

Abstract
Age-related Macular Degeneration (AMD) is the leading cause of blindness among the elderly. While excellent treatment has emerged for neovascular disease, treatment for early AMD is lacking due to an incomplete understanding of the early molecular events. Cigarette smoking is the strongest epidemiologic risk factor, yet we do not understand how smoking contributes to AMD. Smoking related oxidative damage during the early phases of AMD may play an important role. This review explores how cigarette smoking and oxidative stress to the retinal pigmented epithelium (RPE) might contribute to AMD, and how the transcription factor Nrf2 can activate a cytoprotective response.
Keywords: Age-related Macular Degeneration, Cigarette smoking, Nrf2, Oxidative stress, Retinal pigmented epithelium
1. Introduction
Age-related Macular Degeneration (AMD) is the leading cause of blindness among the elderly in the United States, representing 54% of legal blindness (Congdon, O’Colmain, et al., 2004). Due to the aging population, the number of people with advanced AMD will increase from 1.75 million now, to 3 million by 2020 (Friedman, O’Colmain, et al., 2004). At present, 7 million people are at risk of developing advanced AMD, and 1 in 3 persons ≥70 years old with early AMD will develop advanced disease within 10 years (Congdon et al., 2004; Mukesh, Dimitrov, et al., 2004). The vast majority of these people have nonneovascular or dry AMD. Since high dose micronutrient vitamins only slows visual loss in dry AMD, the therapeutic benefit is modest at best (2001). As a result, the impact of AMD to both the individual and the general public is devastating. Improving treatments that reverse, prevent, or even delay the onset of dry AMD would have significant benefit to both the individual and society. Most research has concentrated on neovascular (wet) AMD, which has resulted in the development of effective anti-VEGF treatments. Few studies however, have focused on the factors that are important in the development of dry AMD. Aside from chronological aging, cigarette smoking is the strongest epidemiologic risk factor for AMD yet our understanding of how it contributes to AMD is limited at present. The purpose of this review article is to describe the evidence for how cigarette smoking might contribute to the onset of early AMD.
2. The epidemiologic evidence
The strongest environmental risk factor for AMD is cigarette smoking (Smith, Assink, et al., 2001). Epidemiologic data from several large studies indicate that both AMD onset and disease progression are strongly influenced by smoking (Clemons, Milton, et al., 2005; Khan, Thurlby, et al., 2006; Klein, Knudtson, et al., 2008; Smith et al., 2001). The findings from three continents nicely summarizes the dramatic influence that cigarette smoking has on AMD (Tomany, Wang, et al., 2004). After pooling data, current smoking was associated with an increased incidence of geographic atrophy and late AMD (odds ratios [ORs] relative to nonsmokers: 2.83 and 2.35, respectively; ORs relative to past smokers: 2.80 and 1.82, respectively). Thus, current smokers appear at higher risk of AMD than both past smokers and nonsmokers. In addition, a “dose–response” effect has been established since pack-year smoking strongly correlates with AMD while smoking cessation reduces the risk for dry AMD (Khan et al., 2006). These results provide fundamental evidence for a strong epidemiologic association of cigarette smoking and AMD.
With aging and early AMD, the retinal pigmented epithelium (RPE) undergoes progressive degeneration. The RPE transitions from its normal cuboidal morphology to irregularly shaped, and then flattened or atrophic when overlying thick basal laminar deposits, or accumulations of heterogeneous material within Bruch membrane (Green, McDonnell, et al., 1985; Sarks, 1976; van der Schaft, Mooy, et al., 1992). With aging and early AMD, the number of RPE cells in the macula declines more than in the periphery (Harman, Fleming, et al., 1997). Cells that overlie drusen are swollen, rounded, and vacuolated (Anderson, Mullins, et al., 2002), but appear distinct from apoptotic cells that are shrunken with nuclear fragmentation. These changes are reminiscent of a prelethal form of oncosis, a pathway of cell death characterized by increased cell volume (Anderson et al., 2002). The strongest evidence however, indicates that apoptosis is the major pathway for RPE cell death. Del Priore, Kuo, et al. (2002) showed that the proportion of apoptotic RPE cells in the macula increases significantly with age while Dunaief, Dentchev, et al. (2002) showed that maculas with AMD have significant increases in TUNEL-positive RPE cells compared with normal eyes. In geographic atrophy, TUNEL-positive RPE cell nuclei appear at the edges of RPE atrophy, which correlates with clinically observed expansion of atrophic areas during disease progression. Epidemiologic studies of AMD suggest that the RPE is a specific target of cigarette smoke induced injury. The Blue Mountains Eye study (Mitchell, Wang, et al., 2002) showed that smoking was associated with increased RPE abnormalities and the AREDS cohort (2001) found that smoking was correlated with geographic atrophy, which is characterized by progressive RPE atrophy and apoptotic cell death.
3. RPE cell apoptosis and other early features of AMD develop in mice exposed to cigarette smoke
Our laboratory recently showed that mice exposed to chronic cigarette smoke develop features of early AMD (Fujihara, Nagai, et al., 2008). Mice exposed to cigarette smoke developed oxidative damage and ultrastructural degeneration to the RPE and Bruch membrane, as well as RPE cell apoptosis. Two month old C57Bl6 mice were exposed to either filtered air or cigarette smoke in a smoking chamber for 5 h/day, 5 days/week for 6 months. Fig. 1 (Fujihara et al., 2008) shows mice exposed to cigarette smoke had immunolabeling for 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage, in 85 ± 3.7% of RPE cells counted compared to 9.5 ± 3.9% in controls (p < 0.00001). Using regression analysis, the two most pronounced ultrastructural changes (severity grading scale from 0 to 3) seen were a loss of basal infoldings (mean difference in grade = 1.98; p < 0.0001), and an increase in intracellular vacuoles (mean difference in grade = 1.7; p < 0.0001; Fig. 2 (Fujihara et al., 2008)). These ultrastructural changes to the RPE have been identified in AMD. Bruch membrane was thicker in mice exposed to smoke (1086 ± 332 nm) than those raised in air (543 ± 132 nm; p = 0.0069), which is suggestive of accelerated aging. Ultrastructural changes to Bruch membrane in cigarette-smoke exposed mice were smaller in magnitude than changes in the RPE, but consistently demonstrated significantly higher grade injury in cigarette-exposed mice, including basal laminar deposits (mean difference in grade = 0.54; p < 0.0001), increased outer collagenous layer deposits (mean difference in grade = 0.59; p = 0.002), and increased basal laminar deposit continuity (mean difference in grade = 0.4; p < 0.0001). A higher percentage of apoptotic RPE cells from mice exposed to cigarette smoke (average 8.0 ± 1.1%) than room air (average 0 ± 0%; p = 0.043) was identified using TUNEL staining (Fig. 3, Fujihara et al., 2008). We plan to use this model for studying the mechanism of smoke induced changes during early AMD.
Fig. 1.

Immunohistochemistry of 8-OHdG nuclear labeling of the RPE. Two month C57Bl6 mouse exposed to smoke for 6 months showing (A) DAPI labeled nuclei (arrows); (B) 8-OHdG labeled RPE nuclei (arrows); (C) merged image of A and B with the Brightfield image showing violet nuclei (arrows); (D) merged image of DAPI and IgG1 control image with Brightfield image overlay. Eight month old C57Bl6 mouse raised in air showing DAPI labeled RPE nuclei in (E) and 8-OHdG immunostaining in (F); (G) Merged DAPI and 8-OHdG immunostained image with Brightfield image overlay showing blue nuclei in H. Brightfield image. RPE, retinal pigmented epithelium; Ch, choroid. Bar = 15 mm. Figure shows representative images from N = 10 mice (50 samples/mouse).
Fig. 2.

Transmission electron microscopy of the RPE-Bruch membrane-choriocapillaris of mice exposed to air (A) or cigarette smoke (B–D). (A) The RPE has normal cytoplasm and regular basal infoldings (BI) Bruch membrane (BrM) is unthickened and without deposits. The choriocapillaris (CC) has regular fenestrations. (B) A membranous vacuole (V) appears in the cytoplasm and the basal infoldings (BI) are fewer and dilated in the RPE. A small outer collagenous layer (OCL) deposit is seen in Bruch membrane. (C and D) More severe membranous vacuoles appear in the cytoplasm than in B. The basal infoldings are fewer and dilated. The choriocapillaris fenestrations are fewer (arrows). (D) The RPE has multiple, large vacuoles in the cytoplasm and the basal infoldings are fewer and dilated, or absent. Thin basal laminar deposits (*) are seen where the basal infoldings are absent. Bar = 500 n.m
Fig. 3.

TUNEL labeling of RPE cells from mice exposed to cigarette smoke for 6 months. (A) Nuclei are stained with DAPI (blue), as labeled by the arrows. (B) TUNEL labeled (red) RPE nuclei are indicated by the arrows. (C) Merged image of A and B separating TUNEL from DAPI only stained nuclei. (D) Brightfield image of the RPE, choroid (Ch) and sclera (S). (E) Merged image of a mouse raised in air for 6 months. Arrows point to blue DAPI without red TUNEL labeling. Bar = 15 μm.
Espinosa-Heidmann, Suner, et al. (2006) found that a shorter duration, higher concentration of cigarette smoke in 16 month old C57Bl6 mice induced ultrastructural changes to Bruch membrane and the choriocapillaris endothelium that are compatible with early AMD. Interestingly, they did not find compelling ultrastructural evidence of RPE cell injury. However, they did not specifically study apoptosis. In their study, mice were exposed to more severe levels of cigarette smoke over a shorter period (i.e. 2 h per day, 5 days/week over 3.5 months) than the chronic experimental conditions that we selected (5 h/day, 5 days/week over 6 months). We chose a less acute model on the theory that AMD lesions will develop over a long period of time after chronic cigarette smoke exposure. We were guided by the knowledge that a 6 month exposure using our protocol was sufficient to induce emphysema in mice (Rangasamy, Cho, et al., 2004). Given their more acute exposure of higher concentrations of cigarette smoke, it is possible that due to the significant anti-oxidant capability of the RPE, the RPE was able to accommodate this acute oxidative demand. It is possible that a chronic exposure to the oxidants in cigarette smoke might be necessary to cause RPE injury and apoptosis.
4. What is cigarette smoke?
Before exploring how cigarette smoke can contribute to the development of AMD, it is important to understand the composition of cigarette smoke, and how its components could damage tissue. Cigarette smoke is comprised of a gas and tar phase. Each phase contains free radicals. Gas-phase radicals are both inorganic and organic, including reactive oxygen species (ROS), epoxides, peroxides, nitric oxide (NO), nitrogen dioxide, peroxynitrite, peroxynitrates and various other free radicals (Rahman & MacNee, 1996). In all, cigarette smoke contains over 4700 chemicals, with a high concentration of free radicals (Rangasamy et al., 2004; Smith & Hansch, 2000). In fact, each puff of a cigarette contains 1015 free radicals, including O2− and NO, that combine to form peroxynitrite, which is a potent lipid peroxidation (Rahman & MacNee, 1996). In tissues other than the eye, it is clear that chemical oxidants in cigarette smoke deplete tissues of ascorbic acid and protein sulfhydryl groups, causing the oxidation of DNA, lipids and proteins (Cross, O’Neill, et al., 1993; Lykkesfeldt, Christen, et al., 2000; O’Neill, Halliwell, et al., 1994).
5. Oxidative stress and oxidative damage in AMD
Oxidative stress has long been hypothesized to play a major role in the development of AMD due to the high oxidative stress environment of the fundus. Epidemiologic, genetic, and molecular pathologic studies support a role for oxidative stress in AMD. Previously, oxidative stress through light exposure has been linked with AMD (Taylor, Munoz, et al., 1990; Taylor, West, et al., 1992). Compared with age-matched controls, patients with established AMD had high exposure to both blue (400–500 nm) and visible (400–700 nm) light over the preceding 20 years (p = 0.027), but no difference in light exposure at younger ages when compared to age-matched patients without AMD. These data suggest that high levels of exposure to blue and visible light late in life may be important in causing AMD (Taylor et al., 1990). These findings were supported in a follow up study (Taylor et al., 1992). The Age-related Eye Disease Study (AREDS) showed that anti-oxidant micronutrients reduced the progression of intermediate AMD (2001). Interestingly, the ARED study found that reduction in plasma glutathione and cysteine oxidation correlated with benefit from anti-oxidant treatment for intermediate AMD (Moriarty-Craige, Adkison, et al., 2005). Collectively, these epidemiologic studies, along with the studies on cigarette smoking, implicate oxidative stress in the mechanism of AMD.
Several genetic variations associated with AMD susceptibility provide ample evidence of a role for oxidative stress: the mitochondrial DNA polymorphism A4917G (Canter, Olson, et al., 2008), a polymorphism in superoxide dismutase 2 (Kimura, Isashiki, et al., 2000), and a susceptibility locus in or near the hypothetical LOC387715 gene (Jakobsdottir, Conley, et al., 2005; Rivera, Fisher, et al., 2005). This locus is associated with smoking, and the combination of the LOC387715 polymorphism and smoking confers a higher risk for AMD than either factor alone (Schmidt, Hauser, et al., 2006). Kanda, Chen et al. (2007) have indicated that this locus encodes a mitochondrial protein, raising suspicion for a role of the oxidative defense response in this disease. However,Wang, Spencer, et al. (2009) found that ARMS2 was located in the cytosol and not the mitochondrial membrane, as previously reported.
Oxidative damage is seen in all layers of the fundus in AMD. Importantly, these changes indicate that oxidative damage is an important factor in the mechanism of disease development. In the retina, docosohexanoic acid (DHA), the most abundant fatty acid in photoreceptor tips, is oxidatively modified to carboxyethylpyrrole (CEP) (Gu, Meer, et al., 2003). Other oxidized lipids have been identified which “tag” oxidatively damaged photoreceptors in AMD (Sun, Finnemann, et al., 2005). In the RPE, multiple proteins isolated from lipofuscin are oxidatively damaged including malondialdehyde, 4-hydroxynonenal, and AGE modifications (Schutt, Bergmann, et al., 2003; Schutt, Ueberle, et al., 2002). Using proteomic analysis, Schutt et al. (2002) identified 81 proteins from lipofuscin extracted from human RPE cells. Of these proteins, 32 had malondialdehyde, 15 had 4-hydroxynonenal, and 4 had advanced glycation endproduct modifications, which are all indicators of oxidative damage (Schutt et al., 2003). Our lab and others have shown that AGEs accumulate in Bruch membrane including basal deposits and drusen, and CEP adducts appear in drusen isolated from AMD samples (Crabb, Miyagi, et al., 2002; Farboud, Aotaki-Keen, et al., 1999). These molecular modifications are the same as those typically caused by chemical oxidants in cigarette smoke, as outlined above. The ability to defend against oxidative stress by upregulating the anti-oxidant defense response is likely to be a pivotal event that mediates the initiation and progression of AMD. The molecular damage from oxidative modification illustrated here, suggests that the anti-oxidant response in the macula at some point, becomes unable to neutralize oxidative stress.
6. Oxidative stress augments inflammation
In addition to direct oxidative damage to tissue, oxidative free radicals modulate the immune-inflammatory system in part, through enhanced expression of pro-inflammatory genes, as reviewed in Biswas and Rahman (2009). Inflammation in turn, enhances oxidative stress. For example, in emphysema, increased TNF-a and TGF-b induces decreased glutathione synthesis and cellular glutathione, raising the cell’s susceptibility to oxidative damage (Bakin, Stourman, et al., 2005; de Boer, van Schadewijk, et al., 1998; Jardine, MacNee, et al., 2002; Rahman, Antonicelli, et al., 1999). Inflammation therefore, creates a self-perpetuating amplification loop of oxidative stress which in turn, activates a self-perpetuating amplification loop of inflammation. This exaggerated response can induce tissue damage (Rahman, 2005). Neutralizing oxidative stress, therefore, can interrupt this self-perpetuating loop and reduce tissue injury. The discovery of polymorphisms in several complement factors with AMD susceptibility points toward a specific role for complement mediated inflammation in the pathophysiology of AMD (Edwards, Ritter Iii, et al., 2005; Gold, Merriam, et al., 2006; Hageman, Anderson, et al., 2005; Haines, Hauser, et al., 2005; Hughes, Orr, et al., 2006; Klein, Zeiss, et al., 2005; Zareparsi, Branham, et al., 2005). It is unclear at the present, whether oxidative stress can specifically augment complement activation.
7. Brief overview of the anti-oxidant system
Cells are protected from oxidative stress by an elaborate system. Reactive oxygen species (ROS) are generated as by-products of metabolism. In excess, ROS can damage cellular proteins, lipids, and DNA. To limit injury, the cell has evolved enzymatic and small molecular anti-oxidants which can be categorized into two consecutive reactions. Phase I reactions are mediated by the cytochrome P450 mono-oxygenase system which oxidizes and reduces compounds. The Phase II enzymes conjugate Phase I products with hydrophilic molecules such as glutathione. The Phase II system can be divided into “Direct” and “Indirect” anti-oxidants. “Direct” anti-oxidants include superoxide dismutases, for example, because they quench free radicals. The most important small molecules include the thiol containing glutathione and thioredoxin. Both are easily oxidized and rapidly regenerated. Glutathione is the most abundant and effective quencher. “Indirect” enzymes participate in the biosynthesis and recycling of glutathione and thioredoxin, or facilitate removal of oxidized metabolites.
8. The Nrf2 Signaling system
Much of the protective anti-oxidant response is mediated through upregulation and activation of Nuclear factor erythroid-2 related factor 2 (Nrf2), a basic leucine zipper transcription factor. Nrf2 regulates a coordinated transcriptional program that maintains cellular redox homeostasis and protects the cell from oxidative injury (Nguyen, Sherratt, et al., 2003; Rangasamy, et al., 2004; Thimmulappa, Mai, et al., 2002). Other transcription factors such as Nf-κB and AP1, can be induced by oxidative stress, but they do not regulate as comprehensive an anti-oxidant response as Nrf2 (Biswas & Rahman, 2009). Nrf2 activates transcription by specific binding to the anti-oxidant response element (ARE) in the promoters of its target genes (Fig. 4). Nrf2 is normally sequestered in the cytosol by interacting with Kelch-like ECH-associated protein 1 (Keap1), which facilitates proteolysis of Nrf2 by the ubiquitin-proteosome pathway (Yu & Kensler, 2005). Keap1 also functions as a substrate adaptor protein for a Cul3-dependent E3 ubiquitin ligase complex which also helps to maintain steady-state levels of Nrf2 (Adams, Kelso, et al., 2000; Kobayashi, Kang, et al., 2004; Zhang, Lo, et al., 2004). In the absence of stress, Keap1 constitutively suppresses Nrf2 signaling by preventing Nrf2 translocation to the nucleus. Upon exposure to ROS, Keap1 undergoes a conformational change when its multiple cysteine residues interact with ROS, which releases Nrf2, and inhibits Keap1-mediated proteasomal degradation of Nrf2. The released Nrf2 then translocates to the nucleus, where it dimerizes with Maf proteins, and binds to the ARE to initiate transcription of target genes (Dinkova-Kostova, Holtzclaw, et al., 2005; Kobayashi & Yamamoto, 2005; Wakabayashi, Itoh, et al., 2003).
Fig. 4.

Diagram of Nrf2 signaling response. Without oxidative stress, Keap1 keeps Nrf2 sequestered in the cytoplasm. Upon oxidative stress (ROS), Nrf2 translocates to the nucleus where it binds to the anti-oxidant response elements (ARE) along with MAF proteins to activate transcription of cytoprotective enyzymes.
Nrf2 regulates a number of enzymes that are important in the anti-oxidant response (Osburn, Yates, et al., 2008). These include “direct” response enzymes such as catalase or superoxide dismutase, “indirect” enzymes such as heme oxygenase-1, glutathione and thioredoxin generating enzymes including the regulatory and catalytic subunits of glutamate-cysteine ligase (GCLM, GCLC), the rate limiting step in glutathione biosynthesis, and xenobiotic metabolism enzymes that produce reducing equivalents, such as NADPH quinine oxidoreductase (NQO-1).
The baseline expression of many anti-oxidant genes are not appreciably regulated by Nrf2 (Thimmulappa et al., 2002). This finding emphasizes that the main function of Nrf2 signaling is to induce the anti-oxidant response. When evaluating the Nrf2 signaling response, one must keep in mind, the early acute phase through actions of the “direct” enzymes, and a chronic phase through maintenance of cellular glutathione and thioredoxin. When ROS depletes cellular glutathione, cells die from oxidatively mediated apoptosis (Rahman, Biswas, et al., 2005; Walsh, Michaud, et al., 1995; Will, Mahler, et al., 1999). Thus, Nrf2 is an important mechanistic link between managing the anti-oxidant gene expression stress response and cell survival.
9. Evidence for anti-oxidant protection by Nrf2 in the RPE
The RPE has a formidable anti-oxidant defense system that must respond to its high oxidative stress environment. Several important observations suggest that Nrf2 signaling in the RPE is a central component of this response. Glutathione synthesis in the RPE is mediated through Nrf2 signaling and protects against photo-oxidation (Gao & Talalay, 2004). Acute light toxicity is reduced by inducing thioredoxin through Nrf2 signaling in the retina and RPE of mice in vivo (Tanito, Masutani, et al., 2005). Sulforaphane, an Nrf2 activator, protects RPE cells from oxidative injury (Gao & Talalay, 2004; Nelson, Armstrong, et al., 2002; Nelson, Carlson, et al., 1999; Tanito et al., 2005). Oxidant induced apoptosis by RPE cells in vitro is prevented by augmenting cellular glutathione levels through upregulation of two Nrf2 activated genes, glutathione S-transferase and NADPH-quinone reductase (Nelson et al., 2002, 1999). Zinc increases RPE cellular glutathione through induction of the de novo synthesis pathway, which is mediated by Nrf2 signaling (Ha, Chen, et al., 2006).
10. Nrf2 signaling protects against a dysregulated innate immune response including complement
Besides protecting against oxidative stress, Nrf2 also prevents dysregulation of the innate immune response. The most striking example is in septic shock. Members of our group showed that Nrf2 is a critical host factor that determined survival from septic shock by regulating an appropriate innate immune response (Thimmulappa, Lee, et al., 2006). Disrupting Nrf2 signaling increased the death rate by augmenting inflammation. In contrast, Nrf2 signaling maintained cellular glutathione by increasing phase II enzymes involved in the synthesis and maintenance of glutathione.
Recently, we observed a similar response in a murine model of uveitis by giving mice an intraperitoneal injection of lipopolysaccharide (LPS) (Nagai, Thimmulappa, et al., 2009). With Dihydroethidium staining, LPS increased ROS in the retina and iris-ciliary body of wild-type C57Bl6 mice (Nrf2+/+) and mice deficient for Nrf2 (Nrf2−/−). In Nrf2−/− mice, exon V, the DNA binding and dimerization domain, has been exchanged for an NLS (nuclear localization signal)-LacZ (coding β-galactosidase) sequence. After LPS injection, the expression of inflammatory markers in uveitis such as ICAM-1, IL-6, TNF-α, COX-2, iNOS, and MCP-1, were increased more in the retina and iris-ciliary body of Nrf2−/− than Nrf2+/+ mice. NQO-1 and GCLM, two Nrf2 responsive anti-oxidant enzymes, had reduced expression in Nrf2+/+ retinas after LPS injection, but no change in expression in Nrf2−/− mice. The number of FITC-con A labeled leukocytes adherent to the retinal vascular endothelium increased after LPS treatment in both Nrf2+/+ and Nrf2−/− mice compared to control injections, with the more adherent leukocytes in Nrf2−/− than Nrf2+/+ mice. These results suggest that Nrf2 signaling appears to reduce the inflammatory response at a crucial stage during acute inflammation in the eye. Importantly, deficient Nrf2 signaling predisposes to the development of chronic inflammation. For example, while the expression of pro-inflammatory cytokines is reduced at an early stage of repair, deficient Nrf2 signaling at a later stage prolongs the inflammatory response (Braun, Hanselmann, et al., 2002). With the interactions between inflammation, oxidative stress, and management by the Nrf2 signaling system, further investigation into the role that Nrf2 has on regulation of the emerging role for the immune response appears to be an important and worthwhile direction.
Nrf2 signaling also regulates complement activation. In elderly mice, Nrf2 deficiency increases oxidative damage and complement C3 deposition in several organs including the brain (Li, Stein, et al., 2004). On the other hand, Nrf2 activation prevents inflammation mediated oxidative stress. In a model of liver necrosis, Nrf2 activation by synthetic triterpenoids, which are small molecules that activate Nrf2, prevented late phase pro-inflammatory gene expression, and halted the inflammatory amplification loop (Osburn et al., 2008). In initial studies on mice exposed to cigarette smoking, we found immunohistochemical evidence of C3, C5, and MAC complement deposition in Bruch membrane of mice exposed to smoke while mice raised in air showed no immunolabeling. Complement factor H, a major regulator of C3, also showed mild labeling. (Fig. 5) (Wang, Lukas, et al., 2009). Given the emerging importance of the innate immune response and complement in AMD, the additional regulatory role of Nrf2 on the innate immune response suggests its potential protective role is an important factor in the complex changes that occur during the development of AMD.
Fig. 5.

Immunolocalization of complement pathway components in mice exposed to chronic cigarette smoke. These confocal immunofluorescence images were overlaid with the bright field images. (Top left panel) Immunoreactivity to C3a is observed in the area of Bruch’s membrane of mice exposed to chronic cigarette smoke. (Second left panel) Immunoreactivity to C5 is observed in the area of Bruch’s membrane of mice exposed to chronic cigarette smoke. (Third left panel) Immunoreactivity to C5b-9 is observed in the area of Bruch’s membrane of mice exposed to chronic cigarette smoke. (Bottom left panel) Immunoreactivity to CFH is observed in the area of Bruch’s membrane of mice exposed to chronic cigarette smoke. (Right panels) Immunoreactivity to C3a, C5, C5b-9 and CFH is not seen in mice raised in air. Scale bar = 20 mm. Blue: DAPI.
11. What is the anti-oxidant response in the aging fundus?
Multiple studies have investigated what happens to the antioxidant response during chronological aging, to investigate the hypothesis that the anti-oxidant response becomes insufficient with aging. Most of the studies have focused on important, but individual components of the anti-oxidant system, and have evaluated mRNA, protein and enzymatic activity levels. De La Paz, Zhang, et al. (1996) evaluated the effect of age on protective anti-oxidant enzyme activity of normal fresh cadaver human retina of the macula and periphery from 7 to 85 years. Anti-oxidant enzymes assayed included superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase. Interindividual variability was high, and variability increased with age. The difference between the macular and peripheral enzyme activities for glutathione peroxidase tended to decline with increasing donor age (p = 0.025, R2 = 0.33). There was no effect of age on the specific activities of catalase, glucose-6-phosphate dehydrogenase, and glutathione reductase. It appears that age does not have a significant effect on the activity of major anti-oxidant enzymes of the macula in normal human retina in part, due to the high interindividual variability of anti-oxidant enzyme activity. On the other hand, Liles, Newsome, et al. (1991) found that catalase activity decreased with age (p < 0.02) and AMD (p < 0.05) in both macular and peripheral RPE, but that superoxide dismutase activity showed no changes with aging or AMD.
Using a different strategy, Frank, Amin, et al. (1999) showed by quantitative electron microscopic immunocytochemistry that heme oxygenase 1 and 2, and catalase decreased while copper-zinc superoxide dismutase increased with age in macular RPE with aging and neovascular AMD specimens. We also looked at the mRNA and protein distribution of selected anti-oxidant enzymes in the RPE of specimens 27–87 years old (Miyamura, Ogawa, et al., 2004). Using in situ hybridization and immunohistochemistry, we found no definitive age-related decline in HO-1 and catalase in the macular and peripheral RPE. Perhaps the most striking finding was the mosaic expression pattern of these enzymes in the RPE monolayer.
In view of these mixed results, our laboratory attempted a broad based investigation into aging of the macula using microarray analysis of laser capture microdissected RPE cells. We found that the overall expression profiles of macular and peripheral RPE cells from elderly donors were similar, and that patient genotype separated profiles more than topographical location (Ishibashi, Tian, et al., 2004). Of eleven genes were found to be under expressed by macular RPE, glutathione S-transferase M1 (GSTM1), was an anti-oxidant gene of interest. Patients with the GSTM1 null genotype, which is seen in 50% of the white population, do not neutralize photo-oxidative stress, and are highly susceptible to solar keratosis or at increased risk for atherosclerosis from an impaired ability to detoxify tobacco smoke (Carless, Lea, et al., 2002; de Waart, Kok, et al., 2001; Rebbeck, 1997). GSTM1 activity, whether from genetic susceptibility or age-related decline, therefore, could make macular RPE susceptible over time, to either tobacco related or photo-oxidative stress. Importantly, the high throughput approach suggested that a significant, global decline in the anti-oxidant system is lacking in the aging macula without AMD.
Nordgaard, Berg, et al. (2006) showed with proteomic analysis that the RPE from eyes with progressive stages of AMD display proteins from different critical pathways. In early AMD eyes, identified proteins have functions related to protecting the cell from stress-induced protein unfolding and aggregation, mitochondrial trafficking and protein refolding, and regulating apoptosis. This investigation provided the first solid evidence of decreased heat shock protein expression (HSP70, HSC70, HSP60, α-A crystallin) in early AMD. Heat shock proteins in general, are protein chaperones that prevent damage after oxidative stress from unfolded proteins. Since the HSP proteins that were identified are involved in the import and folding of mitochondria proteins, these changes may reflect impaired mitochondrial biogenesis or an inadequate stress response. Several proteins with anti-apoptotic function, such as α-A crystallin, VDAC1, HSP70, GST-π decreased linearly with AMD progression, suggesting that their decrease could promote RPE cell apoptosis. While the authors speculated that the proteins identified in early disease are causal to AMD development, they also recognized that the identified changes likely under-represented the complexity of molecular changes that evolve during disease development due to the stringent conditions that were necessary, and the relative insensitivity of proteomics to low abundant, but important proteins. The content of several anti-oxidant enzymes and specific proteins that facilitate refolding or degradation of oxidatively damaged proteins increased significantly in later stages of dry AMD. These proteins are involved in the primary (copper–zinc superoxide dismutase [CuZnSOD], manganese superoxide dismutase [MnSOD], and catalase) and secondary (heat shock protein [HSP] 27, HSP 90, and proteasome) defense against oxidative damage (Decanini, Nordgaard, et al., 2007). Overall, the results investigating these major anti-oxidant enzymes in the fundus have been mixed, which suggests instead, that the coordinated global anti-oxidant response becomes uncoupled or dysregulated rather than globally insufficient during aging.
12. The Nrf2 signaling response in aging and disease: a new avenue for investigation
While the anti-oxidant response may not globally decline per se, with aging, the Nrf2 signaling response to a stimulus such as cigarette smoking can decline with aging and disease. The impact of cigarette smoke induced oxidative stress may become magnified with aging. Suzuki, Betsuyaku, et al. (2008) showed an age-dependent response in Nrf2 signaling to cigarette smoking. The Nrf2 response from young patients was robust and not affected by smoking. On the other hand, Nrf2 mRNA was down-regulated in macrophages of old smokers compared with old nonsmokers. Importantly, oxidized glutathione and carbonylated albumin levels in bronchoalveolar fluid were inversely correlated with Nrf2 mRNA levels. These results were corroborated in mice, where aging suppressed the ability of Nrf2 and its target genes in alveolar macrophages, to respond to the stress of cigarette smoking (Suzuki, et al., 2008). The Nrf2 transcriptional and nuclear translocational response with aging, however, can be restored to normal by (R)-α-lipoic acid (Suh, Shenvi, et al., 2004). This restoration of Nrf2 after the cigarette smoking insult suggests that pharmacologic induction of Nrf2 is a target for treating age-related oxidative stress.
Nrf2 appears to be declined in some diseases. Nrf2 mRNA and protein, and Nrf2-dependent anti-oxidants are reduced, glutathione levels are reduced, and oxidative stress is increased in human emphysematous compared to normal lungs (Malhotra, Thimmulappa, et al., 2008; Suzuki et al., 2008). Mice deficient in Nrf2 develop emphysema through enhanced oxidative damage from cigarette smoke (Rangasamy et al., 2004). The impaired anti-oxidant response can be reversed by either knocking down Keap1 or increasing Nrf2 (Suzuki et al., 2008), again, suggesting that Nrf2 activation is a possible therapy for oxidative disease. The decline in Nrf2 signaling appears to be a critical, but understudied program in AMD. Given the genetic models at our disposal, and the emerging pharmacological agents (see below), testing the impact of cigarette smoking on AMD phenotype offers a view into the mechanism of this disease and a potential therapeutic target.
13. Preliminary studies that support Nrf2 signaling in mice exposed to cigarette smoke
We recently initiated several pilot studies to investigate whether Nrf2 signaling influences the phenotype of the fundus from chronic cigarette smoke exposure by evaluating mice deficient in Nrf2 signaling (Nrf2−/−). We exposed Nrf2−/− and Nrf2+/+ mice for 5 h/day, as described above, to either cigarette smoke or filtered air. We first evaluated the RPE/choroid for an Nrf2 signaling response by determining whether Nrf2 responsive anti-oxidant genes, NADPH quinine oxidoreductase (NQO-1) and Glutamate-cysteine ligase regulatory unit (GCLM), are induced by acute cigarette smoke exposure in wild type mice. Table 1 shows that GCLM and NQO-1 both are induced by cigarette smoke exposure in the RPE/choroid of Nrf2+/+ mice after one day, and more strongly after 21 days. On the other hand, as expected Nrf2−/− mice do not show any induction of these enzymes.
Table 1.
RT-qPCR analysis showing acute induction of Nrf2 responsive anti-oxidant genes (NQO-1 and GCLM) by the RPE of Nrf2+/+ mice (n = 4 each group) after 1 and 21 day exposure to cigarette smoke. Expression was normalized to β-actin.
| Mouse | Day | NQO-1 |
GCLM |
||
|---|---|---|---|---|---|
| Fold changea | p-value | Fold changea | p-value | ||
| Nrf2+/+ | 1 | 3 | 0.11 | 3.1 | 0.0003 |
| 21 | 2.9 | 0.04 | 1.7 | 0.04 | |
| Nrf2−/− | 1 | 1.4 | 0.19 | 1.6 | 0.17 |
| 21 | 1.6 | 0.27 | 0.42 | 0.48 | |
Ratio of CS/air.
We next evaluated whether there is evidence of oxidative damage in the fundus with chronic cigarette smoke exposure in Nrf2−/− mice. Using the same protocol as described above, Nrf2−/− mice exposed for 6 months in air showed a slight increase (9.5%; n = 3) in immunolabeling of RPE nuclei for 8-OHdG compared to Nrf2+/+ mice in air (0%; p < 0.001; n = 3). When Nrf2−/− mice were exposed to cigarette smoke for 6 months, 85% of RPE cell nuclei were labeled with anti-8-OHdG (p < 0.0001; n = 3). Far more sections per Nrf2−/− mouse exposed to smoke were needed for evaluation since the number of RPE cell nuclei for counting was reduced, presumably from increased cell death.
We subsequently evaluated whether there was evidence of ultrastructural injury to the RPE, Bruch membrane and choriocapillaris in Nrf2+/+ and Nrf2−/− mice exposed to cigarette smoke or air for 6 months. At this age, the ultrastructure of the RPE, Bruch membrane, and choriocapillaris in Nrf2+/+ mice raised in air (n = 3) are healthy (Fig. 6A). Bruch membrane maintained a pentalaminar structure composed of the RPE basement membrane, inner collagenous layer, middle elastic layer, outer collagenous layer, and choriocapillaris basement membrane. The choriocapillaris endothelium appeared healthy with fenestrations. We chose to evaluate RPE basolateral infoldings and cytoplasmic vacuoles as indicators of RPE cell degeneration because loss of basal infoldings is a marker of epithelial injury (Drueke, Hennessen, et al., 1990; Olsen, Burdick, et al., 1989; Olsen, Wassef, et al., 1986) and cytoplasmic vacuoles have been identified in RPE that overlie drusen deposits in AMD (Anderson et al., 2002). Like previously, Nrf2+/+ mice raised in cigarette smoke (n = 3) showed ultrastructural injury with loss of basolateral infoldings and cytoplasmic vacuole formation (Fig. 6B). The RPE of 8 mo old Nrf2−/− mice raised in air (n = 3) also appear relatively healthy, but displayed mild focal loss of normal basolateral infoldings (Fig. 6C). In contrast, Nrf2−/− mice raised in cigarette smoke (n = 3) exhibited profound ultrastructural injury to the RPE-Bruch membrane (Fig. 6D). The RPE basolateral infoldings are dilated and fewer in number, and contain large cytoplasmic vacuoles. These changes appear more severe than Nrf2+/+ mice exposed to smoke. While preliminary, our assessment is that the RPE demonstrated more injury than Bruch membrane or the choriocapillaris.
Fig. 6.

Transmission electron microscopy of the RPE-Bruch membrane-choriocapillaris of Nrf2+/+ (A and B) and Nrf2−/− mice (C and D) exposed to air or cigarette smoke. (A) 8 month old Nrf2+/+ mouse raised in air for 6 months has healthy RPE basolateral infoldings (BL), Bruch membrane (BrM), and choriocapillaris endothelium (CC). (B) An 8 month old Nrf2+/+ mouse that had been exposed to cigarette smoke for 6 months shows loss and dilation of basolateral infoldings, an intracytoplasmic vacuole (V), and an outer collagenous layer deposit (OCL). (C) Eight month old Nrf2−/− mouse that was raised in air for 6 months appears relatively healthy although shows small cytoplasmic vacuoles and mild abnormalities to basolateral infoldings. (D) Eight month old Nrf2−/− mouse exposed to 6 months of cigarette smoke showed marked cytoplasmic vacuoles, and loss of basolateral infoldings. Bar = 500 nm.
The main function of Nrf2 signaling is to induce an anti-oxidant response upon an oxidative stress, as the baseline expression of many anti-oxidant genes are not appreciably regulated by Nrf2 (Thimmulappa et al., 2002). If the anti-oxidant response from reactive oxygen species is insufficient, and ROS depletes cellular glutathione, cells die from oxidatively mediated apoptosis (Rahman et al., 2005; Walsh et al., 1995; Will et al., 1999). We therefore, evaluated whether RPE cell apoptosis is influenced by the Nrf2 signaling response after chronic cigarette smoke exposure. Nrf2−/− mice raised in air for 6 months (n = 3) did not show any RPE cell apoptosis by TUNEL labeling. On the other hand, Nrf2−/− mice exposed to cigarette smoke for 6 months (n = 3), had a 9% RPE cell apoptosis (p < 0.001). Like with the 8-OHdG immunolabeling evaluation, many more sections of RPE/choroid from Nrf2−/− mice exposed to cigarette smoke were needed to identify RPE nuclei than mice raised in air. The need to evaluate more sections suggested to us that many dead cell remnants had already been removed at the time we tested for apoptosis, and hence, in the Nrf2−/− mice, this value is an under-representation of the true degree of apoptosis. These results suggest that there is minimal oxidative stress leading to oxidative damage when mice are raised in air, but that with cigarette smoke exposure, there is significant oxidative DNA damage and increased RPE cell apoptosis.
14. Small molecule activators of Nrf2
As mentioned previously, several compounds have the ability to activate Nrf2 and induce a protective anti-oxidant response. The isothiosynate sulforaphane (1-isothiocyanato-(4R)-(methylsulfinil) butane), isolated from broccoli sprouts, is an Nrf2 activator. Sulforaphane sulfydryl groups interact with the cysteine thiols of Keap1 (Dinkova-Kostova, 2005), resulting in conformational changes that abolish the capacity of Keap1 to suppress Nrf2. The result is nuclear translocation of Nrf2, and in heterodimeric combination with small Maf transcription factors, Nrf2 binds to the ARE transcriptional site to activate cytoprotective gene expression (Kobayashi, 2006; Motohashi, 2004). Sulforaphane induces Nrf2 activation and an anti-oxidant response in both RPE cells and photoreceptors (Gao & Talalay, 2004; Kong, Tanito, et al., 2007; Nelson et al., 2002, 1999; Tanito et al., 2005; Wang, Chen, et al., 2008).
Triterpenoids are steroid-like molecules that occur widely in nature in hundreds of plant species, and have been widely used in Asian medicine. These small molecules are an intriguing potential therapy for AMD because they have both anti-oxidant and anti-inflammatory activity. Since the pharmacological activity of most naturally occurring triterpenoids is weak, their structure has been chemically modified in an attempt to enhance its potency. One triterpenoid, oleanolic acid, with chemical modification, can have its intrinsic anti-inflammatory activity enhanced many thousand-fold (Liby, Yore, et al., 2007). Three of the most useful derivatives synthesized (Liby et al., 2007) from oleanolic acid are 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO), and its methyl ester and imidazolide analogs (CDDO-Me and CDDO-Im). In an important series of studies, these compounds were found to be both strongly anti-inflammatory and exceptionally potent inducers of “Phase II” enzymes, which have robust anti-oxidative and cytoprotective activities (Dinkova-Kostova, Holtzclaw, & Kensler, 2005; Liby, Hock, et al., 2005).
Synthetic triterpenoids can dramatically curtail both oxidative and inflammatory stress in many different cell types (Dinkova-Kostova, Liby, et al., 2005; Liby et al., 2005; Thimmulappa, Scollick, et al., 2006; Yates, Tauchi, et al., 2007) in culture and in vivo (Thimmulappa et al., 2006; Yates et al., 2007). Moreover, the Nrf2 pathway is essential for mediating such cytoprotective activities, since this protection is lost in cells in which Nrf2 has been knocked out (Liby et al., 2005; Thimmulappa et al., 2006; Yates et al., 2007). The mechanism of action by the triterpenoids is mediated by their ability to form Michael adducts with cysteine residues on Keap1. Under basal conditions, Keap 1 forms a tight complex with Nrf2, but upon reaction with Michael reagents such as triterpenoids, Keap1 dissociates and allows Nrf2 translocation to the nucleus, where it upregulates the expression of the genes for Phase 2 enzymes. Importantly, triterpenoids have been shown to be safe in Phase I trial in patients with solid tumors and lymphoid malignancies (Hong D. et al. AACR-NCI-EORTC International Conference, page 188, 2008). Triterpenoids are substantially more potent activators of Nrf2 than sulforaphane or (R)-α-lipoic acid (Yates et al., 2007).
We have preliminary indications that triterpenoids penetrate the blood ocular barrier and show anti-oxidant activity with evidence of improvement in both the uveitis and AMD models. We chose CDDO-Im for initial study because it has demonstrated efficacy in the mouse (Thimmulappa et al., 2006; Yates et al., 2007). In mice given an LPS stimulus to simulate uveitis (Nagai et al., 2009), Nrf2+/+ mice treated with CDDO-Im had increased expression of GCLM and HO-1 in the retina, and NQO-1 and HO-1 in the iris-ciliary body of Nrf2+/+ mice compared to vehicle controls (See Fig. 7 (Nagai et al., 2009); *p < 0.05, **p < 0.01). Nrf2−/2 mice stimulated with LPS and treated with either CDDO-Im or vehicle showed no difference in the expression of HO-1, GLCM, and NQO-1 in Nrf2−/− mice (data not shown). Pretreatment with CDDO-Im also reduced inflammatory mediator expression, and reduced leukocyte adherence to retinal vasculature after LPS treatment in Nrf2+/+ mice, but had no effect on Nrf2−/− mice (Fig. 8, Nagai et al., 2009).
Fig. 7.
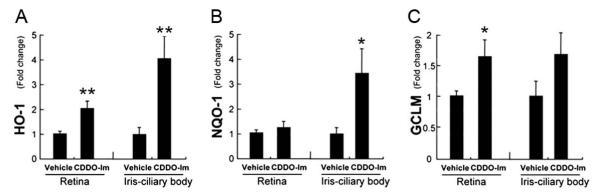
CDDO-Im increases the expression of anti-oxidant enzymes. Nrf2+/+ mice were given CDDO-Im (3 μmol/kg bodyweight; n = 5) or vehicle control (n = 5) once daily for 3 days, and the expression of anti-oxidant enzymes (HO-1 (A), NQO-1 (B) and GCLM (C)) was evaluated by RT-qPCR in the retina and iris-ciliary body. *p < 0.05, **p < 0.01.
Fig. 8.

CDDO-Im decreases leukocyte adherence to retinal vasculature. (A) Flat mounted retinas of Nrf2+/+ mice show FITC-Con A labeled leukocytes that were adherent to the retinal vasculature. Bar = 100 μm. (B) The graph depicts the percent of adherent leukocytes after pretreatment with CDDO-IM to vehicle control for both LPS stimulated Nrf2−/− and Nrf2+/+ mice (n = 5 in each group). The number of adherent leukocytes from Nrf2+/+ mice pretreated with CDDO-Im is decreased compared to vehicle control eyes (*p < 0.01). On the other hand, the number of adherent leukocytes in retinas of Nrf2−/− mice was not different between CDDOIm and vehicle control. The number of adherent leukocytes from CDDO-Im to vehicle control treatment was significantly different between Nrf2+/+ and Nrf2−/− mice (**p < 0.001).
To demonstrate the potential benefit of triterpenoids in early AMD, we were able to examine the eyes of Nrf2+/+ mice that were given CDDO-Im in the diet 3x/week as described above, and exposed to air (n = 3) or CS (n = 3) for 6 months. Fig. 9 shows the RPE of 2 month old mice exposed to CS or air for 6 months. Panel A shows an Nrf2+/+ mouse raised in air had normal ultrastructure of the RPE including healthy basolateral infoldings, and lack of cytoplasmic vacuoles. Bruch membrane is non-thickened and regular in appearance. Panel B shows an Nrf2+/+ mouse raised in air and given CDDO-Im that is also healthy appearing. Panel C shows an Nrf2+/+ mouse raised in cigarette smoke for 6 months and given vehicle control. The RPE infoldings are shortened and fewer in number. Cytoplasmic vacuoles are present. Panel D shows an Nrf2+/+ mouse treated with CDDO-Im and CS for 6 months. The RPE and Bruch membrane appear similar to mice raised in air. These data demonstrate the potential benefit of activating Nrf2 with CDDO-Im on reversing cellular injury to RPE cells in vivo.
Fig. 9.

Transmission electron microscopy of Nrf2+/+ treated with CDDO-Im. (A) Nrf2+/+ mouse raised in air given vehicle had normal ultrastructure of the RPE including healthy basolateral infoldings (BL), mitochondria, and lack of cytoplasmic vacuoles. Bruch membrane (BrM) is non-thickened and the choriocapillaris endothelium (CC) is healthy. (B) Nrf2+/+ mouse raised in air for 6 months and given vehicle control appears healthy. (C) Nrf2+/+ mouse exposed to cigarette smoke for 6 months and given vehicle control demonstrates RPE basolateral infolding abnormalities and cytoplasmic vacuoles. (D) Nrf2+/+ mouse treated with CDDO-Im and exposed to cigarette smoke for 6 months appears healthy. Bar = 500 nm. Bar in (A) also represents scale for (B). Bar in (C) also represents scale for (D).
While these pilot studies are initially promising, many questions remain unanswered. Does the Nrf2 signaling response decrease with aging and/or cigarette smoke during the development of AMD? What are the anti-oxidant genes within the comprehensive anti-oxidant response that are most important to protecting the fundus from oxidative insult? What is the interaction of oxidative stress and complement activation in the fundus with AMD pathogenesis, and does Nrf2 play a modulating role? If the Nrf2 anti-oxidant response system protects the fundus from an oxidative stress and immune mediated AMD onset, can it be rescued by triterpenoids or other small molecule Nrf2 activators and reduce AMD severity? Addressing these questions may uncover additional mechanisms of AMD and provide a framework for effective preventive or treatment for early disease before vision loss.
Acknowledgments
This work is supported by Grants EY019904 (JTH), The Robert Bond Welch Professorship (JTH); NIH HL081205 (SB), NIH/NHLBI SCCOR Grant P50HL084945 (SB), Clinical Innovator award from FAMRI (SB); NIH Grant CA-78814 (MS), and a grant from Reata Pharmaceuticals (MS); and generous gifts from Ric and Sandy Forsythe, the Kwok family, the Merlau family, and Aleda Wright, and Research to Prevent Blindness to Wilmer Eye Institute, Forsythe Foundation (AHN), and unrestricted grants from Research to Prevent Blindness (Northwestern University).
References
- AREDS A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Archives of Ophthalmology. 2001;119(10):1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J, Kelso R, et al. The kelch repeat superfamily of proteins: Propellers of cell function. Trends in Cell Biology. 2000;10(1):17–24. doi: 10.1016/s0962-8924(99)01673-6. [DOI] [PubMed] [Google Scholar]
- Anderson DH, Mullins RF, et al. A role for local inflammation in the formation of drusen in the aging eye. American Journal of Ophthalmology. 2002;134(3):411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Stourman NV, et al. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding Phase II detoxifying proteins. Free Radical Biology and Medicine. 2005;38(3):375–387. doi: 10.1016/j.freeradbiomed.2004.10.033. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Rahman I. Environmental toxicity, redox signaling and lung inflammation: The role of glutathione. Molecular Aspects of Medicine. 2009;30(1–2):60–76. doi: 10.1016/j.mam.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Hanselmann C, et al. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Molecular and Cellular Biology. 2002;22(15):5492–5505. doi: 10.1128/MCB.22.15.5492-5505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canter JA, Olson LM, et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS One. 2008;3(5):e2091. doi: 10.1371/journal.pone.0002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carless MA, Lea RA, et al. The GSTM1 null genotype confers an increased risk for solar keratosis development in an Australian Caucasian population. Journal of Investigative Dermatology. 2002;119(6):1373–1378. doi: 10.1046/j.1523-1747.2002.19646.x. [DOI] [PubMed] [Google Scholar]
- Clemons TE, Milton RC, et al. Risk factors for the incidence of advanced age-related macular degeneration in the age-related eye disease study (AREDS) AREDS report no. 19. Ophthalmology. 2005;112(4):533–539. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon N, O’Colmain B, et al. Causes and prevalence of visual impairment among adults in the United States. Archives of Ophthalmology. 2004;122(4):477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proceedings of the National Academy of Sciences USA. 2002;99(23):14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross CE, O’Neill CA, et al. Cigarette smoke oxidation of human plasma constituents. Annals of the New York Academy of Sciences. 1993;686:72–89. doi: 10.1111/j.1749-6632.1993.tb39157.x. (discussion 89–90) [DOI] [PubMed] [Google Scholar]
- de Boer WI, van Schadewijk A, et al. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 1998;158(6):1951–1957. doi: 10.1164/ajrccm.158.6.9803053. [DOI] [PubMed] [Google Scholar]
- De La Paz MA, Zhang J, et al. Antioxidant enzymes of the human retina: Effect of age on enzyme activity of macula and periphery. Current Eye Research. 1996;15(3):273–278. doi: 10.3109/02713689609007621. [DOI] [PubMed] [Google Scholar]
- de Waart FG, Kok FJ, et al. Effect of glutathione S-transferase M1 genotype on progression of atherosclerosis in lifelong male smokers. Atherosclerosis. 2001;158(1):227–231. doi: 10.1016/s0021-9150(01)00420-8. [DOI] [PubMed] [Google Scholar]
- Decanini A, Nordgaard CL, et al. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. American Journal of Ophthalmology. 2007;143(4):607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Priore LV, Kuo YH, et al. Age-related changes in human RPE cell density and apoptosis proportion in situ. Investigative Ophthalmology and Visual Science. 2002;43(10):3312–3318. [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, et al. The role of Keap1 in cellular protective responses. Chemical Research in Toxicology. 2005;18(12):1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chemical Research in Toxicology. 2005;(18):1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Liby KT, et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proceedings of the National Academy of Sciences USA. 2005;102(12):4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drueke T, Hennessen U, et al. Ultrastructural and functional abnormalities of intestinal and renal epithelium in the SHR. Kidney International. 1990;37(6):1438–1448. doi: 10.1038/ki.1990.134. [DOI] [PubMed] [Google Scholar]
- Dunaief JL, Dentchev T, et al. The role of apoptosis in age-related macular degeneration. Archives of Ophthalmology. 2002;120(11):1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, Iii, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner IJ, et al. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Investigative Ophthalmology and Visual Science. 2006;47(2):729–737. doi: 10.1167/iovs.05-0719. [DOI] [PubMed] [Google Scholar]
- Farboud B, Aotaki-Keen A, et al. Development of a polyclonal antibody with broad epitope specificity for advanced glycation endproducts and localization of these epitopes in Bruch’s membrane of the aging eye. Molecular Vision. 1999;5:11. [PubMed] [Google Scholar]
- Frank RN, Amin RH, et al. Antioxidant enzymes in the macular retinal pigment epithelium of eyes with neovascular age-related macular degeneration. American Journal of Ophthalmology. 1999;127(6):694–709. doi: 10.1016/s0002-9394(99)00032-x. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O’Colmain BJ, et al. Prevalence of age-related macular degeneration in the United States. Archives of Ophthalmology. 2004;122(4):564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Fujihara M, Nagai N, et al. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS One. 2008;3(9):e3119. doi: 10.1371/journal.pone.0003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Talalay P. Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proceedings of the National Academy of Sciences USA. 2004;101(28):10446–10451. doi: 10.1073/pnas.0403886101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B, Merriam JE, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nature Genetics. 2006;38(4):458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WR, McDonnell PJ, et al. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92(5):615–627. [PubMed] [Google Scholar]
- Gu X, Meer SG, et al. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. Journal of Biological Chemistry. 2003;278(43):42027–42035. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- Ha KN, Chen Y, et al. Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: Implication for protection against oxidative stress. Investigative Ophthalmology and Visual Science. 2006;47(6):2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proceedings of the National Academy of Sciences USA. 2005;102(20):7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Harman AM, Fleming PA, et al. Development and aging of cell topography in the human retinal pigment epithelium. Investigative Ophthalmology and Visual Science. 1997;38(10):2016–2026. [PubMed] [Google Scholar]
- Hughes AE, Orr N, et al. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nature Genetics. 2006;38(10):1173–1177. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- Ishibashi K, Tian J, et al. Similarity of mRNA phenotypes of morphologically normal macular and peripheral retinal pigment epithelial cells in older human eyes. Investigative Ophthalmology and Visual Science. 2004;45(9):3291–3301. doi: 10.1167/iovs.04-0168. [DOI] [PubMed] [Google Scholar]
- Jakobsdottir J, Conley YP, et al. Susceptibility genes for age-related maculopathy on chromosome 10q26. American Journal of Human Genetics. 2005;77(3):389–407. doi: 10.1086/444437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine H, MacNee W, et al. Molecular mechanism of transforming growth factor (TGF)-beta1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP-1/ARE and Fra-1. Journal of Biological Chemistry. 2002;277(24):21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- Kanda A, Chen W, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proceedings of the National Academy of Sciences USA. 2007;104(41):16227–16232. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan JC, Thurlby DA, et al. Smoking and age related macular degeneration: The number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. British Journal of Ophthalmology. 2006;90(1):75–80. doi: 10.1136/bjo.2005.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Isashiki Y, et al. Genetic association of manganese superoxide dismutase with exudative age-related macular degeneration. American Journal of Ophthalmology. 2000;130(6):769–773. doi: 10.1016/s0002-9394(00)00552-3. [DOI] [PubMed] [Google Scholar]
- Klein R, Knudtson MD, et al. Further observations on the association between smoking and the long-term incidence and progression of age-related macular degeneration: The Beaver Dam Eye Study. Archives of Ophthalmology. 2008;126(1):115–121. doi: 10.1001/archopht.126.1.115. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Molecular and Cellular Biology. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2–Keap1 pathway of antioxidant gene regulation. Antioxidants Redox Signaling. 2005;7(3–4):385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Advances in Enzyme Regulation. 2006;(46):113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Kong L, Tanito M, et al. Delay of photoreceptor degeneration in tubby mouse by sulforaphane. Journal of Neurochemistry. 2007;101(4):1041–1052. doi: 10.1111/j.1471-4159.2007.04481.x. [DOI] [PubMed] [Google Scholar]
- Li J, Stein TD, et al. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiological Genomics. 2004;18(3):261–272. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- Liby K, Hock T, et al. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Research. 2005;65(11):4789–4798. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- Liby KT, Yore MM, et al. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nature Reviews Cancer. 2007;7(5):357–369. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- Liles MR, Newsome DA, et al. Antioxidant enzymes in the aging human retinal pigment epithelium. Archives of Ophthalmology. 1991;109(9):1285–1288. doi: 10.1001/archopht.1991.01080090111033. [DOI] [PubMed] [Google Scholar]
- Lykkesfeldt J, Christen S, et al. Ascorbate is depleted by smoking and repleted by moderate supplementation: A study in male smokers and nonsmokers with matched dietary antioxidant intakes. American Journal of Clinical Nutrition. 2000;71(2):530–536. doi: 10.1093/ajcn/71.2.530. [DOI] [PubMed] [Google Scholar]
- Malhotra D, Thimmulappa R, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. American Journal of Respiratory and Critical Care Medicine. 2008;178(6):592–604. doi: 10.1164/rccm.200803-380OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mitchell P, Wang JJ, et al. Smoking and the 5-year incidence of age-related maculopathy: The Blue Mountains Eye Study. Archives of Ophthalmology. 2002;120(10):1357–1363. doi: 10.1001/archopht.120.10.1357. [DOI] [PubMed] [Google Scholar]
- Miyamura N, Ogawa T, et al. Topographic and age-dependent expression of heme oxygenase-1 and catalase in the human retinal pigment epithelium. Investigative Ophthalmology and Visual Science. 2004;45(5):1562–1565. doi: 10.1167/iovs.02-0761. [DOI] [PubMed] [Google Scholar]
- Moriarty-Craige SE, Adkison J, et al. Antioxidant supplements prevent oxidation of cysteine/cystine redox in patients with age-related macular degeneration. American Journal of Ophthalmology. 2005;140(6):1020–1026. doi: 10.1016/j.ajo.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends in Molecular Medicine. 2004;8(10):549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Mukesh BN, Dimitrov PN, et al. Five-year incidence of age-related maculopathy: The Visual Impairment Project. Ophthalmology. 2004;111(6):1176–1182. doi: 10.1016/j.ophtha.2003.08.042. [DOI] [PubMed] [Google Scholar]
- Nagai N, Thimmulappa RK, et al. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radical Biology and Medicine. 2009 doi: 10.1016/j.freeradbiomed.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KC, Armstrong JS, et al. Protection of retinal pigment epithelial cells from oxidative damage by oltipraz, a cancer chemopreventive agent. Investigative Ophthalmology and Visual Science. 2002;43(11):3550–3554. [PubMed] [Google Scholar]
- Nelson KC, Carlson JL, et al. Effect of dietary inducer dimethylfumarate on glutathione in cultured human retinal pigment epithelial cells. Investigative Ophthalmology and Visual Science. 1999;40(9):1927–1935. [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, et al. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annual Review of Pharmacology and Toxicology. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Nordgaard CL, Berg KM, et al. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Investigative Ophthalmology and Visual Science. 2006;47(3):815–822. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- O’Neill CA, Halliwell B, et al. Aldehyde-induced protein modifications in human plasma: Protection by glutathione and dihydrolipoic acid. Journal of Laboratory and Clinical Medicine. 1994;124(3):359–370. [PubMed] [Google Scholar]
- Olsen S, Burdick JF, et al. Primary acute renal failure (acute tubular necrosisin the transplanted kidney: Morphology and pathogenesis. Medicine (Baltimore) 1989;68(3):173–187. doi: 10.1097/00005792-198905000-00005. [DOI] [PubMed] [Google Scholar]
- Olsen TS, Wassef NF, et al. Ultrastructure of the kidney in acute interstitial nephritis. Ultrastructural Pathology. 1986;10(1):1–16. doi: 10.3109/01913128609015558. [DOI] [PubMed] [Google Scholar]
- Osburn WO, Yates MS, et al. Genetic or pharmacologic amplification of Nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicological Sciences. 2008;104(1):218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I. Regulation of glutathione in inflammation and chronic lung diseases. Mutation Research. 2005;579(1–2):58–80. doi: 10.1016/j.mrfmmm.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Rahman I, Antonicelli F, et al. Molecular mechanism of the regulation of glutathione synthesis by tumor necrosis factor-alpha and dexamethasone in human alveolar epithelial cells. Journal of Biological Chemistry. 1999;274(8):5088–5096. doi: 10.1074/jbc.274.8.5088. [DOI] [PubMed] [Google Scholar]
- Rahman I, Biswas SK, et al. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxidants Redox Signaling. 2005;7(1–2):42–59. doi: 10.1089/ars.2005.7.42. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radical Biology and Medicine. 1996;21(5):669–681. doi: 10.1016/0891-5849(96)00155-4. [DOI] [PubMed] [Google Scholar]
- Rangasamy T, Cho CY, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. Journal of Clinical Investigation. 2004;114(9):1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck TR. Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiology, Biomarkers and Prevention. 1997;6(9):733–743. [PubMed] [Google Scholar]
- Rivera A, Fisher SA, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Human Molecular Genetics. 2005;14(21):3227–3236. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- Sarks SH. Ageing and degeneration in the macular region: A clinico-pathological study. British Journal of Ophthalmology. 1976;60(5):324–341. doi: 10.1136/bjo.60.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Hauser MA, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. American Journal of Human Genetics. 2006;78(5):852–864. doi: 10.1086/503822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutt F, Bergmann M, et al. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Investigative Ophthalmology and Visual Science. 2003;44(8):3663–3668. doi: 10.1167/iovs.03-0172. [DOI] [PubMed] [Google Scholar]
- Schutt F, Ueberle B, et al. Proteome analysis of lipofuscin in human retinal pigment epithelial cells. FEBS Letters. 2002;528(1–3):217–221. doi: 10.1016/s0014-5793(02)03312-4. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Hansch C. The relative toxicity of compounds in mainstream cigarette smoke condensate. Food and Chemical Toxicology. 2000;38(7):637–646. doi: 10.1016/s0278-6915(00)00051-x. [DOI] [PubMed] [Google Scholar]
- Smith W, Assink J, et al. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology. 2001;108(4):697–704. doi: 10.1016/s0161-6420(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Suh JH, Shenvi SV, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proceedings of the National Academy of Sciences USA. 2004;101(10):3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Finnemann SC, et al. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: A potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. Journal of Biological Chemistry. 2005 doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Betsuyaku T, et al. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology. 2008;39(6):673–682. doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- Tanito M, Masutani H, et al. Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Investigative Ophthalmology and Visual Science. 2005;46(3):979–987. doi: 10.1167/iovs.04-1120. [DOI] [PubMed] [Google Scholar]
- Taylor HR, Munoz B, et al. Visible light and risk of age-related macular degeneration. Transactions of the American Ophthalmological Society. 1990;88:163–173. (discussion 173–178) [PMC free article] [PubMed] [Google Scholar]
- Taylor HR, West S, et al. The long-term effects of visible light on the eye. Archives of Ophthalmology. 1992;110(1):99–104. doi: 10.1001/archopht.1992.01080130101035. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. Journal of Clinical Investigation. 2006;116(4):984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, et al. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Research. 2002;62(18):5196–5203. [PubMed] [Google Scholar]
- Thimmulappa RK, Scollick C, et al. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-imidazolide. Biochemical and Biophysical Research Communications. 2006;351(4):883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomany SC, Wang JJ, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111(7):1280–1287. doi: 10.1016/j.ophtha.2003.11.010. [DOI] [PubMed] [Google Scholar]
- van der Schaft TL, Mooy CM, et al. Histologic features of the early stages of age-related macular degeneration. A statistical analysis. Ophthalmology. 1992;99(2):278–286. doi: 10.1016/s0161-6420(92)31982-7. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Itoh K, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nature Genetics. 2003;35(3):238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- Walsh AC, Michaud SG, et al. Glutathione depletion in human T lymphocytes: Analysis of activation-associated gene expression and the stress response. Toxicology and Applied Pharmacology. 1995;133(2):249–261. doi: 10.1006/taap.1995.1149. [DOI] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, et al. Changes in retinal pigment epithelium related to cigarette smoke: Possible relevance to smoking as a risk factor for age-related macular degeneration. PLoS ONE. 2009;4(4):e5304. doi: 10.1371/journal.pone.0005304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Spencer KL, et al. Localization of age-related macular degeneration-associated ARMS2 in cytosol, not mitochondria. Investigative Ophthalmology and Visual Science. 2009;50(7):3084–3090. doi: 10.1167/iovs.08-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chen Y, et al. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Investigative Ophthalmology and Visual Science. 2008;49(4):1671–1678. doi: 10.1167/iovs.07-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will O, Mahler HC, et al. Influence of glutathione levels and heat-shock on the steady-state levels of oxidative DNA base modifications in mammalian cells. Carcinogenesis. 1999;20(2):333–337. doi: 10.1093/carcin/20.2.333. [DOI] [PubMed] [Google Scholar]
- Yates MS, Tauchi M, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Molecular Cancer Therapeutics. 2007;6(1):154–162. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutation Research. 2005;591(1–2):93–102. doi: 10.1016/j.mrfmmm.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Zareparsi S, Branham KE, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. American Journal of Human Genetics. 2005;77(1):149–153. doi: 10.1086/431426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, et al. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Molecular and Cellular Biology. 2004;24(24):10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]