phosphorylate β-catenin and marks it for proteosomal degradation. In carcinogenic tissue, GSK-3β and β-catenin are disrupted by mutant-APC. As a result GSK-3β can no longer phosphorylate β-catenin. β-Catenin accumulates in the cytoplasm and then translocates to the nucleus forming a heterodimer with its cognate binding partner, TCF/LEF (T-cell factor/lymphoid-enhancing factor). The heterodimer binds to promoter regions of genes and alters expression [2]. Dysregulation of WNT signalling leads to changes in c-MYC gene expression. MYC activation occurs in neuroblastoma, breast and prostate cancers, but its dysregulation in CRC is unique given mutations in the upstream APC gene and rapid cellular turnover.
In APC-dependent carcinogenesis c-MYC activation affects transcription of ODC through binding regions in the promoter. An SNP (single nucleotide polymorphism) occurs in ODC between two MYC-binding regions. Differential repression of ODC occurs by MAD1 binding at the SNP sequence to the A-allele, not the G-allele. In a population-based study of humans with prior colonic adenomas, aspirin use was associated with a 90% reduction in relative risk for development of metachronous adenomas among individuals homozygous for the minor ODC A-allele compared with the non-aspirin users homozygous for the major ODC G-allele [8]. This ODC polymorphism appears to be a genetic marker for CRC risk. Our group has sought to test features of the hypothesis depicted in Figure 1. We have developed a transgenic mouse expressing a mutant Apc and conditional deletion of the c-Myc alleles in the intestinal and colonic mucosa. Conditional suppression of Myc in the intestinal tract was associated with reduced intestinal tumour number, compared with these same ApcMin/+ mice expressing intestinal Myc [9]. Treatment with DFMO (difluoromethylornithine) also suppresses intestinal tumorigenesis in ApcMin/+ mice [10]. These results implicate both Myc and Odc as downstream mediators of APC-dependent intestinal carcinogenesis.
The MYC protein can act as a transcription factor activator when bound to MAX. The MYC–MAX heterodimer can activate the expression of genes through binding on consensus sequences (enhancer box sequences or E-boxes), as well as recruiting HATs (histone acetyltransferases). In contrast, when MAX is bound to MAD1, transcription is repressed. In the presence of normal APC, C-MYC was suppressed whereas MAD1 was elevated. In the presence of mutant-APC, C-MYC was elevated whereas MAD1 was suppressed [2]. This interaction is shown in Figure 2.
Figure 2. ODC regulation occurs through both positive and negative mechanisms.
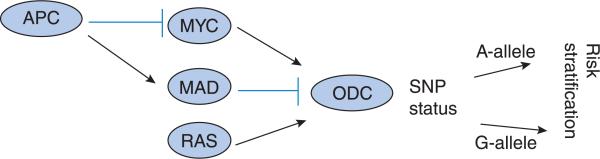
ODC is suppressed by MAD. Transcriptional activation of ODC can occur through MYC, RAS or both. Upstream activation of c-myc may occur via mutant APC. Pharmacogenetic manipulation of ODC occurs through identification of SNP status. The combination of these factors may increase individual risk stratification.
Mutant-APC led to elevations in c-MYC that increased expression of ODC [2]. The results indicated that ODC was a modifier of APC-dependent signalling in models of CRC. Mouse models were employed to determine whether mutant-APC led to elevated polyamines in vivo. Murine models recapitulate this finding, with elevated polyamines found in the small intestine of ApcMin/+ mice [10].
Polyamine-dependent regulation and colon carcinogenesis
Arginine is a component of the urea cycle which converts nitrogenous waste for excretion [11]. Elevated dietary arginine can also increase polyamine levels through its conversion into ornithine in the urea cycle. ApcMin/+ mice fed a diet of elevated arginine had an elevated tumour burden [12]. In patients with CRC, increased meat consumption was a surrogate for arginine and was associated with decreased overall survival [13]. Polyamines are exported, as depicted in Figure 3, via a mechanism which involves an arginine transporter [14].
Figure 3. Polyamine transport is shown schematically via import, export, anabolism and catabolism.

Arginine is imported into the cell and then converted into ornithine which contributes to the polyamine pools. Polyamine pools may be further increased by import or decreased by catabolism and export.
K-RAS mutation is another significant risk factor for CRC. The RAS-family of proteins is a mediator of extracellular signals through the cytoplasm and eventually into the nucleus; the effect of which is to alter gene expression and enhance proliferation. In cell culture models, mutant K-RAS increases ODC transcription and decreases transcription of SAT1 (spermidine/spermine acetyltransferase; also referred to as SSAT in other chapters in this volume), an enzyme important in the catabolism of polyamines. Mutant K-RAS increases polyamine biosynthesis via ODC activation and decreases acetylation of polyamines.
Polyamine pool limitation
One strategy to inhibit polyamine levels is to decrease biosynthesis. Selective inhibition of ODC by the suicide-inhibitor, α-DFMO, was developed at the Merrell Dow Research Center in Strasbourg, France [11]. Although α-DFMO showed promise in cell culture models, compensatory polyamine import limited the success of DFMO in early murine models.
SAT1 is an important factor in polyamine export. As shown in Figure 3, SAT1 can acetylate both spermidine and spermine, targeting them for export. SAT1 can be induced by NSAIDs (non-steroidal anti-inflammatory drugs), including aspirin, sulindac, ibuprofen and indomethacin [15]. SAT1 induction can lead to apoptosis in CRC cell lines [16]. Sulindac induced Sat1, decreased intestinal levels of monoacetylspermidine, spermidine and spermine, and reduced tumour number in the small intestine of mouse models [17]. Dietary putrescine restored tissue polyamine content and partially abrogated the antitumour effects of sulindac, indicating that sulindac was acting via a polyamine-dependent mechanism.
Inflammation and colorectal carcinogenesis
In 1863 Virchow hypothesized a causal interaction between chronic inflammation and cancer mediated via the tumour microenvironment [18]. Within the microenvironment of carcinogenesis both intrinsic and extrinsic cellular factors contribute to a pro-inflammatory state. The intrinsic pro-inflammatory factor NF-κB (nuclear factor κB) is activated by many signals. Activation of NF-κB up-regulates target genes facilitating cancer growth by initiation, promotion and progression. One of the target genes of NF-κB is the enzyme COX. COX-1 is a constitutive gene that mediates homoeostatic functions, whereas inducible COX-2 is associated with inflammation. PGE2 (prostaglandin E2) is downstream of COX-2 and is associated with tumorigenesis (Figure 4). In cell culture models the NSAID sulindac decreased both COX-2- and PGE2 synthase-mediated inflammation. While NSAIDs block COX-2, they also inhibit production of NO (nitric oxide) via inhibition of NOS-2 (inducible nitric oxide synthase). NO is a known activator of inflammation. As shown in Figure 4, arginine can be converted into NO, leading to increased inflammation.
Figure 4. Inflammation within a colonic epithelial cell may occur through multiple mechanisms.

Imported dietary arginine may either be converted into NO or processed in the polyamine pathway. NSAIDs can disrupt these and other pathways via inhibition of NOS-2, up-regulation of polyamine export or inhibition of COX-2. External sources of polyamines and bile acids may also contribute to inflammation.
Clinical data with DFMO
Cancer therapeutic experience
DFMO has been evaluated as a cancer therapeutic agent. It was not especially active as a single agent and its use was associated with ototoxicity at high doses [11,19]. Based on mouse model studies, DFMO was subsequently evaluated as a potential cancer chemopreventive agent [11].
Clinical trials of DFMO for colon cancer prevention
Early studies have confirmed that ODC and polyamine contents were elevated in human colon cancer tissue compared with adjacent normal colorectal mucosa [20]. Measurements of colorectal tissue polyamine contents were subsequently validated as measures of DFMO effects in patients. Validation of these markers allowed for the conduct of clinical trials to assess efficacy of DFMO dose, oral dose delivery and frequency of dosing. Consequently, we were able to conduct a dose de-escalation trial to determine the lowest DFMO dose capable of suppressing colorectal polyamine contents while minimizing toxicities, including ototoxicity [21]. A subsequent trial built on these findings evaluated three oral doses of DFMO given daily for 1 year. Patients were randomized to a control or treatment group with three separate doses of DFMO: 0.075, 0.2 and 0.4 g/m2 per day [22]. The end of trial analysis showed that the 0.2 g/m2 per day dose had similar biological effects compared with the 0.4 g/m2 per day dose, with a decreased report of toxic side effects and decreased drop-out rate. The implications were that a low dose of DFMO inhibited colorectal polyamine content in rectal mucosa while demonstrating a safe toxicity profile.
Sulindac
While a phase IIb/III trial was being considered with DFMO, strong and extensive epidemiological evidence accumulated suggesting that aspirin and other NSAIDs might be effective as colon cancer prevention agents. Sulindac decreased polyp formation in high-risk patients with FAP [23]. It was hypothesized that sulindac and DFMO in combination would have a greater effect than either agent alone on the development of colon polyps, and this point was established in preclinical models [24]. As shown in Figure 5, NSAIDs and DFMO affect multiple targets related to inflammation.
Figure 5. The interaction among mutant-APC, polyamines and inflammation is depicted in a colonic epithelial cell.

Mutant-APC and activated K-RAS lead to increased MYC production and up-regulate ODC which increases the polyamine concentration. Elevated dietary arginine can also contribute to increased polyamine pools. NSAIDs and DFMO can inhibit multiple targets in both the polyamine and inflammatory pathways.
Phase IIb and III DFMO and sulindac trials in non-cancerous patients
Cell culture models indicated that DFMO and sulindac could act at least additively to suppress growth and cell survival [24]. With mounting cell culture, murine model, clinical and epidemiological data, a prospective, randomized, placebo-controlled, phase IIb trial, with the combination of DFMO and sulindac, for 3 years was initiated in patients with prior sporadic colon polyps. The study used 0.2 g of DFMO/m2 per day and was converted from a liquid oral dose into an oral pill form. A dose of 500 mg/day closely approximated the 0.2 g/m2 per day liquid form. The study also used 150 mg of sulindac, 50% of the conventional dose [23]. In addition to the need for efficacy, a major intent was to evaluate potential toxicity. The phase IIb trial of DFMO and sulindac, with biochemical markers as primary endpoints, was subsequently modified and converted into a phase III trial with metachronous adenomas as the primary endpoint of the study.
Baseline and serial audiological tests were performed to assess potential long-term ototoxicity. Participants with greater than 20 dB uncorrectable hearing loss above the age-adjusted norms were ineligible for the phase III trial. The eligible patients were randomized to receive placebo or 500 mg of DFMO plus 150 mg of sulindac. Patients were stratified according to two parameters: the seven clinical sites and low-dose aspirin use (either 81 mg/day or less than 325 mg twice weekly). Safety evaluations of the patients occurred after the 1 month run-in as well as at 3, 6 and 9 months, and every 6 months for the remainder of the phase III trial. Evaluations included physical examination and laboratory evaluations. Pure-tone audiograms were performed at 0, 18 and 36 months. Adenomas removed during any part of the phase III trial were submitted to a central pathology facility with standardized diagnostic objectives. The colorectal polyps were counted, measured and graded according to predetermined criteria. Safety analysis included investigator-reported adverse events and were coded according to the COSTART (Coding Symbols for Thesaurus of Adverse Reaction Terms) Body System [25]. An independent DSMB (Data and Safety Monitoring Board) reviewed the safety and efficacy of the data twice yearly. The investigators were blinded to the results of the DSMB's findings throughout the phase IIb and phase III trials. A pre-specified early stopping point was made based on potential efficacious or futile results. Interim analysis was planned at approx. 60% and 80% of the total accrual of patient information.
Based on the primary and secondary endpoints of the phase III trial, as well as oversight by the DSMB, the blind study was broken at the second interim analysis. The phase III trial intervention of combination DFMO and sulindac was found to be significantly effective. The combination of 500 mg of DFMO and 150 mg of sulindac decreased the number and severity of adenoma recurrence without significant toxicity [25]. The results are presented in Table 1 and summarized here. There was a 70% reduction in metachronous adenomas for those in the treatment arm. In addition, there was a 92% reduction in advanced adenomas and a 95% decrease in the recurrence of multiple adenomas in patients in the treatment arm compared with those in the placebo arm. Sporn and Hong [26] wrote an accompanying editorial with this publication and stated that “the clinical results represent a landmark advance to [reduce the number] of cancer deaths”.
Table 1. Adenoma recurrence as a function of intervention.
The adenomas and pathologies are shown on the left-hand side, while intervention number and type are shown along the top. The risk ratio for development of adenomatous lesions based on intervention strategy is quantified and statistical significance is shown. Data taken from [25].
| Pathology | Placebo (n = 129) | DFMO+sulindac (n = 138) | Risk ratio | 95% Confidence interval | P |
|---|---|---|---|---|---|
| Any adenoma | 53 | 17 | 0.30 | 0.18–0.49 | <0.001 |
| Advanced adenoma | 11 | 1 | 0.085 | 0.011–0.65 | 0.001 |
| Large (≥1 cm) advanced adenoma | 9 | 1 | 0.10 | 0.013–0.81 | 0.004 |
| Multiple adenomas | 17 | 1 | 0.055 | 0.0074–0.41 | <0.001 |
Toxicity
An adjusted, non-significant, mean decrease in hearing threshold of 1.08 dB was detected in the treatment arm compared with placebo [27]. There was no difference in the clinical audiotoxicity between the two arms. Cardiotoxicity was also evaluated in this study. Patients were stratified into low-, moderate-and high-CV (cardiovascular) risk factors. Among high-risk patients, the number of CV events was higher in the treatment than the placebo arm. Excluding the high-risk CV patients, the numbers of CV events were similar between treatment and placebo arms [28].
These clinical results from a phase III trial proved to be consistent with preclinical studies in mouse models. Combination DFMO and sulindac was effective in reducing tumour number by more than 80% when compared with the untreated controls in the ApcMin/+ mouse model (P < 0.0001) [29]. Combination sulindac–DFMO was effective in reducing the number of high-grade adenomas when compared with the sulindac alone (P = 0.003). The clinical implications are twofold: (i) first DFMO is effective in the reduction of adenomatous polyps and (ii) secondly, the combination of DFMO and sulindac can further reduce the risk of CRC through the reduction in the number of high-grade intestinal adenomas. These high-grade adenomas are those lesions most likely to progress to colon cancer.
Phase III DFMO and sulindac in patients with cancer
Although the phase III trial with DFMO and sulindac provided strong evidence for preventing disease recurrence in patients with prior adenomas, the chemoprevention has not been evaluated in higher risk populations. Even after surgical resection and optimal treatment with chemotherapy (when indicated), stage I–III colon cancer patients remain at considerable risk for distant recurrence, secondary colonic tumour formation and subsequent mortality. A phase III trial of these key compounds among surgically resected colon cancer patients is in the planning stages.
Future
Clinical practice
Approx. 30 million patients over the age of 50 years will develop adenomatous polyps each year in the United States. A proportionally similar number will develop these lesions in Western Europe. Approx. 10% will progress to advanced polyps or frank cancer. Genetically at-risk patients along with those with a history of CRC [sporadic, FAP, HNPCC (hereditary nonpolyposis colorectal cancer) or prior CRC] are the target population for chemoprevention. Current techniques for CRC screening include endoscopy at regular intervals with polyps removed. There is a significant risk reduction for CRC through endoscopy and polypectomy, but barriers of cost, preparation and the procedure translates into approx. 50% screening of eligible populations.
Endoscopy is also a poor screening method for right-sided cancers, as well as flat or depressed lesions. Further complicating the role of endoscopy is the frequent overuse by patients with previously diagnosed disease. The resultant gaps in screening with overuse by others necessitate another disease prevention strategy. Chemoprevention clearly has the potential to reduce disease burden.
The results from the phase III trials potentially affect the surveillance of higher risk populations. As previously stated, approx. 50% of CRC patients with Stage I, II or III disease recur. Patients’ and physicians’ anxieties may decrease with combination therapy and lessen the overutilization of endoscopic procedures in patients with a moderate risk of colon cancer. Combination chemoprevention may also have applications in high-risk populations such as those with FAP. The positive results of this combination in the Apc Min/+ model of FAP may lead to trials in this high-risk group. Positive results of such trials could increase the time to surgery in patients with FAP.
Dietary sources of polyamines
A database has been developed to assess dietary polyamine content [30]. It is hypothesized that a reduction in total-body polyamine pools may reduce carcinogenesis. Table 2 presents some of the foods for which the polyamine content has been determined. The intention for the database is to quantify dietary polyamines and qualify them as a risk factor for carcinogenesis.
Table 2. Polyamine content in food.
The three polyamines: putrescine, spermidine and spermine are listed. The amount of polyamines, in nmol/day, is given for multiple food items. Data taken from [30].
| Polyamine | Food item | Amount (nmol/day) |
|---|---|---|
| Putrescine | Orange juice and grapefruit juice | 44441 |
| Oranges, grapefruit and tangerines (excluding juice) | 17613 | |
| Fresh tomatoes | 10042 | |
| Bananas | 7344 | |
| Beer (all types) | 6374 | |
| Spermidine | Green peas | 3283 |
| Cheese, such as American and cheddar | 3124 | |
| Lasagne and pasta with meat sauce | 2900 | |
| Potatoes (boiled, baked and mashed) | 2388 | |
| Burritos, tacos, tostadas and quesadillas | 1890 | |
| Spermine | Ground meat | 2186 |
| Lunch meats (e.g. ham, turkey, bologna and salami) | 1977 | |
| Green peas | 1905 | |
| Lasagne and pasta with meat sauce | 1443 | |
| Peanut butter, peanuts and other nuts and seeds | 1237 |
Conclusions
Epithelia provide a protective barrier from the external environment. It is a mediator of transport. In the process, epithelia are exposed to harmful substances and harbour genetic mutations. In this context, cells are transformed from normal to neoplastic and the carcinogenic process is initiated. For the past several decades, collaborative efforts of scientific research have led to a new paradigm in cancer management: chemoprevention. Chemoprevention is an approach that is not applicable to everyone. It should target people with elevated risks of cancer. The caveat for chemoprevention is that target populations are still relatively healthy compared with patients who currently have cancer. Therefore chemoprevention must have a clear benefit that exceeds the risk of treatment. The development of DFMO and sulindac underscore this sentiment.
Summary
Polyamine pools are elevated in cancerous tissue.
ODC is a committed step, converting ornithine into putrescine.
DFMO is an irreversible, competitive inhibitor of ODC.
In CRC cell culture models, DFMO decreased polyamine levels.
Murine models of CRC with mutant-Apc have increased Myc and Odc activation.
DFMO alone had limited efficacy in therapeutic clinical trials.
NSAIDs decrease COX activity.
NSAIDs can activate polyamine export pathways.
Polyamine pools are dynamically regulated by anabolism, catabolism, import and export.
Low-dose DFMO was evaluated as a chemopreventative agent for CRC by biomarker assays, efficacy, safety, toxicity and dose de-escalation before large clinical trials.
Combination DFMO and sulindac was evaluated in a large, randomized, prospective, multi-centre, double-blind clinical trial.
Combination DFMO and sulindac was effective in reducing adenoma recurrence by number, size and grade.
Ototoxicity was measured in the treatment arm, but without clinical significance.
Patients with baseline high-CV risk had a higher number of CV events.
References
- 1.Russell D, Snyder SH. Amine synthesis in rapidly growing tissues: ornithine decarboxylase activity in regenerating rat liver, chick embryo, and various tumors. Proc. Natl. Acad. Sci. U.S.A. 1968;60:1420–1427. doi: 10.1073/pnas.60.4.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fultz KE, Gerner EW. APC-dependent regulation of ornithine decarboxylase in human colon tumor cells. Mol. Carcinog. 2002;34:10–18. doi: 10.1002/mc.10043. [DOI] [PubMed] [Google Scholar]
- 3.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identifi cation of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 4.Bello-Fernandez C, Packham G, Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7804–7808. doi: 10.1073/pnas.90.16.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identifi cation and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 6.Thun MJ, Namboodiri MM, Heath CW., Jr Aspirin use and reduced risk of fatal colon cancer. N. Engl. J. Med. 1991;325:1593–1596. doi: 10.1056/NEJM199112053252301. [DOI] [PubMed] [Google Scholar]
- 7.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J. Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 8.Martinez ME, O'Brien TG, Fultz KE, Babbar N, Yerushalmi H, Qu N, Guo Y, Boorman D, Einspahr J, Alberts DS, Gerner EW. Pronounced reduction in adenoma recurrence associated with aspirin use and a polymorphism in the ornithine decarboxylase gene. Proc. Natl. Acad. Sci. U.S.A. 2003;100:7859–7864. doi: 10.1073/pnas.1332465100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ignatenko NA, Holubec H, Besselsen DG, Blohm-Mangone KA, Padilla-Torres JL, Nagle RB, de Alboranc IM, Guillen RJ, Gerner EW. Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol. Ther. 2006;5:1658–1664. doi: 10.4161/cbt.5.12.3376. [DOI] [PubMed] [Google Scholar]
- 10.Erdman SH, Ignatenko NA, Powell MB, Blohm-Mangone KA, Holubec H, Guillen-Rodriguez JM, Gerner EW. APC-dependent changes in expression of genes infl uencing polyamine metabolism, and consequences for gastrointestinal carcinogenesis, in the Min mouse. Carcinogenesis. 1999;20:1709–1713. doi: 10.1093/carcin/20.9.1709. [DOI] [PubMed] [Google Scholar]
- 11.Gerner EW, Meyskens FL., Jr Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer. 2004;4:781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 12.Yerushalmi HF, Besselsen DG, Ignatenko NA, Blohm-Mangone KA, Padilla-Torres JL, Stringer DE, Guillen JM, Holubec H, Payne CM, Gerner EW. Role of polyamines in arginine-dependent colon carcinogenesis in ApcMin/+ mice. Mol. Carcinog. 2006;45:764–773. doi: 10.1002/mc.20246. [DOI] [PubMed] [Google Scholar]
- 13.Zell JA, Ignatenko NA, Yerushalmi HF, Ziogas A, Besselsen DG, Gerner EW, Anton-Culver H. Risk and risk reduction involving arginine intake and meat consumption in colorectal tumorigenesis and survival. Int. J. Cancer. 2007;120:459–468. doi: 10.1002/ijc.22311. [DOI] [PubMed] [Google Scholar]
- 14.Uemura T, Yerushalmi HF, Tsaprailis G, Stringer DE, Pastorian KE, Hawel L, III, Byus CV, Gerner EW. Identifi cation and characterization of a diamine exporter in colon epithelial cells. J. Biol. Chem. 2008;283:26428–26435. doi: 10.1074/jbc.M804714200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babbar N, Gerner EW, Casero RA., Jr Induction of spermidine/spermine N1-acetyltransferase (SSAT) by aspirin in Caco-2 colon cancer cells. Biochem. J. 2006;394:317–324. doi: 10.1042/BJ20051298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babbar N, Ignatenko NA, Casero RA, Jr, Gerner EW. Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J. Biol. Chem. 2003;278:47762–47775. doi: 10.1074/jbc.M307265200. [DOI] [PubMed] [Google Scholar]
- 17.Ignatenko NA, Besselsen DG, Roy UK, Stringer DE, Blohm-Mangone KA, Padilla-Torres JL, Guillen RJ, Gerner EW. Dietary putrescine reduces the intestinal anticarcinogenic activity of sulindac in a murine model of familial adenomatous polyposis. Nutr. Cancer. 2006;56:172–181. doi: 10.1207/s15327914nc5602_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coussens LM, Werb Z. Infl ammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croghan MK, Aickin MG, Meyskens FL. Dose-related α-difl uoromethylornithine ototoxicity. Am. J. Clin. Oncol. 1991;14:331–335. doi: 10.1097/00000421-199108000-00012. [DOI] [PubMed] [Google Scholar]
- 20.Hixson LJ, Garewal HS, McGee DL, Sloan D, Fennerty MB, Sampliner RE, Gerner EW. Ornithine decarboxylase and polyamines in colorectal neoplasia and mucosa. Cancer Epidemiol. Biomarkers Prev. 1993;2:369–374. [PubMed] [Google Scholar]
- 21.Meyskens FL, Jr, Emerson SS, Pelot D, Meshkinpour H, Shassetz LR, Einspahr J, Alberts DS, Gerner EW. Dose de-escalation chemoprevention trial of α-difl uoromethylornithine in patients with colon polyps. J. Natl. Cancer Inst. 1994;86:1122–1130. doi: 10.1093/jnci/86.15.1122. [DOI] [PubMed] [Google Scholar]
- 22.Meyskens FL, Jr, Gerner EW, Emerson S, Pelot D, Durbin T, Doyle K, Lagerberg W. Effect of α-difl uoromethylornithine on rectal mucosal levels of polyamines in a randomized, double-blinded trial for colon cancer prevention. J. Natl. Cancer Inst. 1998;90:1212–1218. doi: 10.1093/jnci/90.16.1212. [DOI] [PubMed] [Google Scholar]
- 23.Giardiello FM, Offerhaus JA, Tersmette AC, Hylind LM, Krush AJ, Brensinger JD, Booker SV, Hamilton SR. Sulindac induced regression of colorectal adenomas in familial adenomatous polyposis: evaluation of predictive factors. Gut. 1996;38:578–581. doi: 10.1136/gut.38.4.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawson KR, Ignatenko NA, Piazza GA, Cui H, Gerner EW. Infl uence of K-ras activation on the survival responses of Caco-2 cells to the chemopreventive agents sulindac and difl uoromethylornithine. Cancer Epidemiol. Biomarkers Prev. 2000;9:1155–1162. [PubMed] [Google Scholar]
- 25.Meyskens FL, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, Kelloff G, Lawson MJ, Kidao J, McCracken J, et al. Difl uoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev. Res. 2008;1:9–11. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sporn MB, Hong WK. Clinical prevention of recurrence of colorectal adenomas by the combination of difl uoromethylornithine and sulindac: an important milestone. Cancer Prev. Res. 2008;1:9–11. doi: 10.1158/1940-6207.CAPR-08-0049. [DOI] [PubMed] [Google Scholar]
- 27.McClaren CE, Fujikawa-Brooks S, Chen W, Gillen DL, Pelot D, Gerner EW, Meyskens FL. Longitudinal assessment of air conduction audiograms in a phase III clinical trial of difluoromethylornithine and sulindac for prevention of sporadic colorectal adenomas. Cancer Prev. Res. 2008;1:514–521. doi: 10.1158/1940-6207.CAPR-08-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zell JA, Pelot D, Chen WP, McLaren CE, Gerner EW, Meyskens FL. Risk of cardiovascular events in a randomized placebo-controlled, double-blind trial of difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas. Cancer Prev. Res. 2009;2:209–212. doi: 10.1158/1940-6207.CAPR-08-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ignatenko NA, Besselsen DG, Stringer DE, Blohm-Mangone KA, Cui H, Gerner EW. Combination chemoprevention of intestinal carcinogenesis in a murine model of familial adenomatous polyposis. Nutr. Cancer. 2008;60(Suppl. 1):30–35. doi: 10.1080/01635580802401317. [DOI] [PubMed] [Google Scholar]
- 30.Zoumas-Morse C, Rock CL, Quintana EL, Neuhouser ML, Gerner EW, Meyskens FL., Jr Development of a polyamine database for assessing dietary intake. J. Am. Diet Assoc. 2007;107:1024–1027. doi: 10.1016/j.jada.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]