

Abstract
Pyrethroid insecticides disrupt nerve function by modifying the gating kinetics of transitions between the conducting and nonconducting states of voltage-gated sodium channels. Pyrethroids modify rat Nav1.6 + β1 + β2 channels expressed in Xenopus oocytes in both the resting state and in one or more states that require channel activation by repeated depolarization. The state dependence of modification depends on the pyrethroid examined: deltamethrin modification requires repeated channel activation, tefluthrin modification is significantly enhanced by repeated channel activation, and S-bioallethrin modification is unaffected by repeated activation. Use-dependent modification by deltamethrin and tefluthrin implies that these compounds bind preferentially to open channels. We constructed the rat Nav1.6Q3 cDNA, which contained the IFM/QQQ mutation in the inactivation gate domain that prevents fast inactivation and results in a persistently open channel. We expressed Nav1.6Q3 + β1 + β2 sodium channels in Xenopus oocytes and assessed the modification of open channels by pyrethroids by determining the effect of depolarizing pulse length on the normalized conductance of the pyrethroid-induced sodium tail current. Deltamethrin caused little modification of Nav1.6Q3 following short (10 ms) depolarizations, but prolonged depolarizations (up to 150 ms) caused a progressive increase in channel modification measured as an increase in the conductance of the pyrethroid-induced sodium tail current. Modification by tefluthrin was clearly detectable following short depolarizations and was increased by long depolarizations. By contrast modification by S-bioallethrin following short depolarizations was not altered by prolonged depolarization. These studies provide direct evidence for the preferential binding of deltamethrin and tefluthrin (but not S-bioallethrin) to Nav1.6Q3 channels in the open state and imply that the pyrethroid receptor of resting and open channels occupies different conformations that exhibit distinct structure–activity relationships.
Keywords: Sodium channel, Nav1.6, Use-dependent modification, Deltamethrin, S-bioallethrin, Tefluthrin
1. Introduction
Effects on voltage-gated sodium channels underlie both the insecticidal actions of pyrethroids and their neurotoxicity to nontarget organisms (Soderlund, 2012; Soderlund et al., 2002). Pyrethroids modify the gating of voltage-gated sodium channels, which mediate the transient increase in the sodium permeability of the nerve membrane that underlies the rising phase of the nerve action potential (reviewed in: Bloomquist, 1993; Narahashi, 1996; Soderlund, 1995). Voltage- and patch-clamp analyses of isolated sodium currents show that pyrethroids retard the kinetics of sodium channel activation, inactivation, and deactivation to produce persistently open channels. The hallmark of pyrethroid modification in all experimental systems is the induction of a sodium tail current following membrane depolarization–repolarization cycles under voltage clamp whose persistence is correlated with pyrethroid structure.
The majority of studies of pyrethroid action on sodium currents in neurons under voltage-clamp conditions have been performed by equilibrating preparations with insecticide at hyperpolarized membrane potentials and assessing the effects of pyrethroids upon depolarization (Bloomquist, 1993; Narahashi, 1996; Soderlund, 1995). This approach is biased toward the detection of the modification of channels in the closed state by pyrethroids. Consequently, only a few studies have explored possible use-dependent modification by assessing the impact of trains of depolarizing prepulses on the amplitude of the pyrethroid-induced sodium tail current. Use-dependent enhancement of modification is detected in assays of fenvalerate on sodium currents in crayfish giant axons (Salgado and Narahashi, 1993), assays of deltamethrin on the tetrodotoxin (TTX)-sensitive and TTX-resistant components of the sodium current in rat dorsal root ganglion neurons (Tabarean and Narahashi, 2001), and assays of tetramethrin and permethrin on sodium currents in honeybee antennal olfactory neurons (Kadala et al., 2011). By contrast, repetitive depolarization did not alter the time-dependent increase in the sodium tail current elicited by perfusion of squid giant axon preparations with phenothrin (de Weille et al., 1988).
Most of the evidence for use-dependent sodium channel modification comes from studies of pyrethroid action on cloned sodium channels expressed transiently in oocytes of the frog Xenopus laevis. Modification by some pyrethroids of both insect and mammalian channels expressed in X. laevis oocytes either depends on or is enhanced by trains of high-frequency depolarizing prepulses (reviewed in: Soderlund, 2010). Recent studies of the rat Nav1.6 sodium channel isoform in the Xenopus oocyte system show that the relative importance of resting and use-dependent channel modification depends on the pyrethroid examined (Tan and Soderlund, 2010) and that use-dependent effects require coexpression of the Nav1.6 α subunit isoform with the auxiliary β1 and β2 subunits (Tan and Soderlund, 2011a).
The use-dependent effects of pyrethroids on insect sodium channels expressed in Xenopus oocytes have been interpreted as evidence for the existence of a high-affinity receptor conformation on channels in the open state (Vais et al., 2000). Despite the inferred importance of pyrethroid binding to open channels there is little direct evidence for open-channel modification by pyrethroids. Assays of Drosophila melanogaster Para sodium channels expressed in Xenopus oocytes employed Anemonia sulcata toxin II (ATX-II) to eliminate fast inactivation and document pyrethroid modification of persistently open channels (Vais et al., 2000). However, the allosteric coupling of the ATX-II and pyrethroid binding sites on mammalian sodium channels (Lombet et al., 1988) suggests that the pyrethroid receptor of ATX-II-modified Para channels may exhibit pharmacological properties that differ from those of native channels.
To study pyrethroid interactions with open channels directly in the absence of pharmacological manipulation we introduced mutations in the inactivation gate region of the rat Nav1.6 sodium channel that are known to eliminate fast inactivation (West et al., 1992; Zhou and Goldin, 2004) and employed these inactivation-deficient channels to assess pyrethroid modification in the Xenopus oocyte expression system. We employed three pyrethroids that exhibit compound-specific differences in the relative importance of resting and use-dependent modification of native Nav1.6 sodium channels in the oocyte expression system. Deltamethrin modification requires repeated channel activation, tefluthrin modification is significantly enhanced by repeated channel activation, and S-bioallethrin modification is unaffected by repeated activation (Tan and Soderlund, 2010). Our results provide direct evidence that the differences in resting and use-dependent modification of native Nav1.6 channels among these three pyrethroids result from their differential modification of resting and open channels. Our results imply that the pyrethroid receptor of resting and open channels occupies different conformations that exhibit distinct structure–activity relationships.
2. Materials and methods
2.1. Sodium channel subunit cDNAs
Cloned rat voltage-sensitive sodium channel subunit cDNAs were obtained from the following sources: the Nav1.6α subunit (Dietrich et al., 1998) from L. Sangameswaran (Roche Bioscience, Palo Alto, CA) and the β1 and β2 subunits (Isom et al., 1992, 1995) from W.A. Catterall (University of Washington, Seattle, WA). The cDNA for the Nav1.6Q3 sodium channel α subunit, containing the IFM to QQQ mutation at amino acid sequence positions 1477–1479, was prepared by oligonucleotide-mediated site-directed mutagenesis on the parental Nav1.6 cDNA using a commercial kit (QuikChange XL, Stratagene, La Jolla, CA) and the structure of the Nav1.6Q3 cDNA was confirmed by DNA sequencing. Plasmid cDNAs were digested with restriction enzymes to provide linear templates for cRNA synthesis in vitro using a commercial kit (mMessage mMachine, Ambion, Austin, TX). The integrity of synthesized cRNA was determined by electrophoresis in 1% (w/v) agarose–formaldehyde gels.
2.2. Expression in oocytes
Freshly dissected X. laevis ovaries obtained from a commercial source (Nasco, Ft. Atkinson, WI) were employed as a source of stage V–VI oocytes, which were isolated as described elsewhere (Smith and Soderlund, 2001). Each data set was derived from oocytes isolated from three different ovaries. Oocytes were injected with a 1:1:1 (mass ratio) mixture of α subunit, β1 subunit and β2 subunit cRNAs (0.5–5 ng/oocyte); this mixture provided a ~9-fold molar excess of β1 and β2 cRNAs to ensure the preferential expression of desired binary or ternary α + β complexes (Tan and Soderlund, 2009, 2011b). Injected oocytes were incubated in ND-96 medium (in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES; adjusted to pH 7.6 at room temperature with NaOH) supplemented with 6% horse serum (Sigma–Aldrich, St. Louis, MO), 0.5% streptomycin/penicillin, and 1% sodium pyruvate (Goldin, 1992) at 16 °C for 3–5 days until electrophysiological analysis of sodium currents.
2.3. Electrophysiology
Sodium currents were recorded from oocytes perfused with ND-96 at 21–23 °C in the two-electrode voltage clamp configuration using an Axon Geneclamp 500B amplifier (Molecular Devices, Sunnyvale, CA). Microelectrodes were pulled from borosilicate glass capillary tubes (1.0 mm O.D.; 0.5 mm I.D.; World Precision Instruments Inc., Sarasota, FL) and filled with 3 M KCl. Filled electrodes had resistances of 0.3–1.0 MΩ when immersed in ND-96 medium. Currents were filtered at 2 kHz with a low-pass 4-pole Bessel filter and digitized at 50 kHz (Digidata 1320A; Molecular Devices). To determine the voltage dependence of activation, oocytes were clamped at a membrane potential of −100 mV and currents were measured during test pulses to potentials to 40 mV in 5-mV increments. Maximal peak transient currents were obtained upon depolarization to −10 mV. For determinations of time-dependent modification of Nav1.6Q3 channels, oocytes were clamped at a membrane potential of −100 mV and currents were measured during and after test pulses increasing in duration from 3 ms to 150 ms in 3, 5 or 10 ms increments. All experiments employed 30-s intervals between pulses or pulse trains to permit complete recovery from pyrethroid modification. Capacitive transients and leak currents were subtracted using the P/4 method (Bezanilla and Armstrong, 1977).
2.4. Assays with pyrethroids
S-bioallethrin (92.9% purity) and deltamethrin (99.5%) were obtained from Bayer CropScience (Research Triangle Park, NC) and tefluthrin (98.8%) was obtained from Syngenta (Bracknell, Berks., UK). Pyrethroids were prepared as stock solutions in dimethyl sulfoxide (DMSO) and diluted with ND-96 immediately before use to final concentrations of 10 μM (deltamethrin) or 100 μM (S-bioallethrin and tefluthrin), the highest concentrations achievable in ND-96 perfusion medium for each compound. The final DMSO concentration in the bath did not exceed 0.1%, a concentration that had no effect on sodium currents. Oocytes were perfused at 0.45 ml/min with pyrethroid in ND-96 for 6 min and then with ND-96 for 3 min prior to recording to stabilize the effective concentration and prevent the continued partitioning of the insecticide from the medium to the oocyte membrane during recording (Choi and Soderlund, 2006). Washout of S-bioallethrin, tefluthrin and deltamethrin was negligible under these conditions. All experiments with pyrethroids employed a disposable capillary perfusion system (Tatebayashi and Narahashi, 1994) and custom-fabricated single-use recording chambers (Smith and Soderlund, 2001) to prevent cross-contamination between oocytes. Net sodium currents following pyrethroid exposure were derived by digital subtraction of control current traces prior to pyrethroid exposure from current traces obtained following pyrethroid exposure in the same oocyte.
2.5. Data analysis
Data were acquired and analyzed using pClamp 10.2 (Molecular Devices) and Origin 8.1 (OriginLab Corp., Northampton, MA). The Boltzmann equation [y = (A1 − A2)/(1 + e(x−x0)/dx) + A2] was used to fit conductance–voltage data. Conductances of net pyrethroid sodium tail currents, normalized to the peak current measured prior to insecticide perfusion in the same oocyte using the same pulse protocol, were employed as indices of relative sodium channel modification. Statistical analyses were performed in Prism 5.0 (GraphPad Software, La Jolla, CA). Comparisons of two or more mean values to a common control data set employed one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test for statistical significance. Comparisons of two means employed Student’s t-test.
3. Results
3.1. Modified kinetics and gating of Nav1.6Q3 sodium channels
We modified the sequence of the inactivation gate region of the Nav1.6 sodium channel α subunit, formed by the intracellular linker between the third and fourth homology domains, by site-directed mutagenesis, replacing the hydrophobic isoleucine–phenylalanine–methionine (IFM) motif at amino acid sequence positions 1477–1479 with three positively charged glutamine (QQQ) residues (Fig. 1A). We expressed either the native Nav1.6α subunit or the mutated α subunit (designated Nav1.6Q3) in Xenopus oocytes in combination with the conspecific β1 and β2 auxiliary subunits and recorded sodium currents under voltage-clamp conditions in response to step depolarizations from a hyperpolarized membrane potential. Nav1.6 channels (Fig. 1B) activated and inactivated rapidly within the first 10 ms of a depolarizing pulse and gave no detectable persistent current during depolarization or tail current following repolarization. By contrast Nav1.6Q3 channels (Fig. 1C) activated more slowly than Nav1.6 channels, giving a steady-state current by 15–20 ms after depolarization and remaining persistently open during prolonged depolarizations. Following repolarization to the holding potential, Nav1.6Q3 channels also exhibited a pronounced tail current that decayed completely within the first two to three ms following repolarization. Fits of these tail currents to a single exponential decay function yielded a first-order time constant (τtail) of 1.4 ± 0.3 ms (mean ± SE, n = 9).
Fig. 1.
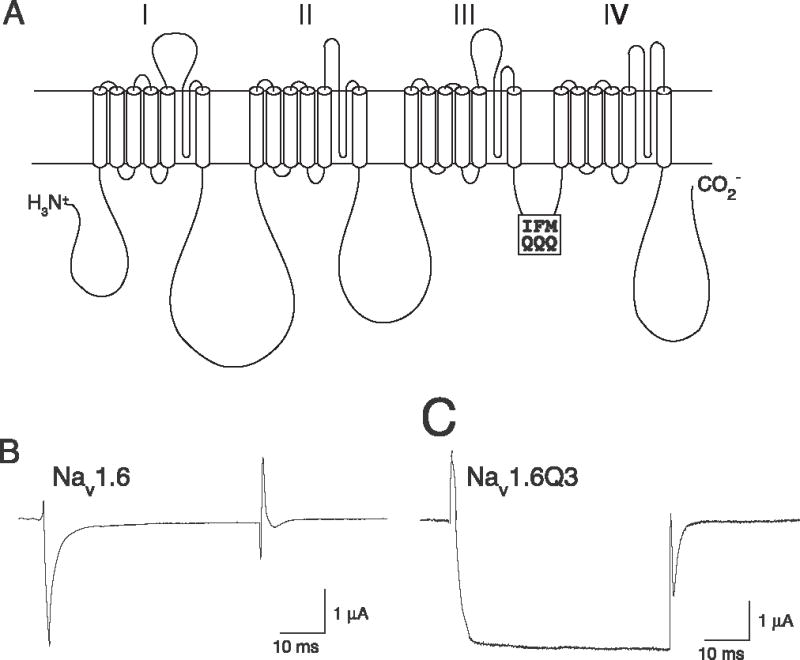
(A) Diagram of the extended transmembrane structure of a voltage-gated sodium channel α subunit protein showing the location of the IFM/QQQ mutation introduced into the Nav1.6Q3 channel. (B) Representative sodium current trace recorded from an oocytes expressing Nav1.6 channels during a 40-ms step depolarization from −100 mV to −10 mV. (C) Representative sodium current trace recorded from an oocytes expressing Nav1.6Q3 channels during a 40-ms step depolarization from −100 mV to −10 mV.
Fig. 2 compares the voltage dependence of activation of Nav1.6 and Nav1.6Q3 channels. Fits of the plots of sodium conductance as a function of test potential from individual experiments with different oocytes to the Boltzmann equation gave a midpoint potential for activation (V0.5) of −22.9 ± 0.2 mV and a slope factor (K) of 6.55 ± 0.14 for Nav1.6 channels (n = 10). Parallel experiments with Nav1.6Q3 channels gave a V0.5 value of −21.0 ± 1.1 mV and a slope factor of 5.66 ± 0.30 (n = 14). The slope factor for the activation of Nav1.6Q3 channels differed significantly from that for Nav1.6 channels (unpaired t-test; P < 0.05).
Fig. 2.
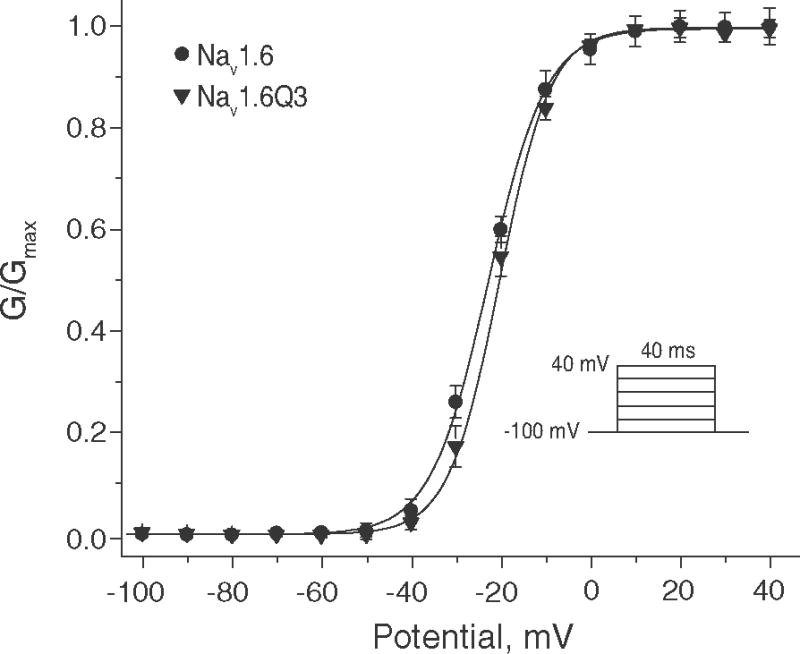
Voltage dependence of activation of Nav1.6 and Nav1.6Q3 sodium channels expressed in Xenopus oocytes. Normalized conductance (G/Gmax) was derived from the current-voltage relationship obtained using the indicated pulse protocol by dividing peak transient current (INa) by the driving force (V − Vrev) and normalizing to the maximum conductance observed in each cell. Values for G/Gmax were plotted as a function of test potential and curves were drawn by fitting the mean values to the Boltzmann equation. Each data point is the mean of 10 (Nav1.6) or 14 (Nav1.6Q3) determinations with different cells; bars show SE values larger than the data point symbols.
3.2. Effects of pyrethroids on the kinetics and gating of Nav1.6Q3 sodium channels
Fig. 3A illustrates the effects of tefluthrin on sodium currents measured in oocytes expressing Nav1.6Q3 channels, which were representative of the effects of all three pyrethroids examined in this study. Specifically, we found no effect of pyrethroids on the rate of onset, magnitude, or persistence of currents carried by Nav1.6Q3 channels during a depolarizing pulse. The sole evidence for modification of Nav1.6Q3 by pyrethroids was the enhancement and prolongation of the sodium tail current measured upon membrane repolarization. Fig. 3B compares representative sodium tail currents recorded from control oocytes and oocytes exposed to S-bioallethrin, tefluthrin or deltamethrin. The tail currents observed in each oocyte following insecticide exposure were composites of the endogenous (control) tail current and the prolonged tail current resulting from pyrethroid modification. As illustrated in Fig. 3B, tail currents caused by the three pyrethroids differed in their decay kinetics. Fits of tail currents induced by S-bioallethrin and tefluthrin to a first-order decay model gave apparent τtail values of 4.0 ± 0.6 and 6.9 ± 0.9 ms, respectively (mean ± SE, n = 9). The τtail values for S-bioallethrin and tefluthrin were significantly greater than the control value (one-way ANOVA with Dunnett’s post hoc test; P < 0.05). By contrast, deltamethrin produced tail currents that were extremely persistent, requiring up to 1 s for complete decay. Fits of deltamethrin-induced tail currents to single or multiple decay models gave highly variable results.
Fig. 3.
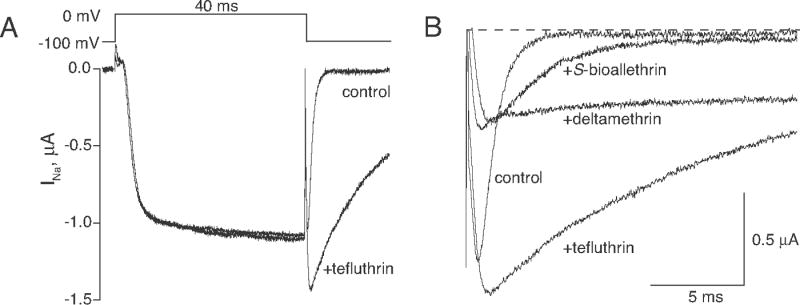
Pyrethroid-modified currents carried by Nav1.6Q3 channels expressed in Xenopus oocytes. (A) Representative sodium currents recorded from a single oocyte before (control) and after equilibration with 100 μM tefluthrin using the indicated pulse protocol. (B) Comparison of representative sodium tail currents recorded from different oocytes either before pyrethroid exposure (control) or after equilibration with S-bioallethrin (100 μM), tefluthrin (100 μM) or deltamethrin (10 μM). Currents were recorded for 20 ms immediately following a 40-ms step depolarization from −100 mV to −10 mV. The dashed line indicates zero current.
Fig. 4 illustrates the effects of S-bioallethrin, tefluthrin, and deltamethrin on the voltage dependence of activation of Nav1.6Q3 channels and Table 1 summarizes the statistical analyses of these data. Deltamethrin and S-bioallethrin appeared to shift the threshold for channel activation slightly in the direction of hyperpolarization, but V0.5 and K values measured in the presence of each of the three pyrethroids did not differ significantly from controls.
Fig. 4.
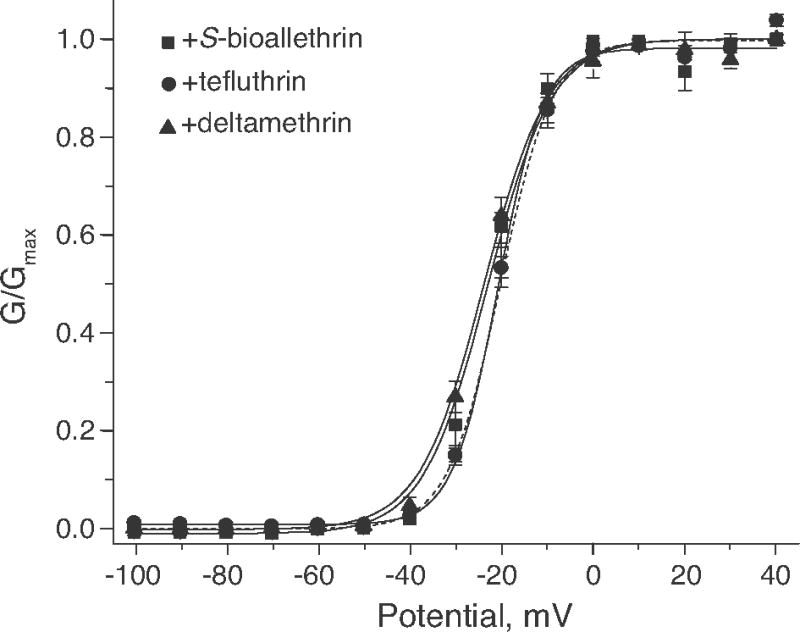
Effects of S-bioallethrin (100 μM), tefluthrin (100 μM), and deltamethrin (10 μM) on the voltage dependence of activation of Nav1.6Q3 sodium channels expressed in Xenopus oocytes. Normalized conductance (G/Gmax) values were plotted as a function of test potential and curves were drawn by fitting the mean values to the Boltzmann equation. Each data point is the mean of 5 (S-bioallethrin), 9 (tefluthrin) or 6 (deltamethrin) determinations with different cells; bars show SE values larger than the data point symbols. The dashed line shows the curve (from Fig. 2) obtained by fitting mean control values to the Boltzmann equation.
Table 1.
Effects of S-bioallethrin, tefluthrin, and deltamethrin on the voltage dependence of activation of rat a Nav1.6Q3 sodium channels expressed in Xenopus oocytes.a
| Condition | V0.5 | K | n |
|---|---|---|---|
| Control | −21.0 ± 1.1 | 5.66 ± 0.30 | 14 |
| +S-bioallethrin (100 μM) | −23.0 ± 0.4 | 5.16 ± 0.27 | 5 |
| +Tefluthrin (100 μM) | −21.4 ± 1.0 | 5.39 ± 0.26 | 9 |
| +Deltamethrin (10 μM) | −23.8 ± 0.4 | 6.54 ± 1.11 | 6 |
Values calculated from fits of the data from the indicated number of individual experiments to the Boltzmann equation; V0.5, midpoint potential (mV) for voltage-dependent activation; K, slope factor.
3.3. Effect of depolarization time on the extent of modification of Nav1.6Q3 channels by pyrethroids
We equilibrated oocytes with S-bioallethrin, tefluthrin or deltamethrin at a hyperpolarized membrane potential and then assessing the effect of depolarizations of varied length on the amplitudes of the sodium tail current observed following repolarization. Fig. 5 shows superimposed current traces obtained from an oocyte before and after equilibration with tefluthrin (100 μM) that were recorded during and after a series of depolarizations ranging from 5 ms to 25 ms. In the absence of insecticide both the maximum steady-state current during depolarization and the tail current following repolarization increased with depolarization time for depolarizations up to 20 ms and then remained constant for up to 150 ms (data for long depolarizations not shown). By contrast, composite tail currents measured following tefluthrin exposure increased in amplitude relative to the peak current during depolarization as a function of depolarization time. The traces shown in Fig. 5 also illustrate the slower decay of tefluthrin-induced tail currents compared to control tail currents at all depolarization times.
Fig. 5.
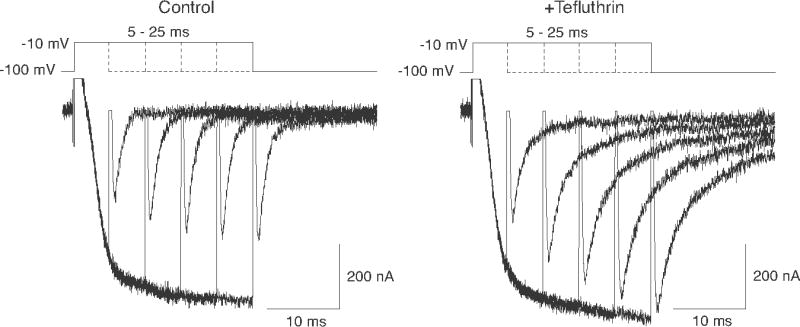
Effect of increasing depolarization time on the amplitude of control (left) and tefluthrin-induced (right) sodium tail currents. Representative traces were recorded from a single oocyte expressing Nav1.6Q3 channels either before or after equilibration with tefluthrin (100 μM) during and after depolarizations of 5–25 ms in 5-ms increments from a holding potential of −100 mV to a test potential of −10 mV.
Fig. 6 summarizes data from multiple experiments such as that shown in Fig. 5 for depolarizations of 10–150 ms. Conductances of control tail currents, normalized to the maximal steady-state sodium current conductance obtained during the immediately preceding depolarization, did not vary over the range of depolarizations examined. By contrast, normalized conductances of tefluthrin-induced composite tail currents increased progressively with the duration of depolarization up to ~100 ms.
Fig. 6.
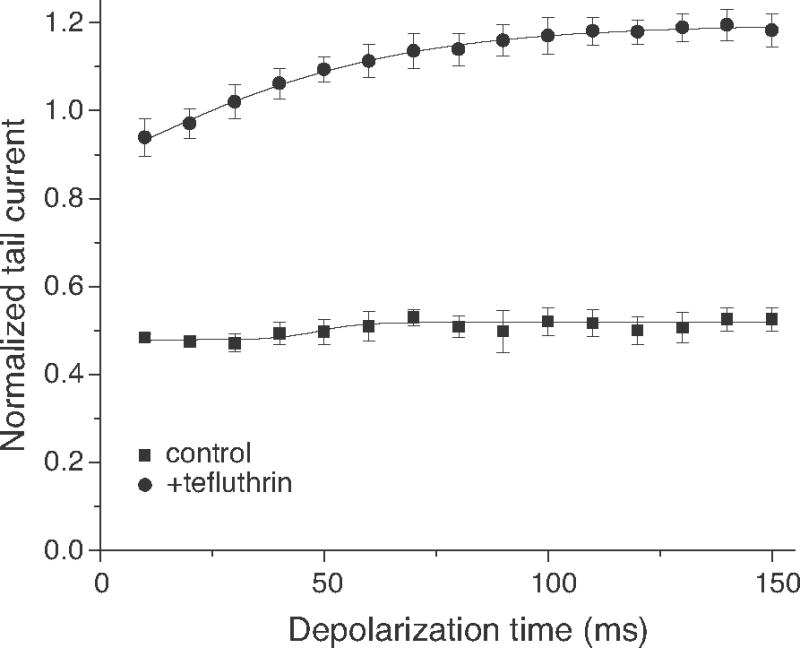
Effect of increasing depolarization time on sodium tail currents recorded from oocytes expressing Nav1.6Q3 sodium channels before (control) or after equilibration with tefluthrin (100 μM). Each data point is the mean of 6 determinations with different cells; bars show SE values larger than the data point symbols.
We calculated the net conductance of each pyrethroid-induced tail current after subtraction of the corresponding control tail current, measured in the same oocyte prior to pyrethroid exposure, and normalized those values to the maximal sodium conductance measured during the immediately preceding depolarization. Fig. 7A summarizes net pyrethroid-specific tail currents measured in oocytes following exposure to tefluthrin (100 μM), S-bioalle-thrin (100 μM) or deltamethrin (10 μM). Consistent with the results of shown in Fig. 6, net tefluthrin-induced tail currents were clearly detectable following short (10 ms) depolarizations and increased progressively with depolarization time up to 100 ms (Fig. 7A). At 150 ms, the normalized conductance of the tefluthrin-specific component of tail currents were approximately 2.6-fold greater than those measured at 10 ms. Net deltamethrin-induced tail currents were smaller than those measured for tefluthrin following a 10-ms depolarization but also increased progressively with depolarization times up to 100 ms. At 150 ms, the normalized conductance of the deltamethrin-induced tail currents were approximately 3.5-fold greater than those measured at 10 ms. By contrast, depolarizations of up to 150 ms did not increase the amplitude of the net tail currents induced by S-bioallethrin (Fig. 7A).
Fig. 7.
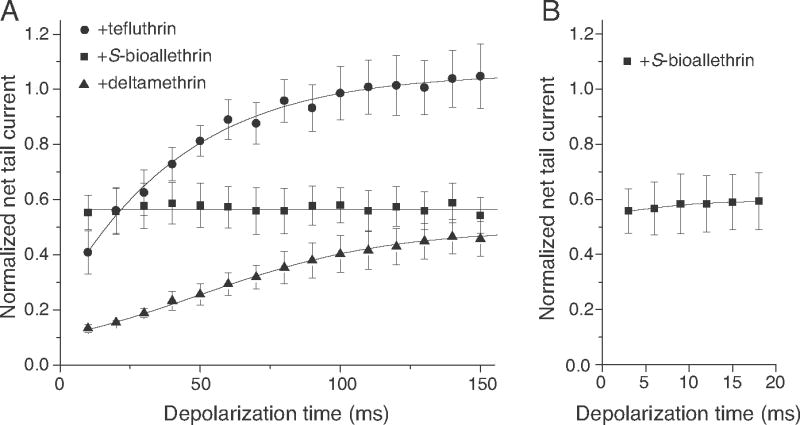
Effect of increasing depolarization time on the pyrethroid-specific component of sodium tail currents recorded from oocytes expressing Nav1.6Q3 sodium channels. (A) Normalized net tail currents measured following equilibration with S-bioallethrin (100 μM), tefluthrin (100 μM) or deltamethrin (10 μM) and step depolarizations of 10–150 ms in 10-ms increments. (B) Normalized net tail currents measured following equilibration with S-bioallethrin (100 μM) and step depolarizations of 3–18 ms in 3-ms increments. Values are means of 6 determinations with different cells and are corrected for control tail currents measured prior to pyrethroid exposure in the same oocyte; bars show SE values larger than the data point symbols.
To determine whether depolarization time-dependent modification by S-bioallethrin had already reached a steady-state level by 10 ms, the shortest depolarization time used in the experiments shown in Fig. 7A, we assessed the impact of 100 μM S-bioallethrin on the extent of channel modification following depolarization at 3-ms increments. As shown in Fig. 7B, the conductances of S-bioallethrin-induced tail currents also did not vary with depolarization time following very short depolarizations.
4. Discussion
Fast inactivation of voltage-gated sodium channels terminates the transient sodium current associated with membrane depolarization. Fast inactivation involves a ‘hinged lid’ mechanism, in which the conserved intracellular inactivation gate domain between homology domains III and IV occludes the inner pore of the channel (Goldin, 2003). A hydrophobic isoleucine–phenyl-alanine–methionine (IFM) motif in this domain is essential for the operation of the inactivation gate. Replacement of these hydrophobic residues with glutamine (Q) permits the selective decoupling of sodium channel activation and fast inactivation, producing channels that remain open during prolonged depolarizations (West et al., 1992). We inserted the IFM/QQQ mutation in the rat Nav1.6 sequence to create inactivation-deficient rat Nav1.6Q3 sodium channels and employed these channels to explore directly the interaction of pyrethroid insecticides with sodium channels in the open state.
We expressed rat Nav1.6Q3 channels in combination with the rat β1 and β2 auxiliary subunits in Xenopus oocytes to facilitate the direct comparisons to our previous studies of Nav1.6 + β1 + β2 channels (Tan and Soderlund, 2010, 2011a,b). The kinetics and voltage-dependent activation gating of rat Nav1.6Q3 channels in the present study were consistent with the properties of rat Nav1.2Q3, rat Nav1.4Q3, and mouse Nav1.6Q3 sodium channels described in previous studies (Featherstone et al., 1996; West et al., 1992; Zhou and Goldin, 2004). Depolarizing pulses from hyperpolarized holding potentials activated sodium currents that rapidly reached a steady state, which persisted until the membrane was returned to a hyperpolarized potential. In addition to prolonged sodium currents during depolarization, oocytes expressing Nav1.6Q3 channels exhibited a prominent, rapidly decaying sodium tail current upon repolarization of the membrane. This tail current represents open-channel deactivation (i.e., closing of the sodium channel activation gate), a process that is normally obscured with normal channels by the onset of fast inactivation during a depolarizing pulse (Featherstone et al., 1996; Groome et al., 2002).
S-bioallethrin, tefluthrin and deltamethrin enhanced and prolonged the sodium tail currents carried by Nav1.6Q3 channels but had no effect on the kinetic properties of the persistent currents measured during a depolarizing pulse. The decay kinetics of the composite (control plus pyrethroid-induced) tail currents caused by S-bioallethrin and tefluthrin in the present study agreed well with the decay constant of tail currents measured for tefluthrin in assays with Nav1.6 channels (Tan and Soderlund, 2010). However, deltamethrin-induced tail currents measured in assays with Nav1.6Q3 channels was much more persistent than that measured with Nav1.6 channels (Tan and Soderlund, 2010).
We varied the length of depolarizing pulses applied to oocytes expressing Nav1.6Q3 channels to investigate the duration of channel opening as a determinant of the extent of pyrethroid modification. The time-dependent increase in the pyrethroid-specific component of sodium tail currents caused by tefluthrin and deltamethrin provided direct evidence that these compounds bind with higher affinity to the open state of the channel than to the resting state. These results confirm that open-channel modification causes the use-dependent effects of these compounds on Nav1.6 channels (Tan and Soderlund, 2010). Moreover, values for the factor of tail current increase caused by long depolarizations in assays with tefluthrin and deltamethrin (2.6-fold and 3.5-fold, respectively) corresponded closely to the factors of use-dependent enhancement of modification by these compounds in assays with native Nav1.6 channels (2.8-fold and 3.7-fold, respectively) (Tan and Soderlund, 2010). In contrast to the results obtained with tefluthrin and deltamethrin, net S-bioallethrin-induced tail currents measured in assays with Nav1.6Q3 channels did not vary with depolarization time. This result confirms the lack of enhanced binding of S-bioallethrin to open channels that we inferred from the lack of use-dependent modification of Nav1.6 channels by this compound (Tan and Soderlund, 2010).
The three pyrethroids in this study also differed in the extent of channel modification obtained at the shortest depolarization times examined. Although the extent of modification of Nav1.6Q3 channels by tefluthrin and deltamethrin agree generally with the relative resting modification of Nav1.6 channels by the same compounds (Tan and Soderlund, 2010), it is likely that even at short (5–10 ms) depolarizations the modification of Nav1.6Q3 channels represents a mixture of binding of tefluthrin and deltamethrin to closed and open channels. By contrast, S-bioallethrin produced substantially greater modification of Nav1.6Q3 channels following short depolarizations than we found previously for Nav1.6 channels in the resting state (Tan and Soderlund, 2010). Our data showing the lack of effect of very short depolarization times on the extent of Nav1.6Q channel modification by S-bioallethrin tend to rule out rapid open-channel modification as an explanation for the greater sensitivity of Nav1.6Q3 channels to this compound. We therefore hypothesize that the IFM/QQQ mutation somehow alters the access of S-bioallethrin to the pyrethroid receptor or its binding at that site when channels are in the resting state.
Taken together, results of the present study and previous studies provide strong evidence that the pyrethroid receptor on voltage-gated sodium channels occupies different conformations on closed and open channels that exhibit unique structure–activity relationships for pyrethroid binding. It would therefore be of interest to expand current efforts to model the pyrethroid receptor (Du et al., 2009; O’Reilly et al., 2006; Usherwood et al., 2007) to include models of the receptor on closed channels. Docking experiments with pyrethroids that interact preferentially with either closed or open channels would be valuable to identify possible structural determinants of state-dependent binding.
The conclusions drawn here regarding the relative importance of closed- and open-channel modification by pyrethroids are derived solely from studies of channels expressed in Xenopus oocytes. Recently, we reported that use-dependent modification of rat Nav1.6 + β1 + β2 channels expressed in HEK293 cells by tefluthrin and deltamethrin is apparently less significant than in oocytes (He and Soderlund, 2011). In the HEK293 cell environment tefluthrin caused only resting modification whereas deltamethrin caused mixed resting and use-dependent modification. Extension of studies of inactivation-deficient channels such as Nav1.6Q3 to include other cellular expression environments, such as HEK293 cells, are needed to identify the role that cellular context may play in modulating the differential effects of pyrethroids on sodium channels in the resting and open state.
Acknowledgments
This work was supported in part by a grant (R01-ES013686) from the National Institute of Environmental Health Sciences, National Institutes of Health. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Environmental Health Sciences. We thank P. Adams and S. Kopatz for technical assistance, and we thank R. Araújo and R. von Stein for critical reviews of the manuscript.
Footnotes
Conflict of interest statement
The authors declare that they have no conflicts of interest with regard to sources of funding for this research or the design and interpretation of the experiments described herein.
References
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. J Gen Physiol. 1977;70:549–66. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomquist JR. Neuroreceptor mechanisms in pyrethroid mode of action and resistance. In: Roe M, Kuhr RJ, editors. Reviews in pesticide toxicology. Raleigh, NC: Toxicology Communications; 1993. pp. 181–226. [Google Scholar]
- Choi J-S, Soderlund DM. Structure–activity relationships for the action of 11 pyrethroid insecticides on rat Nav1.8 sodium channels expressed in Xenopus oocytes. Toxicol Appl Pharmacol. 2006;211:233–44. doi: 10.1016/j.taap.2005.06.022. [DOI] [PubMed] [Google Scholar]
- de Weille JR, Vijverberg HPM, Narahashi T. Interactions of pyrethroids and octylguanidine with sodium channels of squid giant axons. Brain Res. 1988;445:1–11. doi: 10.1016/0006-8993(88)91067-0. [DOI] [PubMed] [Google Scholar]
- Dietrich PS, McGivern JG, Delgado SG, Koch BD, Eglen RM, Hunter JC, et al. Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J Neurochem. 1998;70:2262–72. doi: 10.1046/j.1471-4159.1998.70062262.x. [DOI] [PubMed] [Google Scholar]
- Du Y, Lee J-E, Nomura Y, Zhang T, Zhorov B, Dong K. Identification of a cluster of residues in transmembrane 6 of domain III of the cockroach sodium channel essential for the action of pyrethroid insecticides. Biochem J. 2009;419:377–85. doi: 10.1042/BJ20082082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996;71:3098–109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Maintenance of Xenopus laevis and oocyte injection. Meth Enzymol. 1992;207:266–97. doi: 10.1016/0076-6879(92)07017-i. [DOI] [PubMed] [Google Scholar]
- Goldin AL. Mechanisms of sodium channel inactivation. Curr Opin Neurobiol. 2003;13:284–90. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Groome J, Fujimoto E, Walter L, Ruben P. Outer and central charged residues in DIVS4 of skeletal muscle sodium channels have differing roles in deactivation. Biophys J. 2002;82:1293–307. doi: 10.1016/S0006-3495(02)75485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Soderlund DM. Differential state-dependent modification of rat Nav1.6 sodium channels expressed in human embryonic kidney (HEK293) cells by the pyrethroid insecticides tefluthrin and deltamethrin. Toxicol Appl Pharmacol. 2011;257:377–87. doi: 10.1016/j.taap.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BFX, Offord J, Charbonneau H, et al. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–42. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, DeJongh KS, Westenbroek RE, Reber BFX, Scheuer T, et al. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–42. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- Kadala A, Charreton M, Jakob I, Le Conte Y, Collet C. A use-dependent sodium current modification induced by type I pyrethroid insectides in honeybee antennal olfactory neurons. Neurotoxicology. 2011;32:320–30. doi: 10.1016/j.neuro.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Lombet A, Mourre C, Lazdunski M. Interactions of insecticides of the pyrethroid family with specific binding sites on the voltage-dependent sodium channel from mammalian brain. Brain Res. 1988;459:44–53. doi: 10.1016/0006-8993(88)90284-3. [DOI] [PubMed] [Google Scholar]
- Narahashi T. Neuronal ion channels as the target sites of insecticides. Pharmacol Toxicol. 1996;78:1–14. doi: 10.1111/j.1600-0773.1996.tb00234.x. [DOI] [PubMed] [Google Scholar]
- O’Reilly AO, Khambay BPS, Williamson MS, Field LM, Wallace BA, Davies TGE. Modelling insecticide binding sites at the voltage-gated sodium channel. Biochem J. 2006;396:255–63. doi: 10.1042/BJ20051925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado VL, Narahashi T. Immobilization of sodium channel gating charge in crayfish giant axons by the insecticide fenvalerate. Mol Pharmacol. 1993;43:626–34. [PubMed] [Google Scholar]
- Smith TJ, Soderlund DM. Potent actions of the pyrethroid insecticides cismethrin and cypermethrin on rat tetrodotoxin-resistant peripheral nerve (SNS/PN3) sodium channels expressed in Xenopus oocytes. Pestic Biochem Physiol. 2001;70:52–61. [Google Scholar]
- Soderlund DM. Mode of action of pyrethrins and pyrethroids. In: Casida JE, Quistad GB, editors. Pyrethrum flowers: production, chemistry, toxicology, and uses. New York: Oxford University Press; 1995. pp. 217–33. [Google Scholar]
- Soderlund DM. State-dependent modification of voltage-gated sodium channels by pyrethroids. Pestic Biochem Physiol. 2010;97:78–86. doi: 10.1016/j.pestbp.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund DM. Molecular mechanisms of pyrethroid insecticide neurotoxicity: recent advances. Arch Toxicol. 2012;86:165–81. doi: 10.1007/s00204-011-0726-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund DM, Clark JM, Sheets LP, Mullin LS, Piccirillo VJ, Sargent D, et al. Mechanisms of pyrethroid toxicity: implications for cumulative risk assessment. Toxicology. 2002;171:3–59. doi: 10.1016/s0300-483x(01)00569-8. [DOI] [PubMed] [Google Scholar]
- Tabarean IV, Narahashi T. Kinetics of modulation of tetrodotoxin-sensitive and tetro-dotoxin-resistant sodium channels by tetramethrin and deltamethrin. J Pharmacol Exp Ther. 2001;299:988–97. [PubMed] [Google Scholar]
- Tan J, Soderlund DM. Human and rat Nav1.3 voltage-gated sodium channels differ in inactivation properties and sensitivity to the pyrethroid insecticide tefluthrin. Neurotoxicology. 2009;30:81–9. doi: 10.1016/j.neuro.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Soderlund DM. Divergent actions of the pyrethroid insecticides S-bioallethrin, tefluthrin, and deltamethrin on rat Nav1.6 sodium channels. Toxicol Appl Pharmacol. 2010;247:229–37. doi: 10.1016/j.taap.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Soderlund DM. Coexpression with auxiliary β subunits mdoulates the action of tefluthrin on rat Nav1.6 and Nav1.3 sodium channels. Pestic Biochem Physiol. 2011a;101:256–64. doi: 10.1016/j.pestbp.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Soderlund DM. Independent and joint modulation of rat Nav1.6 voltage-gated sodium channels by coexpression with the auxiliary β1 and β2 subunits. Biochem Biophys Res Commun. 2011b;407:788–92. doi: 10.1016/j.bbrc.2011.03.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebayashi H, Narahashi T. Differential mechanism of action of the pyrethroid tetramethrin on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J Pharmacol Exp Ther. 1994;270:595–603. [PubMed] [Google Scholar]
- Usherwood PNR, Davies TGE, Mellor IR, O’Reilly AO, Peng F, Vais H, et al. Mutations in DIIS5 and the DIIS4-5 linker of Drosophila melanogaster sodium channel define binding domains for pyrethroids and DDT. FEBS Lett. 2007;581:5485–92. doi: 10.1016/j.febslet.2007.10.057. [DOI] [PubMed] [Google Scholar]
- Vais H, Williamson MS, Goodson SJ, Devonshire AL, Warmke JW, Usherwood PNR, et al. Activation of Drosophila sodium channels promotes modification by deltamethrin: reductions in affinity caused by knock-down resistance mutations. J Gen Physiol. 2000;115:305–18. doi: 10.1085/jgp.115.3.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–4. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Goldin AL. Use-dependent potentiation of the Nav1.6 sodium channel. Biophys J. 2004;87:3862–72. doi: 10.1529/biophysj.104.045963. [DOI] [PMC free article] [PubMed] [Google Scholar]