

Abstract
Poly(ethylene glycol) (PEG) and polylactic acid (PLA)-based copolymeric nanoparticles were synthesized and investigated as a carrier for prolonged delivery of insulin via the parenteral route. Insulin loading was simultaneously achieved with particle synthesis using a double emulsion solvent evaporation technique, and the effect of varied PEG chain lengths on particle size and insulin loading efficiency was determined. The synthesized copolymer and nanoparticles were analyzed by standard polymer characterization techniques of gel permeation chromatography, dynamic light scattering, nuclear magnetic resonance, and transmission electron microscopy. In vitro insulin release studies performed under simulated conditions provided a near zero-order release pattern up to 10 days. In vivo animal studies were undertaken with varied insulin loads of nanoparticles administered subcutaneously to fed diabetic rabbits and, of all doses administered, nanoparticles containing 50 IU of insulin load per kg body weight controlled the blood glucose level within the physiologically normal range of 90–140 mg/dL, and had a prolonged effect for more than 7 days. Histopathological evaluation of tissue samples from the site of injection showed no signs of inflammation or aggregation, and established the nontoxic nature of the prepared copolymeric nanoparticles. Further, the reaction profiles for PLA-COOH and NH2-PEGDA-NH2 were elucidated using molecular mechanics energy relationships in vacuum and in a solvated system by exploring the spatial disposition of various concentrations of polymers with respect to each other. Incorporation of insulin within the polymeric matrix was modeled using Connolly molecular surfaces. The computational results corroborated the experimental and analytical data. The ability to control blood glucose levels effectively coupled with the nontoxic behavior of the nanoparticles indicates that these nanoparticles are a potential candidate for insulin delivery.
Keywords: parenteral delivery, insulin, nanoparticles, poly(lactide-ethylene glycol) diblock copolymer, molecular mechanics energy relationship
Introduction
Considerable interest has been generated over the past few decades in the development of effective drug delivery systems for peptides and proteins such as insulin. However, multiple injection therapy is still the preferred route for insulin delivery, and attempts are being made to explore various noninvasive routes for insulin dosing, such as oral, pulmonary, buccal, nasal, and ocular routes. However, thus far, researchers have not yet developed a successful formulation for clinical application due to the inherent limitations associated with each route. Insulin is poorly suited to oral administration because of extensive proteolytic degradation by intestinal enzymes and insufficient membrane permeability owing to the high molecular weight and complex structure of insulin.1–4 Buccal delivery of insulin is incompetent due to its low permeability across the buccal mucosal epithelium and the fact that it is easily washed out with saliva. Delivery via the nasal and ocular route is not favorable because of the degradation of insulin by enzymes within the nasal mucosa and eye tissue.5
A number of research groups are working in this area in order to overcome these challenges. However, there is still no commercial product available for efficient delivery of insulin through either of these routes. Therefore, parenteral delivery of insulin remains the most suitable and preferred route. There are challenges associated with the parenteral mode of delivery. Insulin injected parenterally can only be maintained for a few hours due to faster consumption/ degradation. Moreover, high doses of insulin may result in drastic reduction of glucose levels, leading to other major complications and ultimately death. Multiple daily injection therapy with insulin is required to maintain potential effectiveness, which has associated limitations, including local tissue necrosis, tenderness, and nerve damage, along with pain that results in poor patient compliance.6
To avoid administering daily multiple injections to diabetic patients, researchers are focusing on development of controlled-release parenteral insulin delivery systems. Formulation of such solid polymer systems or hydrogels has been achieved through in situ crosslinking by different polymerization mechanisms, such as photoinitiation, ionic interaction, and radical-initiated polymerization, or by crosslinking of low molecular weight oligomers.7–9 Precipitation of polymers at the injection site is accomplished by replacement of the solvent by body fluids. Furthermore, a few polymers when injected as highly concentrated solutions deposit as gels upon thermal induction.10,11 Major challenges in insulin delivery are its stability during particle formation, controlled release in optimum concentrations, and the low absorption of insulin in the small intestine.
Biodegradable microparticles based on the polymers, poly(lactic-co-glycolic acid) (PLGA) and poly(lactic acid) (PLA), have been investigated extensively for the delivery of therapeutic proteins and vaccines.12,13 For proteins to be used as drugs, delivery via polymer particles overcomes many of the associated unfavorable pharmacokinetics and side effects.14 Multiple emulsion solvent evaporation methods are considered to be simple and versatile, so have been widely used for the preparation of protein-entrapped polymer particles.15,16 However, major limitations for successful utilization of this modality of protein delivery include the instability of proteins during formulation, lyophilization, storage, and polymer degradation.17–20 In addition, the in situ encapsulation of protein molecules in polymeric microspheres exposes the former to harsh organic solvents, heat, and surface tension that may lead to protein breakdown, aggregation, and denaturation.21 This results in loss of functionality from the active protein. Insulin delivery based on microparticle technology using various types of biodegradable polymers has been reported previously.
Biodegradable polymers that are used in microsphere and nanosphere preparation include PLGA,22–25 PLA,26–28 polyphosphazene,29 polycaprolactone,23,30 poly(fumaric-co-sebacic anhydride),6,31 poly(acryloyl-L-proline methyl ester),32 and PLA-poly(ethylene glycol) (PEG).33,34 Because of its amphiphilic nature, the copolymer PLA-PEG has been of interest for the delivery of various peptides and proteins of therapeutic value, including insulin. Ring opening polymerization of PEG and lactide is a popular method,34,35 but major concerns about the process include the fact that the initiator used in this reaction is stannous octate which is highly toxic and not suitable for synthesizing materials with biomedical application,36 and the synthesis of PEG–PLA block copolymers by this method is very expensive and not suitable for large-scale production.35 Copolymers synthesized by ring opening polymerization have nevertheless been used for the preparation of nanospheres and microspheres, but a lower encapsulation efficiency has been reported with such systems. In addition, the in vitro release kinetics exhibited either a burst-release or a short duration of release.33,34,37,38 Such release behavior is not practically adaptable for further in vivo studies in a diabetic animal model. For successful delivery of insulin with therapeutic efficiency and efficacy, these challenges needs to be addressed.
Therefore, in this study, an attempt was made to synthesize PLA-PEG copolymers by an alternative method utilizing conjugation chemistry to avoid the use of toxic initiators and the other limitations posed by ring opening polymerization. Nanoparticles prepared from the synthesized copolymer were used further to develop a biodegradable parenteral depot for insulin delivery. The nanoparticles were subsequently evaluated in terms of the insulin encapsulation efficiency and release profile in vitro. The effect of a single subcutaneous dose of the insulin-loaded nanoparticles was also evaluated in vivo in terms of longevity in controlling blood glucose levels in the diabetes-induced experimental rabbit model.
Materials and methods
Materials
PLA (molecular weight 60,000 g/mol) was purchased from Birmingham Polymer Inc (Birmingham, AL, USA). Poloxamer and insulin were sourced from Sigma-Aldrich (St Louis, MO, USA). PEG, alloxan monohydrate, methacrylic acid, and methane sulfonic acid were supplied by the Central Drug House (New Delhi, India). Acetonitrile, dichloromethane, toluene, and hexane were purchased from Rankem (New Delhi, India). Male New Zealand White rabbits weighing 2.5–3.0 kg were provided by the experimental animal facility of the Institute of Nuclear Medicine and Allied Sciences (New Delhi, India). The study protocol was reviewed and approved by the Institutional Animal Ethical Committee of the Institute of Nuclear Medicine and Allied Sciences, Delhi, India.
Synthesis of PLA-PEG copolymer
Synthesis of PEG diacrylate
In a standard experiment, one mole of PEG 575, 2000, and 4000 were mixed separately with 2.5 moles of acrylic acid each in a three-neck round-bottom flask equipped with a mechanical stirrer and Dean-Stark apparatus. The esterification reaction was performed at 80°C–90°C for 7 hours at 200 rpm. Methane sulfonic acid (1.4%) as a catalyst, hydroquinone (0.15%) as an inhibitor, and toluene (22%) as an azotropic solvent were used for the esterification reaction. The quantities of catalyst, inhibitor, and solvent were computed as the (w/w) percentage of the total monomer. The synthesized PEG diacrylate (PEGDA) was then neutralized with 5% sodium bicarbonate (Figure 1A and B), precipitated from the solution by adding ice-cold hexane and drying under vacuum at 60°C for 24 hours.39,40
Figure 1.

Stepwise synthesis of PLA-PEG copolymer showing (A) PEG, (B) PEGDA, (C) PEGDA amine, (D) activated PLA, and (E) PLA-PEG copolymer.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid; PEGDA, PEG diacrylate.
Amination of PEGDA
The synthesized PEGDA was then modified with diamine to generate functional NH2 groups to be utilized for conjugation with PLA. The amination reaction between 1 mole of PEGDA and 2 moles of ethylene diamine was performed in a shaking incubator at 37°C for 24 hours. The reaction involved replacement of a double bond in PEGDA by the amino group of ethylene diamine, resulting in the formation of PEGDA diamine (known as the Michael addition reaction), as shown in Figure 1C.
Activation of PLA
PLA was then activated by 1-ethyl-3-(-3-diethylaminopropyl)-carbodiimide (EDC) and N-hydroxy succinamide (NHS) using the method described by Cheng et al.41 Briefly, 0.28 mmol of PLA was dissolved in 10 mL of methylene chloride and converted to PLA-NHS using excess NHS (1.1 mmol) in the presence of EDC (1.2 mmol). This was undertaken by incubation at room temperature for one hour in a shaking incubator. PLA-NHS was then precipitated from the reaction mixture using ethyl ether and repeatedly washed with an ice-cold mixture of ethyl ether and methanol to remove the residual NHS and finally dried under vacuum.
Conjugation of activated PLA with PEGDA diamine
The activated PLA was then conjugated with PEGDA amine using an adaption of the procedure described by Cheng et al.41 Briefly, 1 mole of activated PLA was added to 1.2 moles of PEGDA diamine after dissolving in chloroform and kept at room temperature for 12 hours under constant shaking (Figure 1D). The resulting PLA-PEG copolymer was precipitated with cold methanol to remove unreacted PEGDA diamine (Figure 1E). The synthesized PLA-PEG copolymer was dried under vacuum before being used for further characterization and preparation of insulin-loaded nanoparticles.
Preparation of insulin-loaded PLA and PLA-PEG nanoparticles
Insulin-loaded PLA and PLA-PEG nanoparticles were prepared using a double emulsion solvent evaporation method. For preparation of the nanoparticles, PLA and PLA-PEG copolymers (1 g) with different PEG chain lengths of 575, 2000, and 4000 were separately dissolved in 15 mL of acetonitrile. Insulin 500 IU was then added to the solution, with brief sonication for 15 seconds to produce a primary emulsion. The resulting primary emulsion was then added dropwise to the aqueous phase comprising Poloxamer 187 (150 mg) in distilled water (100 mL) and stirred magnetically at room temperature for 6–8 hours in order to facilitate solvent evaporation and nanoparticle stabilization. The nanoparticles formed were then collected by centrifugation at 21,000 rpm (for 10 minutes) and washed three times in distilled water. The synthesized PLA and PLA-PEG nanoparticles were lyophilized and stored at 4°C until further use.
Characterization of synthesized PLA and PLA-PEG nanoparticles
Gel permeation chromatography
Gel permeation chromatography was performed at room temperature using a Viscotek RI apparatus (GPCmax with 270 dual detector; Malvern Instruments Ltd, Malvern, UK) equipped with a light scattering detector (right angle and low angle), refractive index detector, and columns consisting of 2 × Plgel 5 μm (300 × 7.5 mm) mix-bed columns (Agilent, Santa Clara, CA, USA). Tetrahydrofuran was used as the mobile phase at a flow rate of 1 mL per minute. The sample concentration was 4 mg/mL, and 25 μL samples were injected for each analysis. Calibration of the light scattering detector was performed using the instrument internal standard.
Nuclear magnetic resonance spectroscopy
1H nuclear magnetic resonance (1H NMR) spectra for the synthesized copolymer samples were recorded on a Bruker AC300 MHz spectrometer in CDCl3 solvent at room temperature using tetramethylsilane as the internal standard.
Dynamic light scattering
The nanoparticle size (mean diameter) was determined using a particle size analyzer (ZetaSizer Nano ZS; Malvern Instruments, Malvern, UK). Light scattering measurements were performed for 200 seconds per sample after suspending the nanoparticles in buffer solution for 2 hours at 37°C, and the data obtained were analyzed.
Morphological characterization of nanoparticles
In order to investigate the morphology and size of the prepared nanoparticles, transmission electron microscopy was performed (Model CM12; Philips, Eindhoven, The Netherlands) at an acceleration voltage of 100 kV and a magnification of 110,000×. Carbon-coated copper grids were dipped into a water dispersion of PLA-PEG nanoparticles to deposit nanoparticles onto the grid and dried at room temperature for preparation of samples for transmission electron microscopy.
Assessment of insulin encapsulation efficiency within nanoparticles
The encapsulation efficiency of the nanoparticles was computed by reverse phase high performance liquid chromatography. Insulin-loaded nanoparticles (10 mg) were dissolved in 5 mL of acetonitrile, and 10 mL of phosphate-buffered saline was then added. In order to extract insulin from the organic phase, the phosphate-buffered saline mixture was stirred vigorously using a magnetic stirrer at a speed of 500 rpm for one hour. The samples were centrifuged at 21,000 rpm for 10 minutes. Aqueous phosphate-buffered saline solution was withdrawn and insulin was quantified utilizing chromatographic conditions and a previously reported high performance liquid chromatography method.42 A Kromasil C18 column was used and the wavelength of the instrument detector was set at 214 nm. The mobile phase comprised a mixture of acetonitrile and sodium sulfate buffer (pH 2.3) at a ratio of 24:76 with a flow rate of 1.0 mL per minute. The percentage encapsulation efficiency was expressed as the ratio of actual to theoretical insulin content. The encapsulation efficiency was computed as follows in Equation 1:
| (1) |
In vitro insulin release studies
In vitro insulin release studies were performed by placing 1 g of insulin-loaded PLA and PLA-PEG nanoparticles with varying PEG chain lengths separately in 10 mL of phosphate-buffered saline (37°C, 200 rpm). At various time intervals, phosphate-buffered saline samples were collected as supernatant after centrifugation (21,000 rpm) of the nanoparticles and resuspended in the same volume of fresh phosphate-buffered saline. Each sample was analyzed by reverse phase high performance liquid chromatography, and the quantity of insulin released was computed by means of a standard calibration curve.
Preclinical in vivo studies in the New Zealand White rabbit diabetic model
Experimental rabbits were made diabetic by injecting a single intravenous dose of alloxan (150 mg/kg body weight) dissolved in sterilized water. There is a potential risk of fatal hypoglycemia due to massive insulin release from the pancreas on exposure to alloxan. Therefore, the animals were treated with 20% v/v glucose orally every 4 hours for the first 24 hours following alloxan administration, and after 48 hours, their glucose levels were checked for diabetic status. Only those animals with glucose levels ≥300 mg/dL were used in subsequent studies.43 The study protocol was reviewed and approved by the Institutional Animal Ethics Committee of the Institute of Nuclear Medicine and Allied Sciences, Delhi, India.
Subcutaneous delivery of insulin-loaded PLA-PEG nanoparticles as a parenteral depot
Twelve diabetic rabbits were selected randomly and divided into three groups. Each group had four rabbits that were housed in a single cage and provided a standard diet and water ad libitum. The insulin-loaded PLA-PEG nanoparticles were then suspended in normal sterile saline and administered subcutaneously using a 26-gauge needle and a 2 mL syringe. Group 1 was used as a control to which placebo (insulin-free) PLA-PEG nanoparticles were administered. Groups 2 and 3 were subcutaneously injected with a suspension of insulin-loaded PLA-PEG nanoparticles corresponding to 25 IU and 50 IU of insulin per kg body weight of the animals, respectively. Blood samples were taken from the ear vein of each experimental rabbit and their glucose levels were measured using an Accucheck® (Roche Diagnostics, Indianapolis, IN, USA) blood glucose testing kit.
Toxicity studies
Eight healthy rats (300 ± 30 g body weight) were selected randomly and divided in two groups of four rats per group for the toxicity studies. Animals in group 1 were subcutaneously injected with a 500 mg/kg dose of PLA-PEG nanoparticles, while group 2 served as a control and were not administered any nanoparticles. After 7 days, following anesthesia, a skin sample (5 × 10 mm) was taken from each rat at the injection site for histopathological studies to assess the potential toxicity of the PLA-PEG nanoparticles.
Histopathological evaluation
Histopathological evaluation was completed under the supervision of Dr AK Mukherjee, Safdarganj Development Enclave, New Delhi, India. Histopathological testing of all samples was performed as described below.
Preparation of tissue samples for microtomy
All skin samples were kept in separate vials containing 10 mL of formalin solution (HCHO 10% w/v) for 24 hours to ensure tissue fixation. After fixation and washing in tap water for 2 hours, the samples were dehydrated by immersion in increasing strengths of alcohol ranging from 50% to 95% v/v for 2 hours and finally immersing the samples in absolute alcohol overnight. The samples were then removed from the absolute alcohol and immersed in xylene. After 2 hours, when the alcohol was completely replaced by xylene, the samples acquired a translucent appearance. Wax was impregnated into the samples via a mixture of xylene and wax in 3:1 ratio, then a 1:1 ratio, and finally pure wax for 2 hours at a temperature not exceeding 45°C. For embedding of the skin samples, the wax was melted completely at <45°C and poured between two Leukhart’s “L” pieces. When fully impregnated with wax, the skin samples were then placed carefully in the molten wax inside the “L” pieces and placed in a refrigerator for solidification of the blocks. After solidification, the paraffin wax-embedded skin blocks were carefully trimmed and labeled.
Microtomy and staining of skin samples
The trimmed paraffin wax-embedded skin blocks were then mounted on molds fitted on a rotary rocking microtome to which was attached a microtome knife adjusted to 15 μm. Ribbons of skin sections were carefully collected and transferred to clean slides containing albumin as an adhesive. These slides were dried over a hot plate at 45°C. Prior to staining, the skin sections were rehydrated by passing the slides through xylene and then decreasing strengths of alcohol ranging from 90% to 30% v/v, and finally in water for 5 minutes. The sections were then kept in hematoxylin staining dye for 20 minutes, followed by three dips in acid alcohol (1% HCl and 70% ethanol) and washing in running water to remove any extra staining dye. The sections were neutralized with five dips in ammonia water and washed under running tap water. These sections were then given six dips in eosin dye and washed again to remove extra staining dye. The sections were subsequently dehydrated once more by passing serially through 70% alcohol (3–4 dips), 90% alcohol (6–7 dips), absolute alcohol (for 5 minutes), and finally xylene (for 15 minutes). The stained slides were observed under an inverted microscope ([100×] Olympus Deutschland GmbH, Hamburg, Germany) equipped with a manual camera.
Static lattice atomistic simulations
Molecular simulations were performed using the HyperChem™ 8.0.8 molecular modeling system (Hypercube Inc, Gainesville, FL, USA) and ChemBio3D Ultra 11.0 (Cambridge Soft Corporation, Cambridge, UK). The structures of PEGDA-amine with varying monomer lengths (hereafter referred to as PEG for molecular modeling) and PLA were built in their syndiotactic stereochemistry as three-dimensional models, while the structure of the insulin active sequence was generated using a built-in sequence editor module on HyperChem. The models were energy-minimized using a progressive convergence strategy, where initially the MM+ force field was used, followed by energy minimization with an Amber (Assisted Model Building and Energy Refinements) 3 force field. The conformer having the lowest energy was used to create the polymer-polymer and polymer-protein complexes. A complex of one polymer molecule with another was assembled by disposing the molecules in parallel, and the same procedure of energy minimization was repeated to generate the final models, ie, PLA, PEGDA-amine, PLA-PEG, and PLA-PEG-insulin. Full geometrical optimization was performed in vacuum employing the Polak-Ribiere conjugate-gradient algorithm until a root mean square gradient of 0.001 kcal/mol was reached.
For molecular mechanics computations in vacuum, the force fields were utilized with a distance-dependent dielectric constant scaled by a factor of one. The 1–4 scale factors were electrostatic 0.5 and van der Waals 0.5.44 To generate final models in a solvated system, molecular mechanics simulations were performed for cubic periodic boxes with the polymer-polymer at the center of the cubic box and the remaining free space occupied with water molecules. The same procedure of energy minimization was repeated to generate the solvated models, except that the force fields were utilized with a distance-independent dielectric constant and no scaling (Table 1). PLA-PEG1-H2O, PLA-PEG2-H2O, and PLA-PEG4-H2O represented copolymeric nanoparticles with PEG chain lengths of 575, 2000, and 4000, respectively. In addition, the force field options in the AMBER (with explicit solvent) were extended to incorporate limits to the inner and outer options with nearest-image periodic boundary conditions, and the outer and inner limits were to ensure that there were no discontinuities in the potential surface45 (Table 1).
Table 1.
Particle size and encapsulation efficiency of synthesized PLA-PEG nanoparticles with different PEG chain lengths
| Particle composition | Particle size (nm) | Total insulin (IU/g) | Loading of particles (IU/g) | Insulin encapsulation efficiency (%) |
|---|---|---|---|---|
| PLA | 150.2 | 500 | 165.1 | 33.4 |
| PLA-PEG(575) | 168.5 | 500 | 190.8 | 38.4 |
| PLA-PEG(2000) | 175.2 | 500 | 230.5 | 46.7 |
| PLA-PEG(4000) | 181.9 | 500 | 313.4 | 58.5 |
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
Results and discussion
Characterization of PLA and PLA-PEG conjugate
Gel permeation chromatography
The molecular weight distribution curve for the PLA-PEG copolymer is shown in Figure 2. The molecular weight of the synthesized copolymer was about 72,000 g/mol, which confirmed conjugation of PLA with PEG.
Figure 2.

Molecular mass distribution of PLA-PEG copolymer determined by gel permeation chromatography.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
NMR spectroscopy
1H NMR spectroscopy confirmed the composition of the synthesized monomers and copolymers. Figure 3A shows the proton NMR spectrum of PEGDA, with characteristic peaks at 3.6 ppm and 4.3 ppm corresponding to (–O–CH2–) and (–COO–CH2), respectively. The other peaks at 5.8 ppm and 6.4 ppm are indicative of vinyl protons (CH2=CH–). In the NMR spectrum of aminated PEGDA (Figure 3B), these vinyl peaks were absent and there was an appearance of proton peaks at 2.7 ppm and 2.9 ppm, confirming synthesis of aminated PEGDA by the Michael addition reaction involving the vinyl groups of PEGDA. NMR spectra of PLA (Figure 3C) showed characteristic peaks at 1.6 ppm and 5.2 ppm corresponding to the –CH3 and (–CH) proton, respectively. Figure 3D is the NMR spectrum of the PLA-PEG copolymer in which a peak was observed at 3.6–3.7 ppm attributed to the (–CH2) proton of PEGDA, and the signature protons of the PLA fraction were observed at 1.6 ppm (CH3) and 5.1 ppm (–CH). A very small peak at 4.2 ppm was attributed to the methylene proton (–COO–CH2) of PEGDA. This confirmed the conjugation of PLA with PEG.
Figure 3.

Nuclear magnetic resonance spectra of (A) PEGDA, (B) PEGDA aminated, (C) PLA, and (D) PLA-PEG.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid; PEGDA, PEG diacrylate.
Particle size and surface morphology of PLA-PEG nanoparticles
The hydrodynamic diameter and polydispersity index values for the synthesized nanoparticles are shown in Table 2. At a constant PLA chain length (60,000 g/mol), the particle size increased with increasing chain length of PEG, possibly due to an increase in the viscosity of the organic phase during particle synthesis and to an increase of the extending PEG “brush” on the final particles.46,47 Transmission electron micrographs of the polymeric nanoparticles are shown in Figure 4 and demonstrate that the particles had a spherical morphology.
Table 2.
Computational parameters used to construct aqueous-phase model building and simulations
| S no | Parameter | Description |
|---|---|---|
| 1 | Periodic box dimensions | 15 × 15 × 20Å |
| 2 | Cut-offs | Switched |
| 3 | Dielectric (epsilon) | Constant |
| 4 | 1–4 scale factors | Electrostatic: 0.5 van der Waals: 0.5 |
| 5 | Outer radius | 7.5Å |
| 6 | Inner radius | 3.5Å |
| 7 | Water molecules | 149 |
| 8 | Solvent/polymer distance | 2.3Å |
Figure 4.

Transmission electron micrograph of PLA-PEG nanoparticles.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
Insulin encapsulation efficiency within PLA-PEG nanoparticles
At a constant PLA chain length, the effect of varying molecular weights of PEG (575, 2000, and 4000 g/mol) on insulin encapsulation efficiency within the respective PLA-PEG nanoparticles was investigated. Insulin encapsulation efficiency as recorded for the PLA nanoparticles was 33%, while that of the copolymeric PLA-PEG nanoparticles increased from 38% to 58% with an increase in the PEG chain length from 575 to 4000 g/mol (Table 2). Similar results have been reported by Akbarzadeh et al, who studied the encapsulation efficiency of doxorubicin in Fe3O4-PLGA-PEG nanoparticles synthesized with PEG 2000, 3000, and 4000 g/mol.48 This enhancement in insulin encapsulation efficiency, as observed for nanoparticles with low molecular weight PEG compared with that of PLA nanoparticles, occurred due to the fact that PEG 575 introduced flexible chains into the copolymer. However, the chain length of incorporated PEG 575 was short and insufficient to improve the hydrophilicity of the nanoparticles to any considerable extent, whereas an increase in PEG chain length from PEG 575 to PEG 4000 significantly improved the hydrophilicity and flexibility of the copolymers and resulted in superior entrapment of insulin by the copolymer.47
In vitro insulin release
PLA-PEG nanoparticles with PEG chain lengths of 575 g/mol, 2000 g/mol, and 4000 g/mol provided a cumulative insulin release of 74.4 IU (39%), 110.6 IU (48%), and 185.0 IU (59%) over a period of 10 days, respectively (Figure 5). The mechanism of insulin release from the PLA-PEG nanoparticles can be divided into two phases, ie, a burst release phase and a controlled release phase. In the burst release phase, appreciable release occurred in the first 24 hours, corresponding to the insulin adsorbed or attached by weak bonding onto the surface of the nanoparticles. In the controlled release phase, the nanoparticles showed pseudo zero-order insulin release up to 10 days. The rate and quantity of insulin released increased with the increasing PEG chain lengths used to prepare the PLA-PEG nanoparticles. An increase in flexibility and hydrophilicity of the copolymeric nanoparticles with a related increase in PEG chain length consequently increased the rate of diffusion of insulin across the matrix. This resulted in higher insulin release from the nanoparticles when the molecular weight of PEG in the polymeric backbone was higher.46 Therefore, superior insulin release was observed with the PLA-PEG4000 nanoparticles. These nanoparticles were used further for the preclinical in vivo animal studies in the diabetic rabbit model. Akbarzadeh et al,48 who studied a similar trend of doxorubicin release from Fe3O4-PLGA-PEG nanoparticles, did not elaborate on the inherent mechanism underlining this interesting observation. Further elucidation of the conformational profile of the PEG chains with regard to entrapment efficiency and insulin release is shown later in this paper using detailed molecular mechanics simulations.
Figure 5.

In vitro release of insulin from PLA-PEG4000 nanoparticles (n = 3) at pH 7.4.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
In vivo release of insulin from PLA-PEG nanoparticles in the diabetic rabbit model
PLA-PEG4000 insulin-loaded nanoparticles were evaluated in experimental rabbits. The hypoglycemic effect of two insulin doses (25 IU and 50 IU/kg body weight) administered subcutaneously to diabetic rabbits was studied. Nanoparticles containing 50 IU/kg body weight controlled blood glucose levels within the normal physiological range of 90–140 mg/dL, and the effect was prolonged for 8 days before showing a gradual increase in blood glucose levels (Figure 6). Insulin formulations of 25 IU/kg body weight were observed to be clinically ineffective in maintaining blood glucose levels for an appreciable length of time. The PLA-PEG4000 nanoparticles formed a depot at the site of injection and released the entrapped insulin by gradual diffusion across the polymeric chains, as well as slow degradation of the copolymer matrix. Glucose levels did not revert back to the original diabetic level (500 mg/dL) even after 8 days, indicating that the nanoparticles had the ability to hold and release insulin in a sustained manner for more than 7 days.
Figure 6.

Hypoglycemic effect of PLA-PEG4000 nanoparticles parenteral depot administered to diabetic rabbits (n = 4) at a dose of 25 and 50 IU/kg body weight of insulin.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
Toxicity studies and histopathological evaluation
No change was observed in body weight or behavior of the rabbits injected with PLA-PEG nanoparticles for the entire study duration (7 days). The behavior of the experimental rabbits was closely observed and found to be similar to that of the control animals. Histopathological analysis of tissue samples from the injection site were prepared for toxicity studies and the results are presented in Figures 7 and 8. Tissue samples from the experimental animals (after 7 days of injection) showed no abnormal features and the skin had a normal histological appearance, similar to that of the skin from the control group. Low powered photomicrographs of sections of skin from the site of injection showed that the epidermis and dermis were both within normal limits (Figure 7). High powered photomicrographs of the same section showed normal superficial dermis and epidermis (Figure 8).
Figure 7.

Low intensity photomicrograph of a skin section from (A) a control animal and (B) an animal subcutaneously administered PLA-PEG nanoparticles.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
Figure 8.

High intensity photomicrograph of a skin section from (A) a control animal and (B) an animal subcutaneously administered PLA-PEG nanoparticles.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
Molecular mechanics-assisted model building and energy refinements
A molecular mechanics conformational search procedure was used to acquire the data for the statistical mechanics analysis to obtain differential binding energies of a Polak-Ribiere algorithm and to permit potential application to polymer composite assemblies. MM+ is a HyperChem modification and extension of Allinger’s molecular mechanics program, MM2,49 whereas AMBER applies molecular mechanics, normal mode analysis, molecular dynamics, and free energy computations to simulate the structural and energetic properties of molecules.50
Molecular mechanics energy relationships, a method for analytico-mathematical representation of potential energy surfaces, was used to provide data regarding the contributions of valence terms, noncovalent coulombic terms, and noncovalent van der Waals interactions for the polymer-polymer-insulin complexes. The molecular mechanics energy relationships model for the potential energy factor in various molecular complexes can be written as Equation 2:
| (2) |
where V∑ is related to total steric energy for an optimized structure, Vb corresponds to bond stretching contributions, Vθ denotes bond angle contributions, Vϕ represents torsional contribution arising from deviations from optimum dihedral angles, Vij incorporates van der Waals interactions due to non-bonded interatomic distances, Vhb symbolizes hydrogen-bond energy function, and Vel stands for electrostatic energy.
In addition, the total potential energy deviation, ΔETotal, was computed as the difference between the total potential energy of the complex system and the sum of the potential energies of isolated individual molecules using equation 3:
| (3) |
The molecular stability could be estimated by comparing the total potential energies of the isolated and complexed systems (Equations 4–10). If the total potential energy of the complex is smaller than the sum of the potential energies of isolated individual molecules in the same conformation, the complexed form is more stable and its formation is favored.51
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
Formation of polymeric assemblies
The PEGDA-amines (NH2-PEGDA-NH2) were modeled in the form of PEGDA1, PEGDA2, and PEGDA4 with chain lengths of 1, 2, and 4 monomer contents representing PEG575, PEG2000, and PEG4000, respectively. This was performed for superior efficiency in terms of computational time and modeling space. The energetic profiles for the formation of polymeric assemblies, ie, PLA-PEG1, PLA-PEG2, and PLA-PEG4, in vacuum, are represented by the energy equations 4–10, and the conformational profiles are depicted in Figure 9. The energy equations demonstrated that the polymeric systems were desirable in terms of their respective bonding and nonbonding energy factors. The energy stabilization with negative steric energies in all three cases suggested superior compatibility and miscibility. In addition, the geometric conformation and H bonding suggested involvement of the –COO functionality of PLA with the –NH2 functionality of PEGDA-amine. The geometric stabilization followed a specific trend, showing an increase in ΔE with an increase in PEG chain length: ΔE =16.606 kcal/mol, 19.756 kcal/mol, and 24.028 kcal/mol for PLA-PEG1, PEGDA2, and PEGDA4, respectively. The complexes were stabilized by generalized energy times in terms of torsional contributions (bonding energies) as well as London dispersion forces and H bonding (nonbonding interactions). Interestingly, the introduction of an increased monomer content introduced deviations from optimum dihedral angles, with values in the range of about 5 kcal/mol, which led to a stabilized molecular entity. The van der Waals interactions added further to the stabilization and retrieved high negative values which may be due to PEG acting as a filler in the space lattice of the binary system (Figure 9). Furthermore, the H bonding, although not contributing as much as van der Waals forces, displayed exponentially lowered energies due to the H bonds introduced, as explained earlier. This nonbonding, from H bonding to van der Waals forces, is due to the hydrophobic interactions arising from the inclusion of amphiphilic PEG, which further led to formation of nanoparticles in excess of aqueous Pluronic solution.
Figure 9.

Visualization of geometrical preferences of (A) PLA-PEG1, (B) PLA-PEG2, and (C) PLA-PEG4 after molecular simulation in vacuum.
Notes: Elements are color coded: cyan = C; red = O; blue = N; yellow = P; white = H; brown = Zn.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
Amphiphilic properties involving nanoparticle formation, insulin entrapment, and release
Modeling was performed in the absence (vacuum) and presence (periodic box) of water molecules corresponding to the lipophilic/hydrophobic and hydrophilic phases, respectively. It was assumed that the more the complex was stabilized in vacuum, the more it leaned towards lipophilicity. Inversely, the more the complex was stabilized in water, the more it contributed to hydrophilicity. In accordance with the minimization values obtained in vacuum (simulated as the hydrophobic phase) from equations 4–10, an increase in amphiphilic PEG chain length led to a shift in preference towards the hydrophobic phase, acetonitrile (internal phase) compared with the aqueous (external) phase, thereby forming more stabilized nanostructures at higher levels:
| (11) |
| (12) |
| (13) |
This explained the higher entrapment values of insulin during nanoparticle formation because a more stabilized structure (PLA-PEG4000) entrapped more insulin in the presence of a hydrophobic phase. Given that PLA is a hydrophobic polymer, this led to a highly hydrophobic proposition, resulting in decreased leaching of insulin into the aqueous external phase. Considering this, it is expected that the highly stabilized PLA-PEG4 system would release less insulin at a slower rate over a given time period. However, it was observed that a higher and faster release in the case of PLA-PEG4000 was obtained compared with the other polymeric systems. To investigate this finding further, the three systems were individually modeled within a periodic box setup, where the energy minimizations were performed in the presence of water molecules. Interestingly, PLA-PEG1 showed the highest stabilized system with the lowest energy (−1408.530 kcal/mol) compared with PLA-PEG2 (−1220.868 kcal/mol) and PLA-PEG4 (−1145.627 kcal/mol). We assume here that, given the aqueous phase (as there was no nonaqueous phase used during in vitro release studies), the PEG chains contribute towards the hydrophilicity of the matrix – thereby acting against the hydrophobic PLA part, making the system less stable – and hence tend to release more drug at a faster rate. Stabilization of the electrostatic interactions in the case of PLA-PEG1-H2O (about −1400 kcal/mol) further confirmed the role of hydrophilicity.52 Furthermore, more indepth investigation of the energy-minimized structures in Figures 9 and 10 showed that PLA-PEG4 (vacuum) and PLA-PEG1-H2O (aqueous) were denser than their respective counterparts, leading to enhanced entrapment and slower release of insulin, respectively. The presence of the –COOH and –NH2 chemistry on the polymeric assemblies interacted with the incorporated insulin. Therefore, insulin with PLA and PEG was modeled in stoichiometric quantities relative to the concentration used in the preparation of the insulin-loaded nanoparticles. Because the mass of the polymers used was constant (PEG 1 ×4 = PEGDA 2 ×2 = PEGDA 4 ×1), these were modeled as insulin-PLA-PEG14, insulin-PLA-PEG22, and insulin-PLA-PEG41 for stoichiometric similarity:
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
| (20) |
Figure 10.
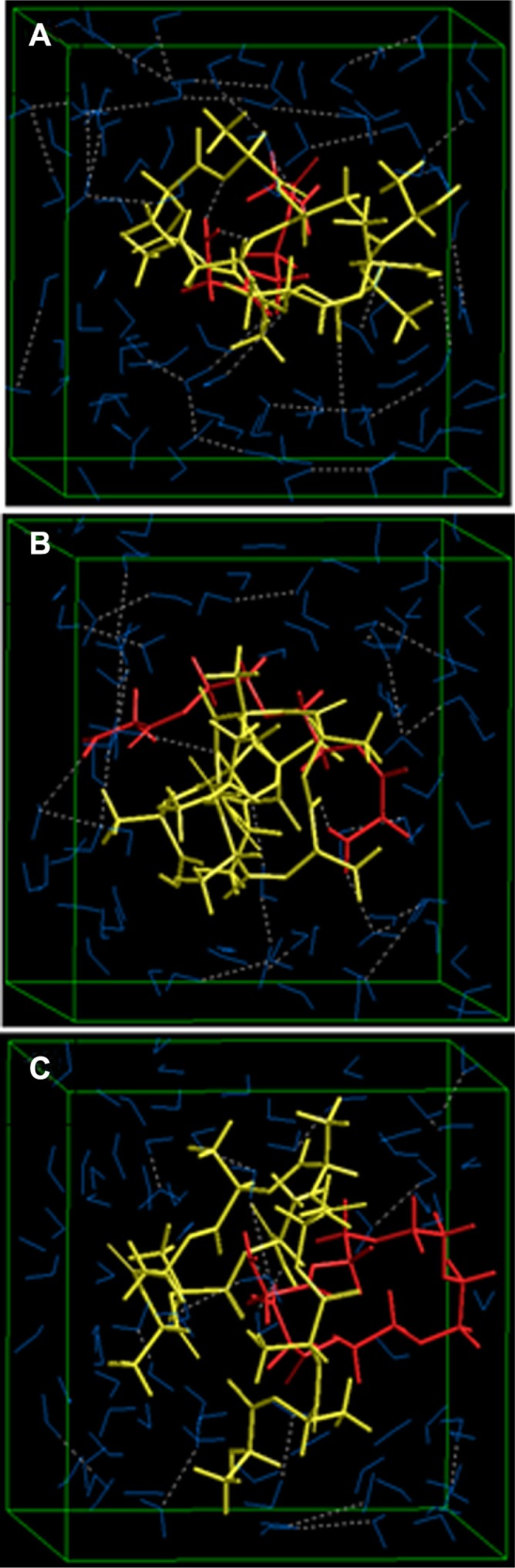
Visualization of geometrical preferences of (A) PLA-PEG1, (B) PLA-PEG2, and (C) PLA-PEG4 after molecular simulation in a solvated system consisting of 149 water molecules (blue molecules).
Note: The PLA (yellow) and PEG (red) are rendered in tube display.
Abbreviations: PEG, poly(ethylene glycol); PLA, polylactic acid.
As shown in Figure 11, PLA-PEG14 was highly conjugated with insulin because there was a greater number of –NH2 end groups (per given mass stoichiometrically) to interact with the –COOH groups of insulin, leading to an energy stabilization of about 39 kcal/mol. H bonding in addition to electrostatic stabilization of the insulin-PLA-PEG14 complex compared with insulin-PLA-PEG22 and insulin-PLA-PEG41 (Equations 14–20) led to a polymer-protein conjugated system, further densifying the polymeric matrix. The higher and rapid release of insulin from the PLA-PEG4000 nanoparticles was due to the loose binding of insulin onto the polymeric matrix compared with PLA-PEG575. The Connolly molecular surfaces shown in Figure 11 displayed the in silico shape, morphology, and network structure of the insulin-loaded nanostructures. It was evident that PLA-PEG41 developed a well oriented and regular matrix conformation compared with its lower chain length analogs (even though stoichiometrically equal in quantity). This was consistent with the morphological analysis on transmission electron microscopy. With reference to the results from the vacuum, solvated system, and protein polymer simulations, PLA-PEG4000 displayed superior release and high entrapment of insulin because of the following:
Affinity of PEG chains toward the hydrophobic phase (in the presence of the hydrophilic phase) during nanoparticle formation led to higher entrapment.
Affinity of PEG chains toward the hydrophilic phase (in the absence of the hydrophobic phase) during in vitro insulin release led to more rapid release.
Dense network structure during nanomatrix formation and sparse network structure during in vitro release led to higher entrapment and rapid release, respectively.
Lower polymer-protein interaction in the case of PLA-PEG4000 (<NH2 groups) compared with lower chain lengths (>NH2 functionality) led to increased insulin release.
Insulin-loaded PLA-PEG4000 nanoparticles displayed regular anisotropy and periodic arrangement and thereby provided the desired release profile.
Figure 11.

Visualization of geometrical preferences of the insulin molecule in complexation with (A) PLA-PEG14, (B) PLA-PEG22, and (C) PLA-PEG41 after molecular simulations in vacuum.
Notes: The peptide molecules are rendered in tube (elements color coded) and thin-ribbon secondary structures (violet). Color codes for insulin tube rendering: C (cyan), O (red), H (white), and P (yellow). The respective Connolly molecular electrostatic potential surfaces for the nanoparticulate matrix in transparent display mode are also shown.
Conclusion
PLA-PEG copolymers were synthesized with varying chain lengths of PEG (575–4000), and their corresponding nanoparticles were prepared using the double emulsion polymerization technique. The conjugation reaction between PLA and PEG was confirmed by NMR and gel permeation chromatography, and morphology studies revealed the nanoparticles to be spherical in shape. Particle size was measured, and with an increase in molecular mass of PEG from 575 g/mol to 4000 g/mol in the reaction mixture, the copolymeric particle size increased from 168 nm to 181 nm. The highest insulin encapsulation efficiency of 58% was achieved with PLA-PEG4000 nanoparticles, which also recorded the highest cumulative insulin release of 185.0 IU (59%) over 10 days. In vivo animal studies using the diabetic rabbit model demonstrated that a single subcutaneous injection of PLA-PEG4000 nanoparticles containing 50 IU of insulin load/kg body weight was effective in maintaining blood glucose levels in the physiologically normal range of 90–140 mg/dL for more than 7 days. Molecular mechanics iterations performed by static lattice atomistic simulations for determining the reaction profile and molecular energy dynamics of the insulin-loaded nanoparticles generated results that were consistent with the experimental data, that further supported the nanoparticles being suitable as a carrier for parenteral insulin therapy.
Acknowledgments
The authors acknowledge the financial support of the National Research Foundation of South Africa and the Department of Biotechnology, Ministry of Science and Technology, India.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Onuki Y, Morishita M, Takayama K. Formulation optimization of water-in-oil-water multiple emulsion for intestinal insulin delivery. J Control Release. 2004;97:91–98. doi: 10.1016/j.jconrel.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Park K, Kwon IC, Park K. Oral protein delivery: current status and future prospect. React Funct Polym. 2011;71:280–287. [Google Scholar]
- 3.Sullivan CO, Birkinshaw C. In vitro degradation of insulin-loaded poly (n-butylcyanoacrylate) nanoparticles. Biomaterials. 2004;25:4375–4382. doi: 10.1016/j.biomaterials.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Wadher K, Kalsait R, Umekar M. Oral insulin delivery: facts, developments and challenges. Der Pharmacia Lettre. 2009;1:121–129. [Google Scholar]
- 5.Kushwaha SKS, Keshari RK, Rai AK. Advances in nasal trans-mucosal drug delivery. Journal of Applied Pharmaceutical Science. 2011;1:21–28. [Google Scholar]
- 6.Furtado S, Simkhay L, Wobbekind D, Mathiowitz E. Subcutaneous delivery of insulin loaded poly(fumaric-co-sebacic anhydride) microspheres to type 1 diabetic rats. Eur J Pharm Biopharm. 2006;63:229–236. doi: 10.1016/j.ejpb.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Payne RG, McGonogle JS, Yaszemski MJ, Yasko AW, Mikos AG. Development of an injectable, in situ crosslinkable, degradable polymeric carrier for osteogenic cell populations. Part 2. Viability of encapsulated marrow stromal osteoblasts cultured on crosslinking. Biomaterials. 2002;23:4373–4380. doi: 10.1016/s0142-9612(02)00185-0. [DOI] [PubMed] [Google Scholar]
- 8.Qiu B, Stefanos S, Ma J, et al. A hydrogel prepared by in situ cross-linking of a thiol-containing poly(ethylene glycol)-based copolymer: a new biomaterial for protein drug delivery. Biomaterials. 2003;24:11–18. doi: 10.1016/s0142-9612(02)00227-2. [DOI] [PubMed] [Google Scholar]
- 9.Saeedi M, Akbari J, Enayatifard R, Morteza-Semnani K, Tahernia M, Valizadeh H. In situ cross-linking of polyanionic polymers to sustain the drug release from theophylline tablets. Iran J Pharm Res. 2009;8:241–249. [Google Scholar]
- 10.Ravivarapu HB, Moyer KL, Dunn RL. Parameters affecting the efficacy of a sustained release polymeric implant of leuprolide. Int J Pharm. 2000;194:181–191. doi: 10.1016/s0378-5173(99)00371-3. [DOI] [PubMed] [Google Scholar]
- 11.Yong CS, Choi JS, Quan QZ, et al. Effect of sodium chloride on the gelation temperature, gel strength and bioadhesive force of poloxamer gels containing diclofenac sodium. Int J Pharm. 2001;226:195–205. doi: 10.1016/s0378-5173(01)00809-2. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Gao Y, Lv W, et al. Preparation of bleomycin A2-PLGA microspheres and related in vitro and in vivo studies. J Pharm Sci. 2011;100:2790–2800. doi: 10.1002/jps.22514. [DOI] [PubMed] [Google Scholar]
- 13.Shenoy DB, D’Souza RJ, Tiwari SB, Udupa N. Potential applications of polymeric microsphere suspension as subcutaneous depot for insulin. Drug Dev Ind Pharm. 2003;29:555–563. doi: 10.1081/ddc-120018644. [DOI] [PubMed] [Google Scholar]
- 14.Takenaga M, Yamaguchi Y, Kitagawa A, Ogawa Y, Mizushima Y, Igarashi R. A novel insulin formulation can keep providing steady levels of insulin for much longer periods in-vivo. J Pharm Pharmacol. 2002;54:1189–1194. doi: 10.1211/002235702320402026. [DOI] [PubMed] [Google Scholar]
- 15.Takenaga M, Yamaguchi Y, Kitagawa A, Ogawa Y, Mizushima Y, Igarashi RA. Novel sustained-release formulation of insulin with dramatic reduction in initial rapid release. J Control Release. 2002;79:81–91. doi: 10.1016/s0168-3659(01)00518-1. [DOI] [PubMed] [Google Scholar]
- 16.Hu YQ, Guo JX, Wang LJ, Tan R, Zhen LY. Preparation and evaluation of insulin-loaded polylactide microspheres using factorial design. Drug Dev Ind Pharm. 2000;26:1309–1313. doi: 10.1081/ddc-100102314. [DOI] [PubMed] [Google Scholar]
- 17.Ibrahim MA, Ismail A, Fetouh MI, Göpferich A. Stability of insulin during the erosion of poly(lactic acid) and poly(lactic-co-glycolic acid) microspheres. J Control Release. 2005;106:241–252. doi: 10.1016/j.jconrel.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 18.Uchida T, Nagareya N, Sakakibara S, et al. Preparation and characterization of polylactic acid microspheres containing bovine insulin by a w/o/w emulsion solvent evaporation method. Chem Pharm Bull (Tokyo) 1997;45:1539–1543. doi: 10.1248/cpb.45.1539. [DOI] [PubMed] [Google Scholar]
- 19.Caliceti P, Veronese FM, Lora S. Polyphosphazene microspheres for insulin delivery. Int J Pharm. 2000;211:57–65. doi: 10.1016/s0378-5173(00)00588-3. [DOI] [PubMed] [Google Scholar]
- 20.Mukerjee A, Sinha VR, Pruthi V. Preparation and characterization of poly-ε-caprolactone particles for controlled insulin delivery. J Biomed Pharm Eng. 2007;1:40–44. [Google Scholar]
- 21.Furtado S, Abramson D, Burrill R, et al. Oral delivery of insulin loaded poly(fumaric-co-sebacic) anhydride microspheres. Int J Pharm. 2008;347:149–155. doi: 10.1016/j.ijpharm.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 22.Caliceti P, Salmaso S, Lante A, et al. Controlled release of biomolecules from temperature-sensitive hydrogels prepared by radiation polymerization. J Control Release. 2001;75:173–181. doi: 10.1016/s0168-3659(01)00380-7. [DOI] [PubMed] [Google Scholar]
- 23.Sonaje K, Italia JL, Sharma G, Bhardwaj V, Tikoo K, Kumar MN. Development of biodegradable nanoparticles for oral delivery of ellagic acid and evaluation of their antioxidant efficacy against cyclosporine A-induced nephrotoxicity in rats. Pharm Res. 2007;24:899–908. doi: 10.1007/s11095-006-9207-y. [DOI] [PubMed] [Google Scholar]
- 24.O’Hagan DT, Singh M, Ulmer JB. Microparticles for the delivery of DNA vaccines. Immunol Rev. 2004;199:191–200. doi: 10.1111/j.0105-2896.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- 25.Hye JL. Protein drug oral delivery: the recent progress. Arch Pharm Res. 2002;25:572–584. doi: 10.1007/BF02976925. [DOI] [PubMed] [Google Scholar]
- 26.Tiwari S, Verma P. Microencapsulation technique by solvent evaporation method (study of effect of process variables) Int J Pharm Life Sci. 2011;2:998–1005. [Google Scholar]
- 27.Vila A, Sánchez A, Tobío M, Calvo P, Alonso MJ. Design of biodegradable particles for protein delivery. J Control Release. 2002;78:15–24. doi: 10.1016/s0168-3659(01)00486-2. [DOI] [PubMed] [Google Scholar]
- 28.van de Weert M, Hennink WE, Jiskoot W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm Res. 2000;17:1159–1167. doi: 10.1023/a:1026498209874. [DOI] [PubMed] [Google Scholar]
- 29.Tamber H, Johansen P, Merkle HP, Gander B. Formulation aspects of biodegradable polymeric microspheres for antigen delivery. Adv Drug Deliv Rev. 2005;57:357–376. doi: 10.1016/j.addr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 2000;17:100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Chua KM, Wang CH. Stabilization and encapsulation of human immunoglobulin G into biodegradable microspheres. J Colloid Interface Sci. 2004;271:92–101. doi: 10.1016/j.jcis.2003.08.072. [DOI] [PubMed] [Google Scholar]
- 32.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit Rev Ther Drug Carrier Syst. 2002;19:73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 33.Elvassore N, Bertucco A, Caliceti P. Production of insulin-loaded poly(ethylene glycol)/poly(l-lactide) (PEG/PLA) nanoparticles by gas antisolvent techniques. J Pharm Sci. 2001;90:1628–1636. doi: 10.1002/jps.1113. [DOI] [PubMed] [Google Scholar]
- 34.Vila A, Sánchez A, Evora C, Soriano I, McCallion O, Alonso MJ. PLA-PEG particles as nasal protein carriers: the influence of the particle size. Int J Pharm. 2005;292:43–52. doi: 10.1016/j.ijpharm.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Xiao RZ, Zeng ZW, Zhou GL, Ang JJ, Li FZ, Wang AM. Recent advances in PEG-PLA block copolymer nanoparticles. Int J Nanomedicine. 2010;5:1057–1065. doi: 10.2147/IJN.S14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem Rev. 2004;104:6147–6176. doi: 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- 37.Sheshala R, Peh KK, Darwis Y. Preparation, characterization and in vivo evaluation of insulin-loaded PLA-PEG microspheres for controlled parenteral drug delivery. Drug Dev Ind Pharm. 2009;35:1364–1374. doi: 10.3109/03639040902939213. [DOI] [PubMed] [Google Scholar]
- 38.Ibrahim MA. Assessment of insulin stability inside diblock copolymer PEG-PLA microspheres. Sci Pharm. 2010;78:493–505. doi: 10.3797/scipharm.1002-01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomar LK, Tyagi C, Lahiri SS, Singh H. Poly(PEGDMA-MAA) copolymeric micro and nanoparticles for oral insulin delivery. Polym Adv Technol. 2011;22:1760–1767. [Google Scholar]
- 40.Lowman AM, Morishita M, Kajita M, Nagai T, Peppas NA. Oral delivery of insulin using pH-responsive complexation gels. J Pharm Sci. 1999;88:933–937. doi: 10.1021/js980337n. [DOI] [PubMed] [Google Scholar]
- 41.Cheng FY, Wang SPH, Su CH, et al. Synthesis, functionalization, and biomedical applications of multifunctional magnetic nanoparticles. Biomaterials. 2008;29:2104–2112. [Google Scholar]
- 42.Yomota C, Yoshii Y, Takahata T, Okada S. Separation of B-3 monode-samidoinsulin from human insulin by high-performance liquid chromatography under alkaline conditions. J Chromatogr A. 1996;721:89–96. doi: 10.1016/0021-9673(95)00761-x. [DOI] [PubMed] [Google Scholar]
- 43.Al-Azzawie HF, Alhamdani MS. Hypoglycemic and antioxidant effect of oleuropein in alloxan-diabetic rabbits. Life Sci. 2006;78:1371–1377. doi: 10.1016/j.lfs.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 44.Kumar P, Pillay V, Choonara YE, Modi G, Naidoo D, du Toit LC. In silico theoretical molecular modeling for Alzheimer’s disease: the nicotine-curcumin paradigm in neuroprotection and neurotherapy. Int J Mol Sci. 2011;12:694–724. doi: 10.3390/ijms12010694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choonara YE, Pillay V, Ndesendo VMK, et al. Design of polymeric nanoparticles by synthetic wet chemical processing strategies for the sustained delivery of anti-tuberculosis drugs. Colloids Surf B Biointerfaces. 2011;87:243–254. doi: 10.1016/j.colsurfb.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 46.Gref R, Lück M, Quellec P, et al. Stealth corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18:301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 47.Ren J, Yu X, Ren T, Hong H. Preparation and characterization of fenofibrate-loaded PLA-PEG microspheres. J Mater Sci Mater Med. 2007;18:1481–1487. doi: 10.1007/s10856-007-0132-3. [DOI] [PubMed] [Google Scholar]
- 48.Akbarzadeh A, Mikaeili H, Zarghami N, Mohammad R, Barkhordari A, Davaran S. Preparation and in vitro evaluation of doxorubicin-loaded Fe3O4 magnetic nanoparticles modified with biocompatible copolymers. Int J Nanomedicine. 2012;7:511–526. doi: 10.2147/IJN.S24326. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Warhurst DC, Craig JC, Adagu IS, Meyer DJ, Lee SY. The relationship of physico-chemical properties and structure to the differential antiplasmodial activity of the cinchona alkaloids. Malaria J. 2003;2:26–39. doi: 10.1186/1475-2875-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pearlman DA, Case DA, Caldwell JW, et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- 51.Yu BY, Chung JW, Kwak S-Y. Reduced migration from flexible poly(vinyl chloride) of a plasticizer containing β-cyclodextrin derivative. Environ Sci Technol. 2008;42:7522–7527. doi: 10.1021/es800895x. [DOI] [PubMed] [Google Scholar]
- 52.Knapp EW. Computation of electrostatic energies for complex molecular systems. Available from: http://agknapp.chemie.fu-berlin.de/agknapp/files/downloads/Sfb765_electrostatic-talk_knapp.pdf. Accessed May 17, 2012.