

Summary
Cancer-associated inflammation is thought to be a barrier to immune surveillance, particularly in pancreatic ductal adenocarcinoma (PDA). Gr-1+ CD11b+ cells are a key feature of cancer inflammation in PDA, but remain poorly understood. Using a genetically engineered mouse model of PDA, we show that tumor-derived GM-CSF is necessary and sufficient to drive the development of Gr-1+ CD11b+ cells that suppressed antigen-specific T cells. In vivo, abrogation of tumor-derived GM-CSF inhibited the recruitment of Gr-1+ CD11b+ cells to the tumor microenvironment and blocked tumor development - a finding that was dependent on CD8+ T cells. In humans, PDA tumor cells prominently expressed GM-CSF in vivo. Thus, tumor-derived GM-CSF is an important regulator of inflammation and immune suppression within the tumor microenvironment.
Keywords: myeloid cells, granulocyte-macrophage colony stimulating factor, pancreas, carcinoma, T lymphocyte
INTRODUCTION
Host inflammatory responses often accompany and even promote tumor development. Although the prevailing theory of immunosurveillance holds that the adaptive arm of the immune system can mount productive anti-tumor responses that eliminate malignant cells (Schreiber et al., 2011), the developing tumor will often co-opt the host immune system for its own tumor-promoting purposes. This phenomenon is particularly notable in the development of pancreatic ductal adenocarcinoma (PDA), a highly lethal malignancy associated with a striking desmoplastic reaction and a marked infiltration of leukocytes into the stromal compartment (Clark et al., 2007). Leukocytes infiltrating PDA lesions are predominantly derived from the innate immune system and coordinate a network of local immune suppression (Clark et al., 2009; Clark et al., 2007). Nevertheless, these immunosuppressive cells exhibit dynamic motility and function, such that a detailed understanding of inflammatory cellular mechanisms in this disease has revealed therapeutic targets that can be translated to clinical success in patients (Beatty et al., 2011).
Although multiple mechanisms are thought to contribute to immune suppression in tumor-bearing hosts, tumor-associated changes in myelopoiesis that result in an abnormal accumulation of immature myeloid cells in the tumor are thought to play a critical immunosuppressive role (Gabrilovich and Nagaraj, 2009; Ostrand-Rosenberg and Sinha, 2009; Peranzoni et al., 2010). In mice, these immature myeloid cells, often called myeloid-derived suppressor cells (MDSC), co-express the markers Gr-1 and CD11b and represent a heterogeneous population of cells comprised of precursors to macrophages, dendritic cells, and granulocytes at earlier stages of differentiation (Gabrilovich and Nagaraj, 2009; Ostrand-Rosenberg and Sinha, 2009; Peranzoni et al., 2010). Normal mouse bone marrow contains 20%-30% of Gr-1+ CD11b+ cells, although these cells only make up 2-4% of spleen cells and are virtually absent from the lymph nodes (Gabrilovich and Nagaraj, 2009). Numerous studies have reported the expansion of Gr-1+ CD11b+ cells in a variety of implantable tumor models and have demonstrated the ability of these myeloid cells to impair T cell responses in vitro (Bronte et al., 2003; Kusmartsev et al., 2004; Sinha et al., 2005). In genetically defined mouse models of cancer – which more closely recapitulate the tumor microenvironments observed in patients with the same disease – the role of these cells remains controversial particularly with regard to the in vivo relevance of the T cell suppressive qualities that are typically demonstrated in vitro (Andreu et al., 2010; Clark et al., 2007; Melani et al., 2003; Stairs et al., 2011). Moreover, the soluble factors that drive the accumulation of Gr-1+ CD11b+ cells in genetically defined models of cancer, particularly in PDA, remain largely unknown.
To understand the role of Gr-1+ CD11b+ cells in PDA, we focused on the KPC mouse model of spontaneous PDA in which expression of oncogenic KrasG12D and mutant p53R172H is targeted to the pancreas by Cre recombinase under the control of the pancreatic specific promoter Pdx-1 (Hingorani et al., 2005). This model is fully penetrant with regard to tumor formation, and KPC mice develop primary PDA lesions that faithfully recapitulate the salient clinical, histopathological, and molecular features of the human disease, including progression from preinvasive pancreatic intraepithelial neoplasia (PanIN) to invasive cancer to metastatic disease (Hingorani et al., 2005). Using the KPC model, we evaluate a mechanism of tumor-induced immune modulation that is critical to maintaining the local immune suppressive network characteristic of PDA.
RESULTS
The dense desmoplasia and leukocytic infiltration classically observed in the tumor stroma of patients with PDA is reproduced with high fidelity in the KPC murine model of the disease (Beatty et al., 2011; Clark et al., 2009; Hingorani et al., 2005). In tumor-bearing KPC mice, we found that Gr-1+ CD11b+ cells prominently accumulated in both the tumor and the spleen compared to normal controls, comprising 20%-30% of all leukocytes in these tissues (Figure 1A). By immunohistochemistry, Gr-1+ CD11b+ cells were evident in close proximity to tumor cells (Figure 1B). Gr-1+ CD11b+ cells were also prominently associated with metastatic lesions in this model, but were not found in other tissues that lacked metastases (Figure 1C). Sorted Gr-1+ CD11b+ leukocytes from the spleen or tumor of KPC mice comprised a heterogeneous population of myeloid cells including myelocytes, metamyelocytes, band and segmented neutrophils, and monocytoid cells (Figure 1D), consistent with previous observations of cancer-associated Gr-1+ CD11b+ cells (Andreu et al., 2010; Bronte et al., 2000; Stairs et al., 2011).
Figure 1. Gr-1+ CD11b+ cells accumulate in the spleen and tumor of KPC mice bearing PDA.
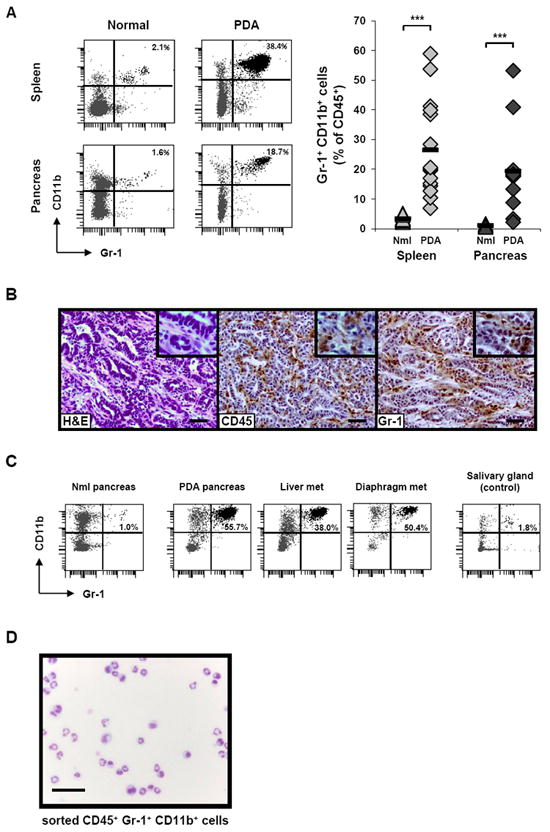
(A) Gr-1+ CD11b+ cells from the spleen and pancreas of tumor-bearing KPC mice vs. normal control mice were evaluated by flow cytometry. Representative flow plots for one mouse in each category (left) and summary data (right) are shown. Cells were gated for CD45 expression and then analyzed. Numbers in top right corner of the plots represent the percentage of Gr-1+ CD11b+ cells among total CD45+ cells. For summary data, each symbol represents an individual mouse: spleen (light gray), pancreas (dark gray), normal mice (triangles) and PDA mice (diamonds). Black horizontal bars represent the mean for each group (n = 10 for normal spleen, n = 15 for PDA spleen, n = 3 for normal pancreas, and n = 13 for PDA pancreas). Nml = normal. *** indicates p < 0.001 for the comparisons shown.
(B) H&E stain (left) and immunohistochemistry for CD45 (middle) or Gr-1 (right) on a representative PDA lesion. Scale bars, 50 μM. Higher magnification images are shown in the top right corner of each panel.
(C) Metastatic lesions on the liver and diaphragm of a representative KPC mouse were evaluated by flow cytometry for Gr-1+ CD11b+ cells (after gating for CD45 expression), compared to salivary gland cells as a control.
(D) Diff-Quick stain of cytospin preparation of sorted CD45+ Gr-1+ CD11b+ cells from the spleen of a tumor-bearing KPC mouse. Scale bar, 20 μM.
Various terms have been used in the literature to describe Gr-1+ CD11b+ cells, including immature myeloid cells (Kusmartsev and Gabrilovich, 2006) or myeloid derived suppressor cells (MDSC) (Gabrilovich et al., 2007). Because the latter term suggests that Gr-1+ CD11b+ cells act to suppress T cell activation, we sought to understand the potential immunosuppressive function of Gr-1+ CD11b+ cells in the KPC model. We therefore evaluated the capacity of highly enriched or sorted Gr-1+ CD11b+ cells from tumor-bearing mice to suppress in vitro proliferation of ovalbumin-specific CD8+ T cells from OT-1 transgenic mice in response to cognate peptide antigen. We found that Gr-1+ CD11b+ cells isolated from either the spleen or pancreas of tumor-bearing KPC mice potently suppressed proliferation (Figure 2A, Figure S1) as well as IFN-γ production by OT-1 cells (Figure 2B). Gr-1+ CD11b+ cells from tumor-bearing KPC mice also suppressed the proliferation of splenic T cells from normal mice stimulated polyclonally with anti-CD3 plus anti-CD28 antibodies (Figure S1). Moreover, Gr-1+ CD11b+ cells exhibited high levels of arginase activity (Figure 2C) and produced high levels of nitrite upon culture (Figure 2D), suggesting expression of inducible nitric oxide synthase (iNOS); both arginase and iNOS have been previously linked to immunosuppression by Gr-1+ CD11b+ cells in tumor-bearing mice (Bronte and Zanovello, 2005; Ma et al., 2011). We found that inhibition of iNOS and, to a lesser extent, arginase abrogated the capacity of Gr-1+ CD11b+ cells to suppress T cell proliferation (Figure 2E). This set of properties of Gr-1+ CD11b+ cells in KPC mice are consistent with described properties of MDSC (Gabrilovich et al., 2007).
Figure 2. Gr-1+ CD11b+ cells isolated from tumor-bearing KPC mice suppress antigen-specific T cell responses in vitro.
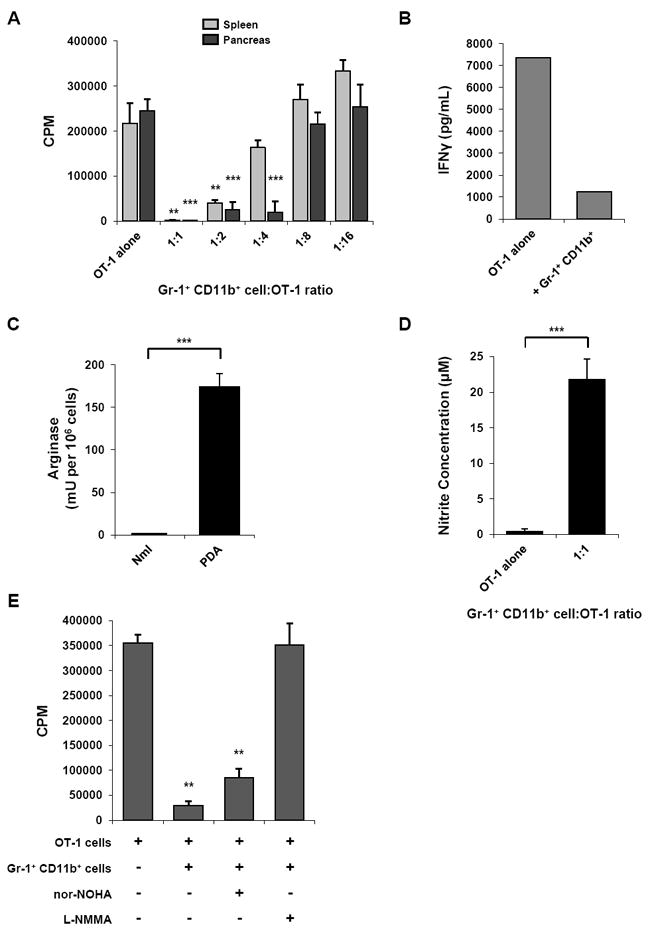
(A) Gr-1+ CD11b+ cells isolated from either the spleen (light gray bars) or pancreas (dark gray bars) of tumor-bearing KPC mice were cocultured with peptide-stimulated OT-1 splenocytes and proliferation was evaluated by 3[H]-thymidine incorporation. Assay was performed in triplicate; data are mean ± SD, representative of 5-10 independent experiments. ** indicates p < 0.01, *** p < 0.001, compared to OT-1 alone.
(B) IFN-γ in supernatant from coculture of Gr-1+ CD11b+ cells with OT-1 T cells (1:1 ratio) was measured by cytokine bead array. Data are representative of 2 independent experiments.
(C) Arginase activity was evaluated in the Gr-1+ fraction of normal spleens and tumor-bearing KPC spleens. Assay was performed in triplicate; data are mean ± SD, representative of 2 independent experiments. *** indicates p < 0.001.
(D) Nitrite concentration in supernatant from coculture of Gr-1+ CD11b+ cells with OT-1 T cells (1:1 ratio) was measured by the Griess reaction as an indicator of iNOS activity. Data are mean ± SD, representative of 3 independent experiments. *** indicates p < 0.001.
(E) Small molecule inhibition of iNOS relieves antigen-specific suppression of T cell proliferation mediated by Gr-1+ CD11b+ cells. Proliferation assay was performed in triplicate at 1:1 ratio of Gr-1+ CD11b+ cells to OT-1 cells; data are mean ± SD. nor-NOHA (arginase inhibitor) = Nω-Hydroxy-nor-L-arginine; L-NMMA (iNOS inhibitor) = NG-Monomethyl-L-arginine. ** indicates p < 0.01, compared to OT-1 alone.
See also Figure S1.
We then examined the potential origin of Gr-1+ CD11b+ cells. We noted that unlike normal controls, tumor-bearing KPC mice developed splenomegaly as a result of extramedullary hematopoiesis (Figure 3A, B) and concomitant to accumulation of Gr-1+ CD11b+ cells in the spleen (Figure 1A). Splenocytes from tumor-bearing KPC mice exhibited a distinct c-kit+ population, higher in percentage than found in splenocytes from normal mice and similar to the percentage of c-kit+ precursors found in bone marrow (Figure 3B). Given the link between the tumor microenvironment and accumulation of functional Gr-1+ CD11b+ cells in the KPC model, we hypothesized that a tumor-derived factor might drive the generation of Gr-1+ CD11b+ cells from c-kit+ cells in the spleen. We therefore isolated c-kit+ Gr-1neg CD11bneg lineageneg cells from the spleens of tumor-bearing KPC mice and incubated them with conditioned media obtained from cultured PDA tumor cells that had been previously isolated. Conditioned media from tumor cells, but not control media, triggered proliferation of c-kit+ splenocytes, as evidenced by CFSE dilution (Figure 3C) or BrdU incorporation (Figure S2). In contrast, Gr-1+ CD11b+ splenocytes isolated from the same mice did not proliferate in response to tumor cell conditioned media (Figure 3C). Importantly, after 5 days of co-culture, c-kit+ cells expressed high levels of Gr-1 and CD11b (Figure 3C), exhibited arginase and iNOS activity (Figure 3D), and potently suppressed T cell proliferation in the OT-1 T cell suppression assay (Figure 3E).
Figure 3. c-kit+ lineageneg splenocytes are highly proliferative precursors of Gr-1+ CD11b+ cells and are driven to proliferate via tumor-derived factors.
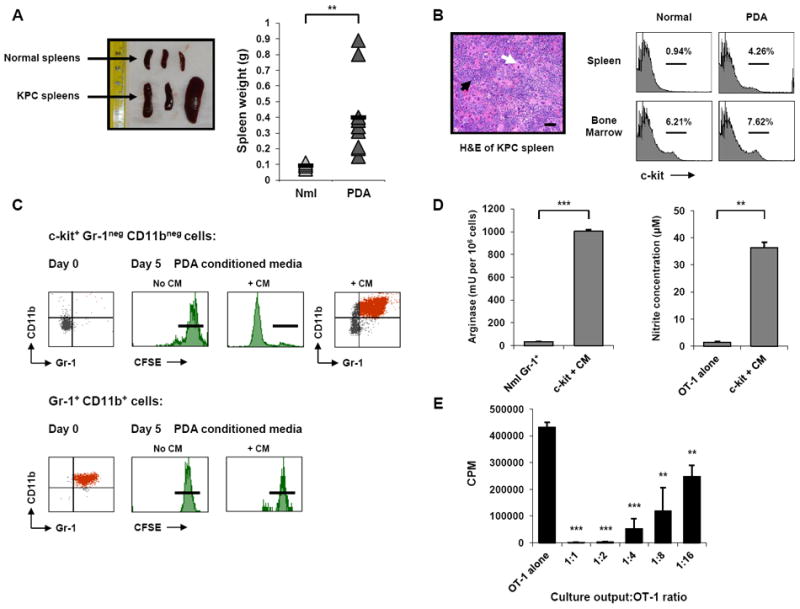
(A) Tumor-bearing KPC mice exhibit splenomegaly as indicated by spleen size of normal mice and tumor-bearing KPC mice (left) and spleen weight of normal and KPC mice (right). Black horizontal bars represent the mean for each group (n = 5 for normal mice, n = 12 for KPC mice). Nml = normal. ** indicates p < 0.01.
(B) H&E of tumor-bearing KPC spleen (left) reveals marked disruption of normal splenic architecture and classic findings of extramedullary hematopoiesis. Note the abundance of megakaryocytes (white arrow) and areas of erythropoiesis (black arrow). Scale bars, 50 μM. Spleen and bone marrow from normal and tumor-bearing KPC mice (right) were evaluated by flow cytometry for c-kit+ cells.
(C) c-kit+ Gr-1neg CD11bneg cells (top) and Gr-1+ CD11b+ cells (bottom) from the spleens of tumor-bearing KPC mice were labeled with CFSE and cultured in the presence of either unconditioned control media (No CM) or PDA conditioned culture media from KPC tumor lines (+ CM). Gr-1+ CD11b+ phenotype is shown at baseline post-isolation (left). Flow cytometric analysis of CFSE dilution on day 5 (middle) demonstrates that Gr-1neg CD11bneg c-kit+ cells proliferate in response to tumor conditioned media, but not control media, whereas freshly isolated Gr-1+ CD11b+ MDSC do not proliferate in either condition. Black horizontal bars indicate undiluted CFSE intensity on day 5. The day 5 output of the proliferative Gr-1neg CD11bneg c-kit+ culture was evaluated by flow cytometry for surface expression of Gr-1 and CD11b (far right). Data are representative of 5-10 independent experiments.
(D) The Gr-1+ CD11b+ cells generated from c-kit+ cells after 5 days with CM were evaluated for arginase (left) and iNOS (right) activity. Data are mean ± SD from one of 2 independent experiments; ** indicates p < 0.01, *** p < 0.001.
(E) Suppressive capacity of the Gr-1+ CD11b+ cells generated from c-kit+ cells after 5 days with CM was assessed in the OT-1 T cell suppression assay, performed in triplicate. Data are mean ± SD. ** indicates p < 0.01, *** p < 0.001, compared to OT-1 alone.
See also Figure S2.
To identify tumor-derived soluble factors contributing to the generation of Gr-1+ CD11b+ cells from c-kit+ precursors, we measured a set of secreted proteins in conditioned media from a panel of PDA tumor cell lines and compared the results to those for conditioned media from benign pancreatic ductal cells from normal control mice. Conditioned media from every PDA line supported proliferation of c-kit+ cells into Gr-1+ CD11b+ cells whereas media from none of the normal pancreatic ductal cells supported c-kit+ cell proliferation (Figure 4A). Among 11 proteins examined, only GM-CSF was expressed at high levels by every PDA line but not by any of the normal pancreatic ductal lines (Figure 4A), suggesting that tumor-derived GM-CSF might be linked to Gr-1+ CD11b+ cell generation. We therefore tested recombinant GM-CSF in our in vitro assays and found that GM-CSF drove proliferation and differentiation of c-kit+ Gr-1neg CD11bneg splenocytes isolated from tumor-bearing mice into functional MDSC (Figure 4B). Proliferation and differentiation of c-kit+ Gr-1neg CD11bneg splenocytes was not observed with recombinant vascular endothelial cell growth factor (VEGF) or stem cell factor (SCF, also known as c-kit ligand) and only minimally with IL-6 (Figure S3). Intermediate proliferation was observed with only high concentrations of recombinant macrophage colony stimulating factor (M-CSF). In each case, neutralization of GM-CSF in tumor cell conditioned media by the addition of anti-GM-CSF mAb completely abrogated generation of Gr-1+ CD11b+ cells from c-kit+ precursors (Figure 4C and Figure S3). Neutralizing antibodies to other cytokines such as anti-SCF, anti-IL-6, or anti-IL-1β had no effect (Figure S3), suggesting that GM-CSF is both necessary and sufficient for in vitro generation of functional, immunosuppressive Gr-1+ CD11b+ cells.
Figure 4. Tumor-derived GM-CSF induces proliferation and differentiation of c-kit+ lineageneg precursors into functional MDSC.
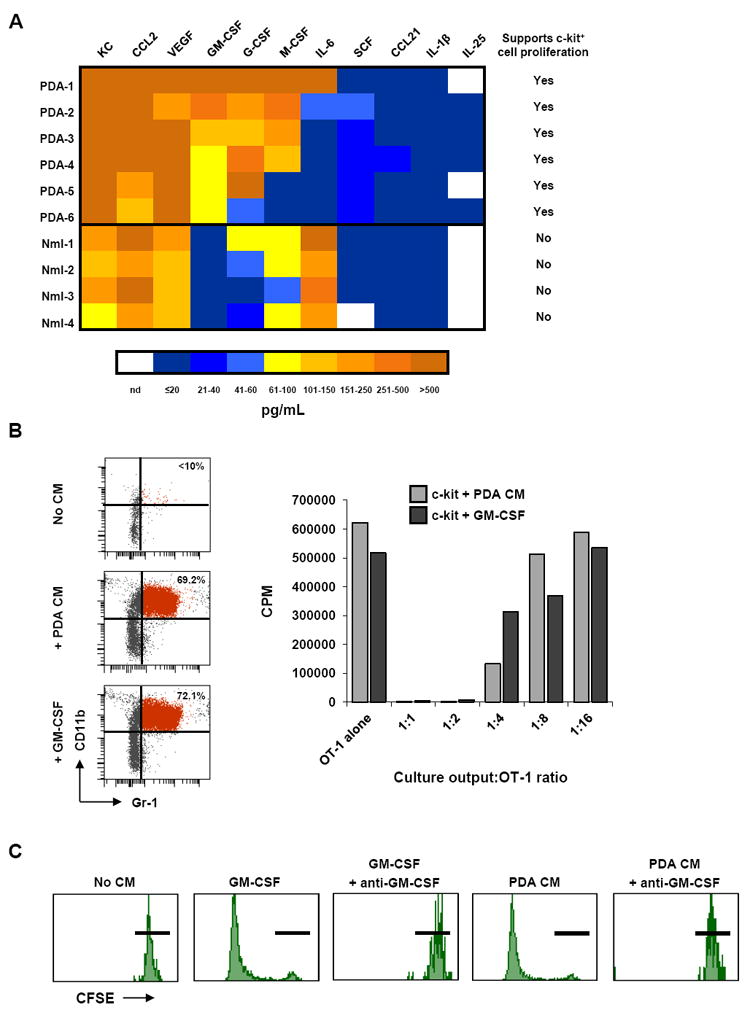
(A) Cytokines produced by primary PDA lines derived from KPC tumors and normal pancreatic ductal cell lines are compared in a heat map. Color scale and corresponding cytokine values (pg/mL) are shown below. Last column indicates whether conditioned media from that cell line was found to drive proliferation of c-kit+ lineageneg cells in a CFSE proliferation assay. Nml = normal; nd = not determined.
(B) Recombinant GM-CSF is sufficient to induce proliferation and differentiation of c-kit+ lineageneg cells into MDSC. Day 5 FACS analysis (top) was performed to evaluate Gr-1 and CD11b expression on c-kit+ lineageneg splenocytes cultured with control media (No CM), CM from PDA lines (+ PDA CM), or recombinant GM-CSF (200 pg/ml). Suppressive capacity of culture outputs (bottom) was evaluated in the OT-1 T cell suppression assay. Data are representative of at least 3 independent experiments.
(C) CFSE dilution of c-kit+ lineageneg cultures is shown for day 5. Neutralization of GM-CSF by anti-GM-CSF mAb (100 μg/mL) in PDA-1 conditioned media blocks proliferation of c-kit+ lineageneg cells. Black horizontal bars indicate undiluted CFSE intensity on day 5. Data are representative of independent experiments using conditioned media from 3 different PDA cell lines. As a control, the neutralizing capability of this anti-GM-CSF mAb was shown by its effect on recombinant GM-CSF in this assay.
See also Figure S3
To understand the relevance of tumor-derived GM-CSF in vivo, first we isolated fresh pancreatic tissue from tumor-bearing KPC mice vs. normal controls and measured GM-CSF concentrations in tissue supernatant. Higher amounts of GM-CSF (>1 log) per gram of tissue were detected in pancreata from tumor-bearing KPC mice relative to pancreata from normal mice (Figure 5A). (Chemokines KC and CCL2 were also elevated in tumor tissue, consistent with the cell line cytokine data). To determine which cells produce GM-CSF within tumors from genetically engineered mice, we utilized a lineage marking approach (Figure 5B). A conditional Rosa26-Lox-STOP-Lox-YFP (RosaYFP) allele was bred onto the KrasG12D; p53fl/+; Pdx-1-Cre background to permanently label the pancreatic epithelial compartment during embryonic development. This approach led to sensitive (>95% of all pancreatic epithelial cells) and specific labeling of pancreas epithelial-derived cells, with no stromal cells labeled (Rhim et al., 2012). Tumors from these mice and normal pancreata from lineage labeled wild type mice (Pdx-1-Cre; RosaYFP) were harvested, dissociated, and single cells were directly sorted based on YFP fluorescence. Subsequent transcriptional analysis revealed that YFP+ cells from tumor-bearing mice expressed high levels of GM-CSF mRNA compared to YFPneg cells from the same mice (Figure 5B). GM-CSF mRNA was undetectable in either YFP+ or YFPneg cells from control mice. To investigate GM-CSF expression by human tumors in vivo, we performed immunohistochemistry for GM-CSF on 20 PDA tumor samples obtained from patients who had undergone surgical resection of a primary PDA tumor. In 19 of these cases, tumor cells prominently expressed GM-CSF (>50% of tumor cells positive) (Figure 5C). For the positive samples, the percentage of tumor cells that expressed GM-CSF was 84.2% ± 13.8% (mean ± SD). Tumor cells that lacked GM-CSF expression were observed scattered throughout the tumor without any association to tumor grade and were only occasionally found as clusters of cells.
Figure 5. PDA cells produce GM-CSF and other factors in vivo in KPC mice.
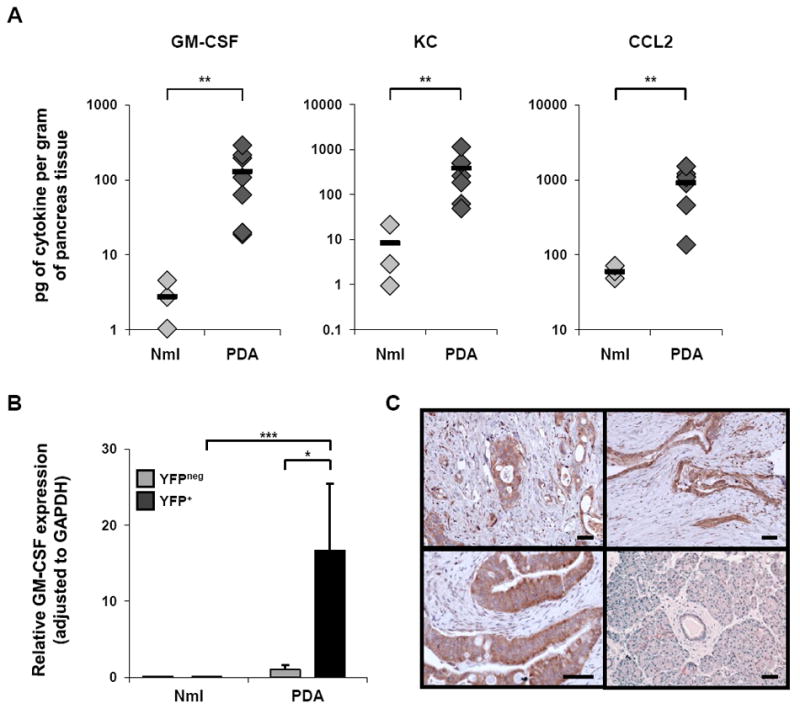
(A) Cytokines in fresh PDA supernatant (directly ex vivo) vs. normal pancreas supernatant were compared by cytokine bead array. y-axes are on log scales; each symbol represents an individual mouse: normal mice (light gray diamonds) and PDA mice (dark gray diamonds). Black horizontal bars represent the mean for each group (n = 3 for normal mice, n = 7 for PDA mice). ** indicates p < 0.01.
(B) Transcriptional analysis was used to evaluate GM-CSF mRNA expression of YFP+ (epithelial) cells vs. YFPneg (stromal) cells sorted from tumors of KrasG12D; p53fl/+; Pdx-1-Cre; RosaYFP mice (n=3) or normal pancreata from lineage labeled wild type mice (Pdx-1-Cre; RosaYFP) (n=3). Data are mean ± SD. * indicates p < 0.05, *** p < 0.001.
(C) Immunohistochemistry for GM-CSF was performed on surgical pancreas samples from 20 patients with PDA. PDA tumors from three representative patients are shown (upper left, upper right, lower left), and adjacent pancreatic tissue without invasive tumor is shown for one patient (lower right). The lower left panel is shown at higher power magnification. Scale bars, 50 μM.
We then studied the role of GM-CSF on tumor growth and inflammation in vivo. PDA tumor cells that secreted GM-CSF (cell line PDA-1) were injected subcutaneously in a matrigel plug containing neutralizing anti-GM-CSF mAb (vs. purified rat IgG2a as a control) into the flanks of normal control mice. This matrigel subcutaneous assay was developed to allow for GM-CSF neutralization within the local tumor microenvironment given that systemic administration of anti-GM-CSF antibody was unable to achieve sufficient concentrations within the tumor microenvironment to neutralize GM-CSF effectively (data not shown). Six days after implantation, PDA-1 cells injected with IgG2a established tumors, but PDA-1 cells injected with anti-GM-CSF failed to grow and the injected plugs, upon resection, weighed statistically significantly less (Figure 6A). Histologically, PDA-1 cells injected with anti-GM-CSF mAb appeared necrotic after 6 days compared to viable tumor cell nests in the control (Figure 6A). Moreover, the infiltration of CD45+ Gr-1+ CD11b+ cells observed in control tumors was reduced in tumor cells implanted with anti-GM-CSF mAb (Figure 6B, C). Weight, histology, and leukocyte infiltration on day 6 was the same for PDA-1 cells injected with anti-GM-CSF mAb as for lethally irradiated (10 Gy) PDA-1 cells injected with anti-GM-CSF mAb (Figure 6A, C).
Figure 6. GM-CSF neutralization with anti-GM-CSF antibody limits tumor growth and recruitment of Gr-1+ CD11b+ cells in vivo.
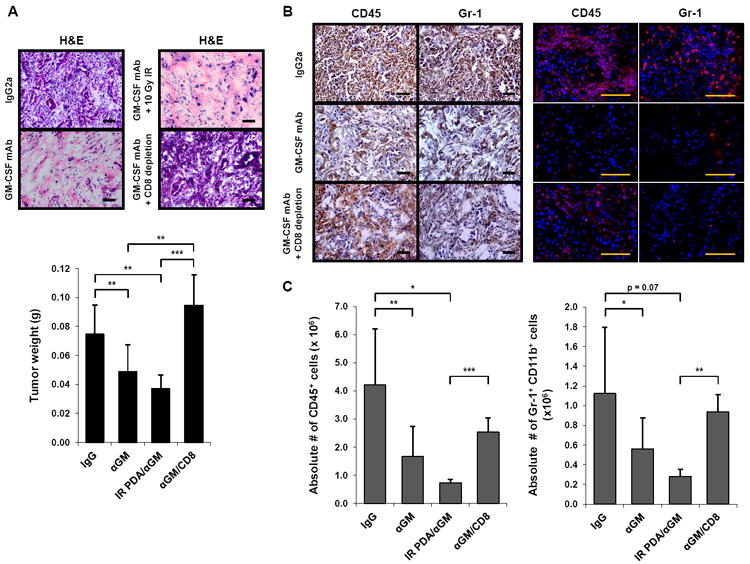
(A) Normal control mice (6-14 weeks) were implanted subcutaneously with the GM-CSF-producing cell line PDA-1 in matrigel with either a neutralizing anti-GM-CSF mAb (αGM) or isotype control antibody, with or without tumor cell irradiation (IR), and with or without intraperitoneal administration of a depleting CD8 mAb. On day 6, tumor growth was evaluated by H&E (top; images of one representative mouse per group are shown) and tumor weight (bottom; data are mean ± SD). n = 3 to 11 mice per group. Scale bars, 50 μM. ** indicates p < 0.01, *** p < 0.001.
(B) The immune infiltrate was evaluated by immunohistochemistry (left) and immunofluorescence (right) for CD45 and Gr-1. Images of one representative mouse per group are shown (3 to 11 mice per group). Scale bars, 50 μM.
(C) Absolute numbers of CD45+ cells (left) and Gr-1+ CD11b+ cells (right) were quantified by flow cytometry. Data are mean ± SD. * indicates p < 0.05, ** p < 0.01, *** p < 0.001.
See also Figure S4.
To determine whether the GM-CSF that was required for tumor formation in this assay was tumor-derived, we knocked down GM-CSF expression in PDA-1 cells using short-hairpin RNA (shRNA) technology. Compared to vector only (mock PDA-1 cells), shGM-CSF PDA-1 cells expressed >90% less GM-CSF, whereas expression of other cytokines such as VEGF and KC was not affected (Figure S5). shGM-CSF PDA-1 cells or mock PDA-1 cells were then implanted subcutaneously in matrigel into the flanks of normal control mice and observed for 6 days. Similar to the experiments with anti-GM-CSF, mock PDA-1 cells established tumors in vivo, but shGM-CSF PDA-1 cells failed to grow and the resected plugs weighed significantly less (Figure 7A). Histologically, shGM-CSF PDA-1 cells appeared necrotic after 6 days compared to viable tumor cell nests in the control (Figure 7A). There was a significant reduction in the inflammatory infiltrate in experiments with shGM-CSF PDA-1 cells, including fewer Gr-1+ CD11b+ cells (Figure 7B, C). Weight, histology, and leukocyte infiltration on day 6 was the same for shGM-CSF PDA-1 cells as these parameters were for lethally irradiated (10 Gy) shGM-CSF PDA-1 cells (Figure 7A, C).
Figure 7. Genetic knockdown of GM-CSF demonstrates that tumor-derived GM-CSF regulates tumor growth and recruitment of Gr-1+ CD11b+ cells in vivo.
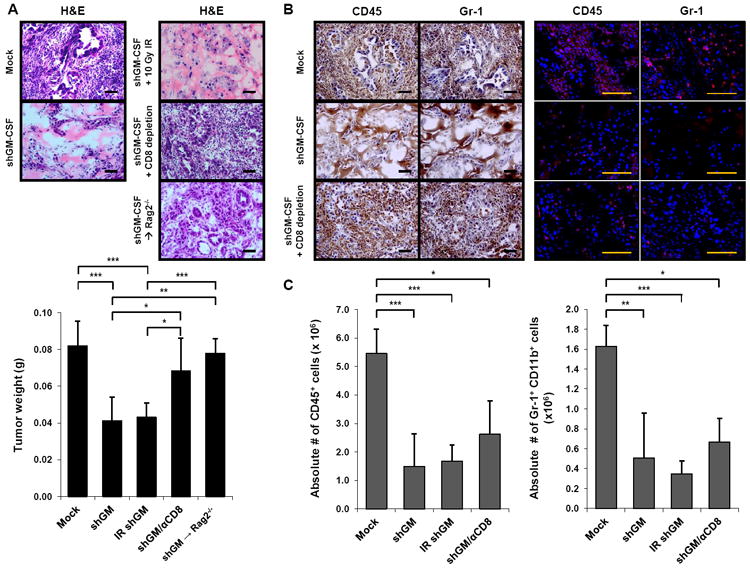
(A) Normal control mice or Rag2-/- mice (6-14 weeks) were implanted subcutaneously with either shGM-CSF PDA-1 cells (shGM) or vector only PDA-1 cells (Mock) in matrigel with or without tumor cell irradiation (IR), and with or without intraperitoneal administration of a depleting CD8 mAb. On day 6, tumor growth was evaluated by H&E (top; images of one representative mouse per group are shown) and tumor weight (bottom; data are mean ± SD). n = 3 to 7 mice per group. Scale bars, 50 μM. * indicates p < 0.05, ** p < 0.01, *** p < 0.001.
(B) The immune infiltrate was evaluated by immunohistochemistry (left) and immunofluorescence (right) for CD45 and Gr-1. Images of one representative mouse per group are shown (3 to 7 mice per group). Scale bars, 50 μM.
(C) Absolute numbers of CD45+ cells (left) and Gr-1+ CD11b+ cells (right) were quantified by flow cytometry. Data are mean ± SD. * indicates p < 0.05, ** p < 0.01, *** p < 0.001.
See also Figure S5.
We also examined the effect of GM-CSF blockade and knockdown on Gr-1neg CD11b+ monocytes/macrophages in the implanted tumor model. In each experimental system (i.e., anti-GM-CSF antibody or shGM-CSF), we observed a statistically significant decrease in tumor infiltrating Gr-1neg CD11b+ cells compared to control tumors (Figure S4). Importantly, a major subset of Gr-1+ CD11b+ cells were found to express high levels of Ly6C, and among this population, >90% expressed the macrophage marker F4/80 (data not shown), underscoring a potential biological continuum of monocytes, macrophages, and immature myeloid cells. We were unable to identify an adequate method of depleting macrophages without impacting Gr-1+ CD11b+ cells in our mice. For example, we found that administration of clodronate encapsulated liposomes (CEL) depleted Gr-1neg CD11b+ F4/80+ macrophages as well as Ly6Chigh Gr-1+ CD11b+ cells (data not shown). We therefore repeated our tumor implantation experiments with anti-Gr-1 antibody vs. isotype control and found that administration of anti-Gr-1 antibody depletes Gr-1+ CD11b+ myeloid cells but not Gr-1neg CD11b+ macrophages (Figure S4). Importantly, coinjection of anti-Gr-1 antibody with PDA-1 cells recapitulated our findings with anti-GM-CSF such that tumor implants failed to grow (Figure S4). Thus, depletion of Gr-1+ CD11b+ cells alone is sufficient for impaired tumor growth in this model.
We hypothesized that GM-CSF produced by tumor cells may drive the generation of Gr-1+ CD11b+ cells which suppress anti-tumor T cell immunity. We therefore examined whether tumor rejection observed in the setting of GM-CSF neutralization or genetic knockdown was mediated by CD8+ T cells. We repeated the 6 day implantation experiments in the setting of in vivo CD8 depletion (Figure S4). Strikingly, CD8 depletion restored tumor growth for both PDA-1 cells implanted with anti-GM-CSF and shGM-CSF PDA-1 cells (Figures 6A and 7A), linking tumor-derived GM-CSF to Gr-1+ CD11b+ cell accumulation and CD8+ T cell suppression. To confirm this, we used a genetic approach and repeated experiments in Rag2-/- mice. We found that shGM-CSF PDA-1 tumor cells grew vigorously in Rag2-/- mice, even though they failed to grow in T cell replete mice (Figure 7A). In addition, we found that CD8+ T cells infiltrated tumors more vigorously after GM-CSF knockdown. This finding was demonstrated by immunofluorescence for CD8 expression and quantified by flow cytometry (Figure S5). To understand if CD8+ T cells from GM-CSF knockdown tumors exhibited effector functions, we evaluated tumor-infiltrating CD8+ T cells for their capacity to secrete IFN-γ and mobilize CD107a, the latter serving as a biomarker of lytic function (Betts et al., 2003). We found that compared to CD8+ T cells from mock PDA-1 tumors, CD8+ T cells from shGM-CSF PDA-1 tumors exhibited a higher percentage of IFN-γ+ CD107a+ cells (Figure S5). Importantly, CD8 depletion did not rescue infiltration of Gr-1+ CD11b+ cells into shGM-CSF PDA-1 tumors, suggesting that PDA-1 tumor cells can grow in the setting of minimal Gr-1+ CD11b+ cells, provided CD8+ T cells are absent (Figure 7).
DISCUSSION
It has become widely appreciated that tumor-associated inflammation is an enabling characteristic of cancer and contributes to tumor escape from immune destruction (Hanahan and Weinberg, 2011). Here, focusing on pancreatic ductal adenocarcinoma in mice as a model of the human disease, we demonstrate that tumor cell-derived GM-CSF can orchestrate an immunosuppressive crosstalk between Gr-1+ CD11b+ immature myeloid cells and CD8+ T cells. Overall, our data support a model in which myeloid inflammatory cells negatively regulate CD8+ T cells via tumor-derived GM-CSF (Figure 8).
Figure 8. Working model links tumor-derived GM-CSF, MDSC generation, and CD8+ T cell suppression in PDA.
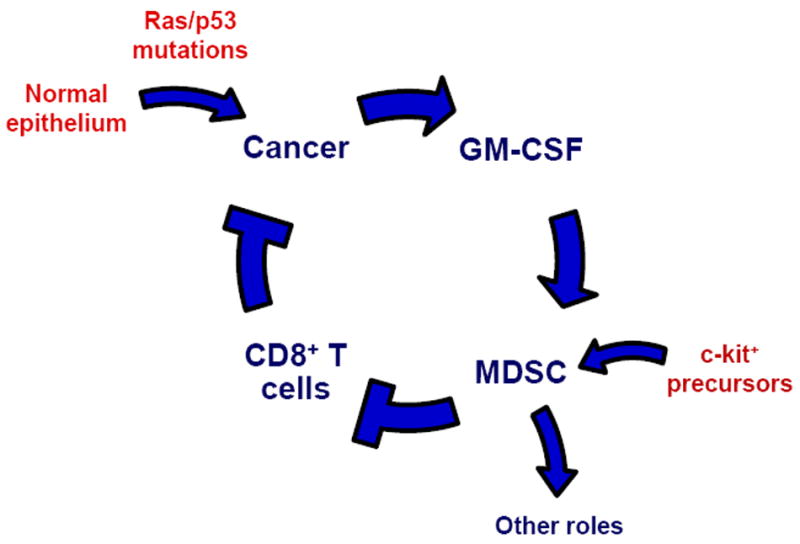
Although GM-CSF clearly regulates hematopoiesis as a growth factor, its role as an immunomodulatory cytokine has become increasingly appreciated. Depending on the setting, these effects can either promote or suppress cellular immune responses. In studies of implantable tumor models, GM-CSF has been linked to the generation of Gr-1+ CD11b+ cells with immunosuppressive features (Bronte et al., 1999; Dolcetti et al., 2010; Morales et al., 2010; Serafini et al., 2004). For example, GM-CSF-transduced melanoma cells induce the systemic expansion of Gr-1+ CD11b+ cells and inhibit memory CD8+ T cells (Bronte et al., 1999). Knockdown of GM-CSF in tumor cell lines has been shown to alter the subset distribution of Gr-1+ CD11b+ myeloid cells following implantation in mice, primarily reducing the Gr-1low and Gr-1int populations which are the most suppressive in these models (Dolcetti et al., 2010). Here, we show that GM-CSF is produced in vivo in a spontaneous tumor that is characterized by prominent infiltration of suppressive Gr-1+ CD11b+ cells. We also demonstrate that human pancreatic tumors cells express GM-CSF in vivo; therefore, these findings are significantly relevant to understanding the human disease.
GM-CSF may not be critical for the development of Gr-1+ CD11b+ cells in every tumor model or histology. In mice with combined loss of IFN-γ, IL-3, and GM-CSF, lung carcinomas develop at a high frequency and exhibit an infiltrate of Gr-1+ CD11b+ leukocytes (Dougan et al., 2011). Moreover, cytokines and growth factors other than GM-CSF have been implicated in the pathophysiology of Gr-1+ CD11b+ leukocytes in other mouse models -- including IL-1β IL-6, and VEGF (Melani et al., 2003) -- but none of these factors explains the effects we observed regarding the generation of suppressive Gr-1+ CD11b+ cells in the PDA model. For example, the pro-inflammatory cytokine IL-1β has been implicated in MDSC pathophysiology such that fibrosarcoma or 4T1 mammary carcinoma cell lines engineered to secrete IL-1β grow more rapidly than control tumors upon implantation due to enhanced accumulation of Gr-1+ CD11b+ and concomitant T cell suppression (Bunt et al., 2006; Song et al., 2005). Mice lacking the IL-1 receptor exhibit delayed accumulation of MDSC and slower progression of implanted 4T1 mammary tumors and interestingly, these effects are partially restored by exogenous administration of IL-6, indicating that IL-6 may contribute independently to MDSC expansion (Bunt et al., 2007). Stomach-specific expression of human IL-1β in transgenic mice leads to spontaneous gastric inflammation, invasive cancer, and mobilization of MDSC (Tu et al., 2008). In the KPC model of PDA, however, we observed accumulation of Gr-1+ CD11b+ cells in the spleen and tumor despite the observations that (i) PDA tumor cells did not secrete IL-1β and (ii) the addition of neutralizing anti-IL-1β antibody to PDA conditioned media failed to attenuate the proliferation and induction of MDSC from precursor cells. Although recombinant IL-6 did trigger low level proliferation of c-kit+ splenocytes, PDA tumors cells (with one exception) did not produce IL-6 and the addition of neutralizing anti-IL-6 antibody to PDA conditioned media had no effect in these assays. More likely in PDA, tumor-associated macrophages release IL-6 that directly impacts the epithelium and promotes PanIN progression and PDA development in vivo (Beatty et al., 2011; Lesina et al., 2011). Indeed, our data are consistent with previous studies demonstrating IL-6 production primarily by stromal cells, not the tumor, in mouse models (Lesina et al., 2011) as well as human PDA (Bellone et al., 2006; Lesina et al., 2011). In mice, IL-6 production can be induced in PDA tumor cells, or in pancreas epithelium harboring mutant Kras, but only after an acute inflammatory insult (Fukuda et al., 2011). IL-6 production has been reported in some, but not all, human PDA cell lines (e.g. Bellone et al., 2006). In addition, IL-6 expression in tumors may be silenced by DNA methylation (Dandrea et al., 2009). For IL-1β, variable expression by human PDA cell lines has been described (Bellone et al., 2006), and in mice, IL-1β can be induced in pancreas epithelium harboring mutant Kras but only after an acute inflammatory insult (Fukuda et al., 2011). Overexpression of IL-1β in the mouse pancreas results in pancreatitis but interestingly, not PanIN or PDA (Marrache et al., 2008).
To begin to understand if GM-CSF production is an early event in oncogenesis, we generated PanIN epithelial cell lines from PanIN only-bearing KPC mice and found low level production of GM-CSF. Supernatant from PanIN lines supported the proliferation of c-kit+ splenocytes into Gr-1+ CD11b+ cells, but not to the extent of supernatant from PDA lines (data not shown). This proliferation, however, could be fully blocked by anti-GM-CSF antibody. A caveat from our studies is that we cannot determine the grade of the PanIN lesions from which the lines were derived. Importantly, our results are consistent with data described by Pylayeva-Gupta et al. (Pylayeva-Gupta et al., 2012).
VEGF and G-CSF have also been implicated in MDSC development (Gabrilovich et al., 1998; Huang et al., 2007; Roland et al., 2009; Dolcetti et al., 2010). In our model of PDA, however, recombinant VEGF had little or no impact on the generation of Gr-1+ CD11b+ myeloid cells from precursor cells isolated from PDA-bearing KPC mice even though our PDA lines produce high levels of VEGF. Furthermore, normal pancreatic ductal cells also secreted VEGF (albeit somewhat less so than PDA cells) yet Gr-1+ CD11b+ cells did not accumulate around normal ducts in vivo and conditioned media from normal ductal cells failed to support proliferation of c-kit+ splenocytes into MDSC. We found that most PDA lines produced G-CSF; however, supernatant from a G-CSF negative line supported proliferation of c-kit+ splenocytes. Some of the normal ductal cell lines also produced G-CSF yet supernatant from these normal lines did not support c-kit+ cell expansion.
Recent studies have also linked the chemokine CCL21 to the appearance of MDSC in the B16-F10 implantable model of melanoma (Shields et al., 2010), and other investigators have uncovered that IL-25 elicits a multipotent progenitor cell population in mucosal tissue that can differentiate into cells of various myeloid lineages (Saenz et al., 2010), but neither CCL21 nor IL-25 is produced by PDA in our model. Since all PDA lines produce high levels of CCL2 and KC, we also evaluated these factors in our in vitro assays, but neither supported the proliferation of precursors into MDSC, and moreover, these factors were produced by normal pancreatic epithelial cells. Rather, GM-CSF was the only factor that met all the criteria of a tumor-associated driving factor for MDSC generation in our experimental system (i.e., secreted by PDA, not secreted by normal pancreatic epithelial cells, and contributed necessarily to the generation of suppressive MDSC from c-kit+ splenocytes).
Disruption of CD8 immunity by elements of the tumor microenvironment is thought to be a major mechanism of tumor immune escape (Schreiber et al., 2011). Our data demonstrate the critical role Gr-1+ CD11b+ cells play in suppressing T cell immune responses in vitro and in vivo in PDA. In particular, abrogation of tumor-derived GM-CSF reduced Gr-1+ CD11b+ cell infiltration and implanted tumors failed to grow; however, CD8+ T cell depletion fully rescued tumor growth, even though Gr-1+ CD11b+ cell infiltration was not completely restored. These data suggest that Gr-1+ CD11b+ cells can play a primary immune suppressive role. Other potential roles of Gr-1+ CD11b+ cells in regulating tumor growth (Yang et al., 2004; Yang et al., 2008; Yang et al., 2011) appear therefore to be secondary or non-contributory to tumor growth, at least in this assay. This may explain our previously observation that in genetically engineered murine models of PDA, the infiltration of CD8+ T cells and Gr-1+ CD11b+ cells is mutually exclusive (Clark et al., 2007).
In summary, we propose that the secretion of GM-CSF by transformed pancreatic epithelial cells is critically involved in the regulation of inflammation associated with PDA. A major component of this inflammation is the generation of Gr-1+ CD11b+ immature myeloid cells from c-kit+ lineageneg precursor cells. We show that these myeloid cells can suppress anti-tumor T cell immunity as a primary function and suggest that MDSC contribute to the failure of T cell immunosurveillance in PDA-bearing hosts. In patients, nearly every tumor studied had high GM-CSF expression such that we found it not practical to correlate levels of tumor GM-CSF with either Kras mutational status. Our findings carry important implications for the design of novel therapies for patients with PDA and highlight the potential for disrupting the crosstalk of tumor cells with the immune system by targeting Gr-1+ CD11b+ cells or the cytokines that regulate their differentiation.
EXPERIMENTAL PROCEDURES
Mice
All animal protocols were reviewed and approved by the Institute of Animal Care and Use Committee of the University of Pennsylvania. KrasLSL-G12D/+, Trp53LSL-R172H/+, Pdx1-Cre (KPC) mice have been previously described (Hingorani et al., 2005). Normal healthy Trp53LSL-R172H/+, Pdx1-Cre mice or Pdx1-Cre mice were used as controls or hosts for implantable tumor studies. Rag2-/- mice were used as hosts in some implantable tumor studies. KrasG12D; p53fl/+; Pdx1-Cre; RosaYFP and Pdx1-Cre; RosaYFP mice (Rhim et al., 2012) were used for lineage tracing studies.
Collection of Tissue Samples from Mice
The entire pancreas containing tumor, peripancreatic lymph nodes and normal pancreas tissue was washed in PBS, minced into small fragments, and incubated in collagenase solution (1 mg/mL collagenase V in RPMI-1640) at 37°C for 45 min. Dissociated cells were passed through a 70 μM cell strainer and washed three times in RPMI-1640 supplemented with 10% FCS and 0.05 mM 2-mercaptoethanol, gentamicin and L-glutamine (complete media). Spleens were homogenized and passed through a 70 μM cell strainer to achieve single cell suspensions. Red blood cells were lysed using ACK Lysis Buffer (Cambrex/BioWhittaker).
Antibodies
The following monoclonal antibodies were used in flow cytometry: anti-CD45 (30-F11; PE, PE-Cy7, APC), anti-Gr-1 (RB6-8C5; APC-Cy7), anti-CD11b (M1/70; PE, PerCP-Cy5.5), anti-CD3 (145-2C11; FITC), anti-CD8 (Ly-2; PE-Cy7, APC-Cy7), anti-CD4 (RM4-5; PerCP-Cy5.5), anti-CD19 (1D3; PerCP-Cy5.5), CD107a (1D4B; FITC), IFN-γ (XMG1.2; PE) (all from BD Biosciences), and anti-CD11c (eBioscience; N418; PerCP-Cy5.5). The viability marker 7-aminoactinomycin D (7-AAD) was from BD Biosciences. For in vivo studies, endotoxin-free antibodies were used: anti-GM-CSF (Biolegend; MP1-22E9), anti-CD8 (BioXcell; 2.43), anti-Gr-1 (BioXcell; RB6-8C5), isotype control rat IgG2a (BioXcell; 2A3), and isotype control rat IgG2b (BioXcell; LTF-2).
Flow Cytometry
Single cell suspensions were stained with fluorochrome-labeled antibodies at 4°C for 15 min in PBS/1% FCS. Cells were analyzed on a FACSCanto flow cytometer (BD Biosciences) using BD FACSDiva software (BD Biosciences Immunocytochemistry).
Cytology of Gr-1+ CD11b+ Cells
CD45+ Gr-1+ CD11b+ cells were sorted (>98%) from spleens and pancreata of tumor-bearing KPC mice using a BD FACSAria flow cytometer. Cytospin preparations were stained with Diff-Quick modified Giemsa reagent (Polysciences).
Isolation of Gr-1+ CD11b+ Cells and Functional T cell Suppression Assay
Gr-1+ CD11b+ cells were isolated from the spleen and pancreas of KPC mice by cell sorting or by high-gradient magnetic cell separation (MACS; Miltenyi Biotec, Bergisch-Gladbach, Germany). For magnetic separation, single cell suspensions were incubated with Gr-1 biotinylated mAb (RB6-8C5) followed by anti-biotin mAb coupled to magnetic beads (Miltenyi Biotec) and sorted using MS MACS columns and the MidiMACS system (Miltenyi Biotec) (>90% Gr-1+ CD11b+ cells by flow cytometry). Antigen-specific and polyclonal suppression of CD8+ T cells was evaluated in a co-culture assay in which splenocytes from either OT-1 transgenic mice (antigen-specific assay) or normal mice (polyclonal assay) were seeded in triplicates in 96-well round bottom plates (8 × 105/well). Splenocytes were cultured in the presence of increasing ratios of Gr-1+ CD11b+ cells and stimulated with either cognate antigen, OVA-derived peptide SIINFEKL (1 μg/mL; New England Peptide) (antigen-specific assay), or anti-CD3 (0.5 μg/mL; BD Biosciences; 145-2C11) and anti-CD28 (1 μg/mL; BD Biosciences; 37.51) (polyclonal assay). On day 3 of the co-culture, cells were pulsed with 3[H]-thymidine (1 μCi per well; Amersham Biosciences) and 18 hours later 3[H]-thymidine incorporation was assessed. In some experiments, the OT-1 splenocytes were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE). OT-1 CD8+ T cells were evaluated on day 3 by flow cytometry for CFSE dilution.
CFSE Labeling of Mouse Cells
Cells were resuspended at 10-20×106 per mL in a minimum volume of 500 μL PBS and incubated with CFSE (0.5 μM) at RT for 8 min with constant swirling. The reaction was then quenched with media containing 10% FCS.
Arginase Production
Gr-1+ CD11b+ cells (106) were isolated, resuspended in 50 μL lysis buffer (0.1% Triton X-100 with 100 μg/mL Pepstatin A, Aprotinin, and Antipain), and shaken for 30 min at 37°C. Following lysis, 50 μL of 50 mM Tris-HCl / 10 mM MnCl2 was added and the mixture was heated for 10 min at 56°C. The lysate was incubated with 100 μL of 0.5 M L-arginine for 120 min at 37°C. The reaction was stopped with 800 μL of H2SO4 (96%) / H3PO4 (85%) / H20 (1:3:7 by volume). Subsequently, 40 μL of 9% (by weight) α-isonitrosopropiophenone (ISPF) in 100% ethanol was added, followed by heating at 95°C for 30 min. After 10 min in the dark, urea concentration was measured at 540 nm and compared to a standard curve generated by serial dilution of 750 μg/mL urea. Arginase activity (units) was determined by the amount of urea (μg) formed per minute.
Nitric Oxide Production
Nitric oxide production by Gr-1+ CD11b+ cells was evaluated in culture supernatant from the OT-1 T cell suppression assay using the Griess Reagent System (Promega). Equal volumes of supernatant (40 μL) and 1% sulfanilamide in 5% phosphoric acid were incubated at RT for 10 min, followed by addition of 0.1% N-1-naphthylethylenediamine dihydrochloride in water. After 10 min at RT, absorbance at 550 nm was measured. Nitrite concentrations were determined by comparing absorbance values to a standard curve generated by serial dilution of 0.1 mM sodium nitrite.
BrdU Labeling and Analysis
c-kit+ lineageneg cells on day 4 of culture were labeled with bromodeoxyuridine (BrdU) (BD Biosciences; 10 μM) for 2 hours at 37°C and prepared for flow cytometric analysis according to the FITC BrdU Flow Kit (BD Biosciences).
Cell Lines
PDA cell lines from KPC mice were derived by from single cell suspensions of PDA tissue. Dissociated cells were plated in a 6-well dish with serum free DMEM. After 2 weeks, media was changed to DMEM + 10% FCS. After 3-6 passages, cells were used in experiments. Cell lines established from PDA tumors of KPC mice with confirmed genotypes were injected into normal mice to confirm tumorigenicity. At necropsy, transplanted tumors were inspected pathologically by H&E staining and invasive carcinoma was confirmed.
Fresh Tumor Supernatant Collection
Tumors were excised, washed in PBS, weighed, and minced into small fragments with sterile scissors. Tissue supernatant was harvested by adding 400 μL complete media and compressing tumor fragments with the plunger of a 3 mL syringe. The tumor tissue solution was centrifuged at 13,000 rpm × 10 min, and the supernatant carefully collected.
Cytokine Analysis
Cytometric bead array (BD Biosciences) and ELISA (R&D systems) were used to quantify cytokines in supernatant from PDA lines and wild-type pancreatic ductal cell lines cultured in DMEM + 10% FCS and in supernatant harvested from freshly isolated KPC tumors. For both methods, quantification of cytokines was determined by reference to recombinant murine standards.
Pancreas Cell Sorting and Transcriptional Analysis
Single pancreas cells from tumor-bearing KrasG12D; p53fl/+; Pdx1-Cre; RosaYFP mice and non-tumor bearing Pdx1-Cre; RosaYFP control mice were sorted based on YFP fluorescence into tubes with chilled RLT buffer (Qiagen). The sorted YFP+ and YFPneg populations are referred to as pancreatic epithelial and stromal compartments, respectively. RNA was extracted from samples using the RNEasy kit (Qiagen). First strand cDNA was synthesized using SuperScript III (Invitrogen). qPCR was performed using Taqman probes for GAPDH and GM-CSF (Applied Biosystems). Relative expression was determined after adjusting for GAPDH.
shRNA Knockdown of GM-CSF in PDA-1 Cells
GIPZ lentiviral shRNAmir targeting GM-CSF (Clone ID: V2LMM_64126) and GIPZ non-silencing control viral particles (Catalog # RHS4348) were purchased from OpenBiosystems. PDA-1 cells were infected with high-titer concentrated lentiviral suspensions and selected with 10 μg/mL Puromycin. GFP-positive single cells were sorted using a MoFlo cell sorter and clonal PDA-1 cell lines were derived. In vitro growth kinetics for shGM-CSF PDA-1 cells or mock PDA-1 cells did not differ from the parental PDA-1 cell line.
Short-Term PDA Implantation Studies
PDA-1 tumor cells (106) were injected subcutaneously with anti-GM-CSF mAb (BioLegend; 250 μg) or purified rat IgG2a (BioXcell; 250 μg) imbedded as plugs with matrigel matrix (Collaborative Biomedical Products) into the flanks of normal control mice for 6 days. For GM-CSF knockdown studies, mock PDA-1 cells (106) or shGM-CSF PDA-1 cells (106) were injected subcutaneously with matrigel matrix for 6 days. In some experiments, tumor cells were lethally gamma-irradiated (10 Gy) prior to implantation. For depletion of CD8+ T cells or Gr-1+ cells, anti-CD8 mAb (BioXcell; 200 μg), anti-Gr-1 mAb (BioXcell; 500 μg), or as a control purified isotype control rat IgG2b (BioXcell; 200 μg) was injected intraperitoneally on days -1, 0, 1, and 3 with respect to tumor cell inoculation.
Immunohistochemistry and Histopathology
Immunohistochemistry and histopathology were performed on frozen tissue sections. Sections were fixed in 3% formaldehyde for analysis of CD45 (BD Biosciences; 3-F11; 1:50) and Gr-1 (BioXCell; RB6-8C5; 15 μg/mL). Endogenous peroxidases were quenched in 0.3% H2O2 in water for 10 min. Sections were blocked with 10% goat serum in PBS + 0.1% Tween-20 for 30 min followed by labeling with primary antibody overnight at 4°C. Sections were washed and incubated with goat anti-rat biotinylated secondary antibody (BD Biosciences; 1:200) for 1 hour at RT. Remaining steps were performed using Vectastain ABC kits (Vector Labs). Slides were counterstained with hematoxylin.
Human surgical material was obtained after informed consent using an Institutional Review Board approved protocol. Formalin-fixed paraffin-embedded tissues were sectioned for immunohistochemistry. After rehydration, sections were incubated in 1% hydrogen peroxide/methanol then with 10 mM sodium citrate/0.05% Tween-20 (pH 6.0) in a microwave oven for antigen retrieval. Sections were then blocked with 10% serum/1% BSA/0.5% Tween-20 for 1 hr at RT, then incubated with rabbit anti-GM-CSF (Novus Biologicals) overnight at 4°C. Secondary antibody was biotinylated goat anti-rabbit IgG (Vector Laboratories).
Immunofluorescence
Immunofluorescence was performed on frozen tissue sections. Section were fixed in 3% formaldehyde for analysis of CD45 (BD Biosciences; 3-F11; 1:50) or -20°C methanol for analysis of Gr-1 (BioXCell; RB6-8C5; 15 μg/mL) and CD8 (BioXCell; 2.43; 15 μg/mL). Sections were blocked with 10% goat serum in PBS + 0.1% Tween-20 for 30 min followed by labeling with primary antibody overnight at 4°C. Sections were washed and incubated with Alexa Fluor 568 goat anti-rat secondary antibody (Invitrogen; 1:500) along with DAPI (Invitrogen; 1:1000) for 1 hour at RT protected from the light. Sections were mounted with Aqua-Poly/Mount (Polysciences).
CD8+ T cell IFN-γ/CD107a Analysis
Single cell suspensions of matrigel tumor plugs were mixed with anti-CD107a FITC (1:100) and plated in a 96-well round bottom plate with GolgiStop protein transport inhibitor (containing monensin) (BD Biosciences) and complete media or media containing anti-CD3 (0.5 μg/mL) and anti-CD28 (1 μg/mL) as a positive control. Cells were incubated at 37°C for 5 hours, then were washed and stained for surface molecules. Cells were fixed and permeabilized (eBiosciences) followed by intracellular cytokine staining with anti-IFN-γ PE (1:50; BD Biosciences) prepared in permeabilization buffer for 20 min at 4°C. Cells were washed and analyzed on a FACSCanto flow cytometer (BD Biosciences) for IFN-γ and CD107a expression on CD45+ CD8+ CD11bneg CD11cneg CD4neg CD19neg cells.
Statistical Analyses
All statistical analyses were determined by Student’s t test using GraphPad Prism software.
Supplementary Material
Significance.
Pancreatic ductal adenocarcinoma (PDA) carries a dire prognosis for which novel therapeutic strategies are urgently needed. An inflammatory desmoplastic stromal reaction is characteristic of PDA and likely contributes to disease progression. Here, using a spontaneous murine model of PDA, we demonstrate that tumor-derived GM-CSF drives the accumulation of Gr-1+ CD11b+ myeloid cells as part of the cancer-associated inflammatory reaction, which in turn suppresses anti-tumor T cell immunity. In humans, PDA tumor cells prominently expressed GM-CSF in vivo. Our findings suggest a therapeutic potential for disrupting the crosstalk between tumor cells and the immune system by targeting Gr-1+ CD11b+ cells or the cytokines that regulate their differentiation.
Highlights.
Tumor-derived GM-CSF drives immunosuppressive inflammation in pancreatic carcinoma
Abrogation of GM-CSF blocks tumor development in a T-cell dependent fashion
In humans, PDA tumor cells prominently express GM-CSF in vivo.
Findings suggest a therapeutic mechanism to target inflammation in pancreatic cancer
Acknowledgments
We thank Carmine Carpenito, Qian-Chun Yu, Hong Wei, Nicole Aiello, and Emily Mirek for important discussions. This work was supported by the Abramson Family Cancer Research Institute (R.H.V., B.Z.S.) and by grants from the Cancer Research Institute (to L.J.B.), the National Institutes of Health (grants K12 CA076931 to G.L.B. and K08 088945 to A.D.R.), the Gabrielle’s Angel Foundation for Cancer Research (to G.L.B.), the American Gastroenterological Association (to A.D.R.), and the Pennsylvania Department of Health (to R.H.V., B.Z.S) The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, Korets L, Lam J, Tawfik D, DeNardo DG, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17:121–134. doi: 10.1016/j.ccr.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone G, Carbone A, Smirne C, Scirelli T, Buffolino A, Novarino A, Stacchini A, Bertetto O, Palestro G, Sorio C, et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J Immunol. 2006;177:3448–3460. doi: 10.4049/jimmunol.177.5.3448. [DOI] [PubMed] [Google Scholar]
- Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–3846. [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–5737. [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–278. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279:1–7. doi: 10.1016/j.canlet.2008.09.037. [DOI] [PubMed] [Google Scholar]
- Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- Dandrea M, Donadelli M, Costanzo C, Scarpa A, Palmieri M. MeCP2/H3meK9 are involved in IL-6 gene silencing in pancreatic adenocarcinoma cell lines. Nucleic Acids Res. 2009;37:6681–6690. doi: 10.1093/nar/gkp723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40:22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- Dougan M, Li D, Neuberg D, Mihm M, Googe P, Wong KK, Dranoff G. A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J Clin Invest. 2011;121:2436–2446. doi: 10.1172/JCI44796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A, Wang SC, Morris JPt, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, Hebrok M. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19:441–455. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Huang Y, Chen X, Dikov MM, Novitskiy SV, Mosse CA, Yang L, Carbone DP. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood. 2007;110:624–631. doi: 10.1182/blood-2007-01-065714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–245. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Ma G, Pan PY, Eisenstein S, Divino CM, Lowell CA, Takai T, Chen SH. Paired Immunoglobin-like Receptor-B Regulates the Suppressive Function and Fate of Myeloid-Derived Suppressor Cells. Immunity. 2011;34:385–395. doi: 10.1016/j.immuni.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrache F, Tu SP, Bhagat G, Pendyala S, Osterreicher CH, Gordon S, Ramanathan V, Penz-Osterreicher M, Betz KS, Song Z, Wang TC. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology. 2008;135:1277–1287. doi: 10.1053/j.gastro.2008.06.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–2145. doi: 10.1182/blood-2003-01-0190. [DOI] [PubMed] [Google Scholar]
- Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010;123:39–49. doi: 10.1007/s10549-009-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic KRas-induced GM-CSF production supports the development of pancreatic neoplasia. Cancer Cell. 2012 doi: 10.1016/j.ccr.2012.04.024. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS One. 2009;4:e7669. doi: 10.1371/journal.pone.0007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF, Jr, Tocker JE, Budelsky AL, Kleinschek MA, Kastelein RA, Kambayashi T, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464:1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328:749–752. doi: 10.1126/science.1185837. [DOI] [PubMed] [Google Scholar]
- Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol. 2005;175:8200–8208. doi: 10.4049/jimmunol.175.12.8200. [DOI] [PubMed] [Google Scholar]
- Stairs DB, Bayne LJ, Rhoades B, Vega ME, Waldron TJ, Kalabis J, Klein-Szanto A, Lee JS, Katz JP, Diehl JA, et al. Deletion of p120-Catenin Results in a Tumor Microenvironment with Inflammation and Cancer that Establishes It as a Tumor Suppressor Gene. Cancer Cell. 2011;19:470–483. doi: 10.1016/j.ccr.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, Wang TC. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XD, Ai W, Asfaha S, Bhagat G, Friedman RA, Jin G, Park H, Shykind B, Diacovo TG, Falus A, Wang TC. Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nat Med. 2011;17:87–95. doi: 10.1038/nm.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.