

Abstract
Background
Wolbachia pipientis, a diverse group of α-proteobacteria, can alter arthropod host reproduction and confer a reproductive advantage to Wolbachia-infected females (cytoplasmic incompatibility (CI)). This advantage can alter host population genetics because Wolbachia-infected females produce more offspring with their own mitochondrial DNA (mtDNA) haplotypes than uninfected females. Thus, these host haplotypes become common or fixed (selective sweep). Although simulations suggest that for a CI-mediated sweep to occur, there must be a transient phase with repeated initial infections of multiple individual hosts by different Wolbachia strains, this has not been observed empirically. Wolbachia has been found in the tsetse fly, Glossina fuscipes fuscipes, but it is not limited to a single host haplotype, suggesting that CI did not impact its population structure. However, host population genetic differentiation could have been generated if multiple Wolbachia strains interacted in some populations. Here, we investigated Wolbachia genetic variation in G. f. fuscipes populations of known host genetic composition in Uganda. We tested for the presence of multiple Wolbachia strains using Multi-Locus Sequence Typing (MLST) and for an association between geographic region and host mtDNA haplotype using Wolbachia DNA sequence from a variable locus, groEL (heat shock protein 60).
Results
MLST demonstrated that some G. f. fuscipes carry Wolbachia strains from two lineages. GroEL revealed high levels of sequence diversity within and between individuals (Haplotype diversity = 0.945). We found Wolbachia associated with 26 host mtDNA haplotypes, an unprecedented result. We observed a geographical association of one Wolbachia lineage with southern host mtDNA haplotypes, but it was non-significant (p = 0.16). Though most Wolbachia-infected host haplotypes were those found in the contact region between host mtDNA groups, this association was non-significant (p = 0.17).
Conclusions
High Wolbachia sequence diversity and the association of Wolbachia with multiple host haplotypes suggest that different Wolbachia strains infected G. f. fuscipes multiple times independently. We suggest that these observations reflect a transient phase in Wolbachia evolution that is influenced by the long gestation and low reproductive output of tsetse. Although G. f. fuscipes is superinfected with Wolbachia, our data does not support that bidirectional CI has influenced host genetic diversity in Uganda.
Keywords: Wolbachia, Population structure, Sequence diversity, groEL, MLST
Background
Wolbachia pipientis is a diverse group of α-proteobacteria found to infect many terrestrial arthropods and filarial nematodes [1], with new hosts being discovered constantly [2,3]. Wolbachia is currently divided into eight monophyletic “supergroup” lineages (A-H) [4], based on Multi-Locus Sequence Typing (MLST) [5].
Although this bacterium may positively influence host physiology [6,7], Wolbachia is best known for parasitism that alters host reproductive success, including cytoplasmic incompatibility (CI)) [8]. CI is the most studied reproductive modification induced by Wolbachia and results in embryonic lethality when uninfected females are crossed with Wolbachia-infected males. In a population composed of infected and uninfected individuals, only infected females can mate successfully with infected and uninfected males [8]. When two Wolbachia strains exist in a population, bidirectional CI can result in incompatibility between individuals carrying different strains, whereas individual females infected with multiple strains (superinfected) can mate with all males and produce infected progeny [9]. In both CI types, Wolbachia is expected to sweep through populations due to higher reproductive fitness because of the higher proportion of successful matings between infected or superinfected females relative to the uninfected ones. However, not all Wolbachia strains cause CI and strength of CI expression (penetrance) can be altered by Wolbachia density or transmission efficiency (maternal transmission fidelity) [10].
Given the influence of Wolbachia on host fitness, the potential impact of Wolbachia on host population genetic variability and geographical patterns is substantial. Since Wolbachia is maternally transmitted, other maternally transmitted organelles (e.g., mitochondria) hitchhike with Wolbachia infections [11,12]. Even though simulations indicate that CI-based spread of Wolbachia sweeps are more likely to involve repeated initial infections via horizontal transmission [13,14], most studies of CI-associated Wolbachia sweeps find it associated with low mitochondrial DNA (mtDNA) variation and with many hosts infected [15,16]. Theoretical models suggest that host dispersal or migration, and genetic background [17,18] can influence these sweeps [9,19-21]. Factors that control Wolbachia density, such as nutrient availability or temperature [22,23], indirectly influence CI-based sweeps, because at high Wolbachia densities maternal transmission fidelity and CI expression are stronger than those at low Wolbachia densities.
The evolutionary dynamics of Wolbachia spread are important for strategies aimed at vector control. In tsetse (Glossina spp.), the sole vectors of trypanosomes that cause Human African Trypanosomiasis (HAT), Wolbachia infections are common [24-26] and can cause CI [27]. In Uganda, the primary HAT vector is G. fuscipes fuscipes. Nuclear microsatellites and mtDNA data in this species identified discrete genetic populations and unexpected genetic breaks with complex patterns of gene flow in relatively continuous landscapes [28,29]. In Ugandan G. f. fuscipes, Wolbachia prevalence varies in most sites (6.7%-100%) with low within-individual Wolbachia density [26]. Contrary to expectations, Wolbachia prevalence was correlated to host groups defined by nuclear microsatellite data rather than mtDNA, suggesting that its spread may depend largely on host gene flow and dispersal, and not a selective sweep [26]. A possible explanation for the observed discrepancy between Wolbachia and host mtDNA is that there were multiple undetected Wolbachia strains present in each host individual, which would go undetected when only testing for infection presence.
To understand Wolbachia evolutionary dynamics and their relationship with host mtDNA, we used G. f. fuscipes from Ugandan populations of known genetic background and Wolbachia infection status [28-30]. We tested for the presence of multiple Wolbachia infections (superinfections) in single flies using four MLST loci. To test the relationship between Wolbachia infections and genetic variation within and among host individuals and to examine its evolutionary origin, we used sequence data from a known variable locus, groEL (heat shock protein 60) [31]. We tested the hypothesis that Wolbachia-induced CI has shaped geographic patterns of host genetic variation and discuss how the unique life history traits of tsetse may generate unique patterns of genetic diversity in Wolbachia.
Methods
Sampling, locus selection and laboratory methods
Samples were selected from Wolbachia-positive flies with known host mtDNA and microsatellite genotypes (Figure 1, Table 1, See Additional file 1: Table S1) [26]. Details on the host genetic make-up can be found in Additional file 1. DNA was extracted from whole bodies or from ovarian tissue using Qiagen DNEasy extraction kits (Qiagen, Inc.). For the MLST examination of superinfection, four individuals (BK08033, BV10, JN6, JN8) were used from three sampling localities (BK, BV, JN, Figure 1). Between two and eight Wolbachia-infected G. f. fuscipes individuals from each of 10 Ugandan sites were selected for groEL for a total of 47 flies. All individuals except BK08033 were sequenced for both MLST and groEL. Due to low-density Wolbachia infections [26], two consecutive PCRs were performed for all target genes (See Additional file 1 for details). PCR products were cleaned and prepared for cloning using the QiaQuick Gel extraction protocol (Qiagen, Inc). For MLST, eight clones were sequenced for each individual and locus. For groEL, between two and 20 clones were used for sequencing for each individual fly.
Figure 1.

Geographic distribution and collection sites for G. f. fuscipes . Distribution, collection site and genetic group assignment based on host mitochondrial (mtDNA) and nuclear (microsatellites) data. Light gray shading illustrates the approximate geographic distribution of tsetse in Uganda. Dark gray shading indicates major water bodies. Sampling localities are illustrated as site abbreviations (See Table 1 for collection details). Circles next to sites show assignments to host mtDNA and microsatellite genetic groups as indicated by symbols in the map key. Inset map on the top left corner shows Uganda location within Africa.
Table 1.
Summary of G. f. fuscipes collection sites, number of individuals used for Wolbachia sequencing, infection prevalence, and sequence diversity
| Collection Site | Site Code | Latitude/ Longitude | No. of Host Individuals | Infection Prevalence (%)1 | Average No. of Nucleotide Differences | No. of Haplotypes | Hd |
|---|---|---|---|---|---|---|---|
| Pallisa |
BK |
1.02/33.88 |
2 |
53.6 (N = 69) |
4.5 |
6 |
1.000 |
| Bunghazi |
BN |
0.93/33.98 |
5 |
6.7 (N = 15) |
4.9 |
10 |
0.970 |
| Busime |
BU |
0.25/33.97 |
3 |
29.4 (N = 34) |
23.4 |
5 |
1.000 |
| Buvuma |
BV |
0.31/33.30 |
5 |
60 (N = 35) |
19.1 |
8 |
0.927 |
| Junda |
JN |
1.33/32.74 |
8 |
65.1 (N = 43) |
11.2 |
32 |
0.995 |
| Kakoga |
KK |
0.37/30.28 |
3 |
35.7 (N = 28) |
4.8 |
8 |
0.972 |
| Masindi |
MS |
1.63/31.69 |
6 |
6.3 (N = 16) |
6.0 |
14 |
1.000 |
| Murchison Falls |
MF |
2.28/31.56 |
3 |
90 (N=) |
7.4 |
19 |
0.983 |
| Osuguro |
OS |
1.53/33.50 |
2 |
20 (N = 20) |
5.3 |
3 |
1.000 |
| Lukaya River |
DRC |
−4.48/15.31 |
5 |
N/A |
N/A |
N/A |
N/A |
| Gfq Colony2 | GF | N/A | 5 | N/A | N/A | N/A | N/A |
1Infection prevalence data from [26].
2Collection site origin is unknown for the colony maintained at the Slovakia Academy of Science, Bratislava, Slovakia. Tissues were kindly provided by Peter Takac.
3Only the complete dataset was used to calculate diversity statistics in this table because the conservative dataset eliminated individuals.
Measures of Wolbachia groEL nucleotide differences and genetic diversity were calculated using the complete dataset3. Hd refers to groEL haplotype diversity within a site.
Artifact and recombinant removal for groEL dataset
Artificial inflation of variation in DNA sequences can arise as PCR artifact in sequence data due to infidelity and error introduced by Taq DNA polymerase. To minimize the effects of PCR artifact in the groEL dataset (no variation was detected among MLST clones), each consecutive PCR amplification served as a “reconditioning PCR” [32], using a small aliquot from an initial PCR. For three individuals from a single geographic locality (JN), we amplified the same template from two independent PCR to verify strain haplotypes [32]. After sequencing, potential PCR artifacts were identified and removed using a statistical approach [33] that incorporated the GoTaq Error rate (1–7 × 10–4 per base pair per cycle (Promega)). We then retained all “common” sequences that were found in multiple copies within or among individuals, as their co-occurrence suggest that they are natural, rather than artificial polymorphisms (See Additional file 1 for details).
Given the high sequence diversity in the groEL dataset, we used GENECONV [34], a statistical software package that detects potential recombinants by measuring whether a large proportion of DNA sequence in a pairwise comparison is more similar than expected by chance. Since we were not directly interested in recombinants, we removed any sequence from the dataset that could be a recombinant (See Additional file 1 for details). Within an individual, some recombinants may be naturally occurring rather than an artificial product arising from laboratory methods (e.g., PCR and cloning). However, there is no way to discriminate between natural and artificial recombinants, so we chose a conservative approach and excluded them, as this removal does not impact the testing of our hypotheses. As a result, our diversity measures may be underestimates, since some recombination is likely to occur between strains within a single host fly.
Excluding all groEL sequences that did not occur more than once eliminated entire individual flies from our dataset and lowered the power to detect patterns of geographic variation. Complete removal of singleton sequences should not imply that Wolbachia is absent from that individual, but that its groEL sequence is not precisely known. As such we used two groEL datasets, a “complete” dataset (all sequences that could not be considered artifacts by the above methods, even if they appeared only once in the dataset), and a “conservative” dataset (only sequences found two or more times in one or more individuals). We used the complete dataset to infer the groEL phylogeny, identify groEL haplogroups (groups of related haplotypes), and examine the possibility of bidirectional CI. We used the conservative dataset to examine host population genetic expectations for bidirectional CI, build a haplotype network and estimate within-individual Wolbachia genetic diversity.
Phylogenetic analysis and sequence diversity
For each individual fly, all clones (N = 8) from each MLST gene were identical (See Additional file 1: Table S2). Thus, we used one sequence per individual and a concatenated dataset of the four MLST genes (1635 bp) to identify Wolbachia superinfections. Glossina fuscipes fuscipes Wolbachia samples were aligned to those from [25] and from the Wolbachia MLST database [35] using MEGA 5.0 [36]. Phylogenetic analysis was performed using Bayesian inference and maximum likelihood. For groEL, Wolbachia sequences from G. f. fuscipes were aligned to ones from other insects [37] (See Additional file 1: Table S3) in MacClade 4.08 [38,39]. jModeltest [39,40] was used to select the model of DNA sequence evolution. Phylogenetic trees were generated by maximum likelihood, as implemented in Garli 0.96b8 [41]. Branch support was generated using 1000 bootstrap replicates. Tree topologies were examined to confirm superinfections in G. f. fuscipes. For details of the phylogenetic analysis, see Additional file 1.
Diversity statistics were calculated on the complete and the conservative groEL datasets using DNAsp [42]. To understand evolutionary relationships within G. f. fuscipes Wolbachia lineages at different hierarchical levels (individual flies, collection sites, and the entire dataset), we constructed haplotype networks using parsimony (TCS v1.21 [43]). Based on genetic distances within and between the networks generated from the complete dataset, we defined two groEL haplogroups, “Group 1” and “Group 2” that correspond to the lineages identified in the phylogeny and divided Group 1 into subgroups (See Additional file 1: Figure S2).
Association of Wolbachia haplogroups to geography and host background
We used Analysis of Molecular Variance (AMOVA [44]) on the complete and conservative datasets to evaluate the partitioning of groEL genetic diversity within and among host mtDNA haplogroups and to examine whether bidirectional CI could have generated the divergent host haplogroups. Specifically, we used AMOVA to test if the diversity in groEL was differentially distributed with respect to host mtDNA haplogroups. Under bidirectional CI Wolbachia sequences within mtDNA haplogroups should be more closely related than between haplogroups, and variance among host haplotype groups should be large and significant. In addition, we tested whether there was an association of Wolbachia Group 2 with southern host mtDNA haplogroups using randomization tests. Specifically, we randomly assigned the presence of Wolbachia Group 2 to the 47 individual flies used in our dataset to generate 100 random datasets. Then, we compared the observed number of individuals with Wolbachia Group 2 and the southern host mtDNA haplogroup to that in the randomized dataset.
To understand geographic relationships between host haplotypes and Wolbachia infection, we compared host mtDNA haplotypes and Wolbachia infection status. We examined G. f. fuscipes Wolbachia groups relative to host haplotypes and compared these samples to individuals screened for host mtDNA haplotypes [28,29] and for Wolbachia prevalence [26]. We specifically tested whether there was an association between host mtDNA haplotype and Wolbachia infection status by examining whether Wolbachia infected individuals were more often associated with a host mtDNA haplotype found in the contact zone between the north and south host mtDNA haplogroups. For this test, we randomly assigned Wolbachia infection status to the individuals screened [26] for which we had host haplotype (N = 366) to generate 100 random datasets. Then, we examined the observed proportion of flies with host haplotypes from the contact zone relative to the randomized datasets.
Results
Clone variation and final dataset composition
For each MLST locus from a single individual, all eight clones yielded identical sequences. In contrast, many clones yielded different sequences for the groEL dataset. Following the removal of PCR and cloning artifacts, the complete dataset consisted of 47 individuals with a total of 102 groEL haplotypes. The conservative dataset consisted of 37 individual flies and a total of 21 groEL haplotypes. GenBank accession numbers for the 102 haplotypes are KC493415 - KC493553.
Phylogenetic analyses for MLST and groEL
Phylogenetic tree topologies inferred from the MLST and groEL datasets are shown in Figure 2. The MLST dataset identified two phylogenetic Wolbachia lineages and suggests that one belongs to supergroup A, a group with Wolbachia from many insects, including other Glossina species [25]. The second lineage is placed outside of supergroup A, but does not align with any previously described group (Figures 2, See Additional file 1: Figure S1). Our data do not include all five Wolbachia MLST loci [35], and therefore do not provide a complete picture of supergroup assignment.
Figure 2.

Rooted maximum likelihood gene tree for groEL and the tree inferred from four MLST loci (inset). Arrows point to the branch that leads to each supergroup (A-F) and are labeled accordingly for both phylogenies. Shaded clades indicate the position of G. f. fuscipes Wolbachia sequences. Light gray shading shows the position of Group 1 sequences within supergroup A. Dark gray shading shows the position of Group 2 sequences. Individual hosts that belong to each clade are shown in the additional file. (See Additional file 1: Table S1 and Figure S1). The groEL tree includes Wolbachia strains from Glossina fuscipes fuscipes and representatives from most known supergroups. For the groEL tree, the outgroup was selected arbitrarily to reflect relationships in [25]. Numbers above branches are bootstrap support percentages that are above 65%. Within supergroups for groEL, bootstrap support was below 65% and thus not reported. Inset (Upper right): A generalized version of the MLST phylogeny (See Additional file 1: Figure S1). An open circle above the branch indicates a Bayesian posterior probability of 1.0 and a closed circle below the branch is a bootstrap of 90% and above. The shaded boxes correspond to the shading of branches in the groEL phylogeny. Individuals analyzed for both groEL and MLST are indicated with an *.
The groEL phylogeny similarly identified two lineages, suggesting one belongs to supergroup A, but the placement of the second lineage is not supported (Figure 2). As a marker, groEL evolves much more rapidly than MLST markers and may not be as informative for phylogenetic inference. We cannot directly compare the tree topology of the MLST and groEL datasets to address the taxonomic placement of Wolbachia from G. f. fuscipes as our sampling differs between the datasets. However, the MLST data confirms the presence of multiple strains within G. f. fuscipes populations, as sequences were identical within individuals, and the groEL data reveal the presence of multiple infections within individual hosts and populations.
groEL haplotype relationships and genetic diversity
TCS generated two parsimony networks for both the complete and conservative datasets (Figures 3, See Additional file 1: Figure S4) corresponding to the two lineages (Group 1 and Group 2) in the phylogenetic analysis (Figure 2). Individuals within supergroup A are found in a large network (Group 1), while those outside of supergroup A are found in a smaller network (Group 2). Group 1 is subdivided into three subgroups (Subgroups 1a, 1b, 1c) for the complete dataset (For subgroup assignment details, see Additional file 1).
Figure 3.

Parsimony network and geographic locations of different haplogroups for Wolbachia groEL. Relationships among groEL haplotypes, group subdivisions, and geographic distribution for the conservative dataset. A. TCS parsimony network for the groEL haplotypes. Each circle shows one haplotype. Solid black circles (nodes) indicate number of nucleotide differences between haplotypes. Circle size represents haplotype frequency. Each branch shows one nucleotide change. Shading highlights different network subdivisions and corresponds to the haplotypes in rounded rectangles. B. Geographic distribution of groEL haplogroups relative to previously defined genetic groups (Figure 1). Small, filled circles indicate the Wolbachia groEL haplogroups represented at each site with shading corresponding to group assignment as in Figure 3A.
Measures of sequence diversity are shown in Table 2 (See Additional file 1: Table S4). In the conservative dataset, groEL haplotype diversity (Hd) is close to 1 (Table 2), ranging between 0.600 and 0.905 in the different subgroups. Nucleotide diversity (π) is highest in Subgroup 1a (π=0.0069), and approximately equal for the other groups (π=0.0018-0.00535). GroEL haplotype and nucleotide diversity statistics for the complete dataset are comparable to those calculated for the conservative dataset (See Additional file 1: Table S4).
Table 2.
Haplotype and genetic diversity estimates for Wolbachia groEL sequence groups for the conservative dataset
| Group | Haplotype Diversity (Hd) | Nucleotide Diversity (Π) | Haplotypes (N) | Sequences (N) | Segregating Sites (N) |
|---|---|---|---|---|---|
| Entire Dataset |
0.945 |
0.02569 |
21 |
56 |
52 |
| Group “1” |
0.938 |
0.01276 |
19 |
52 |
24 |
| Subgroup “1a” |
0.905 |
0.00691 |
13 |
36 |
15 |
| Subgroup “1b” |
0.644 |
0.00535 |
4 |
10 |
7 |
| Subgroup “1c” |
0.600 |
0.00188 |
2 |
6 |
1 |
| Group “2” |
0.667 |
0.00208 |
2 |
4 |
1 |
| mtDNA N |
0.957 |
0.01142 |
14 |
22 |
18 |
| mtDNA S | 0.937 | 0.03197 | 14 | 27 | 46 |
Association of Wolbachia groEL haplogroups to geography and host background
We performed an AMOVA to look for evidence of bidirectional CI in the G. f. fuscipes host. If bidirectional CI shaped host mtDNA diversity, we would expect to see that Wolbachia sequences within mtDNA haplogroups should be more closely related than between host haplogroups. The results of this analysis on the complete dataset suggest that most Wolbachia groEL variation is found within (98.43%) rather than between (1.57%) tsetse host mtDNA haplogroups (p = 0.07, Table 3). The non-significant AMOVA result indicates that the host mtDNA N and mtDNA S groups are not supported by the Wolbachia data, a result that conflicts with the idea of bidirectional CI shaping host genetic groups. This result is also confirmed by the conservative dataset (AMOVA, p = 0.79, Table 3), as host mtDNA haplogroups have identical numbers of Wolbachia groEL haplotypes (Table 2).
Table 3.
Wolbachia groEL AMOVA results that examine the hypothesis of bidirectional CI using groups (North and South) defined by G. f. fuscipes mtDNA
| |
Complete dataset |
Conservative dataset |
||||||
|---|---|---|---|---|---|---|---|---|
| Variance components | Percentage of Variation (%) | Variance components | Percentage of Variation (%) | |||||
| Group Comparison |
Among Group |
Within Group |
Among Group |
Within Group |
Among Group |
Within Group |
Among Group |
Within Group |
| North vs. South | 0.085 | 5.323 | 1.57 | 98.43 | −0.0472 | 1.9411 | −2.49 | 102.49 |
North and South refer to host mtDNA haplogroups.
The relationship between groEL haplotypes, their group assignment, and their relationship to the host genetic variation is shown in Figure 3B. Group 1 and all three of its subdivisions (subgroups 1a-1c) occur in nearly all sites. Group 2 is mostly limited to sites with southern host mtDNA haplotypes, but it also included groEL sequences from two sites (JN, MS), where both southern and northern host mtDNA haplotypes co-occur.
Since Wolbachia is transmitted maternally, Wolbachia sequences should be linked with specific host mtDNA haplotypes. Most of the flies screened for Wolbachia had mtDNA haplotypes found in the sites with mixed host haplotypes (Figure 1, 3, 4, See Additional file 1: Table S1). However, these mtDNA haplotypes were also found in other sites. Only samples from KK, DRC and the colony, which have unique groEL haplotypes, do not follow this pattern. We tested for an association between infection status and host mtDNA haplotypes in the mixed region. In 100 randomizations, we found that the proportion of infected individuals observed to have a host mtDNA haplotype found in the mixed region was not significantly different than when we randomly assigned infected status to host mtDNA haplotype (p = 0.17). GroEL sequences from Group 1 are associated with all sampled host haplotypes (Figure 3B, See Additional file 1: Table S1), and thus, both host mtDNA haplogroups. In Group 2, groEL sequences are only associated with four host mtDNA haplotypes: most flies had southern mtDNA haplotypes, but two flies (JN6, JN18) had the same northern mtDNA haplotype. We also tested whether the association of Group 2 with southern mtDNA host haplotypes was non-random and found a non-significant association (p = 0.16).
Figure 4.
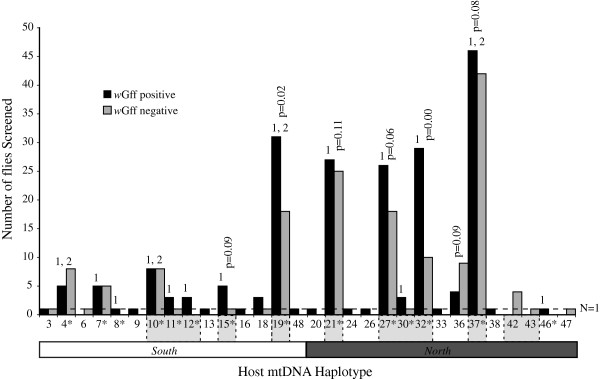
Association between Wolbachia prevalence and mtDNA haplotype of the G. f. fuscipes host. Association of groELfrom Wolbachia in G. f. fuscipes (wGff) groups with mtDNA haplotype and tsetse infection status from Ugandan populations. The Y-axis shows the number of infected and uninfected flies (black and gray bars, respectively). The X-axis shows the host mtDNA haplotypes (numbers) and haplogropus (white and black bars for the southern and northern mtDNA haplogroups, respectively). An asterisk next to the haplotype number indicates an mtDNA haplotype associated with wGff groEL sequence. Gray shaded mtDNA haplotype numbers indicate those haplotypes found in, but not limited to, collection sites where both northern and southern haplotypes co-occur. A number above the bars identifies the wGff group (1 and 2). P-values above haplotypes indicate whether there are more individuals infected than uninfected for a given haplotype (Binomial test).
Figure 4 shows the association between Wolbachia infection status and G. f. fuscipes host mtDNA from a dataset from the same region [26]. The most common host mtDNA haplotypes (19, 27 and 37) have more Wolbachia infected individuals than other haplotypes. However, these haplotypes also occur in uninfected individuals. In G. f. fuscipes, many Wolbachia-infected individuals have host haplotypes found in the geographic region with mixed host mtDNA haplogroups, though this result is not significant (p = 0.17). In the mixed region, more individuals are Wolbachia positive than negative, although only three mtDNA haplotypes have significantly more infected individuals in the mixed region (black vs. grey bars, Figure 4). This and the association with the most common host haplotypes were consistent with the notion that infected rather than uninfected hosts should occur at higher frequency if Wolbachia induces CI.
Discussion
Intra- and inter-individual genetic diversity in G. f. fuscipes Wolbachia lineages
Based on groEL, G. f. fuscipes Wolbachia lineages are diverse (Tables 1, 2), with groEL haplotype diversity varying only slightly (Hd = 0.905-1.0) regardless of scale (e.g., dataset, lineage, group subdivision or collection site, Figure 3, 4, See Additional file 1: Table S1). When not superinfected (infected only with Group 1), groEL haplotype diversity is still high (Hd = 0.938, Table 2). There are 102 and 21 unique groEL haplotypes in the complete and in the conservative dataset, respectively. Of the 21 unique groEL haplotypes, 13 were found in many individuals and sites (See Additional file 1: Table S4 and Figure S4).
Finding Wolbachia strain diversity in wild host populations is not unprecedented, but it has not been reported within individuals at this geographic scale. In European cherry fruit flies, Rhagoletis, multiple strains were found using the wsp gene (Wolbachia surface protein), but multiple strains within an individual were not reported [45]. Similarly in planthoppers, Perkinsiella, a number of B and F supergroup strains were identified using wsp, with F strains inferred to be potential recombinants [46]. While these and other studies identified high levels of Wolbachia diversity among individuals [47], our study found similar high levels of sequence variation at a much smaller scale, within individuals (See Additional file 1: Figure S2).
We suggest that the unique Glossina life history traits facilitate the identification of a transient phase of Wolbachia infection dynamics. Tsetse flies have a viviparous reproductive biology, where one oocyte matures and a single larva is nourished in an intrauterine environment. Females reproduce over their 3–4 month life span, producing 8–10 progeny. The low Wolbachia densities in G. f. fuscipes may reflect this viviparous biology of tsetse, since the few oocytes present in tsetse females may not necessitate retention of high Wolbachia densities that are required in oviparous females. However, Wolbachia densities in G. f. fuscipes were significantly lower than in the laboratory line G. morsitans morsitans, a species with similar life history traits. Environmental influences on Wolbachia densities in natural populations may be relevant and should be tested in other natural Glossina populations. Since G. f. fuscipes Wolbachia densities are very low, if a new Wolbachia variant arises within an individual, it is more likely to be observed and to have a proportionally larger impact on the overall genetic diversity in an individual than in a high-density Wolbachia infection. Thus, the peculiar tsetse life history may indirectly shape Wolbachia diversity within an individual host and allow the identification of variants that would otherwise not be detected.
Origin of G. f. fuscipes Wolbachia infections
The observed patterns and levels of genetic diversity of the two supergroups and their co-occurrence with any one host mtDNA haplotype suggests that the origin of G. f. fuscipes Wolbachia (hereafter wGff refers to any Wolbachia strain found in G. f. fuscipes) infections is complex and different from Wolbachia infection patterns reported in other studies. In insect populations that have undergone a selective sweep due to CI, Wolbachia infections are often associated with a single mtDNA haplotype from one or a few females [16,48]. In G. f. fuscipes, wGff are associated with at least 26 host mtDNA haplotypes (Figure 4, See Additional file 1: Table S1) with only 15 of these host haplotypes carrying the observed wGff sequence diversity. In addition, these mtDNA haplotypes are found in all wGff groups. These observations could suggest that the infection in this tsetse species is ancient with unprecedented horizontal and imperfect transmission. Although this scenario is possible, it is a less likely explanation, because even with horizontal transmission we would expect to see a geographic break in the Wolbachia groEL haplotypes, as we see for the host mtDNA haplotypes. An alternative hypothesis is that multiple females with different mtDNA haplotypes were initially infected. Although tests of the association between infection and mixed region host haplotypes were not significant (Randomization test, p = 0.17), all but four of these host mtDNA haplotypes are found in, but not only in, the region where northern and southern host mtDNA haplotypes co-occur (See Additional file 1: Figure S4, Table S1). This suggests that the wGff Group 1 infection in Uganda may have started in the region where we observe mixed host haplotypes. Since these haplotypes are found throughout Uganda, the wGff infection may have spread from there via host dispersal and subsequent gene flow. Our genetic data support this hypothesis as wGff differentiation between fly populations with the two mtDNA haplogroups is low (Table 3). Furthermore, wGff prevalence is associated with host genetic groups defined by microsatellite loci [26].
While our data are suggestive of a mixed region origin, the mechanism is unclear. Maternal transmission of Wolbachia implies independent infection of each host mtDNA haplotype. Thus our data suggest that wGff in Uganda were repeatedly infected with Wolbachia (See Additional file 1: Table S1), a condition also supported by simulation studies as an initial transient phase in Wolbachia establishment in a new species [13,14], but never before observed empirically. Moreover, it is unlikely that flies dispersing from the mixed region are the sole source of Wolbachia infections in Uganda, as we find wGff with unique host mtDNA haplotypes (KK; Figure 1) from a distinct western Uganda tsetse group defined by nuclear microsatellite data ( [29], See Additional file 1: Table S2). This suggests a second infection potentially from western Uganda. Interestingly, tsetse in Kakoga (KK) and in the Lake Victoria region (Figure 1) carrying rare mtDNA haplotypes are Wolbachia-infected, suggesting a relatively recent infection with a closely related wGff. Since these are rare host mtDNA haplotypes, too few wGff sequences are available to test this hypothesis.
The inclusion of G. f. fuscipes from a distant geographic location (DRC) with extremely divergent mtDNA from the Ugandan flies and from a colony population of a different subspecies (G. f. quanzensis) [49,50] infected by the two Wolbachia lineages (Figure 2) allows us to discuss three possible scenarios for the origin of the Wobachia infection(s). First, infection by both Wolbachia lineages was widespread and pre-dates the sub-species divergence. This would lead to a correlation between Wolbachia and host mtDNA divergence [16,51], as we see in the maternally transmitted obligate symbiont, Wigglesworthia in the Ugandan G. f. fuscipes from the same regions [52]. We do not see this in wGff. Second, Wolbachia is shared between geographically disparate samples because there is extensive dispersal (and subsequent gene flow) among G. f. fuscipes populations. In such a case, we would expect to see wGff haplotypes associated with at least one widespread host haplotype. We find wGff associated with the most common host mtDNA haplotypes, but these host haplotypes are not widespread (Figure 4). Further, wGff is not associated with host genetic groups defined by mtDNA variation, but with those defined by nuclear variation, whose patterns likely originated via genetic drift, not gene flow from geographically distant populations [26,28,29]. Third, it is possible that there were multiple independent infections in G. f. quanzensis in DRC and in G. f. fuscipes in Uganda. Although our data appear to only support the last of these hypotheses, our sampling design does not permit us to specifically test any of these hypotheses. However, these hypotheses warrant investigation to understand Wolbachia infection dynamics in this species, as it can shed light on the general evolutionary dynamics of Wolbachia infections, which are not possible to address in other systems that do not have the viviparous life history traits of Glossina species.
Relevance to CI
In the presence of CI, Wolbachia is expected to be associated with few high frequency host mtDNA haplotypes [9]. In combination with data from [26], we found Wolbachia associated with 26 host mtDNA haplotypes. Of the approximately 40 mtDNA haplotypes found in Ugandan G. f. fuscipes[28,29], more than half are infected with wGff Group 1, and only three with wGff Group 2. Indeed, the host mtDNA haplotypes infected with Group 1 are some of the most common in Uganda (Figure 4), but the low sample size for some host mtDNA haplotypes, due to low infection density, makes it difficult to draw inferences about this pattern. Interestingly, in nearly all of the high frequency mtDNA haplotypes [28,29], wGff-infected individuals are more common than those that are not infected. Although these differences are not all significant, it suggests some fitness advantage for infected flies (Figure 4), consistent with occurrence of CI in G. f. fuscipes.
Unexpectedly, our data found wGff associated with rare mtDNA haplotypes, a result also supported by Wolbachia prevalence data (See Additional file 1: Figure S4, [29]): in 365 flies with known host mtDNA haplotype, wGff was associated with 12 extremely rare mtDNA haplotypes. CI-causing Wolbachia are expected to have higher fitness, driving associated host haplotypes to high frequency, as seen in some of the common groEL haplotypes in our dataset (See Additional file 1: Figure S4). Contrary to the typical observation of a single female driving an infection in insect populations, theoretical studies suggest that before CI can sweep Wolbachia through a population, multiple independent infections must occur [13,14]. It is possible that sweeps occur in other insects too rapidly to observe these multiple, independent infections and we may have captured Wolbachia, even in rare groEL haplotypes, due to the unique host life history.
Our genetic data do not provide evidence that bidirectional CI has shaped genetic variability in G. f. fuscipes[28,29]. Although Group 2 is primarily limited to southern host mtDNA haplotypes (Figure 3, See Additional file 1: Table S2), we found that this association was not significant (p = 0.16). Furthermore, two superinfected individuals have northern host mtDNA haplotypes (JN18, JN6; groEL haplotype 37). This result is unexpected, if we assume solely maternal Wolbachia transmission, and suggests that Group 2 infections have either independently arisen in the northern host mtDNA haplotype lineage, or there is some horizontal transfer of Group 2 infections from southern G. f. fuscipes to individuals found in the northern mtDNA haplogroup. Horizontal transfer among different insect species must occur for Wolbachia to infect novel hosts, but horizontal transfer among different host species with closely related Wolbachia has rarely been empirically documented [53]. Since either horizontal transmission or independent infections appear to be common in G. f. fuscipes, genetic data may not be the ideal method to detect any form of CI as these processes may obscure host genetic patterns induced by Wolbachia. Furthermore, very few of our samples seem to be infected with Group 2 Wolbachia, potentially reducing our power to detect patterns. In contrast, we see some evidence that wGff is associated with the most common haplotypes (Figure 4), suggesting a potential fitness advantage of wGff. Thus, we suggest that it is crucial to examine transmission efficiency and perform laboratory mating experiments before excluding the possibility of bidirectional CI in G. f. fuscipes.
Conclusions
We investigated Wolbachia (wGff) genetic variability in the tsetse fly, Glossina fuscipes fuscipes, populations with known genetic composition and Wolbachia infection status in Uganda. Using four MLST loci, we identified two Wolbachia lineages, indicating superinfection of G. f. fuscipes. Using the variable groEL gene, we confirmed the occurrence of superinfections and uncovered unprecedented sequence diversity within and between individuals. However, we do not find evidence that Wolbachia has influenced patterns of genetic diversity in Ugandan G. f. fuscipes populations through mechanisms like cytoplasmic incompatibility (CI). When compared to the host mtDNA, we found wGff associated with several host mtDNA haplotypes, suggesting independent acquisition of wGff infections. We hypothesize that high genetic variability in wGff may be a consequence of low-density Wolbachia infections and the observation of multiple independent infections may be associated with the unique tsetse life history.
Abbreviations
wGff: Any Wolbachia pipientis strain from Glossina fuscipes fuscipes; groEL: gene for heat shock protein 60; CI: Cytoplasmic incompatibility; MLST: Multi Locus Sequence Typing; mtDNA: mitochondrial DNA; HAT: Human African Trypanosomiasis; G. f. fuscipes: Glossina fuscipes fuscipes; gatB: gene for aspartyl/glutamyl-tRNA(Gln) amidotransferase, subunit B; coxA: gene for cytochrome c oxidase, subunit I; fbpA: gene for fructose-bisphosphatealdolase; ftsZ: gene for cell division protein; AMOVA: Analysis of Molecular Variance; G. austeni: Glossina austeni; G. brevipalpis: Glossina brevipalpis; wsp: gene for Wolbachia surface protein.; Hd: Haplotype diversity; G. f. quanzensis: Glossina fuscipes quanzensis; bp: base pairs.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RES-Wrote and revised the manuscript and performed and interpreted data analysis, UA-carried out the laboratory analyses and collected the data. CB-Collected and analyzed the MLST data, and commented on the manuscript, YW-Extracted DNA and helped collect the molecular data, RE-Provided samples, LMO-Provided samples and facilitated sample collection, SA-Conceived the project and helped write the manuscript, AC-Helped write the manuscript and provided support for the analyses. All authors read and approved the final manuscript.
Supplementary Material
Includes the expanded methodology for laboratory methods and data analysis. Tables of detailed sample information for G. f. fuscipes and other insects, primers and diversity statistics for the complete dataset. Figures show MLST phylogeny and the haplotype networks for individuals, sampling sites and the complete dataset.
Contributor Information
Rebecca E Symula, Email: resymula@olemiss.edu.
Uzma Alam, Email: uzma.alam@yale.edu.
Corey Brelsfoard, Email: clbrelsfoard@uky.edu.
Yineng Wu, Email: yineng.wu@yale.edu.
Richard Echodu, Email: richard_echodu@yahoo.co.uk.
Loyce M Okedi, Email: lmokedi@gmail.com.
Serap Aksoy, Email: serap.aksoy@yale.edu.
Adalgisa Caccone, Email: adalgisa.caccone@yale.edu.
Acknowledgments
This study was supported by NIH AI068932) and the Fogarty Center D43TW007391 and R03 TW008755 grants. Peter Takac (Slovakia Academy of Science, Bratislava, Slovakia) provided G. f. quanzensis samples.
References
- Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren JH. How many species are infected with Wolbachia?–A statistical analysis of current data. FEMS Microbiol Lett. 2008;281:215–220. doi: 10.1111/j.1574-6968.2008.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez CJ, Stouthamer R, Jeong G, Morse JG. Discovery of a CI-inducing Wolbachia and its associated fitness costs in the biological control agent Aphytis melinus DeBach (Hymenoptera: Aphelinidae) Biol Control. 2011;58:192–198. [Google Scholar]
- Wang Z-y, Deng C, Yun Y-l, Jian C, Peng Y. Molecular detection and the phylogenetics of Wolbachia in Chinese spiders (Araneae) J Arachnol. 2010;38:237–241. [Google Scholar]
- Lo N, Paraskevopoulos C, Bourtzis K, O’Neill SL, Werren JH, Bordenstein SR, Bandi C. Taxonomic status of the intracellular bacterium Wolbachia pipientis. Int J Syst Evol Microbiol. 2007;57:654–657. doi: 10.1099/ijs.0.64515-0. [DOI] [PubMed] [Google Scholar]
- Baldo L, Dunning Hotopp JC, Jolley KA, Bordenstein SR, Biber SA, Choudhury RR, Hayashi C, Maiden MC, Tettelin H, Werren JH. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 2006;72:7098–7110. doi: 10.1128/AEM.00731-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlie JC, Cass BN, Riegler M, Witsenburg JJ, Iturbe-Ormaetxe I, McGraw EA, O’Neill SL. Evidence for metabolic provisioning by a common invertebrate endosymbiont, Wolbachia pipientis, during periods of nutritional stress. PLoS Pathog. 2009;5:e1000368. doi: 10.1371/journal.ppat.1000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambris Z, Blagborough AM, Pinto SB, Blagrove MS, Godfray HC, Sinden RE, Sinkins SP. Wolbachia stimulates immune gene expression and inhibits plasmodium development in Anopheles gambiae. PLoS Pathog. 2010;6:e1001143. doi: 10.1371/journal.ppat.1001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–751. doi: 10.1038/nrmicro1969. [DOI] [PubMed] [Google Scholar]
- Engelstadter J, Telschow A. Cytoplasmic incompatibility and host population structure. Heredity (Edinb) 2009;103:196–207. doi: 10.1038/hdy.2009.53. [DOI] [PubMed] [Google Scholar]
- Unckless RL, Boelio LM, Herren JK, Jaenike J. Wolbachia as populations within individual insects: causes and consequences of density variation in natural populations. Proceedings of the Royal Society B: Biological Sciences. 2009;276:2805–2811. doi: 10.1098/rspb.2009.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Hoffmann AA, McKechnie SW. Dynamics of cytoplasmic incompatibility and mtDNA variation in natural Drosophila simulans populations. Genetics. 1992;132:713–723. doi: 10.1093/genetics/132.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasgon JL, Cornel AJ, Scott TW. Evolutionary history of a mosquito endosymbiont revealed through mitochondrial hitchhiking. Proceedings of the Royal Society B: Biological Sciences. 2006;273:1603–1611. doi: 10.1098/rspb.2006.3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egas M, Vala F, Breeuwer JA. On the evolution of cytoplasmic incompatibility in haplodiploid species. Evolution. 2002;56:1101–1109. doi: 10.1111/j.0014-3820.2002.tb01424.x. [DOI] [PubMed] [Google Scholar]
- Jansen VA, Turelli M, Godfray HC. Stochastic spread of Wolbachia. Proc Biol Sci. 2008;275:2769–2776. doi: 10.1098/rspb.2008.0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes MD, Nolte V, Schlotterer C. Nonrandom Wolbachia infection status of Drosophila melanogaster strains with different mtDNA haplotypes. Mol Biol Evol. 2008;25:2493–2498. doi: 10.1093/molbev/msn199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Hoffmann AA. Rapid spread of an inherited incompatibility factor in California Drosophila. Nature. 1991;353:440–442. doi: 10.1038/353440a0. [DOI] [PubMed] [Google Scholar]
- Mouton L, Henri H, Charif D, Bouletreau M, Vavre F. Interaction between host genotype and environmental conditions affects bacterial density in Wolbachia symbiosis. Biol Lett. 2007;3:210–213. doi: 10.1098/rsbl.2006.0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duron O, Fort P, Weill M. Influence of aging on cytoplasmic incompatibility, sperm modification and Wolbachia density in Culex pipiens mosquitoes. Heredity (Edinb) 2007;98:368–374. doi: 10.1038/sj.hdy.6800948. [DOI] [PubMed] [Google Scholar]
- Keeling MJ, Jiggins FM, Read JM. The invasion and coexistence of competing Wolbachia strains. Heredity (Edinb) 2003;91:382–388. doi: 10.1038/sj.hdy.6800343. [DOI] [PubMed] [Google Scholar]
- Flor M, Hammerstein P, Telschow A. Wolbachia-induced unidirectional cytoplasmic incompatibility and the stability of infection polymorphism in parapatric host populations. J Evol Biol. 2007;20:696–706. doi: 10.1111/j.1420-9101.2006.01252.x. [DOI] [PubMed] [Google Scholar]
- Telschow A, Yamamura N, Werren JH. Bidirectional cytoplasmic incompatibility and the stable coexistence of two Wolbachia strains in parapatric host populations. J Theor Biol. 2005;235:265–274. doi: 10.1016/j.jtbi.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Dutton TJ, Sinkins SP. Strain-specific quantification of Wolbachia density in Aedes albopictus and effects of larval rearing conditions. Insect Mol Biol. 2004;13:317–322. doi: 10.1111/j.0962-1075.2004.00490.x. [DOI] [PubMed] [Google Scholar]
- Hurst GD, Johnson AP, Schulenburg JH, Fuyama Y. Male-killing Wolbachia in drosophila: a temperature-sensitive trait with a threshold bacterial density. Genetics. 2000;156:699–709. doi: 10.1093/genetics/156.2.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Ruel TD, Zhou W, Moloo SK, Majiwa P, O’Neill SL, Aksoy S. Tissue distribution and prevalence of Wolbachia infections in tsetse flies, Glossina spp. Med Vet Entomol. 2000;14:44–50. doi: 10.1046/j.1365-2915.2000.00202.x. [DOI] [PubMed] [Google Scholar]
- Doudoumis V, Tsiamis G, Wamwiri F, Brelsfoard C, Alam U, Aksoy E, Dalaperas S, Abd-Alla A, Ouma J, Takac P. et al. Detection and characterization of Wolbachia infections in laboratory and natural populations of different species of tsetse flies (genus Glossina) BMC Microbiol. 2012;12:S3. doi: 10.1186/1471-2180-12-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam UH, Chaz H, Symula RE, Brelsfoard C, Wu Y, Kruglov O, Okedi LM, Caccone A, Aksoy S. Microfauna-host interactions: implications for trypanosome transmission dynamics in Glossina fuscipes fuscipes in Uganda. Appl Environ Microbiol. 2012;78:4627–4637. doi: 10.1128/AEM.00806-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam U, Medlock J, Brelsfoard C, Pais R, Lohs C, Balmand S, Carnogursky J, Heddi A, Takac P, Galvani A, Aksoy S. Wolbachia Symbiont infections induce strong cytoplasmic incompatibility in the tsetse Fly Glossina morsitans. PLoS Pathog. 2011;7:e1002415. doi: 10.1371/journal.ppat.1002415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abila PP, Slotman MA, Parmakelis A, Dion KB, Robinson AS, Muwanika VB, Enyaru JC, Okedi LM, Aksoy S, Caccone A. High levels of genetic differentiation between Ugandan Glossina fuscipes fuscipes populations separated by Lake Kyoga. PLoS Negl Trop Dis. 2008;2:e242. doi: 10.1371/journal.pntd.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadell JS, Hyseni C, Abila PP, Azabo R, Enyaru JC, Ouma JO, Mohammed YO, Okedi LM, Aksoy S, Caccone A. Phylogeography and population structure of Glossina fuscipes fuscipes in Uganda: implications for control of tsetse. PLoS Negl Trop Dis. 2010;4:e636. doi: 10.1371/journal.pntd.0000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echodu R, Beadell JS, Okedi LM, Hyseni C, Aksoy S, Caccone A. Temporal stability of Glossina fuscipes fuscipes populations in Uganda. Parasites and Vectors. 2011;4 doi: 10.1186/1756-3305-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Bordenstein S, Wernegreen JJ, Werren JH. Widespread recombination throughout Wolbachia genomes. Mol Biol Evol. 2006;23:437–449. doi: 10.1093/molbev/msj049. [DOI] [PubMed] [Google Scholar]
- Thompson JR, Marcelino LA, Polz MF. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR’. Nucleic Acids Res. 2002;30:2083–2088. doi: 10.1093/nar/30.9.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings SM, McMullan M, Joyce DA, van Oosterhout C. Solutions for PCR, cloning and sequencing errors in population genetic analysis. Conserv Genet. 2010;11:1095–1097. [Google Scholar]
- Sawyer SA. Book GENECONV: A computer package for the statistical detection of gene conversion. 181a 1999. GENECONV: A computer package for the statistical detection of gene conversion. [Google Scholar]
- Jolley KA, Chan MS, Maiden MC. mlstdbNet - distributed multi-locus sequence typing (MLST) databases. BMC Bioinforma. 2004;5:86. doi: 10.1186/1471-2105-5-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiraghi M, Bordenstein SR, Baldo L, Lo N, Beninati T, Wernegreen JJ, Werren JH, Bandi C. Phylogeny of Wolbachia pipientis based on gltA, groEL and ftsZ gene sequences: clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiology. 2005;151:4015–4022. doi: 10.1099/mic.0.28313-0. [DOI] [PubMed] [Google Scholar]
- Maddison DRaMWP. Book MacClade4: Analysis of phylogeny and character evolution. 408. City: Sinauer Associates; 2004. MacClade4: Analysis of phylogeny and character evolution. [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Zwickl D. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. The University of Texas at Austin, Section of Integrative Biology; 2006. [Google Scholar]
- Rozas J. DNA sequence polymorphism analysis using DnaSP. Methods Mol Biol. 2009;537:337–350. doi: 10.1007/978-1-59745-251-9_17. [DOI] [PubMed] [Google Scholar]
- Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthofer W, Riegler M, Schneider D, Krammer M, Miller WJ, Stauffer C. Hidden Wolbachia diversity in field populations of the European cherry fruit fly, Rhagoletis cerasi (Diptera, tephritidae) Mol Ecol. 2009;18:3816–3830. doi: 10.1111/j.1365-294X.2009.04321.x. [DOI] [PubMed] [Google Scholar]
- Hughes GL, Allsopp PG, Brumbley SM, Woolfit M, McGraw EA, O’Neill SL. Variable infection frequency and high diversity of multiple strains of Wolbachia pipientis in Perkinsiella planthoppers. Appl Environ Microb. 2011;77:2165–2168. doi: 10.1128/AEM.02878-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atyame CM, Delsuc F, Pasteur N, Weill M, Duron O. Diversification of Wolbachia Endosymbiont in the Culex pipiens Mosquito. Molecular Biology and Evolution; 2011. [DOI] [PubMed] [Google Scholar]
- Sun X, Cui L, Li Z. Diversity and phylogeny of Wolbachia infecting Bactrocera dorsalis (Diptera: tephritidae) populations from China. Environ Entomol. 2007;36:1283–1289. doi: 10.1603/0046-225x(2007)36[1283:dapowi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Dyer NA, Lawton SP, Ravel S, Choi KS, Lehane MJ, Robinson AS, Okedi LM, Hall MJ, Solano P, Donnelly MJ. Molecular phylogenetics of tsetse flies (Diptera: Glossinidae) based on mitochondrial (COI, 16S, ND2) and nuclear ribosomal DNA sequences, with an emphasis on the palpalis group. Mol Phylogenet Evol. 2008;49:227–239. doi: 10.1016/j.ympev.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Dyer NA, Ravel S, Choi K-S, Darby AC, Causse S, Kapitano B, Hall MJR, Steen K, Lutumba P, Madinga J. et al. Cryptic diversity within the major trypanosomiasis vector Glossina fuscipes revealed by molecular markers. PLoS Negl Trop Dis. 2011;5:e1266. doi: 10.1371/journal.pntd.0001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker DD, Machado CA, Molbo D, Werren JH, Windsor DM, Herre EA. The distribution of Wolbachia in fig wasps: correlations with host phylogeny, ecology and population structure. Proc R Soc London, Ser B. 2002;269:2257–2267. doi: 10.1098/rspb.2002.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symula RE, Marpuri I, Bjornson RD, Okedi L, Beadell J, Alam U, Aksoy S, Caccone A. Influence of host phylogeographic patterns and incomplete lineage sorting on within-species genetic variability in Wigglesworthia species, obligate symbionts of Tsetse flies. Appl Environ Microb. 2011;77:8400–8408. doi: 10.1128/AEM.05688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA, Goldman-Huertas B, Moreau CS, Baldo L, Stahlhut JK, Werren JH, Pierce NE. Specialization and geographic isolation among Wolbachia symbionts from ants and lycaenid butterflies. Evolution. 2009;63:624–640. doi: 10.1111/j.1558-5646.2008.00579.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Includes the expanded methodology for laboratory methods and data analysis. Tables of detailed sample information for G. f. fuscipes and other insects, primers and diversity statistics for the complete dataset. Figures show MLST phylogeny and the haplotype networks for individuals, sampling sites and the complete dataset.