

Abstract
N,S-diprotected L-thiothreonine and L-allo-thiothreonine derivatives were synthesized using a novel chemical strategy, and used for esterification of the dinucleotide pdCpA. The aminoacylated dinucleotides were then employed for the preparation of activated suppressor tRNACUA transcripts. Thiothreonine and allo-thiothreonine were incorporated into a predetermined position of a catalytically competent dihydrofolate reductase (DHFR) analogue lacking cysteine, and the elaborated proteins were derivatized site-specifically at the thiothreonine residue with a fluorophore.
Keywords: Sulfur-containing amino acids, Aminoacylation, Protein synthesis, Fluorescence labeling
1. Introduction
Nonproteinogenic amino acids having functionalized side chains are constituents of biologically important peptides and peptidomimetics. As a result, significant effort has been expended for the development of synthetic routes useful for preparing such compounds.1 One example is β-methylcysteine (thiothreonine) which has attracted considerable attention based on its presence as a constituent of peptides called lantibiotics.2 Peptides of this class are of current interest due to their potent antimicrobial activity and their novel mode of action.3 Lantibiotics are ribosomally synthesized, posttranslationally modified peptide antibiotics produced by Gram-positive bacteria. They are characterized by lanthionine and methyl lanthionine residues that are connected via thioether bridges to create ring structures critical for their bioactivity.2,3 One such peptide, microbisporicin, is currently undergoing preclinical development as a consequence of its superior efficacy in animal models of multidrug resistant infections compared with the antibiotics of last resort, linezolid and vancomycin.4 Further, of the proteinogenic amino acids, those containing key side chain functional groups are especially interesting for study since they often have functional roles in proteins. For example, cysteine appears in the active sites of enzymes such as cysteine proteases,5 and can be critical structurally in proteins via its participation in disulfide bridges.6 As part of our ongoing efforts to enable the synthesis of modified proteins containing mechanistically interesting non-proteinogenic amino acids,7 we focused on the synthesis of β-methylcysteine (thiothreonine). Thus, the syntheses of L-thiothreonine and L-allo-thiothreonine (Figure 1) have been undertaken utilizing a novel strategy.
Figure 1.
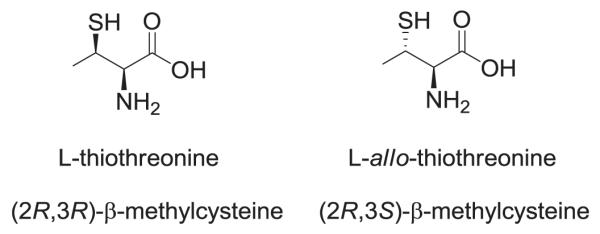
Structures of the β-methylcysteine (thiothreonine) stereoisomers synthesized.
The use of reagents that react with specific amino acid side chains is a common way to realize post-translational protein modification. The introduction of a limited number of non-natural amino acids into predetermined positions in proteins can be an effective method to target specific sites for modification. In particular, a non-natural amino acid having a single thiol functional group can be introduced into a target protein and subsequently transformed by reagents that react with S atoms. The thiothreonines prepared in this study were incorporated into a modified DHFR protein lacking cysteine residues (csDHFR), facilitating site-specific protein modification at introduced thiothreonine and allo-thiothreonine residues via treatment with a thiol specific fluorophore, 7-diethylamino-3-(4′ -maleimidylphenyl)-4-methylcoumarin (CPM). The fluorescent prosthetic group was introduced at the level of pdCpA, suppressor tRNA, or protein to define the optimal methodology for introducing specific functionalities into proteins.
2. Results and discussion
2.1. Chemistry
Several methods have been reported for the preparation of β-methylcysteine derivatives, including displacement of an O-tosylated threonine derivative with potassium thioacetate,8 conjugate addition of a thiol to an oxazolone derivative of an α,β-dehydroamino acid to yield an S-benzyl-β-alkylcysteine upon hydrolysis9 and acid hydrolysis of a threonine-derived thiazolidine.10 However, while the first method gave a single stereoisomeric β-methylcysteine derivative, the latter two methods required laborious separations to access the individual diastereoisomers. The stereospecific ring opening of an aziridine with thiobenzoic acid in the presence of a Lewis acid for the synthesis of threo-β-methylcysteine from D-threonine was originally reported by Wakamiya et al.11 It involved a double inversion at the β-carbon but suffered from the formation of O-acylated by-product in addition to the desired S-acyl product. An optimized variant of this strategy was used in the synthesis of orthogonally protected β-methyllanthionine by ring opening of aziridines with several thiol nucleophiles (Figure 2).12 The introduction of sulfur at the 2-position of threonine by ring opening of cyclic threonine sulfamidate with trityl thiol and Boc-protected cysteine was recently reported for the preparation of stereochemically pure β-methylcysteine and methyllanthionine derivatives.13 Presently, we report the preparation of L-thiothreonine and L-allo-thiothreonine stereoisomers utilizing a novel ring opening reaction involving threonine-derived lactones. The ring opening of such lactones is well documented but the required attack of the nucleophilic sulfur atom at the hindered β-position was expected to be difficult, as reported by Pansare and Vederas who studied the nucleophilic ring opening of sulfonamide derivatives of the lactone (Figure 2).14 Lactone ring opening using LiSH and NaSH as nucleophiles gave products derived exclusively from attack at the carbonyl carbon; an exception was thiourea, which attacked the β-carbon atom affording the β-isothiouronium salt, a plausible precursor to a β-methylcysteine derivative. We applied this strategy for the synthesis of NVOC protected β-methylcysteine using potassium thioacetate as the nucleophile for the key ring opening of the β-lactone at the β-carbon atom (Scheme 1). For the protected stereoisomers of thiothreonine studied (Figure 3), β-ring opening proceeded in yields of 85% and 82%, respectively. The synthesis of protected L-thiothreonines (1a and 1b) commenced with the preparation of NVOC-L-allo-threonine (3) by treatment of L-allo-threonine with 6-nitroveratryl chloroformate (NVOCCl), the latter of which was prepared according to the procedure of Katritzky et al.15 Thus, L-allo-threonine was dissolved in a 1:1 mixture of dioxane and water in the presence of sodium bicarbonate and NVOCCl was added. Upon stirring for 17 hours at room temperature, NVOC-protected L-allo-threonine (3) was obtained following work-up in 60% yield. Although serine lactones can be easily prepared from N-protected serines through a Mitsunobu reaction utilizing diisopropyl azodicarboxylate and triphenylphosphine, the corresponding threonine counterparts are much more difficult to prepare under the same conditions, resulting in decarboxylative elimination.14,16 Therefore, lactone formation through carboxyl group activation using BOP reagent or HBTU was envisaged.17 Thus, compound 3, dissolved in dichloromethane, was treated with triethylamine and HBTU and the reaction mixture was allowed to stir at room temperature under argon for 20 hours. Extractive work-up followed by silica gel column chromatography gave the desired lactone 4 in 60% yield. Next, the ring opening of the lactone with thioacetate anion was studied, constituting the crucial step in the synthesis of protected L-thiothreonine. Gratifyingly, treatment of 4 with potassium thioacetate in DMF at room temperature for 23 h afforded thioacetate 5 regio- and stereospecifically in 85% yield after purification. Since the acetate group used for the protection of the thiol is not easily removed under conditions compatible with the stability of aminoacyl-tRNAs, we sought to replace it with a protecting group such as butyldisulfide or 4,4′-dimethoxytrityl (DMT) group, both of which can be cleaved under neutral conditions. The first attempt to remove the S-acetyl group using potassium carbonate, followed by reprotection of the SH group as a mixed disulfide via the agency of butyl 1-thiobutane-1-sulfinate,18 gave the desired butyldisulfide 1a, albeit in low yield. The use of 1 eq. of sodium methoxide in methanol to remove the acetate group followed by mixed disulfide formation afforded orthogonally protected L-thiothreonine 1a in 72% yield. The introduction of the DMT group was performed via a two step process. Thus, the acetate group was hydrolyzed using lithium hydroxide and the resulting free thiol was treated with 4,4′-dimethoxytrityl chloride in the presence of triethylamine to give the orthogonally protected L-thiothreonine 1b in 51% yield. The successful implementation of the regio- and stereoselective ring opening reaction promoted by the thioacetate anion prompted us to extend this methodology to the synthesis of protected L-allo-thiothreonine (Scheme 2). L-Threonine was employed as starting material and was treated initially with NVOCCl, leading to N-protection and affording compound 6 in 85% yield. Treatment with HBTU in the presence of triethylamine in dichloromethane for 20 h gave β-lactone derivative 7 in 65% yield. Subsequent treatment of compound 7 with potassium thioacetate in DMF resulted in opening of the β-lactone ring and the formation of thioacetate derivative 8, which was obtained in 82% yield. Deprotection of the acetate group of compound 8 was achieved using sodium methoxide in methanol, followed by treatment with butyl 1-thiobutane-1-sulfinate in the same reaction flask. Following work-up and column chromatographic purification, compound 2a was obtained in 90% yield. The DMT protected L-allo-thiothreonine 2b was obtained after deacetylation using lithium hydroxide in aqueous methanol followed by trapping the resulting free thiol with 4,4′-dimethoxytrityl chloride. Compound 2b was obtained in 84% yield after purification. The conversion of protected compounds 1 and 2 to the corresponding cyanomethyl esters was attempted using 1a and 1b, as well as 2a and 2b. The conversions were found to proceed significantly more cleanly with the S-tritylated precursors (1b and 2b). Optimal results with 1b and 2b were achieved by treatment with chloroacetonitrile in dry acetonitrile in the presence of triethylamine. Cyanomethyl esters 9 and 11 were obtained in 67 and 85% yields, respectively (Scheme 3). The treatment of the active esters 9 and 11 with the tris-(tetrabutylammonium) salt of pdCpA in anhydrous DMF gave the corresponding aminoacylated pdCpAs 10 and 12 in 21 and 47% yields, respectively. The removal of the DMT group at the pdCpA level was performed on protected L-allo-thiothreonyl pdCpA ester 12 using silver nitrate in aqueous acetonitrile followed by treatment with dithiothreitol (Scheme 4). The detritylation was complete within 30 minutes and NVOC-protected L-allo-thiothreonyl pdCpA ester 13 was obtained in 82% yield. The dinucleotide was derivatized on the sulfur atom with a coumarin fluorophore [7-diethylamino-3-(4′-maleimidophenyl)-4-methylcoumarin] in acetonitrile/water and the fluorescent dinucleotide 14 was obtained in 64% yield after purification by HPLC (Figure S1, Supplementary data).
Figure 2.
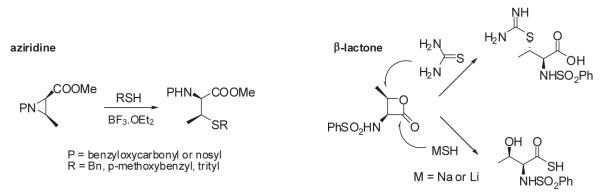
Strategies reported for the synthesis of β-methylcysteine derivatives.
Scheme 1.

Synthesis of N,S-diprotected L-thiothreonine derivatives 1a and 1b.
Figure 3.
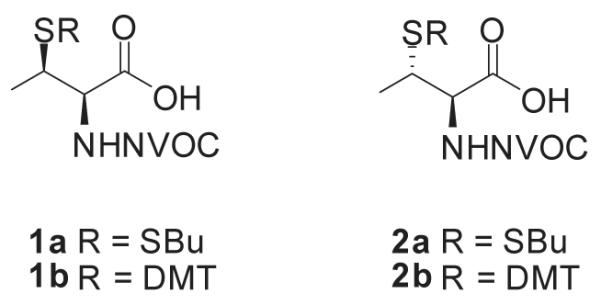
N,S-diprotected derivatives of L-thiothreonine (1) and L-allo-thiothreonine (2).
Scheme 2.

Synthesis of N,S-diprotected L-allo-thiothreonine derivatives 2a and 2b.
Scheme 3.
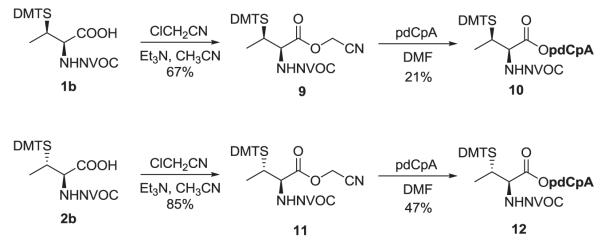
Synthesis of N,S-diprotected L-thiothreonine and L-allo-thiothreonine derivatives of pdCpA.
Scheme 4.

Synthesis of thiothreonyl- and allo-thiothreonyl-tRNACUAs.
2.2. Synthesis of tRNACUAs activated with L-thiothreonines and coupling with CPM
Suppressor tRNACUAs activated either with L-thiothreonine or allo-L-thiothreonine were prepared as outlined in Scheme 4. The aminoacylated pdCpAs were ligated to an abbreviated suppressor tRNACUA transcript (tRNACUA-COH, lacking the terminal cytidine and adenosine moieties present at the 3′-terminus of all tRNAs) via a T4 RNA ligase mediated ligation reaction to obtain the N and S-protected thiothreonyl-tRNACUAs. The ligation efficiencies were evaluated via denaturing PAGE analysis as shown in Figure 4.19 The N-protected aminoacylated tRNAs were then irradiated with UV light at 0 °C for 5 minutes to afford the activated tRNAs having amino acids with free α-amines. Evaluation of the deprotection efficiency of the DMT group was investigated prior to photolysis. A portion of the fully protected tRNAs were subjected to treatment with silver nitrate, followed by treatment with dithiothreitol, affording thiothreonyl-tRNACUAs having free sulfhydryl groups. Removal of the DMT group was verified via coupling with the coumarin fluorophore, and visualized by irradiating with 365 nm wavelength light (Figure 5).
Figure 4.
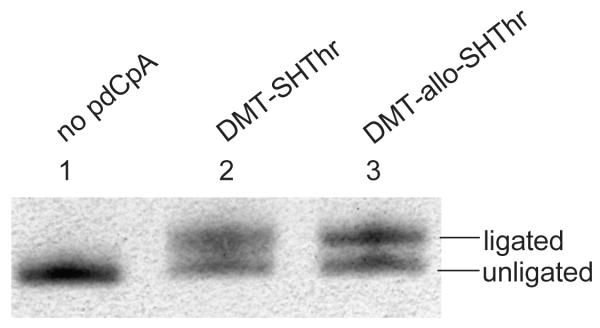
Ligation of thiothreonyl-pdCpA derivatives to tRNACUA-COH. The efficiency of ligation was monitored by 8% denaturing PAGE, pH 5.2, and the gel was stained with methylene blue. Lane 1, abbreviated tRNACUA (1 μg) without pdCpA; lane 2, ligation of DMT-thiothreonyl-pdCpA with tRNACUA (1 μg); lane 3, ligation of DMT-allo-thiothreonyl-pdCpA with tRNACUA (1 μg).
Figure 5.

Coupling of the NVOC-protected thiothreonyl-tRNA analogues with CPM after DMT group removal. The fluorescence was monitored at ~470 nm following irradiation at 365 nm. Sample 1, CPM treated suppressor tRNA (3 μg/μL); sample 2, CPM treated DMT-thiothreonyl-tRNA (3 μg/μL); sample 3, CPM coupled thiothreonyl-tRNA (3 μg/μL); sample 4, CPM treated DMT-allo-thiothreonyl-tRNA (3 μg/μL); sample 5, CPM coupled allo-thiothreonyl-tRNA (3 μg/μL).
Allo-thiothreonyl-tRNACUA derivatized with the same fluorophore was also prepared by the T4 RNA ligase-mediated coupling of dinucleotide 14 with tRNA-COH (Scheme 4). Following irradiation with UV light to remove the NVOC protecting group, the CPM-derivatized allo-thiothreonyl-tRNACUA was employed in a protein synthesizing system, as described below.
2.3. Participation of the activated tRNAs in protein synthesis and post-translational modification of csDHFR
The ability of thiothreonyl-tRNACUAs to participate in protein synthesis was evaluated in a bacterial cell free protein synthesizing system. A plasmid encoding a modified E. coli DHFR in which the Cys85 and Cys152 codons had been replaced with serine codons was used. This modified DHFR was found to retain essentially full enzymatic activity (Figure S2, Supplementary data). Additionally, a stop codon (TAG) was incorporated into the plasmid at position 10 of DHFR to facilitate incorporation of the thiothreonine derivatives from the misacylated suppressor tRNAs (Scheme 5). Both L-thiothreonyl-tRNACUA and L-allo-thiothreonyl-tRNACUA were found to participate in protein synthesis, affording DHFRs with 43% and 45% suppression efficiencies, respectively, relative to the amount of csDHFR produced from the comparable mRNA lacking any stop codon (Figure 6). The DMT-protected thiothreonine and allo-thiothreonine were incorporated in yields of 28% and 26%, respectively. In comparison, the incorporation of the fluorescently labeled allo-thiothreonine from CPM-derivatized allo-thiothreonyl-tRNACUA (prepared by ligation of 14 and tRNA-COH) proceeded only in low (~ 5%) yield. In replicate experiments, the incorporation of CPM-allo-thiothreonine into csDHFR ranged from 2–8%. All of the proteins were purified successively on Ni-NTA and then DEAE-Sepharose columns, as illustrated for the thiothreonine-containing DHFR (Figure S3, Supplementary data). Coupling of the purified protein containing thiothreonine and allo-thiothreonine in position 10 with the coumarin fluorophore was visualized by irradiating with 365 nm wavelength light (Figure 7). As is clear from Figure 7, the fluorophore could be introduced readily into the nascent csDHFRs containing thiothreonine and allo-thiothreonine. As anticipated, the csDHFRs elaborated with DMT-protected thiothreonine and allo-thiothreonine did not react detectably with the coumarin fluorophore.
Scheme 5.

Strategy employed for incorporation of thiothreonines into csDHFR and coupling with CPM.
Figure 6.
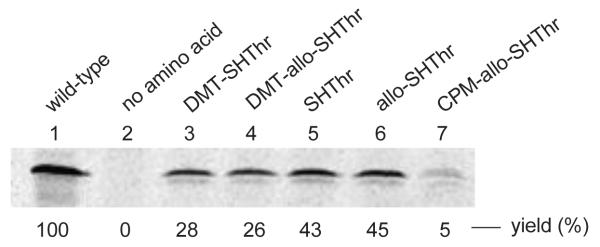
In vitro incorporation of thiothreonine derivatives into csDHFR at position 10. The samples were analyzed by 15% SDS-PAGE at 100 V for 2 h. Phosphorimager analysis was performed using an Amersham Biosciences Storm 820 equipped with ImageQuant version 5.2 software from Molecular Dynamics. Lane 1, wild-type csDHFR; lane 2, incorporation in the absence of any amino acid; lane 3, incorporation of DMT-thiothreonine; lane 4, incorporation of DMT-allo-thiothreonine; lane 5, incorporation of thiothreonine; lane 6, incorporation of allo-thiothreonine; lane 7, incorporation of CPM-allo-thiothreonine.
Figure 7.
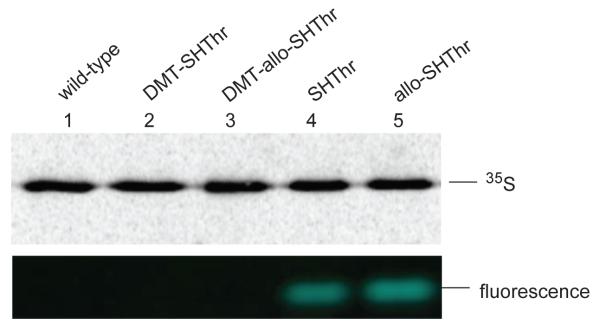
Coupling of thiothreonyl-csDHFR analogues with CPM. All of the protein samples were purified chromatographically (cf Figure 7) prior to treatment with the fluorophore. The samples were analyzed by 15% SDS-PAGE at 100 V for 2 h. The 35S-methionine labeled protein bands were obtained through phosphorimager analysis, and the fluorescence was monitored at ~470 nm following irradiation at 365 nm. Lane 1, CPM treated wild-type csDHFR (1 μg); lane 2, CPM treated DMT-thiothreonyl-csDHFR (1 μg); lane 3, CPM treated DMT-allo-thiothreonyl-csDHFR (1 μg); lane 4, CPM treated thiothreonyl-csDHFR (1 μg); lane 5, CPM treated allo-thiothreonyl-csDHFR (1 μg).
In the present study, we used the DMT group to protect the thiol groups of L-thiothreonine and L-allo-thiothreonine. The DMT group could be removed readily at the pdCpA and tRNA levels, and both the DMT-protected and unprotected thiothreonyl-and allo-thiothreonyl-tRNACUAs were used to incorporate the thiolated amino acids into position 10 of csDHFR. The nascent csDHFRs containing unprotected thiothreonine and allo-thiothreonine reacted readily with CPM, affording protein fluorescently labeled at a single position. However, the DMT group could not be removed from the thiothreonines at the protein level due to protein precipitation under the conditions used for deprotection. The free thiol groups of thiothreonine and allo-thiothreonine could be modified with the fluorophore CPM at the pdCpA, tRNA and protein levels. However, the CPM-protected allo-thiothreonyl-tRNACUA proved to be a poor substrate in the peptidyltransferase reaction, such that the preferred method for elaborating the fluorescent protein was by direct derivatization of the initially formed csDHFR containing a free SH group in the amino acid at position 10.
3. Conclusions
This study has addressed the synthesis of the two stereoisomers of L-thiothreonine in five steps using a novel approach. The key step in these syntheses involved the formation of an intermediate L-threonyl-β-lactone, which was opened using thioacetate as a nucleophile to afford the desired stereoisomers of L-thiothreonine. This synthesis provided access to L-thiothreonine and L-allo-thiothreonine cleanly and in good yields. The two isomers so prepared were used to esterify pdCpA and then ligated to tRNACUA-COH.
To illustrate specific coupling between the thiothreonine amino acids and a thiol-reactive fluorophore, we employed a modified DHFR having serines in lieu of the Cys85 and Cys152 in wild-type DHFR. The mRNA used to direct the syntheses of DHFR had a stop (UAG) codon corresponding to position 10 of the protein.
Thiothreonyl- and allo-thiothreonyl-tRNACUAs functioned well in the cell free protein synthesizing systems employed, effecting suppression of the UAG codon at position 10 in csDHFR in 43 and 45% yields, respectively. The derived proteins both reacted with the thiol-specific fluorophore, while proteins elaborated with the S-tritylated amino acids as a control did not react with the fluorophore detectably.
4. Experimental
4.1. General Materials and Methods
Reagents and solvents for chemical synthesis were purchased from Aldrich Chemical Co. or Sigma Chemical Co. and used without further purification. All reactions involving air- or moisture-sensitive reagents or intermediates were performed under argon. Flash chromatography was performed using Silicycle silica gel (40–60 mesh). Analytical TLC was performed using EM silica gel 60 F254 plates (0.25 mm) and was visualized by UV irradiation (254 nm). 1H and 13C NMR spectra were obtained using a 400 MHz Varian NMR instrument. Chemical shifts are reported in parts per million (ppm, δ) referenced to the residual 1H resonance of the solvent (CDCl3, δ 7.26; CD3OD, δ 3.31, DMSO-d6, δ 2.50). 13C NMR spectra were referenced to the residual 13C resonance of the solvent (CDCl3, δ 77.16; CD3OD, δ 49.00, DMSO-d6, δ 39.52). Splitting patterns are designated as follows: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; q, quartet; quint, quintet; m, multiplet; br, broad. High resolution mass spectra were obtained at the Arizona State University CLAS High Resolution Mass Spectrometry Laboratory or at Michigan State University Mass Spectrometry Facility.
Ni-NTA agarose was obtained from Qiagen Inc. (Valencia, CA). DNA oligonucleotides were ordered from Integrated DNA Technologies (Coralville, IA). DEAE-Sepharose, ammonium persulfate, acrylamide, N, N′-methylene-bis-acrylamide, acetic acid, potassium glutamate, ammonium acetate, dithiothreitol, magnesium acetate, phospho(enol)pyruvate, Escherichia coli tRNA, isopropyl β-D-thiogalactopyranoside (IPTG), ATP, GTP, CTP, UTP, cAMP, amino acids, rifampicin, formamide and 7-diethylamino-3-(4′-maleimidophenyl)-4-methylcoumarin (CPM) were obtained from Sigma-Aldrich (St. Louis, MO). Tris and SDS were obtained from Bio-Rad Laboratories (Hercules, CA). [35S]-methionine (1000 Ci/mmol, 10 μCi/ μL) was purchased from PerkinElmer Inc. (Boston, MA). Protease inhibitor (complete, EDTA-free) was obtained from Boehringer Mannheim Corp. (Indianapolis, IN). T4 RNA ligase and T4 polynucleotide kinase were purchased from New England Biolabs Inc. (Ipswich, MA). Phosphorimager analysis was performed using an Amersham Biosciences Storm 820 equipped with ImageQuant version 5.2 software from Molecular Dynamics. UV spectral measurements were made using a Perkin-Elmer Lamdba 20 UV/vis spectrometer. Fluorescence was monitored through 365 nm UV light. The mutant csDHFR analogues modified at position 10 with thiothreonine were prepared using the general strategy shown in Scheme 5.7,20
4.2. Synthesis of L-thiothreonines and their pdCpA derivatives
4.2.1.N-(6-Nitroveratryloxycarbonyl)-L-allo-threonine (3)
To a solution containing 0.50 g (4.19 mmol) of L-allo-threonine in 20 mL of 1:1 dioxane/H2O was added 0.70 g (8.38 mmol) of NaHCO3. This was followed by the addition of 1.38 g (5.03 mmol) of 6-nitroveratryl chloroformate. The reaction mixture was stirred at room temperature for 17 h. Twenty mL of CHCl3 was then added to the reaction mixture, and the biphasic mixture was separated. The chloroform layer was discarded and the aqueous layer was acidified to pH ~4 by the addition of 1 N NaHSO4. The aqueous solution was then washed with three 30-mL portions of ethyl acetate. The combined organic layer was dried (MgSO4) and the solvent was concentrated under diminished pressure. The crude residue was purified on a silica gel column (15 × 4 cm); elution with 9:1:0.01 CH2Cl2/MeOH/AcOH afforded N-(6-nitroveratryloxycarbonyl)-L-allo-threonine (3) as a yellow foam: yield 0.90 g (60%); silica gel TLC Rf 0.20 (9:1:0.01 CH2Cl2/MeOH/AcOH); [α]24D-7.1 (c 1.0, MeOH); 1H NMR (CD3OD) δ 1.24 (d, 3H, J = 6.4 Hz), 3.89 (s, 3H), 3.96 (s, 3H), 4.14 (m, 1H), 4.27 (d, 1H, J = 5.2 Hz), 5.38, 5.42 (ABq, 2H, J = 15.6 Hz), 7.18 (s, 1H), and 7.71 (s, 1H); 13C NMR (CD3OD) δ 19.2, 56.8, 57.0, 61.3, 64.6, 68.6, 109.2, 110.6, 129.8, 140.5, 149.4, 155.4, 158.3, and 173.6; mass spectrum (APCI), m/z 359.1083 (M+H)+ (C14H19N2O9 requires m/z 359.1091).
4.2.2. N-(6-Nitroveratryloxycarbonyl)-L-allo-threonyl-β-lactone (4)
A sample of 0.87 g (2.42 mmol) of compound 3 was dissolved in 25 mL of dichloromethane and 1.35 mL (0.98 g, 9.68 mmol) of triethylamine was added. To this solution was added 1.83 g (4.84 mmol) of HBTU (O-benzotriazole-N,N,N′,N′-tetramethyluroniumhexafluorophosphate) and the reaction mixture was stirred at room temperature for 20 h under argon. The solvent was concentrated under diminished pressure and the crude product was purified by flash column chromatography on a silica gel column (15 × 4 cm) using 1:1 hexanes/EtOAc for elution. The appropriate fractions were collected and the solvent was concentrated under diminished pressure to give the desired product 4 as a pale yellow solid: yield 498 mg (60%); mp 140-142 °C; silica gel TLC Rf 0.20 (1:1 hexanes/ethyl acetate); [α]24D-58.5 (c 1.0, acetone); 1H NMR (DMSO-d6) δ 1.49 (d, 3H, J = 5.6 Hz), 3.87 (s, 3H), 3.90 (s, 3H), 4.77 (m, 2H), 5.37 (s, 2H), 7.19 (s, 1H), 7.70 (s, 1H), and 8.41 (d, 1H, J = 7.2 Hz); 13C NMR (DMSO-d6) δ 18.2, 56.1, 56.2, 63.26, 63.29, 75.4, 108.2, 111.1, 126.7, 139.5, 147.9, 153.3, 155.0, and 168.9; mass spectrum (APCI), m/z 341.1005 (M+H)+ (C14H17N2O8 requires 341.0985).
4.2.3. S-Acetyl-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine (5)
A solution containing 0.25 g (0.73 mmol) of compound 4 in 8 mL of DMF was treated with 105 mg (0.92 mmol) of potassium thioacetate. The reaction mixture was stirred at room temperature for 23 h under argon. Twenty-five mL of ethyl acetate was added followed by 20 mL of 3% aq HCl. The organic layer was washed with 20 mL of 3% aq HCl. The organic layer was then washed with two 15-mL portions of 5% aq LiCl. The organic layer was dried (MgSO4) and concentrated to dryness under diminished pressure. The crude solid was purified by flash column chromatography on a silica gel column (15 × 3 cm) using 9:1:0.01 CH2Cl2/MeOH/AcOH for elution. The appropriate fractions were collected and concentrated under diminished pressure to give 5 as a yellow foam: yield 262 mg (85%); [α]24D +12.7 (c 1.0, MeOH); 1H NMR (CDCl3) δ 1.41 (d, 3H, J = 6.8 Hz), 2.30 (s, 3H), 3.93 (s, 3H), 3.97 (s, 3H), 4.12 (m, 1H), 4.56 (dd, 1H, J = 8.8 and 4.2 Hz), 5.49, 5.55 (ABq, 2H, J = 15.0 Hz), 5.75 (d, 1H, J = 8.8 Hz), 6.99 (s, 1H), and 7.69 (s, 1H); 13C NMR (CDCl3) δ 16.6, 30.7, 41.0, 56.4, 56.7, 57.6, 64.1, 108.1, 109.7, 127.9, 139.5, 148.1, 153.7, 155.8, 173.6, and 195.2; mass spectrum (APCI), m/z 417.0967 (M+H)+ (C16H21N2O9S requires m/z 417.0966).
4.2.4. N-(6-Nitroveratryloxycarbonyl)-L-thiothreonine n-butyl disulfide (1a)
A solution containing 189 mg (0.45 mmol) of S-acetyl-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine (5) in 10 mL of anhydrous MeOH was purged repeatedly by subjecting it to vacuum and then argon. A solution of 98 μL (25% w/v, 0.45 mmol) of NaOMe in MeOH was added and the reaction mixture was stirred at room temperature under argon for 1 h. Butyl 1-thiobutane-1-sulfinate (94 mg, 0.48 mmol) in 0.8 mL of MeOH was added and stirring was continued for another 2 h. The solvent was concentrated under diminished pressure and residue was partitioned between 10 mL of EtOAc and 8 mL of 1 N aq NaHSO4. The organic layer was washed with 10 mL of water, dried (MgSO4) and concentrated under diminished pressure. The crude product was purified on a silica gel column (14 × 4 cm) using 9:1:0.01 CH2Cl2/MeOH/AcOH for elution. N-(6-Nitroveratryloxycarbonyl)-L-thiothreonine n-butyl disulfide (1a) was obtained as a yellow syrup: yield 152 mg (72%); silica gel TLC Rf 0.55 (9:1:0.01 CH2Cl2/MeOH/AcOH); [α]24D +39.9 (c 1.0, CH2Cl2); 1H NMR (CDCl3) δ 0.90 (t, 3H, J = 7.4 Hz), 1.38 (m, 2H), 1.42 (d, 3H, J = 7.2 Hz), 1.62 (quint, 2H, J = 7.4 Hz), 2.69 (t, 2H, J = 7.4 Hz), 3.59 (m, 1H), 3.94 (s, 3H), 3.98 (s, 3H), 4.61 (dd, 1H, J = 9.2 and 3.2 Hz), 5.52, 5.60 (ABq, 2H, J = 15.2 Hz), 5.78 (d, 1H, J = 9.2 Hz), 7.01 (s, 1H), and 7.71 (s, 1H); 13C NMR (CDCl3) δ 13.8, 18.6, 21.7, 31.1, 39.5, 47.1, 56.5, 56.6, 58.2, 64.3, 108.3, 109.5, 128.3, 139.6, 148.2, 153.9, 156.1, and 175.3; mass spectrum (APCI), m/z 463.1212 (M+H)+ (C18H27N2O8S2 requires m/z 463.1209).
4.2.5. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine (1b)
A solution containing 335 mg of S-acetyl-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine (5) (0.80 mmol) in 10 mL of MeOH was purged repeatedly by subjecting it to vacuum and then argon. A degassed solution of 5 mL of 1 N aq LiOH was added and the reaction mixture was stirred at room temperature under argon for 1 h. The reaction mixture was diluted with 10 mL of satd. aq NaHCO3 and the methanol was removed under diminished pressure. The aqueous residue was extracted with 10 mL of Et2O and the ethereal solution was discarded. The pH of the aqueous phase was adjusted to ~ 3 with 1 N HCl and then extracted with two 15-mL portions of EtOAc. The organic layer was dried (MgSO4) and concentrated under diminished pressure to give the crude thiol product as a yellow syrup. The crude product was dissolved in 10 mL of DMF under argon and 241 μL (1.73 mmol) of triethylamine was added. Argon was bubbled through the mixture and then 325 mg (0.96 mmol) of 4,4′-dimethoxytrityl chloride was added. The reaction mixture was stirred at room temperature for 18 h. The solvent was concentrated and the residue was purified on a silica gel column (22 × 3 cm) using a stepwise gradient of acetone in hexanes (40 → 70%) for elution. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine (1b) was obtained as a yellow foam: yield 277 mg (51%); 1H NMR (CDCl3) δ 1.00 (d, 3H, J = 6.8 Hz), 3.76 (s, 7H), 3.94 (s, 6H), 4.37 (m, 1H), 5.48-5.59 (m, 3H), 6.78 (d, 4H, J = 8.0 Hz), 6.98 (s, 1H), 7.17 (m, 1H), 7.26 (m, 2H), 7.37 (m, 4H), 7.46 (d, 2H, J = 7.2 Hz), and 7.71 (s, 1H); 13C NMR (CDCl3) δ 29.4, 53.8, 55.4, 56.5, 56.7, 59.3, 108.3, 109.6, 113.3, 113.4, 126.9, 127.2, 127.9, 128.0, 128.1, 129.3, 129.4, 130.8, 136.7, 139.6, 145.1, 148.2, 153.9, 158.3, and 173.6; mass spectrum (APCI), m/z 675.2023 (M-H)− (C35H35N2O10S requires m/z 675.2012).
4.2.6. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine cyanomethyl ester (9)
To a solution containing 259 mg (0.38 mmol) of S-(4,4′-dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine (1b) in 8 mL of anhydrous CH3CN was added 267 μL (193 mg, 1.91 mmol) of Et3N followed by 242 μL (288 mg, 3.82 mmol) of chloroacetonitrile. The reaction mixture was stirred at room temperature under argon for 24 h. The solvent was concentrated and the residue was partitioned between 15 mL of EtOAc and 10 mL of 1 N NaHSO4. The organic layer was separated, washed with 10 mL of brine, dried (MgSO4) and concentrated under diminished pressure. The crude product was purified on a silica gel column (20 × 2.5 cm), eluting with 1:1 hexanes/EtOAc. N-(6-Nitroveratryloxycarbonyl)-S-(4,4′-dimethoxytrityl)-L-thiothreonine cyanomethyl ester (9) was obtained as a yellow foam: yield 189 mg (67%); silica gel TLC Rf 0.24 (3:2 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 0.98 (d, 3H, J = 6.8 Hz), 2.72 (m, 1H), 3.72 (s, 6H), 3.89 (s, 3H), 3.93 (s, 3H), 4.34 (m, 1H), 4.40, 4.56 (ABq, 2H, J = 15.6 Hz), 5.47 (m, 3H), 6.75 (d, 4H, J = 9.0 Hz), 6.92 (s, 1H), 7.09-7.23 (m, 3H), 7.30 (dd, 4H, J = 9.0 and 3.0 Hz), 7.39 (d, 2H, J = 7.6 Hz), and 7.65 (s, 1H); 13C NMR (CDCl3) δ 42.5, 49.3, 49.5, 55.4, 56.6, 56.7, 59.5, 64.3, 108.3, 108.4, 109.8, 110.0, 113.3, 113.4, 127.0, 127.2 127.9, 128.08, 128.13, 129.3, 129.4, 130.8, 136.5, 136.6, 139.6, 145.1, 153.9, 155.8, 158.4, 158.8, and 169.2; mass spectrum (APCI), m/z 738.2091 (M+Na)+ (C37H37NaN3O10S requires m/z 738.2097).
4.2.7. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-thiothreonyl pdCpA ester (10)
To a conical vial containing 16.0 mg (22.4 μmol) S-(4,4′-dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-thiothreonine cyanomethyl ester (9) was added a solution containing 5.8 mg (4.3 μmol) of the tris-(tetrabutylammonium) salt of pdCpA21 in 90 μL of anhydrous DMF followed by 10 μL of triethylamine. The reaction mixture was stirred at room temperature under argon atmosphere for 24 h. A 5-μL aliquot of the reaction mixture was diluted with 55 μL of 1:1 CH3CN/50 mM NH4OAc at pH 4.5 and was analyzed by HPLC on a C18 reversed phase column (250 × 10 mm). The column was washed with 1 → 65% CH3CN in 50 mM NH4OAc at pH 4.5 over a period of 45 min at a flow rate of 3.5 mL/min (monitoring at 260 nm). The remaining reaction mixture was diluted to a total volume of 0.6 mL with CH3CN and purified using the same C18 reversed-phase column. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-thiothreonyl pdCpA ester (10) (retention time 33.8 and 34.4 min) was recovered from the appropriate fractions as a colorless solid by lyophilization: yield 1.2 mg (21%); mass spectrum (ESI), m/z 1295.3136 (M+H)+ (C54H61N10O22P2S requires m/z 1295.3158).
4.2.8. N-(6-Nitroveratryloxycarbonyl)-L-threonine (6)
To a solution containing 0.50 g (4.19 mmol) of L-threonine in 20 mL of 1:1 dioxane/H2O was added 0.70 g (8.38 mmol) of NaHCO3. This was followed by the addition of 1.38 g (5.03 mmol) of 6-nitroveratryl chloroformate. The reaction mixture was stirred at room temperature for 17 h. Twenty mL of chloroform was then added to the reaction mixture and the biphasic mixture was separated. The chloroform layer was discarded and the aqueous layer was acidified to pH ~4 by the addition of 1 N NaHSO4. The aqueous solution was then washed with three 30-mL portions of ethyl acetate. The combined organic layer was dried (MgSO4) and the solvent was concentrated under diminished pressure. The crude residue was purified on a silica gel column (15 × 4 cm), elution with 9:1:0.01 CH2Cl2/MeOH/AcOH afforded N-(6-nitroveratryloxycarbonyl)-L-threonine (6) as a solid yellow foam: yield 1.28 g (85%); silica gel TLC Rf 0.16 (4:1 CH2Cl /MeOH); [α]24D-5.5 (c 1.0, MeOH); 1H NMR (CD3OD) δ 1.24 (d, 3H, J = 6.4 Hz), 3.89 (s, 3H), 3.96 (s, 3H), 4.18 (d, 1H, J = 2.8 Hz), 4.34 (m, 1H, J = 3.6 and 2.8 Hz), 5.41, 5.54 (ABq, 2H, J = 15.6 Hz), 7.19 (s, 1H), and 7.71 (s, 1H); 13C NMR (CD3OD) δ 20.5, 56.8, 57.0, 61.1, 64.6, 68.3, 109.2, 110.5, 129.8, 140.5, 149.4, 155.4, 158.6, and 174.0; mass spectrum (APCI), m/z 359.1083 (M+H)+ (C14H19N2O9 requires m/z 359.1091).
4.2.9. N-(6-Nitroveratryloxycarbonyl)-L-threonyl-β-lactone (7)
A sample of 1.20 g (3.34 mmol) of compound 6 was dissolved in 35 mL of dichloromethane and 1.86 mL (1.35 g, 13.4 mmol) of triethylamine was added. To this solution was added 1.90 g (5.02 mmol) of HBTU (O-benzotriazole-N,N,N′,N′-tetramethyluroniumhexafluorophosphate) and the reaction mixture was stirred at room temperature for 20 h under argon. The solvent was concentrated under diminished pressure and the crude product was purified by flash column chromatography on a silica gel column (15 × 4 cm) using 1:1 hexanes/EtOAc for elution. The appropriate fractions were collected and the solvent was concentrated under diminished pressure to give the desired product 7 as a pale yellow solid: yield 741 mg (65%); mp 165-168 °C; silica gel TLC Rf 0.25 (1:1 hexanes/ethyl acetate); [α]24D +6.4 (c 1.0, acetone); 1H NMR (acetone-d6) δ 1.48 (d, 3H, J = 6.4 Hz), 3.94 (s, 3H), 3.95 (s, 3H), 4.94 (q, 1H, J = 6.4 Hz), 5.48 (br s, 2H), 5.56 (dd, 1H, J = 9.6 and 6.0 Hz), 7.20 (s, 1H), 7.67 (d, 1H, J = 8.4 Hz), and 7.71 (s, 1H); 13C NMR (acetone-d6) δ 15.1, 56.6, 56.7, 61.4, 64.5, 75.3, 109.1, 111.4, 128.1, 140.8, 149.4, 154.8, 156.2, and 169.8; mass spectrum (APCI), m/z 341.0992 (M+H)+ (C14H17N2O8 requires m/z 341.0985).
4.2.10. S-Acetyl-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine (8)
A solution containing 0.20 g (0.58 mmol) of compound 7 in 6 mL of DMF was treated with 84 mg (0.73 mmol) of potassium thioacetate. The reaction mixture was stirred at room temperature for 2 h under argon. Fifteen mL of ethyl acetate was added followed by 20 mL of 3% aq HCl. The organic layer was washed with 20 mL of 3% aq HCl. The organic layer was then washed with two 15-mL portions of 5% aq LiCl. The organic layer was dried (MgSO4) and concentrated to dryness under diminished pressure. The crude solid was purified by flash column chromatography on a silica gel column (15 × 4 cm) using 9:1:0.01 CH2Cl2/MeOH/AcOH for elution. The appropriate fractions were collected and concentrated under diminished pressure to give 8 as a light yellow foam: yield 0.78 g (82%); silica gel TLC Rf 0.43 (9:1:0.01 CH2Cl2/MeOH/AcOH); [α]24D-24.2 (c 1.0, MeOH); 1H NMR (CD3OD) δ 1.30 (d, 3H, J = 7.2 Hz), 2.28 (s, 3H), 3.90 (s, 3H), 3.98 (s, 3H), 4.15 (m, 1H), 4.60 (d, 1H, J = 4.0 Hz), 5.44-5.51 (ABq, 2H, J = 15.6 Hz), 7.19 (s, 1H), and 7.73 (s, 1H); 13 C NMR (CD3OD) δ 16.1, 30.4, 42.1, 56.8, 57.0, 58.6, 64.6, 109.2, 110.5, 129.9, 140.5, 149.4, 155.4, 158.5, 172.7, and 196.5; mass spectrum (APCI), m/z 417.0967 (M+H)+ (C16H21N2O9S requires m/z 417.0968).
4.2.11. N-(6-Nitroveratryloxycarbonyl)-L-allo-thiothreonine n-butyl disulfide (2a)
A solution containing 0.49 g (1.19 mmol) of compound 8 in 25 mL of methanol was purged repeatedly by subjecting it alternately to vacuum and argon. A solution of 258 μL (25% w/v, 1.19 mmol) of NaOMe in MeOH was added and the reaction mixture was stirred at room temperature under argon for 1 h. Butyl-1-thiobutane-1-sulfinate (0.24 g; 1.25 mmol) in 2 mL of MeOH was then added to the reaction mixture, which was stirred at room temperature for an additional 2 h. The solvent was concentrated under diminished pressure and the crude product was partitioned between 30 mL of ethyl acetate and 30 mL of 1 N NaHSO4. The organic layer was dried (MgSO4) and concentrated and the crude was purified by flash column chromatography on a silica gel column (18 × 4.5 cm), eluting with 9:1:0.01 CH2Cl2/MeOH/AcOH. The appropriate fractions were combined and concentrated to afford 2a as a yellow syrup: yield 0.50 g (90%); silica gel TLC Rf 0.50 (9:1:0.01 CH2Cl2/MeOH/AcOH); [α]24D-68.2 (c 1.0, CH2Cl2); 1H NMR (CDCl3) δ 0.89 (t, 3H, J = 7.4 Hz), 1.30 (d, 3H, J = 7.2 Hz), 1.38 (m, 2H, J = 7.4 Hz), 1.62 (quint, 2H, J = 7.4 Hz), 2.71 (t, 2H, J = 7.4 Hz), 3.44 (m, 1H), 3.94 (s, 3H), 3.97 (s, 3H), 4.86 (dd, 1H, J = 9.0 and 3.8 Hz), 5.55 (d, 2H, J = 7.6 Hz), 5.59 (d, 1H, J = 9.0 Hz), 6.99 (s, 1H), and 7.70 (s, 1H); 13C NMR (CDCl3) δ 13.8, 15.4, 21.7, 31.4, 39.3, 47.2, 56.5, 56.6, 64.2, 108.2, 109.6, 128.4, 139.6, 148.1, 153.9, 155.9, and 174.9; mass spectrum (APCI), m/z 463.1217 (M+H)+ (C18H27N2O8S2 requires m/z 463.1209).
4.2.12. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine (2b)
A solution containing 540 mg (1.29 mmol) of S-acetyl-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine (8) in 17 mL of MeOH was purged repeatedly by subjecting it to vacuum and then argon. A solution of 8.5 mL (8.5 mmol) of 1 N aq LiOH was added and the reaction mixture was stirred at room temperature for 1 h, while bubbling argon continuously through the solution. The reaction mixture was diluted with 15 mL of satd. aq NaHCO3 and methanol was concentrated under diminished pressure. The aqueous phase was extracted with 10 mL of ether and the organic layer was discarded. The aqueous phase was acidified to pH ~1 with 2 N HCl and again extracted with two 20-mL portions of EtOAc. The organic layer was dried (MgSO4) and concentrated under diminished pressure. The crude product was immediately dissolved in 15 mL of anhydrous DMF and 0.39 mL (283 mg, 2.79 mmol) of triethylamine was added followed by 524 mg (1.54 mmol) of dimethoxytrityl chloride, while bubbling argon continuously through the reaction mixture. The reaction mixture was then stirred under an argon atmosphere at room temperature for 18 h. DMF was concentrated under diminished pressure and the residue was dissolved in 25 mL of EtOAc. The organic layer was washed with 10% aq citric acid, dried (MgSO4) and concentrated under diminished pressure. The crude product was purified on a silica gel column (18 × 3 cm) eluting with 7:3 hexanes/acetone then 1:1 hexanes/acetone. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine (2b) was obtained as a yellow foam: yield 739 mg (84%); 1H NMR (CDCl3) β 1.06 (d, 3H, J = 7.6 Hz), 3.76 (s, 7H), 3.89 (s, 3H), 3.93 (s, 3H), 4.05 (dd, 1H, J = 9.2 and 2.8 Hz), 5.35 (d, 1H, J = 9.2 Hz), 5.53 (s, 2H), 6.79 (d, 4H, J = 8.8 Hz), 6.97 (s, 1H), 7.17 (m, 1H), 7.26 (m, 2H), 7.37 (d, 4H, J = 8.8 Hz), 7.45 (d, 2H, J = 7.6 Hz), and 7.71 (s, 1H); 13C NMR (CDCl3) δ 17.9, 41.7, 53.3, 56.5, 56.7, 58.4, 63.9, 67.4, 108.2, 109.2, 113.4, 126.8, 128.1, 128.9, 129.4, 130.7, 136.9, 139.3, 145.1, 148.0, 153.9, 155.8, 158.2, and 173.6; mass spectrum (APCI), m/z 675.2002 (M-H)− (C35H35N2O10S requires m/z 675.2012).
4.2.13. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine cyanomethyl ester (11)
To a solution containing 600 mg (0.88 mmol) of S-(4,4′-dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine (2b) in 16 mL of anhydrous CH3CN was added 613 μL (445 mg, 4.39 mmol) of Et3N followed by 558 μL (663 mg, 8.8 mmol) of chloroacetonitrile. The reaction mixture was stirred at room temperature under argon for 19 h. The solvent was concentrated and the residue was dissolved in 25 mL of EtOAc and 20 mL of 1 N NaHSO4. The organic layer was separated, washed with 20 mL of brine, dried (MgSO4) and concentrated under diminished pressure. The crude product was purified on a silica gel column (16 × 3 cm), eluting with 3:2 hexanes/EtOAc. S-(4,4′-dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine cyanomethyl ester (11) was obtained as a yellow foam: yield 539 mg (85%); silica gel TLC Rf 0.23 (3:2 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.16 (d, 3H, J = 7.6 Hz), 2.86 (m, 1H), 3.78 (s, 6H), 3.92 (s, 3H), 3.94 (s, 3H), 4.57, 4.66 (ABq, 2H, J = 15.6 Hz), 5.29 (d, 1H, J = 8.8 Hz), 5.52 (q, 2H, J = 15.2 Hz), 6.81 (d, 4H, J = 8.8 Hz), 6.95 (s, 1H), 7.20 (t, 1H, J = 7.2 Hz), 7.28 (t, 2H, J = 7.2 Hz), 7.36 (m, 4H), 7.45 (m, 2H), and 7.71 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 41.4, 49.0, 55.4, 56.5, 56.6, 58.0, 64.1, 67.7, 108.2, 109.4, 113.5, 113.8, 127.0, 128.2, 128.4, 129.3, 130.6, 136.5, 139.5, 144.9, 148.1, 153.9, 155.6, 158.4, and 169.2; mass spectrum (APCI), m/z 738.2089 (M+Na)+ (C37H37NaN3O10S requires m/z 738.2097).
4.2.14. S-(4,4′-Dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (12)
To a conical vial containing 16.0 mg (22.4 μmol) of S-(4,4′-dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonine cyanomethyl ester (11) was added a solution of 5.8 mg (4.3 μmol) of the tris-(tetrabutylammonium) salt of pdCpA in 90 μL of anhydrous DMF, followed by 10 μL of triethylamine. The reaction mixture was stirred at room temperature under argon atmosphere for 24 h. A 5-μL aliquot of the reaction mixture was diluted with 55 μL of 1:1 CH3CN/50 mM NH4OAc at pH 4.5 and was analyzed by HPLC on a C18 reversed phase column (250 × 10 mm). The column was washed with 1 → 65% CH3CN in 50 mM NH4OAc at pH 4.5 over a period of 45 min at a flow rate of 3.5 mL/min (monitoring at 260 nm). The remaining reaction mixture was diluted to a total volume of 0.6 mL with CH3CN and purified using the same C18 reversed-phase column. S-(4,4′-dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (12) (retention time 34.0 and 34.5 min) was recovered from the appropriate fractions as a colorless solid by lyophilization: yield 2.6 mg (47%); mass spectrum (ESI), m/z 1295.3203 (M+H)+ (C54H61N10O22P2S requires 1295.3158).
4.2.15. N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (13)
To a solution containing 1.6 mg (1.23 μmol) of S-(4,4′-dimethoxytrityl)-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (12) in 0.60 mL of 3:1 acetonitrile/water (v/v) was added a solution containing 3.13 mg (18.5 μmol) of AgNO3 in 40 μL of water. The reaction mixture was stirred at room temperature for 1 h then 9.5 mg (61.6 μmol) of dithiothreitol in 50 μL of water was added and stirring was continued for 90 min. The mixture was centrifuged at 15,000 × g at 4 °C for 5 min and the supernatant was collected and purified by HPLC on a C18 reversed phase column (250 × 10 mm). The column was washed with 1 → 65% CH3CN in 50 mM NH4OAc at pH 4.5 over a period of 45 min at a flow rate of 3.5 mL/min (monitoring at 260 nm). N-(6-Nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (13) (retention time 22.9 and 23.3 min) was recovered from the appropriate fractions as a colorless solid by lyophilization and was used immediately in the next step: yield 1.0 mg (82%); mass spectrum (MALDI), m/z 993.4 (M+H)+ (theoretical 993.2).
4.2.16. S-[7-Diethylamino-3-(4′-maleimidophenyl)-4-methylcoumarin]-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (14)
To a solution containing 1.0 mg (1.0 μmol) of N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (13) in 375 μL of 2:1 acetonitrile/water (v/v) was added 0.8 mg (2.0 μmol) of 7-diethylamino-3-(4′-maleimidophenyl)-4-methylcoumarin in 75 μL of acetonitrile. The mixture was stirred at room temperature for 3 h then purified by HPLC on a C18 reversed-phase column (250 × 10 mm). The column was washed with 1 → 65% CH3 CN in 50 mM NH4OAc at pH 4.5 over a period of 45 min at a flow rate of 3.5 mL/min (monitoring at 260 nm). S-[7-Diethylamino-3-(4′-maleimidophenyl)-4-methylcoumarin]-N-(6-nitroveratryloxycarbonyl)-L-allo-thiothreonyl pdCpA ester (14) (retention time of 34-35 min) was recovered from the appropriate fractions as a yellow solid by lyophilization: yield 0.9 mg (64%); mass spectrum (MALDI), m/z 1395.6 (M+H)+ (theoretical 1395.3); mass spectrum (ESI), m/z 1394.3382 (M+) (C57H64N12O24P2S requires 1394.3352).
4.3. Ligation of suppressor tRNACUA-COH with thiothreonine analogues and deprotection of DMT and NVOC groups
The yeast suppressor tRNAPheCUA transcript was prepared as previously reported.7i Activation of suppressor tRNACUA was carried out in 100 μL (total volume) of 100 mM Hepes buffer, pH 7.5, containing 2.0 mM ATP, 15 mM MgCl2, 100 μg of suppressor tRNA-COH, 2.0 A260 units of protected aminoacyl-pdCpA (5-10 fold molar excess), 15% DMSO and 200 units of T4 RNA ligase. After incubation at 37 °C for 1 h, the reaction was quenched by the addition of 10 μL of 3 M NaOAc, pH 6.3, followed by 300 μL of ethanol. The reaction mixture was incubated at −20 °C for 30 min, then centrifuged at 15,000 × g at 4 °C for 30 min. The supernatant was carefully decanted and the tRNA pellet was washed with 100 μL of 70% ethanol and dissolved in 100 μL of RNase free H2O. The efficiency of ligation was estimated by 8% denaturing PAGE (pH 5.2).19 The DMT protecting group of N-(6-nitroveratryloxycarbonyl)-thiothreonyl-tRNA or N-(6-nitroveratryloxycarbonyl)-allo-thiothreonyl-tRNA was removed by treatment with 50 mM AgNO3 at room temperature for 30 min, followed by treatment with 60 mM DTT at room temperature for 30 min.22 The reaction mixture was centrifuged at 15,000 × g at 4 °C for 10 min, and then the supernatant was carefully decanted. The precipitated pellet was washed with 100 μL of 0.3 M NaOAc, pH 6.3. The combined aqueous supernatant was treated with 600 μL of ethanol to precipitate the tRNA. The tRNA pellet was washed with 100 μL of 70% ethanol and then dissolved in 30 μL of RNase free H2O. The NVOC-protected aminoacyl-tRNA was cooled to 2 °C and irradiated with a 500 W mercury-xenon lamp for 5 min.23,24 After irradiation, the deblocked aminoacylated suppressor tRNAs were used in in vitro suppression experiments without further purification.
Also prepared were samples of the thiothreonyl- and allo-thiothreonyl-tRNAs in which the DMT group was not removed prior to photolysis of the NVOC protecting group.
4.4. Coupling of NVOC-thiothreonyl-tRNA analogues with CPM
The NVOC-thiothreonyl-tRNA coupling was carried out in 50 μL (total volume) of 100 mM NaOAc, pH 6.3, containing 90 μg of thiothreonyl-tRNA analogue and 1 mM of CPM. The reaction was incubated at room temperature for 1 h and was quenched by the addition of 3 μL of 3 M NaOAc, pH 6.3, followed by 150 μL of ethanol. The reaction mixture was incubated at −20 °C for 30 min, then centrifuged at 15,000 × g at 4 °C for 30 min. The supernatant was carefully decanted and the tRNA pellet was washed with 50 μL of 70% ethanol and dissolved in 30 μL of RNase free H2O. The fluorescence was monitored at ~ 470 nm following irradiation at 365 nm.
4.5 Ligation of suppressor tRNA-COH with CPM-allo-thiothreonyl-pdCpA and deprotection of NVOC group
Suppressor tRNA aminoacylation was carried out in 100 μL (total volume) of 100 mM Hepes buffer, pH 7.5, containing 2.0 mM ATP, 15 mM MgCl2, 100 μg of suppressor tRNA-COH, 2.0 A260 units of NVOC-protected CPM-allo-thiothreonyl-pdCpA, 15% DMSO and 200 units of T4 RNA ligase. After incubation at 37 °C for 1 h, the reaction was quenched by the addition of 10 μL of 3 M NaOAc, pH 5.2, followed by 300 μL of ethanol. The reaction mixture was incubated at −20 °C for 30 min, then centrifuged at 15,000 × g at 4 °C for 30 min. The supernatant was carefully decanted and the tRNA pellet was washed with 100 μL of 70% ethanol, and dissolved in 30 μL of RNase free H2O. The NVOC-protected CPM-allo-thiothreonyl-tRNA was cooled to 2 °C and irradiated with a 500 W mercury-xenon lamp for 5 min.
4.6. In vitro translation of csDHFR analogues
The wild-type csDHFR plasmid was obtained by site-directed mutation as described previously25 using the wild-type DHFR plasmid as the template. The DNA primer for the mutation at position 85 was 5′-GATGAAGCCATCGCGGCGTCTGGTGACGTACCAGAAATC-3′; and the primer for the mutation at position 152 was 5′-CAGAACTCTCACAGCTATAGCTTTGAGATTCTGGAGC-3′. The mutant csDHFR (TAG at position 10) was obtained by the same site-directed mutation using the wild-type csDHFR plasmid as the template and the sequence of 5′-GTCTGATTGCGGCGTTAGCGTAGGATCGCGTTATCGGCATG-3′ as the primer.
The in vitro expression mixture (300 μL total volume) contained 30 μg of mutant DHFR (TAG at position 10) plasmid DNA, 120 μL of premix (35 mM Tris-acetate, pH 7.0, 190 mM potassium glutamate, 30 mM ammonium acetate, 2.0 mM dithiothreitol, 11 mM maganesium acetate, 20 mM phospho(enol)pyruvate, 0.8 mg/mL of E. coli tRNA, 0.8 mM IPTG, 20 mM ATP and GTP, 5 mM CTP and UTP and 4 mM cAMP), 100 μM of each of the 20 amino acids, 30 μCi of [35S]-L-methionine, 10 μg/μL rifampicin, 90 μg of deprotected misacylated tRNACUA and 90 μL of S-30 extract from E. coli strain BL21(DE3). The reaction mixture was incubated at 37 °C for 45 min. Plasmid DNA containing the gene for wild-type csDHFR was used as the positive control, and an abbreviated tRNA (tRNA-COH) lacking any amino acid was used as the negative control. An aliquot containing 2 μL of reaction mixture was removed, treated with 2 μL of loading buffer and heated at 90 °C for 2 min. This was analyzed by 15% SDS-PAGE at 100 V for 2 h.
4.7. Purification of Analogues of csDHFRs
The analogues of csDHFR containing an N-terminal hexahistidine fusion peptide were purified by Ni-NTA chromatography.26 The in vitro translation reaction mixture (300 μL) was diluted with 900 μL of 50 mM Tris-HCl, pH 8.0, containing 300 mM NaCl and 10 mM imidazole, and mixed gently with 100 μL of a 50% slurry of Ni-NTA resin at 4 °C for 2 h. Then the mixture was loaded on a column and washed with 600 μL of 50 mM Tris-HCl, pH 8.0, containing 300 mM NaCl and 20 mM imidazole. Finally, the csDHFR analogue was washed three times with 200 μL of 50 mM Tris-HCl, pH 8.0, containing 30 mM NaCl and 150 mM imidazole. The three elutions from the Ni-NTA column were combined and loaded on a 200 μL DEAE-Sepharose column. The column was washed with 300 μL of 50 mM Tris-HCl, pH 8.0, containing 100 mM NaCl, 300 μL of 50 mM Tris-HCl, pH 8.0, containing 200 mM NaCl, and then three 300-μL portions of 50 mM Tris-HCl, pH 8.0, containing 300 mM NaCl. Aliquots of each fraction were analyzed by 15% SDS-PAGE.
4.8. Testing the activities of wild-type DHFR and csDHFRs at pH 7.0
The enzymatic activities of wild-type DHFR and csDHFR were measured in 1 mL of MTEN buffer (containing 50 mM MES, 25 mM trizma base, 25 mM ethanolamine, 100 mM NaCl, 0.1 mM EDTA and 10 mM β-mercaptoethanol, pH 7.0). MTEN buffer (0.97 mL) was mixed with 10 μL of 10 mM NADPH and 100 ng of protein. The mixture was incubated at 37 °C for 3 min. Then 20 μL of 5 mM dihydrofolate in MTEN buffer, pH 7.0, was added. The OD value at 340 nm was monitored over a period of 6 min.
4.9. Coupling thiothreonyl-csDHFR analogues with CPM
The thiothreonyl-csDHFR coupling was carried out in 30 μL (total volume) of 50 mM Tris-HCl, pH 7.0, containing 100 mM NaCl, 3 μg of thiothreonyl-csDHFR analogue and 1 mM of CPM. The reaction was incubated at room temperature for 5 min and was quenched by extraction with two 30-μL portions of ethyl acetate. The reaction mixtures were analyzed by 15% SDS-PAGE. The fluorescence was monitored at ~470 nm following excitation at 365 nm.
Supplementary Material
Acknowledgement
This study was supported by Research Grant GM 092946 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1 (a).Sardina FJ, Rapoport H. Chem. Rev. 1996;96:1825. doi: 10.1021/cr9300348. [DOI] [PubMed] [Google Scholar]; (b) Fülöp F. Chem. Rev. 2001;101:2181. doi: 10.1021/cr000456z. [DOI] [PubMed] [Google Scholar]; (c) Ager DJ, Fotheringham IG. Curr. Opin. Drug Discov. Devel. 2001;4:800. [PubMed] [Google Scholar]; (d) Beck G. Synlett. 2002:837. [Google Scholar]; (e) Nájera C. Synlett. 2002:1388. [Google Scholar]
- 2 (a).Sahl H-G, Bierbaum G. Annu. Rev. Microbiol. 1998;52:41. doi: 10.1146/annurev.micro.52.1.41. [DOI] [PubMed] [Google Scholar]; (b) van Kraaij C, de Vos WM, Siezen RJ, Kuipers OP. Nat. Prod. Rep. 1999;16:575. doi: 10.1039/a804531c. [DOI] [PubMed] [Google Scholar]; (c) Chatterjee C, Paul M, Xie L, van der Donk WA. Chem. Rev. 2005;105:633. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]; (d) Piper C, Cotter PD, Ross PR, Hill C. Curr. Drug Discov. Tech. 2009;6:1. doi: 10.2174/157016309787581075. [DOI] [PubMed] [Google Scholar]
- 3 (a).Brötz H, Sahl H-GJ. Antimicrob. Agents Chemother. 2000;46:1. doi: 10.1093/jac/46.1.1. [DOI] [PubMed] [Google Scholar]; (b) McAuliffe O, Ross RP, Hill C. FEMS Microbiol. Rev. 2001;25:285. doi: 10.1111/j.1574-6976.2001.tb00579.x. [DOI] [PubMed] [Google Scholar]; (c) Pag U, Sahl H-G. Curr. Pharm. Des. 2002;8:815. doi: 10.2174/1381612023395439. [DOI] [PubMed] [Google Scholar]; (d) Willey JM, van der Donk WA. Annu. Rev. Microbiol. 2007;61:477. doi: 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]
- 4.Jabés D, Brunati C, Candiani G, Riva S, Romanó G, Donadio S. Antimicrob. Agents Chemother. 2011;55:1671. doi: 10.1128/AAC.01288-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5 (a).Dinakarpandian D, Shenoy B, Pusztai-Carey M, Malcolm BA, Carey PR. Biochemistry. 1997;36:4943. doi: 10.1021/bi963148x. [DOI] [PubMed] [Google Scholar]; (b) Nägler DK, Tam W, Storer AC, Krupa JC, Mort JS, Ménard R. Biochemistry. 1999;38:4868. doi: 10.1021/bi982632s. [DOI] [PubMed] [Google Scholar]; (c) Sakamoto T, Tanaka T, Ito Y, Rajesh S, Iwamoto-Sugai M, Kodera Y, Tsuchida N, Shibata T, Kohno T. Biochemistry. 1999;38:11634. doi: 10.1021/bi990310y. [DOI] [PubMed] [Google Scholar]
- 6 (a).Legowska A, Debowski D, Lukajtis R, Wysocka M, Czaplewski C, Lesner A, Rolka K. Bioorg. Med. Chem. 2010;18:8188. doi: 10.1016/j.bmc.2010.10.014. [DOI] [PubMed] [Google Scholar]; (b) Imani M, Hosseinkhani S, Ahmadian S, Nazari M. Photochem. Photobiol. Sci. 2010;9:1167. doi: 10.1039/c0pp00105h. [DOI] [PubMed] [Google Scholar]; (c) Botelho HM, Leal SS, Veith A, Prosinecki V, Bauer C, Frohlich R, Kletzin A, Gomes CM. J. Biol. Inorg. Chem. 2010;15:271. doi: 10.1007/s00775-009-0596-3. [DOI] [PubMed] [Google Scholar]
- 7 (a).Karginov VA, Mamaev SV, An H, Van Cleve MD, Hecht SM, Komatsoulis GA, Abelson JN. J. Am. Chem. Soc. 1997;119:8166. [Google Scholar]; (b) Arslan T, Mamaev SV, Mamaeva NV, Hecht SM. J. Am. Chem. Soc. 1997;119:10877. [Google Scholar]; (c) Killian JA, Van Cleve MD, Shayo YF, Hecht SM. J. Am. Chem. Soc. 1998;120:3032. [Google Scholar]; (d) Short GF, III, Lodder M, Laikhter AL, Arslan T, Hecht SM. J. Am. Chem. Soc. 1999;121:478. [Google Scholar]; (e) Short GF, III, Laikhter AL, Lodder M, Shayo YF, Arslan T, Hecht SM. Biochemistry. 2000;39:8768. doi: 10.1021/bi000214t. [DOI] [PubMed] [Google Scholar]; (f) Baird TT, Jr., Wang B, Lodder M, Hecht SM, Craik CS. Tetrahedron. 2000;56:9477. [Google Scholar]; (g) Wang B, Brown KC, Lodder M, Craik CS, Hecht SM. Biochemistry. 2002;41:2805. doi: 10.1021/bi011762p. [DOI] [PubMed] [Google Scholar]; (h) Anderson RD, Zhou J, Hecht SM. J. Am. Chem. Soc. 2002;124:9674. doi: 10.1021/ja0205939. [DOI] [PubMed] [Google Scholar]; (i) Gao R, Zhang Y, Choudhury AK, Dedkova LM, Hecht SM. J. Am. Chem. Soc. 2005;127:332131. doi: 10.1021/ja044182z. [DOI] [PubMed] [Google Scholar]; (j) Gao R, Zhang Y, Dedkova L, Choudhury AK, Rahier NJ, Hecht SM. Biochemistry. 2006;45:8402. doi: 10.1021/bi0605179. [DOI] [PubMed] [Google Scholar]; (k) Yakovleva L, Chen S, Hecht SM, Shuman S. J. Biol. Chem. 2008;283:16093. doi: 10.1074/jbc.M801595200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Chen S, Hecht SM. Bioorg. Med. Chem. 2008;16:9023. doi: 10.1016/j.bmc.2008.08.036. [DOI] [PubMed] [Google Scholar]; (m) Chen S, Yi Z, Hecht SM. Biochemistry. 2011;50:9340. doi: 10.1021/bi201291p. [DOI] [PubMed] [Google Scholar]
- 8.Morell JL, Fleckenstein P, Gross E. J. Org. Chem. 1977;42:355. doi: 10.1021/jo00422a044. [DOI] [PubMed] [Google Scholar]
- 9.Carter HE, Stevens CM, Ney FL. J. Biol. Chem. 1941;139:247. [Google Scholar]
- 10.Arnstein HRV. Biochem. J. 1958;68:333. doi: 10.1042/bj0680333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakamiya T, Shimbo K, Shiba T, Nakajima K, Neya M, Okawa K. Bull. Chem. Soc. Jpn. 1982;55:3878. [Google Scholar]
- 12 (a).Zhou H, van der Donk WA. Org. Lett. 2002;4:1335. doi: 10.1021/ol025629g. [DOI] [PubMed] [Google Scholar]; (b) Xiong C, Wang W, Hruby VJ. J. Org. Chem. 2002;67:3514. doi: 10.1021/jo011172x. [DOI] [PubMed] [Google Scholar]; (c) Narayan RS, VanNieuwenhze MS. Org. Lett. 2005;7:2655. doi: 10.1021/ol0507930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cobb SL, Vederas JC. Org. Biomol. Chem. 2007;5:1031. doi: 10.1039/b618178c. [DOI] [PubMed] [Google Scholar]
- 14.Pansare SV, Vederas JC. J. Org. Chem. 1989;54:2311. [Google Scholar]
- 15.Katritzky AR, Xu Y-J, Vakulenko AV, Wilcox AL, Bley KR. J. Org. Chem. 2003;68:9100. doi: 10.1021/jo034616t. [DOI] [PubMed] [Google Scholar]
- 16.Moyano A, Pericas MA, Valenti E. J. Org. Chem. 1989;54:573. [Google Scholar]
- 17 (a).Sliedregt KM, Schouten A, Kroon J, Liskamp RMJ. Tetrahedron Lett. 1996;37:4237. [Google Scholar]; (b) Smith ND, Wohlrab AM, Goodman M. Org. Lett. 2005;7:255. doi: 10.1021/ol047761h. [DOI] [PubMed] [Google Scholar]; (c) Valls N, Borregán M, Bonjoch J. Tetrahedron Lett. 2006;47:3701. [Google Scholar]
- 18.Small LVD, Bailey JH, Cavallito CJ. J. Am. Chem. Soc. 1947;69:1710. doi: 10.1021/ja01199a040. [DOI] [PubMed] [Google Scholar]
- 19.Varshney U, Lee CP, RajBhandary UL. J. Biol. Chem. 1991;266:24712. [PubMed] [Google Scholar]
- 20.Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. Science. 1989;244:182. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 21.Robertson SA, Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. Nucleic Acids Res. 1989;17:9649. doi: 10.1093/nar/17.23.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connolly BA, Rider P. Nucleic Acids Res. 1985;13:4485. doi: 10.1093/nar/13.12.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson SA, Ellman JA, Schultz PG. J. Am. Chem. Soc. 1991;113:2722. [Google Scholar]
- 24.Lodder M, Golovine S, Hecht SM. J. Org. Chem. 1997;62:778. doi: 10.1021/jo971692l. [DOI] [PubMed] [Google Scholar]
- 25.Sawano A, Miyawaki A. Nucleic Acids Res. 2000;28:E78. doi: 10.1093/nar/28.16.e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janknecht R, de Martynoff G, Lou J, Hipskind RA, Nordheim A, Stunnenberg HG. Proc. Natl. Acad. Sci. U.S.A. 1991;88:8972. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.