

Abstract
Over the last two decades, we have extensively studied the genetics of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency (CAH) and have performed 8,290 DNA analyses of the CYP21A2 gene on members of 4,857 families at risk for CAH—the largest cohort of CAH patients reported to date. Of the families studied, 1,507 had at least one member affected with one of three known forms of CAH, namely salt wasting, simple virilizing, or nonclassical CAH. Here, we report the genotype and phenotype of each affected patient, as well as the ethnic group and country of origin for each patient. We showed that 21 of 45 genotypes yielded a phenotypic correlation in our patient cohort. In particular, contrary to what is generally reported in the literature, we found that certain mutations, for example, the P30L, I2G, and I172N mutations, yielded different CAH phenotypes. In salt wasting and nonclassical CAH, a phenotype can be attributed to a genotype; however, in simple virilizing CAH, we observe wide phenotypic variability, particularly with the exon 4 I172N mutation. Finally, there was a high frequency of homozygous I2G and V281L mutations in Middle Eastern and Ashkenazi Jewish populations, respectively. By identifying the predominant phenotype for a given genotype, these findings should assist physicians in prenatal diagnosis and genetic counseling of parents who are at risk for having a child with CAH.
Keywords: pseudogene-derived mutations, genotype-phenotype association
The most common cause of congenital adrenal hyperplasia (CAH) is 21-hydroxylase deficiency (1). Phenotypically, CAH can be divided into classical and nonclassical (NC) forms, with the classical form presenting as salt-wasting (SW) or simple-virilizing (SV) CAH (1), both of which can result in genital ambiguity in the affected female. Mutations in the CYP21A2 gene cause varying degrees of loss of 21-hydroxylase activity, resulting in different severities. In vitro studies performed on a relatively limited number of mutations have confirmed a correlation between disease severity and the degree of functional loss of 21-hydroxylase. Mutations resulting in complete inactivation of 21-hydroxylase activity are associated with the SW phenotype. Those that reduce enzyme activity to ∼2% cause the SV phenotype, whereas those that reduce activity to between 10% and 75% result in the mild NC phenotype (2–12).
We recently used computational modeling to correlate disease severity with 113 known mutations on the basis of the extent to which the enzyme is disrupted in silico (13). By humanizing the crystal structure of bovine CYP21A2, we found that mutations that affect critical enzyme functions, such as membrane anchoring, heme binding, and substrate binding, or alter enzyme stability result in a complete loss of functionality and SW disease. In contrast, mutations that affect the transmembrane region or conserved hydrophobic patches result in up to a 98% reduction in enzyme activity and SV disease. Mild NC disease arises from interference in oxidoreductase interactions, salt-bridge and hydrogen bonding networks, and nonconserved hydrophobic clusters. This structural biology information, albeit with limited in vitro validation, should help clinicians assign phenotypes to novel mutations.
Previous reports of genotype–phenotype correlations (14–16) and our recent in silico assignment of protein structure to a genotype (13) suggest that the phenotype of a CAH fetus and the subsequent newborn can be predicted with reasonable certainty by performing prenatal genetic analysis (17). This report explores this hypothesis.
The Adrenal Steroid Disorders Program in New York has the largest cohort of CAH patients. Over the last two decades, we have performed 8,290 DNA analyses of the CYP21A2 gene and report herein the results of the analysis, including data for 1,507 families who have at least one member affected with CAH. The remaining analyses were performed on unaffected subjects with a family history of CAH who were either heterozygous or had no mutations.
Our data add value for several reasons. First, we analyzed a heterogeneous population as opposed to past reports, which were drawn from specific countries (18–20). Second, our data provide a framework for physicians to ascertain the prevalent form of CAH for a given genotype. Third, a catalog of genotype–phenotype correlations will be valuable for prenatal diagnosis of CAH, where hormonal diagnosis is not possible and the physician must rely on genetic data. Finally, the data will assist in the counseling of parents who are heterozygous for the CYP21A2 mutation or those undergoing preimplantation genetic diagnosis.
Results
CYP21A2 mutations arise from either CYP21A1P microconversion or the unequal cross-over in chromosome 6 (Figs. 1 and 2). Of these, we have routinely studied nine common allelic mutations, the frequency of which is shown in Table 1. The frequency values in our patient cohort are ranked from most to least frequent (1–9) and are compared against rankings reported in four previous studies (15, 16, 21, 22). Overall, the rankings were similar, especially with respect to the high frequency of deletion and g.655A/C>G (I2G) mutations and the low frequency of P30L, exon 3 8-bp deletion (E3Δ8bp), and exon 6 I236N, V237E, and M239K cluster (E6) mutations (Table 1). One critical distinction between our data and three previous studies is the enrichment of the exon 7 V281L mutation noted in our cohort. Only one study presents a similar frequency for this mutation (21). This enrichment is likely due to the large number of Ashkenazi Jews within our population, who we find have a significantly higher frequency of this mutation (Table 2).
Fig. 1.
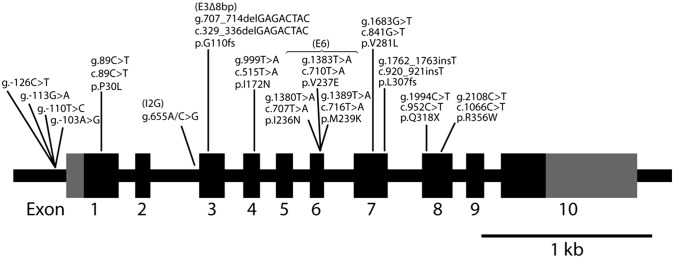
Common CYP21A2 mutations resulted from CYP21A1P gene microconversion. Of 15 disease-causing CYP21A2 mutations arising from CYP21A1P gene microconversion, 4 are in the promoter, 1 is in the intron, and 10 are in the coding region. For the promoter and intronic mutations, nucleotide changes in the genomic sequence are shown. For mutations in the coding region, nucleotide changes in the genomic and cDNA sequence, as well as the amino acid residue changes in the protein sequence, are listed. The coding sequences are in black boxes, and the UTRs are in gray. The g.655A/C>G mutation on intron 2, the g.707_714delGAGACTAC mutation on exon 3, and the p.I236N, p.V237E and p.M239K cluster mutations on exon 6 are designated as I2G, E3∆8bp, and E6, respectively, in this article.
Fig. 2.

CYP21A2 gene deletion. Due to the high homology between the RP1-C4A-CYP21A1P-TNXA and the RP2-C4B-CYP21A2-TNXB regions, gene rearrangement sometimes occurs because of unequal cross-over during meiosis. This rearrangement causes a 30-kb fragment deletion resulting in a CYP21A1P/CYP21A2 chimera. To date, nine types of chimera (CH1–CH9) with different junction sites have been identified. CYP21A1P and CYP21A2 exons are in open and gray boxes, respectively.
Table 1.
Frequency of common allelic mutations in this study and previous reports
| Allelic mutation | Frequency (rank) |
||||
| Current report | Marino et al. (21) | Wedell (16) | Stikkelbroeck et al. (22) | Krone et al. (15) | |
| Deletion and large gene conversions | 20.0% (3) | 11.2% (3) | 32.2% (1) | 31.9% (1) | 29.0% (2) |
| P30L | 2.6% (7) | 0.7% (9) | 1.6% (7) | 0.3% (9) | 2.6% (7) |
| I2G | 22.9% (2) | 20.6% (2) | 26.6% (2) | 28.1% (2) | 30.3% (1) |
| E3Δ8bp | 2.1% (8) | 0.8% (8) | 1.1% (8) | 4.3% (5) | 1.6% (8) |
| I172N | 8.2% (4) | 8.2% (4) | 19.8% (3) | 12.4% (3) | 19.7% (3) |
| E6 | 2.1% (8) | 2.0% (7) | 1.1% (8) | 3.0% (7) | 1.0% (9) |
| V281L | 23.9% (1) | 26.2% (1) | 5.7% (4) | 2.2% (8) | 2.9% (6) |
| Q318X | 3.5% (6) | 6.7% (5) | 2.4% (6) | 3.5% (6) | 4.8% (4) |
| R356W | 3.6% (5) | 4.2% (6) | 3.0% (5) | 8.4% (4) | 4.5% (5) |
| Patient’s ethnicity/country of origin | Heterogeneous | Argentinian | Swedish | Dutch | South German |
| Patient population | 1,507 | 454 | 198 | 155 | |
| Alleles analyzed | 3,005 | 866 | ∼400 | 370 | 310 |
Table 2.
Self-reported ethnicity or country of origin
| Genotype | African American | Ashkenazi Jew | Asian | East Indian | European | Hispanic American | Middle Eastern | Mixed ethnicity | Others | Total |
| Del/Del | 3 | 5 | 1 | 4 | 50 | 15 | 9 | 2 | 27 | 116 (8.7%) |
| Del/P30L | 2 | 6 | 3 | 4 | 15 (1.1%) | |||||
| Del/I2G | 6 | 3 | 5 | 1 | 49 | 9 | 4 | 13 | 27 | 117 (8.7%) |
| Del/E3Δ8bp | 1 | 1 | 5 | 1 | 1 | 8 | 17 (1.3%) | |||
| Del/I172N | 3 | 2 | 28 | 7 | 1 | 7 | 24 | 72 (5.4%) | ||
| Del/E6 | 1 | 3 | 2 | 1 | 3 | 10 (0.7%) | ||||
| Del/V281L | 39 | 23 | 10 | 3 | 20 | 95 (7.1%) | ||||
| Del/Q318X | 1 | 9 | 1 | 3 | 5 | 19 (1.4%) | ||||
| Del/R356W | 1 | 9 | 3 | 1 | 2 | 7 | 23 (1.7%) | |||
| P30L/P30L | 1 | 1 | 1 | 3 (0.2%) | ||||||
| P30L/I2G | 1 | 2 | 1 | 11 | 1 | 2 | 5 | 23 (1.7%) | ||
| P30L/E3Δ8bp | 3 | 1 | 4 (0.3%) | |||||||
| P30L/I172N | 1 | 2 | 1 | 1 | 4 | 9 (0.7%) | ||||
| P30L/E6 | 1 | 1 (0.1%) | ||||||||
| P30L/V281L | 2 | 2 | 2 | 2 | 8 (0.6%) | |||||
| P30L/Q318X | 1 | 1 | 1 | 2 | 5 (0.4%) | |||||
| P30L/R356W | 3 | 1 | 2 | 6 (0.4%) | ||||||
| I2G/I2G | 1 | 8 | 5 | 5 | 40 | 16 | 40 | 7 | 33 | 155 (11.6%) |
| I2G/E3Δ8bp | 1 | 7 | 1 | 1 | 4 | 14 (1.0%) | ||||
| I2G/I172N | 1 | 3 | 1 | 30 | 2 | 2 | 11 | 50 (3.7%) | ||
| I2G/E6 | 6 | 1 | 2 | 5 | 14 (1.0%) | |||||
| I2G/V281L | 37 | 36 | 9 | 6 | 24 | 112 (8.4%) | ||||
| I2G/Q318X | 1 | 1 | 2 | 11 | 5 | 5 | 25 (1.9%) | |||
| I2G/R356W | 1 | 2 | 1 | 10 | 2 | 6 | 22 (1.6%) | |||
| E3Δ8bp/E3Δ8bp | 1 | 3 | 4 (0.3%) | |||||||
| E3Δ8bp/I172N | 2 | 1 | 3 | 6 (0.4%) | ||||||
| E3Δ8bp/E6 | 1 | 1 (0.1%) | ||||||||
| E3Δ8bp/V281L | 1 | 2 | 2 | 1 | 6 (0.4%) | |||||
| E3Δ8bp/Q318X | 1 | 1 | 2 (0.1%) | |||||||
| E3Δ8bp/R356W | 1 | 1 | 2 | 4 (0.3%) | ||||||
| I172N/I172N | 1 | 1 | 2 | 6 | 1 | 5 | 7 | 23 (1.7%) | ||
| I172N/E6 | 1 | 1 | 2 (0.1%) | |||||||
| I172N/V281L | 3 | 12 | 7 | 1 | 8 | 31 (2.3%) | ||||
| I172N/Q318X | 1 | 2 | 5 | 1 | 2 | 11 (0.8%) | ||||
| I172N/R356W | 1 | 1 | 1 | 5 | 2 | 1 | 7 | 18 (1.3%) | ||
| E6/E6 | 1 | 3 | 1 | 4 | 3 | 12 (0.9%) | ||||
| E6/V281L | 2 | 3 | 1 | 2 | 8 (0.6%) | |||||
| E6/Q318X | 1 | 1 (0.1%) | ||||||||
| E6/R356W | 1 | 1 (0.1%) | ||||||||
| V281L/V281L | 135 | 22 | 12 | 3 | 3 | 37 | 212 (15.8%) | |||
| V281L/Q318X | 5 | 1 | 10 | 2 | 3 | 21 (1.6%) | ||||
| V281L/R356W | 4 | 1 | 4 | 4 | 13 (1.0%) | |||||
| Q318X/Q318X | 1 | 2 | 5 | 1 | 9 (0.7%) | |||||
| Q318X/R356W | 1 | 1 | 1 | 3 (0.2%) | ||||||
| R356W/R356W | 1 | 5 | 6 | 1 | 2 | 15 (1.1%) | ||||
| TOTAL | 24 (1.8%) | 260 (19.4%) | 20 (1.5%) | 20 (1.5%) | 427 (31.9%) | 132 (9.9%) | 88 (6.6%) | 61 (4.6%) | 306 (22.9%) | 1,338 (100%) |
The ethnic origin, either reported by the patient or a family member, is listed for the 45 genotypes described. Ethnicities with <10 patients (Native American, Sephardic Jews, Australian, North African, Eskimo, and Russian) and patients with unknown ethnicity or reported themselves as Caucasian without further distinction, were grouped as “Others” in the table.
Fig. 3 and Tables S1–S4 show 45 genotypes representing nine common allelic mutations on one or both alleles, without distinguishing between their maternal or paternal inheritance. The phenotypes are divided into the NC, SV, and SW forms of CAH. The SW form of CAH is prominent in deletion or intronic splice mutations, namely Del/Del (19.1%), I2G/I2G (23.6%), and Del/I2G (18.2%). In contrast, SV CAH was observed most frequently in Del/I172N (28.3%) and I2G/I172N (19.3%) mutations. NC CAH was associated with V281L/V281L (38.5%), I2G/V281L (19.8%), and Del/V281L (17.4%) mutations. Interestingly, of the missense mutations, our in silico analysis (13) attributes the I172N and V281L to the SV and NC forms, respectively, confirming long-term clinical observations.
Fig. 3.

Frequency of CYP21A2 genotypes in 1,507 CAH patients. The number of CAH patients with each of the CYP21A2 genotypes (NC, green; SV, blue; SW, red) is shown. This figure should be used in conjunction with Tables S1–S6.
Computational modeling further predicts that the exon 4 I172N mutation, by causing a loss of the hydrophobic pocket, will cause SV disease (13). This prediction is confirmed by in vitro expression studies, which demonstrate a reduction in 21-hydroxylase activity to ∼2% (5). However, our data show that SV disease occurs in only ∼76% of CAH patients who carry the I172N mutation on one allele and a severe mutation on the other (Fig. 3; Table S1). Approximately 23% of these patients present with SW CAH, indicating a block in aldosterone production. This genotype–phenotype discordance could result from reduced expression of the mutated 21-hydroxylase enzyme arising from subtle variations in transcriptional regulation or downstream protein translation. One patient with a Del/I172N genotype has a NC phenotype. To account for this genotype–phenotype discrepancy, we speculated that the patient had a duplicated CYP21A2 allele with a mild rare mutation. Similar examples of CYP21A2 duplication can be found in unaffected individuals carrying the Q318X mutation in the compound heterozygous form (23–26). We recently reported an erroneous prenatal diagnosis owing to this duplication (26). In this haplotype, the Q318X-mutated allele coexists with a normal CYP21A2 allele on the same chromosome. Because CAH is an autosomal recessive disorder, the phenotype is correlated with the mildest mutation among the three alleles.
Likewise, although the intron 2 I2G splicing mutation results primarily (78.6%) in SW CAH, we noted that ∼20% patients present with SV disease (Fig. 3; Table S2). The mechanism underlying the variation(s) in the clinical phenotype of a single dominant mutation remains unknown. One possible mechanism is that a percentage of patients produce a small amount of the correctly spliced product to decrease the severity of the phenotype. Further, although the P30L mutation causes only 40–70% loss of 21-hydroxylase enzyme activity in vitro and is traditionally associated with the NC form (27), we found that >30% P30L-carrying patients present with classical CAH (Fig. 3; Table S3).
In contrast, the phenotypic outcome of the exon 7 V281L mutation is consistent with in silico predictions, in which enzyme activity is only partially reduced due to steric clashes arising from increased chain length of the mutant Leu residue (13). However, although 98% of cases are NC, a smaller number of patients do present with classical CAH (Fig. 3; Table S4), suggesting that genotype–phenotype or structure–phenotype correlations in CAH are usually not perfect. Sanger sequencing (28) of all nine cases of classical CAH with the V281L mutation showed no other mutation from the proximal promoter to the proximal 3′ UTR region in the CYP21A2 gene to account for the phenotypic variation. We are unclear of the mechanism through which a ∼50% reduction in 21-hydroxylase activity results in classical CAH.
With the variation of phenotype noted in common mutations, we identified in our cohort a select number of mutations that display only SW or NC disease. Our findings indicate that a combination of any of the following mutations, Del, E3Δ8bp, E6 (I236N, V237E, and M239K), Q318X, and R356W, will almost invariably result in SW CAH (Fig. 3; Tables S5 and S6). Although M239K does not affect enzyme structure or function, both I236N and V237E cause a near abrogation of enzyme activity in vitro. Likewise, the R356W mutation causes a disruption of the H-bonding with residue Q389 (which lies within 3.5 Å), resulting in increased steric clashes, changes in tertiary structure, alterations in heme binding, and abrogation of enzyme activity (13). Interestingly, however, patients carrying the V281L/R356W genotype are assigned to the NC disease-only phenotype (Fig. 3; Table S5). In this case, complete loss of function due to R356W is compensated by a partial loss of activity of the V281L-carrying enzyme. Similar compensation by V281L is also seen in the Del/V281L and E3∆8bp/V281L genotypes. In contrast to the select mutations solely causing SW and NC CAH, mutations such as I172N, which have been attributed to SV alone, may also result in SW, or in limited instances, to NC CAH (Fig. 3; Table S6).
Further, we found that each sex is equally represented. In genotypes with V281L mutation on at least one allele, there are more females than males, probably due to the obvious signs of hyperandrogenemia in females. In males, signs of adrenal hyperandrogenemia are difficult to ascertain because most of the testosterone is secreted by the testis and not the adrenals.
Table 2 illustrates the ethnic diversity of CAH genotypes. We note that certain genotypes are more frequent in specific ethnic groups. For example, the I2G/I2G genotype is more prevalent in the Middle Eastern population. As noted before, the exon 7 V281L mutation associated with NC CAH is very frequent in the Ashkenazi Jewish population. We also find that CAH is rare in Asian, African American, and East Indian populations in this cohort.
Finally, 169 patients with genotypes that are not the combination of frequent maternal and paternal allelic mutations are shown in Table S7. Only one member from immediate families with more than one sibling affected with CAH per generation is included. All of these genotypes are very infrequent in our cohort and therefore represent less than five patients for each genotype.
Discussion
This study provides a compendium of the largest number of CAH patients with complete genotype–phenotype correlations (21, 22, 29). In each case, either the phenotype was assigned by hormonal profiles and physical examination by the primary investigator or thoroughly discussed with the referring physician for confirmation. We found that the predictability of a CAH phenotype was less certain than anticipated, which leads to several broad conclusions regarding the diversity of this otherwise monogenic disorder.
First, we found a direct genotype–phenotype correlation in <50% (21 of 45) of the genotypes studied. Using the same 45 genotypes, Krone et al. (15) and Speiser et al. (14) came to a similar conclusion, but with significantly fewer patients. This lack of predictability may be because of the methodologies that we and others have used in clinical practice. In our cohort, patient samples were analyzed using allele-specific PCR designed to identify a specific panel of the nine most frequent mutations. Multiplex ligation-dependent probe amplification (MLPA) (30) is also frequently used by others for locus-specific mutation detection. Select cases of genotype–phenotype discordance were subject to Sanger sequencing, which did not reveal any further mutations that could account for the discordance. Sanger sequencing was also performed on patients with a CAH phenotype, but in whom none of the nine common mutations were identified. These sequencing results yielded a list of previously unreported mutations, as well as rare genotypes attributable to different allelic variations of known mutations (Table S7). We suggest that patients in whom there is genotype–phenotype discordance may benefit from whole-exome sequencing.
Second, we found that certain mutations can cause different CAH phenotypes (Fig. 4). For example, although in most cases the intron 2 I2G mutation is associated with the SW phenotype, some patients present with the SV form. The I2G (g.655A/C>G) mutation activates a cryptic upstream 3′ splice acceptor site and causes aberrant splicing. Its occasional association with the SV form is probably due to the correct splicing of a small number of transcripts. In contrast, although the exon 4 I172N mutation predominantly results in SV CAH, it can also cause SW CAH for unknown reasons. Likewise, whereas P30L has traditionally been associated with the NC category, we report patients with classical disease (Fig. 3; Table S3).
Fig. 4.
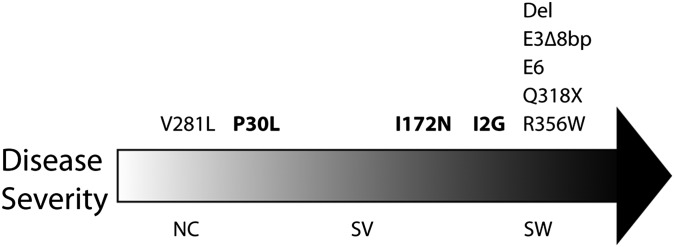
Genotype–phenotype association in CAH caused by 21-hydroxylase deficiency. There is a high genotype–phenotype concordance in the V281L, del, E3∆8bp, E6, Q318X, and R356W mutations. In contrast, whereas the P30L mutation is frequently associated with NC, >30% patients carrying the P30L mutation display classical CAH phenotypes. Similarly, although the I172N mutation is usually associated with SV, it is also seen in 23% of SW CAH patients. Likewise, the I2G mutation is mostly associated with SW but is also observed in 20% of SV CAH patients.
Third, we showed that disease severity can be generally predicted with the genotypes for SW and NC CAH; however, in the SV from of the disorder, we observed wide phenotypic variability. Most cases carrying the exon 7 V281L mutation were of the NC variety, even when the other allele bears a SW mutation, such as R356W. We identified one exception, a patient homozygous for V281L, who was surprisingly a salt waster (serum sodium: 112 mEq/L; serum potassium: 7.6 mEq/L). As this patient was referred from Iran, it is difficult to exclude another mutation without additional sequencing. Unlike the relatively conserved NC phenotype of the V281L mutation, we and others find, in contrast to a previous report, that the exon 4 I172N mutation is associated with both the SV and SW phenotypes (15, 21, 22).
Fourth, we found no significant sex difference for any of the common mutations on the CYP21A2 allele. The frequency of SW CAH is similar for each sex because equally severe enzyme disruption in both males and females lead to a salt-wasting crisis shortly after birth. In contrast, SV CAH is diagnosed more readily in females because only females present with genital ambiguity. However, newborn females carrying the V281L mutation do not have ambiguous genitalia. However, we find a higher frequency of the V281L mutation and NC disease in females, likely reflecting the difficulty in identifying signs of adrenal hyperandrogenemia in males, because androgens are mainly produced in the testes.
Fifth, our data suggest that the homozygous genotype (i.e., identical maternal and paternal mutations) is frequent in populations with high rates of consanguinity, either owing to tradition or religious constraints. Notable is the high frequency of homozygous I2G and V281L mutations in Middle Eastern and Ashkenazi Jewish populations, respectively. It is also clear from our study that CAH is rare in African American, Asian, and East Indian ethnicities. Although African Americans represent 16% of the total population of New York City (NYC), there are very few African Americans represented in our cohort. In contrast, the Ashkenazi Jewish population constitutes a smaller percentage of the population (1,087,000 Jews, 12.5% as of 2011 in NYC and its suburbs). However, our cohort comprises a large number of Ashkenazi Jews, likely accounting for, at least in part, the high frequency of the V281L mutation and NC CAH (1 in 27, with 1 in 3 being heterozygous) (31).
Finally, mutations in the noncoding region of the CYP21A2 gene may be responsible for some of the genotype–phenotype discordance. The pseudogene-derived promoter g.-126C>T, g.-113G>A, g.-110T>C, and g.-103A>G mutations reduce transcriptional activity to 20% (32, 33) and cause NC CAH in patients (34). Likewise, the 3′ UTR g.*13G>A mutation, identified in the NCBI SNP database as rs6447 with 1–4.7% allelic frequency in different populations, is associated with NC CAH (35). When present in cis with another mild mutation on the same allele, the promoter or the 3′ UTR mutation can increase the severity of the phenotype.
Overall, our study, which reports genotype–phenotype correlations in the largest CAH cohort in the world, together with our documentation of structure–phenotype predictions, has profound implications in the diagnosis of CAH patients. These data will be of special interest to physicians performing a prenatal diagnosis of CAH. For prenatal diagnosis, fetal DNA, obtained by amniocentesis or chorionic villus sampling, is used to identify a genotype, which notwithstanding the aforementioned caveats, can be used to predict the fetal phenotype. In the case of a female fetus, this could allow for prenatal therapeutic decisions. This study confirms that prenatal DNA analysis will not predict the newborn phenotype with complete accuracy. Nonetheless, it will still be useful in predicting the predominant phenotype for a particular genotype. Consequently, even with the prenatal genotype being known, careful vigilance is necessary for newborns at risk, particularly for SW CAH. Further, a catalog of genotype–phenotype correlations, as presented here, should assist newborn screening programs. A positive test on newborn screening using 17-OH-progesterone measurements does not differentiate between the SW and SV forms. Genotyping using DNA extracted from newborn blood or from the filter paper used for newborn screening could allow the tentative assignment of a phenotype. Our data may also help with genetic counseling, particularly in instances when both parents are heterozygotes and wish to know the probability of having a child affected with a particular form of CAH. The data would equally be of benefit to affected parents undergoing a preimplantation diagnosis for CAH. Finally, in the case of novel mutations, computational modeling could be used in conjunction with our findings to assess the extent of 21-hydroxylase enzyme disruption, and in doing so, move closer to the prediction of a phenotype.
Methods
For all patients in this study, the phenotypic and genotypic form of CAH was determined. DNA was extracted from blood drawn from patients after obtaining informed consent.
Clinical and hormonal evaluations were used to assign the phenotypic form of CAH. The extent to which serum concentrations of 17-hydroxyprogesterone (17OHP), androstenedione, and testosterone are elevated above basal level corresponds with disease severity. In the SW and SV forms of CAH, affected females are born with ambiguous genitalia. Patients with SW CAH are characterized by an early life adrenal crisis, with hyponatremia and hyperkalemia resulting from impaired aldosterone secretion, despite high renin levels. SV and NC patients do not have aldosterone deficiency and are not at risk for a salt-wasting crisis, although signs of hyperandrogenemia will become evident later in life if left untreated. NC CAH is distinguished from the classical forms using a nomogram that compares baseline and corticotropin-stimulated 17OHP levels (36). However, the nomogram cannot distinguish between the low-responding NC CAH patients and the heterozygous carriers due to overlapping 17OHP levels. When in doubt, genetic testing was carried out. Females affected with the NC form of the disease are diagnosed by signs of hyperandrogenemia. Males are born with normal genitalia in all forms of CAH.
The phenotype was specified either by physical examination and monitoring of hormone values in the New York clinic patients or by discussing with the referring physicians. These discussions included a review of genital examination, Prader score, hormonal results, glucocorticoid and/or salt retaining hormone therapy, salt addition to the diet, and, in NC cases, signs of hyperandrogenemia. Serum aldosterone and plasma renin concentrations were determined to confirm the SW form of CAH. These hormones were not assayed in all referred patients.
To determine the CAH genotypes, DNA analysis was conducted using allele-specific PCR designed against eight common mutation loci in the CYP21A2 gene (37). Large gene deletion (Del) was detected by either Southern blot analysis or by PCR (38). DNA analysis was carried out on the proband, his/her parents, affected/unaffected siblings, and, whenever possible, additional relatives. When there was more than one affected offspring in the family, only the youngest was used in this study to avoid duplication of genotypes. In cases where there were genotype–phenotype discordances, we performed DNA sequencing to identify rare mutations (39). However, it is important to note that sequencing was not routinely done.
Supplementary Material
Acknowledgments
We thank Dr. Jean Wilson for suggestions and guidance. This study is supported by the Children’s Hormone Foundation, the Genesis Foundation, and the National Institute of Child Health and Human Development [Grants 7R37HD00072 (June 1, 1964–May 31, 2008), IT32HD41895 (Pediatric Endocrinology Research Training Program), and 454RR19484 (Natural History of Rare Genetic Steroid Disorders)]. M.Z. and L.S. acknowledge the support of National Institutes of Health Grants DK80459, AG23176, and AG40132.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300057110/-/DCSupplemental.
References
- 1.Wajnrajch MP, New MI. Defects of adrenal steroidogenesis. In: Jameson JL, De Groot LJ, editors. Endocrinology. Philadelphia: Elsevier; 2010. 6th Ed, Vol 2, pp 1897–1920. [Google Scholar]
- 2.Soardi FC, et al. Inhibition of CYP21A2 enzyme activity caused by novel missense mutations identified in Brazilian and Scandinavian patients. J Clin Endocrinol Metab. 2008;93(6):2416–2420. doi: 10.1210/jc.2007-2594. [DOI] [PubMed] [Google Scholar]
- 3.Tardy V, et al. Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patients affected with 21-hydroxylase deficiency and in one carrier. J Clin Endocrinol Metab. 2010;95(3):1288–1300. doi: 10.1210/jc.2009-1202. [DOI] [PubMed] [Google Scholar]
- 4.Chiou SH, Hu MC, Chung BC. A missense mutation at Ile172—Asn or Arg356—Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. 1990;265(6):3549–3552. [PubMed] [Google Scholar]
- 5.Tusie-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265(34):20916–20922. [PubMed] [Google Scholar]
- 6.Amor M, Parker KL, Globerman H, New MI, White PC. Mutation in the CYP21B gene (Ile-172----Asn) causes steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA. 1988;85(5):1600–1604. doi: 10.1073/pnas.85.5.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lajic S, et al. A cluster of missense mutations at Arg356 of human steroid 21-hydroxylase may impair redox partner interaction. Hum Genet. 1997;99(6):704–709. doi: 10.1007/s004390050436. [DOI] [PubMed] [Google Scholar]
- 8.Higashi Y, Fujii-Kuriyama Y. Functional analysis of mutant P450(C21) genes in COS cell expression system. Methods Enzymol. 1991;206:166–173. doi: 10.1016/0076-6879(91)06087-j. [DOI] [PubMed] [Google Scholar]
- 9.Hsu LC, et al. The common I172N mutation causes conformational change of cytochrome P450c21 revealed by systematic mutation, kinetic, and structural studies. J Biol Chem. 1996;271(6):3306–3310. doi: 10.1074/jbc.271.6.3306. [DOI] [PubMed] [Google Scholar]
- 10.Hu MC, Hsu LC, Hsu NC, Chung BC. Function and membrane topology of wild-type and mutated cytochrome P-450c21. Biochem J. 1996;316(Pt 1):325–329. doi: 10.1042/bj3160325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikoshkov A, Lajic S, Holst M, Wedell A, Luthman H. Synergistic effect of partially inactivating mutations in steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1997;82(1):194–199. doi: 10.1210/jcem.82.1.3678. [DOI] [PubMed] [Google Scholar]
- 12.Nikoshkov A, et al. Naturally occurring mutants of human steroid 21-hydroxylase (P450c21) pinpoint residues important for enzyme activity and stability. J Biol Chem. 1998;273(11):6163–6165. doi: 10.1074/jbc.273.11.6163. [DOI] [PubMed] [Google Scholar]
- 13.Haider S, et al. Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc Natl Acad Sci USA. 2013;110:2611–2616. doi: 10.1073/pnas.1221133110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Speiser PW, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992;90(2):584–595. doi: 10.1172/JCI115897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab. 2000;85(3):1059–1065. doi: 10.1210/jcem.85.3.6441. [DOI] [PubMed] [Google Scholar]
- 16.Wedell A. Molecular genetics of congenital adrenal hyperplasia (21-hydroxylase deficiency): Implications for diagnosis, prognosis and treatment. Acta Paediatr. 1998;87(2):159–164. doi: 10.1080/08035259850157598. [DOI] [PubMed] [Google Scholar]
- 17.Wilson RC, Mercado AB, Cheng KC, New MI. Steroid 21-hydroxylase deficiency: Genotype may not predict phenotype. J Clin Endocrinol Metab. 1995;80(8):2322–2329. doi: 10.1210/jcem.80.8.7629224. [DOI] [PubMed] [Google Scholar]
- 18.Loidi L, et al. High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf) 2006;64(3):330–336. doi: 10.1111/j.1365-2265.2006.02465.x. [DOI] [PubMed] [Google Scholar]
- 19.Rabbani B, et al. Mutation analysis of the CYP21A2 gene in the Iranian population. Genet Test Mol Biomarkers. 2012;16(2):82–90. doi: 10.1089/gtmb.2011.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skordis N, et al. Molecular defects of the CYP21A2 gene in Greek-Cypriot patients with congenital adrenal hyperplasia. Horm Res Paediatr. 2011;75(3):180–186. doi: 10.1159/000320040. [DOI] [PubMed] [Google Scholar]
- 21.Marino R, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients: Genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2011;75(4):427–435. doi: 10.1111/j.1365-2265.2011.04123.x. [DOI] [PubMed] [Google Scholar]
- 22.Stikkelbroeck NM, et al. CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: Six novel mutations and a specific cluster of four mutations. J Clin Endocrinol Metab. 2003;88(8):3852–3859. doi: 10.1210/jc.2002-021681. [DOI] [PubMed] [Google Scholar]
- 23.Wedell A, Stengler B, Luthman H. Characterization of mutations on the rare duplicated C4/CYP21 haplotype in steroid 21-hydroxylase deficiency. Hum Genet. 1994;94(1):50–54. doi: 10.1007/BF02272841. [DOI] [PubMed] [Google Scholar]
- 24.Koppens PF, Hoogenboezem T, Degenhart HJ. Duplication of the CYP21A2 gene complicates mutation analysis of steroid 21-hydroxylase deficiency: Characteristics of three unusual haplotypes. Hum Genet. 2002;111(4-5):405–410. doi: 10.1007/s00439-002-0810-7. [DOI] [PubMed] [Google Scholar]
- 25.Kleinle S, et al. Duplications of the functional CYP21A2 gene are primarily restricted to Q318X alleles: Evidence for a founder effect. J Clin Endocrinol Metab. 2009;94(10):3954–3958. doi: 10.1210/jc.2009-0487. [DOI] [PubMed] [Google Scholar]
- 26.Lekarev O, et al. Erroneous prenatal diagnosis of congenital adrenal hyperplasia owing to a duplication of the CYP21A2 gene. J Perinatol. 2013;33(1):76–78. doi: 10.1038/jp.2012.5. [DOI] [PubMed] [Google Scholar]
- 27.Tusie-Luna MT, Speiser PW, Dumic M, New MI, White PC. A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol Endocrinol. 1991;5(5):685–692. doi: 10.1210/mend-5-5-685. [DOI] [PubMed] [Google Scholar]
- 28.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94(3):441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 29.Finkielstain GP, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96(1):E161–E172. doi: 10.1210/jc.2010-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schouten JP, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.New MI. Extensive clinical experience: Nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2006;91(11):4205–4214. doi: 10.1210/jc.2006-1645. [DOI] [PubMed] [Google Scholar]
- 32.Bristow J, Gitelman SE, Tee MK, Staels B, Miller WL. Abundant adrenal-specific transcription of the human P450c21A “pseudogene”. J Biol Chem. 1993;268(17):12919–12924. [PubMed] [Google Scholar]
- 33.Chang SF, Chung BC. Difference in transcriptional activity of two homologous CYP21A genes. Mol Endocrinol. 1995;9(10):1330–1336. doi: 10.1210/mend.9.10.8544841. [DOI] [PubMed] [Google Scholar]
- 34.Araújo RS, et al. Microconversion between CYP21A2 and CYP21A1P promoter regions causes the nonclassical form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92(10):4028–4034. doi: 10.1210/jc.2006-2163. [DOI] [PubMed] [Google Scholar]
- 35.Menabò S, et al. A sequence variation in 3’UTR of CYP21A2 gene correlates with a mild form of congenital adrenal hyperplasia. J Endocrinol Invest. 2012;35(3):298–305. doi: 10.3275/7680. [DOI] [PubMed] [Google Scholar]
- 36.New MI, et al. Genotyping steroid 21-hydroxylase deficiency: Hormonal reference data. J Clin Endocrinol Metab. 1983;57(2):320–326. doi: 10.1210/jcem-57-2-320. [DOI] [PubMed] [Google Scholar]
- 37.Wilson RC, Wei JQ, Cheng KC, Mercado AB, New MI. Rapid deoxyribonucleic acid analysis by allele-specific polymerase chain reaction for detection of mutations in the steroid 21-hydroxylase gene. J Clin Endocrinol Metab. 1995;80(5):1635–1640. doi: 10.1210/jcem.80.5.7745011. [DOI] [PubMed] [Google Scholar]
- 38.Tukel T, et al. A novel semiquantitative polymerase chain reaction/enzyme digestion-based method for detection of large scale deletions/conversions of the CYP21 gene and mutation screening in Turkish families with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2003;88(12):5893–5897. doi: 10.1210/jc.2003-030813. [DOI] [PubMed] [Google Scholar]
- 39.Nimkarn S, et al. Congenital adrenal hyperplasia (21-hydroxylase deficiency) without demonstrable genetic mutations. J Clin Endocrinol Metab. 1999;84(1):378–381. doi: 10.1210/jcem.84.1.5554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.