

Abstract
Rhomboid proteases are evolutionary conserved intramembrane serine proteases. Because of their emerging role in many important biological pathways, rhomboids are potential drug targets. Unfortunately, few chemical tools are available for their study. Here, we describe a mass spectrometry-based assay to measure rhomboid substrate cleavage and inhibition. We have identified isocoumarin inhibitors and developed activity-based probes for rhomboid proteases. The probes can distinguish between active and inactive rhomboids due to covalent, reversible binding of the active-site serine and stable modification of a histidine residue. Finally, the structure of an isocoumarin-based inhibitor with Escherichia coli rhomboid GlpG uncovers an unusual mode of binding at the active site and suggests that the interactions between the 3-substituent on the isocoumarin inhibitor and hydrophobic residues on the protease reflect S′ subsite binding. Overall, these probes represent valuable tools for rhomboid study, and the structural insights may facilitate future inhibitor design.
Keywords: MALDI screening, covalent inhibition, regulated intramembrane proteolysis
Proteolysis controls many important biological processes, such as apoptosis, antigen presentation, and blood coagulation. Selective digestion of protein substrates is possible by a combination of tight posttranslational control of protease activity (1) and the protease’s substrate specificity, which generally is governed by the primary sequence around the scissile bond (2). The use of inhibitors and activity-based probes (ABPs) has led to a tremendous gain in understanding the roles of proteases within physiological and pathological processes (3). ABPs are small molecules that bind only to active enzymes, but not to zymogen or inhibitor-bound forms (4). ABPs generally consist of a detection tag, a spacer, and a “warhead.” The warhead covalently binds to the target enzyme(s) and often is derived from a mechanism-based inhibitor. In the past, ABPs were used to study the activation, localization, and function of soluble proteases in a variety of organisms and disease models (5).
Most proteases are soluble and surrounded by an aqueous environment. However, several families of intramembrane proteases exist (6–8): the metalloprotease family M50 (site-2 protease), the aspartic protease family A22 (signal peptide peptidase and γ-secretase), and the serine protease family S54 [rhomboid; numbering according to the MEROPS database (9)]. Rhomboid was discovered in 2001 as a protease in the EGF receptor signaling pathway in the fruitfly Drosophila melanogaster (10). Interestingly, rhomboid genes occur in all kingdoms of nature and are found in most sequenced organisms (11, 12). Rhomboids appear to have a wide range of physiological functions, including bacterial protein export (13) and invasion by apicomplexan parasites (14, 15), but the roles of many rhomboids remain to be discovered.
Rhomboids catalyze peptide bond hydrolysis using a catalytic dyad formed by a serine residue in transmembrane domain 4 (TM4) and a histidine residue in TM6. Crystal structures of the Escherichia coli rhomboid GlpG have shown that these residues are in close enough proximity to form a hydrogen bond (16, 17). The attack onto the scissile bond of the substrate is proposed to occur at the si-face, opposite that of most other serine proteases (18, 19). Another difference between rhomboids and classical serine proteases is the form in which they are translated. Soluble proteases are produced mainly as inactive zymogens, which need proteolytic activation. Subsequently, the protease activity is tightly controlled by posttranslational processes, such as phosphorylation, ATP binding, and inhibition by endogenous proteins. Although the human rhomboid RHBDL2 is proposed to undergo autocleavage for activation (20), most rhomboids appear to be translated in their active form. Whether rhomboid activity is regulated directly, and how this is achieved mechanistically, currently is unclear.
Only a few serine protease inhibitors work against rhomboids. 3,4-Dichloroisocoumarin (DCI) inhibits Drosophila rhomboid-1 (10) and purified bacterial rhomboids (21, 22), but it lacks potency and selectivity. One other isocoumarin (JLK-6; 20, Table S1) has been reported to inhibit E. coli rhomboid GlpG (23). Sulfonylated β-lactams recently were found to inhibit bacterial rhomboids (24), as well as two fluorophosphonates (25, 26).
In this work, we present a unique rhomboid inhibition assay that monitors the cleavage of a protein substrate by MALDI mass spectrometry (MS). In a screen of small molecules, we discovered inhibitors and ABPs for bacterial rhomboids. The ABPs, which are based on the isocoumarin reactive group, label active rhomboids and may be used in activity-based profiling. Additionally, we provide structural insight into an unusual mode of inhibitor binding at the active site of rhomboids, providing a framework for rational design of inhibitors.
Results
MALDI-Based Quantification of Rhomboid Substrate Cleavage.
Gel-based assays are the most widely used method to detect cleavage of rhomboid substrates, in bacteria (27), in eukaryotic cell culture (10), or by purified rhomboids (21, 22). However, gel analysis is not optimal for identifying inhibitors because of the low throughput. One FRET-based assay for the rhomboid AarA of the Gram-negative bacterium Providencia stuartii has been reported (24); it made use of a 16-mer FRET peptide, but many rhomboids do not cleave this substrate efficiently. The development of small molecule fluorescent reporters for rhomboids is difficult because the details of their substrate specificities still are not well defined. However, various natural and engineered protein substrates are known. We therefore decided to directly monitor the cleavage of a protein substrate by rhomboid proteases in a gel- and label-free analysis method using MS. We chose MALDI-MS because it is much less restricted in the use of salts and buffers compared with electrospray ionization MS, and it requires only minimal sample preparation efforts. Hence, we expressed recombinant E. coli rhomboid GlpG, P. stuartii rhomboid AarA, and its natural substrate TatA in E. coli and purified these in dodecylmaltoside (DDM) micelles. Overexpression of P. stuartii TatA in E. coli led to incomplete deformylation of the initiator N-formylmethionine by peptide deformylase, which is a known problem for recombinantly produced proteins (28) (Fig. 1A). However, the N-terminal formyl group on the substrate protein did not have any influence on the rhomboid cleavage kinetics in comparison with the deformylated substrate (Fig. S1A). Both AarA and GlpG were able to cleave TatA at its known cleavage site (13) and give rise to a product peak corresponding to a loss of the first eight N-terminal amino acids (Fig. 1A). We used the ratio of the signal intensities from the intact substrate and the cleavage product (corrected for the difference in ionizability; Fig. S1B) as a read-out of substrate turnover. We then monitored the substrate cleavage in time (Fig. 1B) and found that full substrate cleavage can be achieved within 1–2.5 h.
Fig. 1.

MALDI-MS detection of rhomboid protein substrate cleavage. (A) Recombinant P. stuartii TatA in its N-terminally formylated (m/z 11416.5) and unformylated (m/z 11388.5) form. The addition of rhomboid protease AarA leads to a cleavage of the substrate and a concurrent reduction in mass corresponding to proteolysis at the natural cleavage site (Δm = 804.8, equal to the N-terminal MESTIATA peptide sequence). (B) Cleavage of TatA (20 μM) by rhomboids AarA (P. stuartii; 0.5 μM) and GlpG (E. coli; 1.5 μM) monitored over time. Cleavage percentage refers to the means ± SE from four independent reactions. TatA cleavage by AarA is faster than by GlpG, probably because TatA is the natural substrate of AarA and better matches its substrate selectivity.
MALDI-Based Inhibition Assay.
For the identification of rhomboid inhibitors, we used an endpoint assay in which the rhomboid first was treated with small molecules and subsequently incubated with the substrate. The percentage of residual substrate then was used as a read-out of inhibition. From eight replicates of positive and negative controls (AarA active-site mutant S150A and wild type, respectively), we determined the Z′-factor (29), which is a statistical parameter for overall assay quality. The high Z′-score of 0.82 shows that this assay setup is sensitive and robust.
We screened a collection of compounds consisting of reactive electrophiles that are known to modify the active-site residues of serine proteases (Table S1). All proteolytic reactions were stopped before 100% of cleavage was achieved, which allows one to observe a decrease as well as an increase in substrate processing. As a positive inhibitor control, we used DCI at 200 μM. Several compounds completely inhibited GlpG or AarA (Fig. S2 A and B), and a duplicate screen showed good reproducibility of the data (Fig. S2C). Besides inhibitors, we also found molecules that led to an enhancement of the TatA cleavage by GlpG or AarA. Enhancers of cleavage have been found before (24), but in view of our search for mechanism-based inhibitors to be used as warheads for ABPs, we focused on the compounds that showed an inhibitory effect.
For GlpG, the hits in the screening were mainly 4-chloro-isocoumarins (ICs) and one peptido sulfonyl fluoride. For AarA, ICs were the best inhibitors. One diphenyl phosphonate showed weak inhibition of AarA but did not react as a mechanism-based inhibitor (see Activity-Based Probes for Rhomboids). For a better quantification of inhibition, we determined the apparent IC50 values of the best hits (Table 1). IC 16 displayed approximately an order of magnitude higher potency against GlpG compared with DCI. For selected inhibitors, we also measured the apparent IC50 values against bovine trypsin and chymotrypsin as two representative examples of the largest family of serine proteases (S1 family). Although IC 6 and 16 achieve good selectivity over trypsin, they also inhibit chymotrypsin, probably as a result of the hydrophobic substituent at the 3-position that can fit into the hydrophobic S1 binding pocket of chymotrypsin.
Table 1.
Apparent IC50 values (μM) of rhomboid hit structures
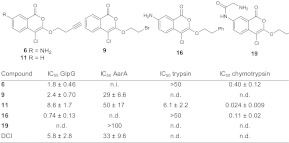 |
Values are calculated from triplicate experiments and given ± SE. n.d., not determined; n.i., no inhibition.
Structural Insights into Inhibitor Binding.
To understand the structural basis of the higher potency of the new isocoumarin inhibitors, we performed soaking studies with preformed crystals of E. coli GlpG. Among the different isocoumarins tested, IC 16 readily reacted with GlpG crystals (Table S2). As expected, the structure of the protease inhibitor complex shows the ring-opened reaction product of IC 16, which forms after the nucleophilic attack of active-site serine on the carbonyl group (Figs. 2A and 3H). Surprisingly, the second nucleophilic attack on the 4-position of IC 16 is performed by H150 (Fig. 2A) rather than the active-site H254, which was observed in a previous structure with the IC inhibitor JLK-6 (23). The carbonyl oxygen points toward the oxyanion hole, hydrogen bonding to N154 side chain, and water molecule. Although the Nπ (ND1) atom of H150 is within hydrogen-bonding distance to the carbonyl oxygen, it is unlikely to play a role in stabilization of the anion as it is covalently bonded to the inhibitor. The amino group of the IC forms hydrogen bonds with the hydroxyl of Y205 and the backbone carbonyl of W236 (Fig. 2B). The hydrophobic substituent on the 3-position of IC 16 makes contact with the side chains of M149 and F153 in TM2 and the side chains of F245 and M247 in L5 (Fig. 2C), an interaction also observed in a recent structure of GlpG in complex with a Cbz-containing phosphonate inhibitor (28). The aromatic ring of IC 16 stacks against the side chain of W236, which in other inhibitor complexes is rotated away toward the bilayer (23, 25, 30). Perhaps it is because of the position of the W236 side chain that a hydrophobic cavity postulated as the S2′ substrate binding site (23) is not observed in this new rhomboid–IC structure (Fig. 2D).
Fig. 2.

Structures of rhomboid in complex with IC 16. (A) 2Fo-Fc map drawn around the inhibitor and residues S201 and H150 at 1σ. (B) Interactions of IC 16 with GlpG. The carbonyl oxygen of IC 16 points toward the oxyanion hole, and hydrogen bonds to N154 (3.05 Å) and a water molecule (2.8 Å). Other polar interactions include the hydrogen bonds between the amino group and the hydroxyl of Y205 (2.87 Å) and main chain carbonyl of W236 (3.1 Å). The NE1 atom from the side chain of W236 hydrogen bonds to the oxygen atom of the ester carbonyl of the inhibitor. The aromatic ring with the amino group of the inhibitor stacks against the side chain of W236. For clarity, the interactions with side chains from L5 are not shown. (C) Loop5 covers the inhibitor and the active site. The side chains of F245, M247, and M249 in L5 are shown in stick representation. Hydrophobic interactions between these residues in L5 and residues in TM2, such as M149 and F153, with the inhibitor are highlighted. (D) Surface representation of GlpG viewed from the periplasm showing the bound inhibitor. The protein molecule is color coded according to the biochemical properties of the amino acids: positive and negatively charged residues are shown in blue and red, polar residues in light blue, and the rest in gray. The active-site residues S201 and H254 are colored green and orange, respectively. Water molecules (red spheres) are found occluded in a cavity that has been postulated as the S1 substrate-binding site. The carbon atoms of the inhibitor are colored in yellow and shown in stick representation.
Fig. 3.

Rhomboid ABPs. (A) Rhomboid GlpG, either in a DDM-micelle environment or in a crude E. coli lysate, can be detected by incubation with probe 6 or 11 (2 μM) and subsequent click chemistry functionalization. The S201A active-site mutant does not react with the probe. (B) The ABP labeling of GlpG can be abolished by preincubation with DCI or the best hit from the screen, IC 16. (C) Structure of the ABP 36, derived from IC 6 and its labeling of purified GlpG. (D) Endogenous GlpG (MW: 31.3 kDa) can be visualized in gel by ABP 36, and was not detected in a ΔglpG control cell strain. Labeling of endogenous GlpG was blocked upon treatment with active-site inhibitor IC 16. (E) Recombinant GlpG was labeled in vivo by ABP 36, and labeling was prevented by preincubation with active-site inhibitor IC 16. (F) Activity-based labeling of GlpG followed over time shows stable modification with probe 6, whereas probe 11 led to a loss of signal over time. (G) A better separation shows that reaction with probe 6 leads to two labeled protein species. The signal intensity of the lower band decreases over time. (H) The binding mechanism of ICs with and without a 7-amino group. The initial step comprises an attack of the serine onto the IC carbonyl group. The 7-amino group on the IC (present in 6, but not in 11) then leads to formation of a quinone imine methide electrophile (Center) that reacts with the active-site histidine. This enables stable, covalent modification of the active site. Compound 11 slowly hydrolyzes from the active site of GlpG because of the instability of the ester bond. (I) An IC probe, when doubly bound to a serine and a histidine, cross-links two TMs and makes the protein more compact, leading to a lower apparent MW during gel electrophoresis.
Overall, the present GlpG-inhibitor structure closely resembles the apoenzyme (average rmsd for all Cα atoms is 0.6 Å). As observed in structures with other inhibitors, TM5 moves slightly away from TM2 and L5 lifts upward (23, 25, 30) (Fig. S3 A and B). The conformation of the L5 loop is slightly different from conformations observed in previous GlpG-inhibitor complexes, but it still covers the ligand. In the active site, the mode of inhibitor binding results in different conformations of S201 and H150 (Fig. S3C). The conformations of residues Y205, W236, and H254, however, closely match those of the apoenzyme rather than the conformations observed in the presence of inhibitors (23, 25, 30) (Fig. S3 D–G).
The position of the IC inhibitor resembles not only that of the Cbz-phosphonate (25) but also that of a lipid molecule previously observed in the structure of GlpG (31) (Fig. S3G). To make a comparison with substrate binding, we covalently docked the tetrahedral intermediate of a tetrapeptide comprising the four amino acids around the cleavage site of TatA into GlpG (Fig. S4A). The phenylalanine side chain in the P2′ position of the substrate shows interactions similar to those of the hydrophobic substituent on IC 16 and the Cbz-group of the phosphonate inhibitor (Fig. S4B), suggesting that the hydrophobic residues in TM2 and L5 play a role in ligand binding.
Activity-Based Probes for Rhomboids.
Some of the inhibitors identified in our screen (Table 1) carry an alkyne functional group amenable to functionalization with a fluorophore by click chemistry (32, 33). In addition, we synthesized an azide analog of the peptido sulfonyl fluoride 32 (Fig. S5A, Scheme S1, and Dataset S1). This azide analog also inhibits substrate processing by GlpG (Fig. S5B), indicating that the slight modification in the structure does not influence the activity of the molecule. The peptido sulfonyl fluoride indeed covalently reacts with GlpG, but it also labels the catalytically inactive S201A mutant (Fig. S5C). Apparently, the molecule sulfonylates amino acid residues other than the expected active-site serine and therefore does not label in an activity-dependent way. Diphenyl phosphonate 22, which showed weak inhibition of AarA, did not yield any labeling of the active AarA protease and probably acts as a competitor for substrate binding inside the active site.
The ICs 6 and 11 label GlpG, but not the GlpG S201A active-site mutant, either in detergent micelles or in crude lysates of E. coli expressing recombinant GlpG (Fig. 3A). Pretreatment of the enzyme with DCI or IC 16 blocks labeling (Fig. 3B). The same is observed for AarA (Fig. S6A). Next, we synthesized ABP 36 (Scheme S2 and Dataset S1), a preclicked version of IC 6, which also labels GlpG (Fig. 3C). Although in crude lysates there is some labeling of other proteins by IC 6, 11, and 36, the labeled rhomboid can be resolved easily on 1D gels (Fig. S6 B and C). We also treated different proteomes not containing bacterial rhomboids with IC 36 (Fig S6D). This only gave rise to the labeling of a few other protein bands, illustrating that the IC probes are suitable for use in complex protein mixtures. With ABP 36, we were able to tag endogenous GlpG in isolated E. coli membranes (Fig. 3D). This labeling was not present in a ΔglpG E. coli cell strain or in the presence of the inhibitor IC 16. ABP 36 also enabled in vivo labeling of recombinantly expressed GlpG in E. coli, which was diminished upon preincubation of the cells with IC 16 (Fig. 3E). Hence, both the alkynylated isocoumarins and fluorophore-conjugated compound 36 act as true ABPs, labeling the rhomboid active site in micelles, lysates, and in vivo. To further illustrate the applicability of ICs on live cells, we incubated P. stuartii bacteria with different AarA inhibitors. We observed the same change in cell morphology as is typical for ΔaarA strains (34), which shows the possibility of functional modulation of rhomboids by small molecules (Fig. S7).
Interestingly, the labeling pattern of GlpG reveals the mechanism of inactivation by ICs. It comprises an initial attack by the active-site serine and—depending on the presence of the 7-amino group—a second attack by a histidine residue (Fig. 3H). Indeed, when GlpG labeling was followed in time, IC 6 showed a robust and permanent signal owing to the stable carbon–nitrogen bond formed between the histidine residue and the 4-position of the IC (Fig. 3F). In contrast, IC 11 binding showed a loss in signal over time, which is the result of slow hydrolysis of the ester bond between the serine and the IC ring. This is in accordance with recent findings for DCI (30). With better gel resolution, labeling of GlpG by IC 6 shows two distinct protein species (Fig. 3G). It is likely that the lower band is GlpG with IC 6 temporarily cross-linked to both serine and histidine side chains. This species has a lower apparent molecular weight (MW) as the result of a more compact protein structure (Fig. 3I). The lower gel band disappears in time, because the ester bond is labile. The IC 6 finally forms a single-bonded probe complex with the 4-position of the IC attached to the histidine residue. As expected, IC 11 shows only the upper (single-bonded) complex.
We then applied our ABPs to evaluate the effect of small molecules on the activity state of rhomboids. To this end, GlpG was reacted with ABP 36 in the presence of maltoside detergents with increasing alkyl tail lengths, varying from 8 to 14 carbon atoms. We found that the labeling kinetics were equal in all detergent micelles except for octyl maltoside, in which the reaction rate was decreased by ∼35% (Fig. 4). An influence of detergent and lipid environments on rhomboid substrate cleavage has been reported (21, 35). Here, we see that the alkyl chain length does not have a significant effect on the rhomboid activity, unless the chains are very short compared with the natural E. coli phospholipids. Overall, this supports the observation that mild, nonionic detergents represent good environments for the reconstitution of bacterial rhomboids.
Fig. 4.

Rhomboid labeling dependent on detergent. Labeling kinetics of GlpG in different detergent environments. GlpG was incubated with 36 in the presence of maltoside detergents with increasing alkyl chain length. At different reaction times, samples were analyzed by SDS/PAGE and the increase of fluorescent gel band intensity was determined. The slope of the linear part of the curve was calculated from duplicate experiments and is given in arbitrary units per minute ± SE. *P < 0.05. DDM, dodecyl maltoside; DM, decyl maltoside; OM, octyl maltoside; TDM, tetradecyl maltoside.
Discussion
For most soluble proteases, fluorogenic or chromogenic peptide substrates are available that are based on the protease substrate specificity (2). Although helix-breaking residues within a TM seem to be an important factor for cleavage (36), it remains largely unclear how the initial substrate recognition by intramembrane proteases takes place. For the rhomboid AarA, a consensus sequence around the scissile bond has been discovered (37), but substrate recognition may extend over a large part of the TM, so small peptide substrates are not cleaved efficiently enough to be used as diagnostic tools in inhibitor screens. Here, we report a label-free MALDI-TOF–based assay for screening potential rhomboid inhibitors. The assay uses a protein substrate with a complete TM, which ensures proper recognition by rhomboids. A good Z′-factor illustrates that this assay is amenable to high-throughput screening. In addition to the detection of inhibition, MALDI-TOF–based analysis of substrate processing may be used in future studies of the cleavage kinetics of rhomboids (Fig. S8). Comparison of parameters such as kcat/KM of different substrates or rhomboid mutants might give further insight into the factors that govern substrate recognition.
In the current assay, we screened a focused library of different electrophiles to find covalent, mechanism-based inhibitors. Several IC derivatives were found that showed higher potency than previously reported IC inhibitors. Interestingly, some inhibitors of GlpG did not show inhibition of AarA, and vice versa. For example, compounds 10 and 22 inhibited AarA and displayed no inhibitory effect on GlpG, whereas compound 20 inhibited GlpG and had almost no effect on AarA. Although GlpG and AarA can cleave the same TatA substrate and apparently have an overlapping substrate recognition motif, the differences in the inhibition profile suggest that there is enough structural variation around the active site to design inhibitors selective for one rhomboid over others. Originally, the IC scaffold was designed for the inhibition of soluble serine proteases (38). The reported IC reagents indeed are not highly selective for rhomboids. Although this is not problematic for use in activity-based protein profiling, other applications may require higher selectivity. Further optimization of the IC scaffold for rhomboids may be aided by structural design based on the crystal structure reported here.
The structural characterization of the most potent hit structure IC 16 showed a binding mode different from the one reported for the IC JLK-6. For soluble serine proteases, multiple modes of IC inhibition have been observed before. In elastase, for example, ICs may form either an acyl enzyme species (39)—i.e., single-bonded to the catalytic serine residue—or a cross-linked species (40) bonded to both serine and histidine catalytic residues. Despite these data supporting the flexible mode of IC binding, it was surprising to see that IC 16 reacts with H150 whereas the related JLK-6 binds to H254. The interactions between the bulky hydrophobic group at the 3-position of IC 16 and hydrophobic residues on TM2 and L5 may bring the electrophilic 4-position closer to H150. The smaller methoxy group on JLK-6 perhaps does not undergo this interaction, promoting reaction with H254. The present mode of IC binding also may explain our observation that the ester bond is hydrolyzed slowly during the labelings with the ABPs 6 and 11 derived from IC 16: this probably happens through the activation of a water molecule by H254 and subsequent attack onto the ester bond between S201 and the IC. The observation that both the histidines in GlpG can perform a nucleophilic attack and form an irreversible complex may be important for the future design of rhomboid-specific inhibitors.
Rhomboids cleave single-pass TM proteins, but at the moment, an enzyme-substrate structure is not available for any rhomboid. Enzyme-inhibitor complexes and simplified substrate models therefore are valuable alternatives to gain insight in rhomboid–ligand interactions. In our structure, the hydrophobic substituent in IC 16 is sufficiently big that the phenyl group protrudes toward the bilayer and interacts with hydrophobic residues on TM2 and L5. A model of a tetrapeptide derived from the TatA substrate suggests that these might be residues involved in primed-site interaction. In general, the P′-residues of the substrate (largely part of the TM helix) are hydrophobic, and the present structure thus provides a further extension in our understanding of how a substrate may interact with the enzyme.
Tagged ICs, either comprising an alkyne “minitag” or directly conjugated to a fluorescent label, act as rhomboid ABPs, can distinguish between the active and inactive forms, and allow detection of endogenous rhomboid. These tools provide a means for future examination of the possible mechanisms of rhomboid activity regulation, which remain largely unknown. The rhomboid ABPs may be used not only on purified preparations, but also in lysates and on whole cells. This will enable the future study of rhomboids in whole proteomes, which may be particularly beneficial for the study of instable rhomboids, such as eukaryotic rhomboids that up to date have not been purified in their active form. ABPs also may facilitate profiling of rhomboid activity in proteomes derived from different cell or tissue types. Altogether, these probes represent valuable tools for the study of rhomboid proteases.
Methods
Protein Purification.
Expression and purification of rhomboids and substrate were performed as previously described (22), with slight modifications (SI Methods).
Substrate Cleavage Assay.
TatA cleavage by the rhomboids GlpG and AarA was performed in buffer (50 mM Hepes, pH 7.4; 10% (vol/vol) glycerol; 0.0125% DDM) at 37 °C. The reaction was stopped by adding 1 volume of 2% (vol/vol) TFA in water and mixed with 2,5-dihydroxyacetophenone MALDI matrix [sample:2% (vol/vol) TFA in water:matrix, 1:1:1]. On a MALDI target plate, 1.5 µL of sample/TFA/matrix solution was spotted, and samples were measured using a Bruker Ultraflex MALDI-TOF/TOF mass spectrometer. The signal intensities of the protein substrate and the rhomboid cleavage product were analyzed using a custom-made software macro and imported into a Microsoft Excel worksheet for further analysis. The Z′-factor was calculated by measuring the substrate to product a ratio of eight independent sets of positive (0.5 µM AarA S201A inactive mutant + 10 µM TatA; no cleavage = full inhibition) and negative control (0.5 µM AarA + 10 µM TatA; full cleavage = no inhibition). The reaction was stopped after 30 min and analyzed by MALDI-MS.
MALDI-Based Inhibitor Screening.
Purified AarA (0.5 µM) or GlpG (1.5 µM) was incubated with small molecules (200 µM; from 10 mM DMSO stocks) for 20 min. The cleavage reaction was started by adding 10 µM TatA and reacted at 37 °C for 30 min (AarA) or 60 min (GlpG). All reactions were done in 10 µL volume and stored at −20 °C until MALDI-MS analysis. Inhibitor screening was performed in duplicate, and every reaction was analyzed in triplicate.
Synthesis of Compounds.
The synthesis of compounds 35 and 36 is described in SI Methods. All other compounds were made as described (41–43) or obtained from commercial suppliers.
Crystallization.
Expression and purification were carried out as described previously (23). Crystals of GlpG in complex with IC 16 were obtained as described in SI Methods.
Click Chemistry.
Complexes between rhomboid and alkyne- or azide-ABPs were coupled to fluorophore using Cu(I)-catalyzed click chemistry. Coupling was done by adding 50 μM tetramethylrhodamine (azido-propyl-TAMRA for alkyne probes, propargyl-TAMRA for azide probes; both from 5 mM DMSO stock), 17 μM Tris-(triazolylbenzylmethyl)-amine (from 1.7 mM stock in H2O), 1 mM CuSO4 (from freshly prepared 100 mM stock in H2O), and 1 mM Tris-(2-carxyethyl)phosphine (from 100 mM stock in H2O). The reaction was incubated for 1 h and stopped by adding 4× sample buffer.
ABP Labeling of Rhomboids.
Purified enzymes and bacterial lysates were incubated with ABPs at 37 °C and analyzed directly by SDS/PAGE (in case of fluorophore-bound ABP 36) or visualized using click chemistry before gel electrophoresis. Samples were separated using 15% Tris-glycine or 10% Bis-Tris gels. Fluorescent gel bands were detected using a Typhoon TRIO+ fluorescent scanner.
Supplementary Material
Acknowledgments
We thank Christian Ried for help with bioinformatic analysis, Matthew Freeman for supplying plasmids for rhomboid and substrate expression, Garib Murshdov for advice on structural refinement, Klaus Neuhaus for the P. stuartii strain, Christine Polte for help with S2 work, and Dieter Langosch and Matt Bogyo for critical reading of the manuscript. We acknowledge financial support from the Deutsche Forschungsgemeinschaft (Emmy Noether program), the Center for Integrated Protein Science Munich (to S.H.L.V.), and a Marie Curie Intra-European fellowship (to K.R.V.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3ZEB).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215076110/-/DCSupplemental.
References
- 1.Turk B, Turk SA, Turk V. Protease signalling: The cutting edge. EMBO J. 2012;31(7):1630–1643. doi: 10.1038/emboj.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poreba M, Drag M. Current strategies for probing substrate specificity of proteases. Curr Med Chem. 2010;17(33):3968–3995. doi: 10.2174/092986710793205381. [DOI] [PubMed] [Google Scholar]
- 3.Deu E, Verdoes M, Bogyo M. New approaches for dissecting protease functions to improve probe development and drug discovery. Nat Struct Mol Biol. 2012;19(1):9–16. doi: 10.1038/nsmb.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 5.Serim S, Haedke U, Verhelst SHL. Activity-based probes for the study of proteases: Recent advances and developments. ChemMedChem. 2012;7(7):1146–1159. doi: 10.1002/cmdc.201200057. [DOI] [PubMed] [Google Scholar]
- 6.Urban S. Taking the plunge: Integrating structural, enzymatic and computational insights into a unified model for membrane-immersed rhomboid proteolysis. Biochem J. 2010;425(3):501–512. doi: 10.1042/BJ20090861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erez E, Fass D, Bibi E. How intramembrane proteases bury hydrolytic reactions in the membrane. Nature. 2009;459(7245):371–378. doi: 10.1038/nature08146. [DOI] [PubMed] [Google Scholar]
- 8.Wolfe MS. Intramembrane proteolysis. Chem Rev. 2009;109(4):1599–1612. doi: 10.1021/cr8004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawlings ND, Morton FR, Kok CY, Kong J, Barrett AJ. MEROPS: The peptidase database. Nucleic Acids Res. 2008;36(Database issue):D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 2001;107(2):173–182. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- 11.Koonin EV, et al. The rhomboids: A nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003;4(3):R19. doi: 10.1186/gb-2003-4-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemberg MK, Freeman M. Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 2007;17(11):1634–1646. doi: 10.1101/gr.6425307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevenson LG, et al. Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc Natl Acad Sci USA. 2007;104(3):1003–1008. doi: 10.1073/pnas.0608140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker RP, Wijetilaka R, Urban S. Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog. 2006;2(10):e113. doi: 10.1371/journal.ppat.0020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Donnell RA, et al. Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J Cell Biol. 2006;174(7):1023–1033. doi: 10.1083/jcb.200604136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444(7116):179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 17.Wu Z, et al. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13(12):1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Ha Y. Open-cap conformation of intramembrane protease GlpG. Proc Natl Acad Sci USA. 2007;104(7):2098–2102. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks CL, Lazareno-Saez C, Lamoureux JS, Mak MW, Lemieux MJ. Insights into substrate gating in H. influenzae rhomboid. J Mol Biol. 2011;407(5):687–697. doi: 10.1016/j.jmb.2011.01.046. [DOI] [PubMed] [Google Scholar]
- 20.Lei X, Li YM. The processing of human rhomboid intramembrane serine protease RHBDL2 is required for its proteolytic activity. J Mol Biol. 2009;394(5):815–825. doi: 10.1016/j.jmb.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 21.Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci USA. 2005;102(6):1883–1888. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemberg MK, et al. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 2005;24(3):464–472. doi: 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vinothkumar KR, et al. The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J. 2010;29(22):3797–3809. doi: 10.1038/emboj.2010.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pierrat OA, et al. Monocyclic β-lactams are selective, mechanism-based inhibitors of rhomboid intramembrane proteases. ACS Chem Biol. 2011;6(4):325–335. doi: 10.1021/cb100314y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue Y, et al. Conformational change in rhomboid protease GlpG induced by inhibitor binding to its S’ subsites. Biochemistry. 2012;51(18):3723–3731. doi: 10.1021/bi300368b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherratt AR, Blais DR, Ghasriani H, Pezacki JP, Goto NK. Activity-based protein profiling of the Escherichia coli GlpG rhomboid protein delineates the catalytic core. Biochemistry. 2012;51(39):7794–7803. doi: 10.1021/bi301087c. [DOI] [PubMed] [Google Scholar]
- 27.Maegawa S, Ito K, Akiyama Y. Proteolytic action of GlpG, a rhomboid protease in the Escherichia coli cytoplasmic membrane. Biochemistry. 2005;44(41):13543–13552. doi: 10.1021/bi051363k. [DOI] [PubMed] [Google Scholar]
- 28.Tang J, Hernández G, LeMaster DM. Increased peptide deformylase activity for N-formylmethionine processing of proteins overexpressed in Escherichia coli: Application to homogeneous rubredoxin production. Protein Expr Purif. 2004;36(1):100–105. doi: 10.1016/j.pep.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 30.Xue Y, Ha Y. Catalytic mechanism of rhomboid protease GlpG probed by 3,4-dichloroisocoumarin and diisopropyl fluorophosphonate. J Biol Chem. 2012;287(5):3099–3107. doi: 10.1074/jbc.M111.310482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci USA. 2007;104(2):462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolb HC, Finn MG, Sharpless KB. Click chemistry: Diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 33.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125(16):4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 34.Rather PN, Orosz E. Characterization of aarA, a pleiotrophic negative regulator of the 2′-N-acetyltransferase in Providencia stuartii. J Bacteriol. 1994;176(16):5140–5144. doi: 10.1128/jb.176.16.5140-5144.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherratt AR, Braganza MV, Nguyen E, Ducat T, Goto NK. Insights into the effect of detergents on the full-length rhomboid protease from Pseudomonas aeruginosa and its cytosolic domain. Biochim Biophys Acta. 2009;1788(11):2444–2453. doi: 10.1016/j.bbamem.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Urban S, Freeman M. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell. 2003;11(6):1425–1434. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- 37.Strisovsky K, Sharpe HJ, Freeman M. Sequence-specific intramembrane proteolysis: Identification of a recognition motif in rhomboid substrates. Mol Cell. 2009;36(6):1048–1059. doi: 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harper JW, Powers JC. 3-Alkoxy-7-amino-4-chloroisocoumarins—a new class of suicide substrates for serine proteases. J Am Chem Soc. 1984;106(24):7618–7619. [Google Scholar]
- 39.Powers JC, Oleksyszyn J, Narasimhan SL, Kam CM. Reaction of porcine pancreatic elastase with 7-substituted 3-alkoxy-4-chloroisocoumarins: Design of potent inhibitors using the crystal structure of the complex formed with 4-chloro-3-ethoxy-7-guanidinoisocoumarin. Biochemistry. 1990;29(12):3108–3118. doi: 10.1021/bi00464a030. [DOI] [PubMed] [Google Scholar]
- 40.Vijayalakshmi J, Meyer EF, Jr, Kam CM, Powers JC. Structural study of porcine pancreatic elastase complexed with 7-amino-3-(2-bromoethoxy)-4-chloroisocoumarin as a nonreactivatable doubly covalent enzyme-inhibitor complex. Biochemistry. 1991;30(8):2175–2183. doi: 10.1021/bi00222a022. [DOI] [PubMed] [Google Scholar]
- 41.Haedke U, Götz M, Baer P, Verhelst SHL. Alkyne derivatives of isocoumarins as clickable activity-based probes for serine proteases. Bioorg Med Chem. 2012;20(2):633–640. doi: 10.1016/j.bmc.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 42.Brouwer AJ, Ceylan T, van der Linden T, Liskamp RMJ. Synthesis of beta-aminoethanesulfonyl fluorides or 2-substituted taurine sulfonyl fluorides as potential protease inhibitors. Tetrahedron Lett. 2009;50(26):3391–3393. [Google Scholar]
- 43.Brouwer AJ, Ceylan T, Jonker AM, van der Linden T, Liskamp RMJ. Synthesis and biological evaluation of novel irreversible serine protease inhibitors using amino acid based sulfonyl fluorides as an electrophilic trap. Bioorg Med Chem. 2011;19(7):2397–2406. doi: 10.1016/j.bmc.2011.02.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.