

Abstract
Ischemia–reperfusion is a major component of injury in vascular occlusion both during liver surgery and during liver transplantation. The pathophysiology of hepatic ischemia–reperfusion includes a number of mechanisms including oxidant stress that contribute to various degrees to the overall organ damage. A large volume of recent research has focused on the use of antioxidants to ameliorate this injury, although results in experimental models have not translated well to the clinic. This review focuses on critical sources and mediators of oxidative stress during hepatic ischemia–reperfusion, the status of current antioxidant interventions, and emerging mechanisms of protection by preconditioning. While recent advances in regulation of antioxidant systems by Nrf2 provide interesting new potential therapeutic targets, an increased focus must be placed on more in-depth mechanistic investigations in hepatic ischemia–reperfusion injury and translational research in order to refine current strategies in disease management.
1. Introduction
Formation of reactive oxygen species (ROS) and oxidant stress are the most invoked disease mechanisms in ischemia–reperfusion injury including hepatic ischemia–reperfusion. However, despite its postulated universal role in the pathogenesis, no drug targeting ROS is in clinical use. The main reason for this discrepancy is the general lack of depth as these mechanisms are considered. In order to successfully target ROS, it is necessary to consider the specific ROS involved, the sources generating ROS in what particular location, at what time in the pathogenesis, and how much oxidant stress is generated. In addition, it is critical to understand by what mechanism ROS are actually causing cell death and organ injury and how relevant the experimental models are for the human conditions. The current review focuses on these different aspects of the pathophysiology and the attempts targeting ROS as strategy to limit reperfusion injury.
2. Sources of postischemic oxidant stress
2.1. Xanthine oxidase
Early investigations on the mechanisms of hepatic ischemia–reperfusion injury focused on intracellular sources of ROS formation, especially xanthine oxidase [1,2]. Even more recently, xanthine oxidase was invoked as a critical source of ROS [3]. This hypothesis was mainly based on indirect evidence for oxidant stress and the protective effect of the xanthine oxidase inhibitor allopurinol [1]. Attempts to directly provide evidence for an intracellular oxidant stress during reperfusion mainly failed [4,5]. In fact, it requires quite extensive ischemic damage in order to find a relevant but still temporary oxidant stress by xanthine oxidase [6,7]. There are multiple reasons for the limited importance of xanthine oxidase under clinically relevant conditions. First, the enzyme exists as xanthine dehydrogenase and the conversion to the ROS-generating oxidase requires lengthy ischemic times [8]. In addition, the ROS formation depends on the substrates xanthine and hypoxanthine. Although these substrates accumulate during ischemia, they are relatively fast metabolized and also flushed out together with other metabolites such as lactate, pyruvate and glucose from the liver during reperfusion [5,6]. Because it requires a substantial and continuous intracellular oxidant stress to cause any liver injury [9], not just a short-term minor ROS formation, xanthine oxidase cannot be a relevant source of the postischemic oxidant stress. Unfortunately, the repeated protection by allopurinol has revived xanthine oxidase several times during the last 30 years. However, allopurinol is only protective if animals are pretreated multiple times with high doses (≥50 mg/kg) [1,10]. Because a single dose of allopurinol (<10 mg/kg) is completely effective in inhibiting xanthine oxidase [11], the high doses used in ischemia–reperfusion injury have off-target effects, which may actually be responsible for the protection. Although the mechanism remains unclear, allopurinol appears to have protective effects against mitochondrial injury [10–12].
2.2. Mitochondria
The function of the mitochondrial electron transport chain involves a number of redox reactions with the transfer of 4 electrons to molecular oxygen, leading ultimately to the formation of water. However, even under physiological conditions, there is a constant, albeit low level leak of electrons from the electron transport chain resulting in the formation of superoxide [13,14]. Under stress conditions, mitochondrial dysfunction may impair the electron flow and enhance superoxide formation. However, in order to trigger a mitochondrial oxidant stress in the liver, it requires prolonged ischemia times [7], which are generally avoided during liver surgery or transplantation. In addition, mitochondria contain Mn-superoxide dismutase (MnSOD), glutathione and glutathione peroxidase, thioredoxin-2, and glutaredoxin, all of which can effectively detoxify ROS in the mitochondria or help repair minor damage [15]. In addition, removal of dysfunctional mitochondria by autophagy can limit the potential negative impact on the cells [16]. In fact, the beneficial effect of activation of autophagy processes has been shown during ischemia–reperfusion injury [17,18]. In assessing the importance of mitochondrial oxidant stress during hepatic ischemia–reperfusion injury, it has to be kept in mind that mitochondria can be a primary target during the very early injury phase, but mitochondria can also be a target of the inflammatory response at later times.
2.3. Kupffer cells
More detailed investigations of ROS formation in vivo indicated that the main oxidant stress after moderate ischemia occurs in the extracellular space of the liver [19]. This conclusion was based on the observation that glutathione is not oxidized intracellularly but in the vascular space [19,20]. Because at the very early time of reperfusion (≤1 h), very few infiltrating leukocytes (neutrophils) were detected, the resident macrophages of the liver (Kupffer cells) became the focus of attention [20–22]. Glutathione disulfide formation in plasma correlated with the activation or inactivation of Kupffer cells and isolation of Kupffer cells during reperfusion confirmed the activation of Kupffer cells, suggesting that these macrophages are the main source of a vascular oxidant stress during the early reperfusion phase after warm [20,22] and cold ischemia [23]. Although the ischemic stress can trigger an oxidant burst in macrophages during reoxygenation [24], the prolonged ROS formation during the early phase of reperfusion in vivo is, at least in part, triggered by activated complement fragments [25]. The critical point is that inhibiting the capacity of Kupffer cells to generate ROS using gadolinium chloride [26] or complement depletion [25] effectively attenuated reperfusion injury. Recent data provided evidence for Kupffer cell activation by damage-associated molecular patterns (DAMPs) such as high-mobility group box-1 protein, which are released during reperfusion [27]. However, DAMPs act mainly through toll-like receptors 4 (tlr4) and others to generate cytokines [27]. However, tlr4 substrates such as endotoxin do not directly trigger ROS formation by Kupffer cells [28]; instead, high doses of endotoxin activate the complement cascade, and activated complement factors induce a Kupffer cell-mediated oxidant stress [29]. Although specific inactivation of Kupffer cells by gadolinium chloride attenuates ROS formation during ischemia–reperfusion [20,26], Kupffer cells are not a relevant target for therapeutic interventions because of their vital host defense function in the hepatic vasculature.
2.4. Polymorphonuclear leukocytes (neutrophils)
Another cell type that is equipped to specifically generate ROS is the neutrophil. NADPH oxidase, specifically NOX2, is the main source of superoxide formation by neutrophils [30]. Neutrophil recruitment into the liver starts during the first few hours after initiation of reperfusion [20,31]. However, these neutrophils are only primed for enhanced ROS formation at that time [22]. A neutrophil-mediated oxidant stress actually is observed at 6 –24 h of reperfusion [32,33] and correlates with a neutrophil-mediated injury phase [31]. Although controversial for some time due to some early hepatocyte–neutrophil coculture experiments [34,35], which did not accurately reflect the in vivo conditions [36], there is now solid evidence that neutrophils kill by ROS in vivo [37,38]. The main cytotoxic ROS species, which are generated during a neutrophil attack and actually end up inside a hepatocyte, are hydrogen peroxide [37] and hypochlorous acid [33,38,39]. This primary intracellular oxidant stress in hepatocytes triggered by neutrophil-derived oxidants leads to a secondary oxidant stress and propagation of the original insult by mitochondria [40]. Thus, similar to Kupffer cells, the ROS are generated outside and diffuse into the hepatocyte. However, these ROS are unable to kill the cell directly but create a severe enough disturbance of the cellular homeostasis to trigger mitochondrial dysfunction and oxidant stress and eventually the MPT, which kills the cell [41,42].
A critical receptor for neutrophil activation and oxidant stress is Mac-1 (CD11b/CD18) [43,44]. Interventions directed against CD11b or CD18 effectively suppress a neutrophil-induced oxidant stress and liver injury during ischemia–reperfusion and other conditions [26,39,45,46]. Alternatively, direct inhibition of NOX-2 is effective in reducing the oxidant stress and liver injury [38,47,48]. Neutrophil priming and activation for ROS formation involve complement factors [25], damage-associated molecular patterns such as HMGB1 and DNA fragments, which either directly through TLR9 [49] or indirectly through cytokine formation by macrophages enhance ROS priming [49–51]. However, similar to the concern with inactivation of Kupffer cells, functionally inactivating neutrophils is an effective strategy to document neutrophil involvement in the pathophysiology, but it impairs the host defense function of these cells and increases the risk for developing sepsis.
3. Mechanisms of oxidant stress-induced liver injury
3.1. Lipid peroxidation
One of the most popular hypotheses of ROS-mediated cell injury is lipid peroxidation (LPO) [52]. This process is initiated by the reductive cleavage of hydrogen peroxide by Fe2+ (Fenton reaction) resulting in formation of a hydroxyl radical, which then initiates a radical chain reaction leading to destruction of polyunsaturated fatty acids. Because these types of radical reactions are very dangerous to cells, a highly effective antioxidant system is in place to counteract these processes at all levels including preventing the formation of the most reactive species, detoxification, and repair of oxidative damage [15]. Nevertheless, some increase in LPO can always be measured under most conditions in hepatic ischemia–reperfusion injury. In addition, numerous interventions that protect against ischemia–reperfusion injury also reduce LPO. The natural conclusion from these data is that LPO caused the injury. However, the same data can also be interpreted that LPO is an epiphenomenon of the injury. Thus, the fundamental question is really “how much LPO is necessary to cause cell death”? We have tried to address this question by infusing high concentrations of tert-butylhydroperoxide into the liver for various times (Fig. 1). After 45 min of massive oxidant stress, there was no injury, but LPO was increased 2.5-fold over baseline. After 90 min, there was moderate liver injury, and LPO was increased by 30–50-fold. In contrast, 45 min of hepatic ischemia followed by 1–24 h of reperfusion resulted in severe liver injury but only a 2-fold increase in LPO (Fig. 1). These data strongly suggest that on a quantitative basis, LPO cannot be the direct cause of cell death after hepatic ischemia–reperfusion injury [53]. Similar limited evidence for LPO was observed in human transplant patients suggesting that there is a postischemic oxidant stress, but LPO is not the direct mechanism of cell injury in humans [54–56]. Thus, various parameters of LPO can be biomarkers for oxidant stress and potentially intracellular signaling molecules [52,57]. In addition, LPO products can be chemotactic factors and thereby participate in the amplification of the inflammatory response during reperfusion [58].
Fig. 1.
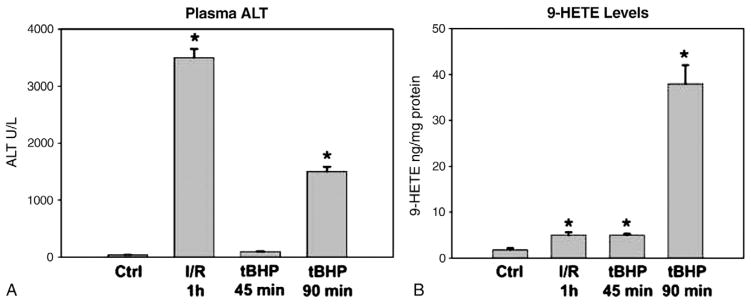
Plasma alanine aminotransferase (ALT) activities (A) and hepatic content of 9-hydroxy eicosatetraenoic acid (HETE) (B) were measured in control livers, after 45-min ischemia to the median and left lobes plus 1-h reperfusion, and after 45-or 90-min continuous infusion of 0.8 μmol tert-butylhydroperoxide/min/g liver into the portal vein. All values represent means±SE of n=4. *p<0.05 (compared to controls). Data adapted from Mathews et al. [53].
3.2. Mitochondria
These cell organelles are central to mechanisms of cell death as they can trigger necrosis mainly after opening of the mitochondrial membrane permeability transition (MPT) pores, which causes the collapse of the mitochondrial membrane potential with cessation of ATP synthesis [59]. The MPT can be regulated by cyclophilin D after a milder insult but may also be cyclophilin D-independent after more severe stress [60]. Inhibitors of cyclophilin D are effective in reducing cell necrosis after warm or cold hepatic ischemia [61,62]. Mitochondria can also release proapoptotic factors like cytochrome c from the intermembrane space through mitochondrial outer-membrane permeabilization (MOMP) induced by bax or matrix swelling and rupture of the outer membrane as a consequence of the MPT [59]. Oxidant stress is a potent trigger of the MPT pore opening in hepatocytes [40]. In fact, moderate extracellular oxidant stress has been shown to effectively induce the MPT through inducing mitochondrial oxidant stress [41]. Interestingly, the iron chelator deferoxamine blocked the mitochondrial oxidant stress and cell killing [42]. More recent findings support these findings and documented that release of lysosomal iron and its uptake into mitochondria through the calcium uniporter can act synergistically with oxidant stress to promote the MPT and cell death [42]. Since hepatic ischemia and reperfusion can trigger lysosomal instability [63,64] and oxidant stress can be produced by mitochondria or extracellularly by leukocytes, which then triggers an intracellular stress, the combination of the lysosomal iron and ROS can induce the MPT and necrosis [42]. The critical importance of mitochondrial dysfunction and the MPT in determining cell death during hepatic warm and cold ischemia–reperfusion injury has been demonstrated by the protective effects of cyclophilin D inhibitors [61,62].
3.3. Apoptosis
It has been postulated that apoptosis is a relevant cell death mechanism during hepatic ischemia–reperfusion injury [65]. In addition, it was reported that oxidant stress induced by menadione can cause apoptotic cell death in cultured rat hepatocytes [66,67]. The mechanisms include the prolonged activation of c-jun-N-terminal kinase (JNK) pathway [68]. However, selective superoxide formation by redox-cycling agents such as diquat does not cause apoptosis but necrotic cell death in vivo [9]. Although there was JNK activation, oxidant stress-induced liver injury did not depend on JNK [9]. This suggested that there could be a difference between cells in culture versus in vivo, especially since high oxygen concentrations in cell culture enhance the oxidant stress [69]. However, menadione also caused necrotic and not apoptotic cell death in cultured mouse hepatocytes (Yan and Jaeschke, unpublished).
The other controversial issue is the role of apoptosis in hepatic ischemia–reperfusion injury [70]. The conclusions about apoptosis are mainly based on the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay, which is not specific for apoptosis. However, based on detailed morphological assessment, caspase activation, and the TUNEL assay together with the lack of relevant protection using a pancaspase inhibitor, it is quite obvious that reperfusion injury does not involve relevant apoptosis but is predominantly caused by necrotic cell death after warm or cold ischemia [71,72]. Most of the controversies about apoptosis are caused by interpretation issues, which could be avoided if a few basic principles including using morphological evidence as gold standard for apoptosis would be considered [70,73]. Another caveat to consider when using caspase inhibitors is the fact that resolution of the inflammatory response may involve neutrophil apoptosis. This may explain why treatment of liver transplant patients during reperfusion had a worse outcome than just exposing the graft during ischemia to the inhibitor [74]. Furthermore, the generally high doses of caspase inhibitors used in vivo may also affect other proteases such as cathepsins and calpains [75], which have been shown to be involved in reperfusion injury [76,77]. Taken together, ROS predominantly causes necrosis, which is the main mechanism of cell death in hepatic ischemia–reperfusion injury.
4. Antioxidant strategies against hepatic ischemia–reperfusion injury
Because of the strong evidence for the importance of ROS in hepatic ischemia–reperfusion injury, a number of different intervention strategies have been successfully used in multiple models of warm and cold ischemia with reperfusion. While some current therapies are beginning clinical trials, more information on specific mechanisms and advanced therapeutic options will be critical to successful disease management.
4.1. Prosurvival genes and antioxidants
Different endogenous genes or gene products have been established as highly protective when induced before and, in some cases, after the start of the ischemic injury. In general, all these components are in some way involved in direct scavenging of ROS or general detoxifying enzymes capable of removing endogenous threats. Many of these genes are controlled by the transcription factor nuclear factor-erythroid 2 p45-related factor 2 (Nrf2)–Kelch-like ECH-associated protein 1 (Keap1) system [78,79]. Nrf2 is normally sequestered in the cytoplasm by Keap1 (Fig. 2); however, conditions such as oxidative stress or hypoxia can result in the activation of Nrf2 and its translocation to the nucleus and consequential altered gene expression [80]. Many of the Nrf2 target genes (e.g., NAD(P)H:quinone oxidoreductase 1, heme oxygenase-1 [Ho-1], glutamate-cysteine ligase [Gcl], microsomal epoxide hydrolase, glutathione-S-transferases, and sulfiredoxin 1) could be potential therapeutic targets to combat ischemia–reperfusion-induced oxidant stress [78]. Individual genes have been evaluated in hepatic ischemia–reperfusion injury models.
Fig. 2.
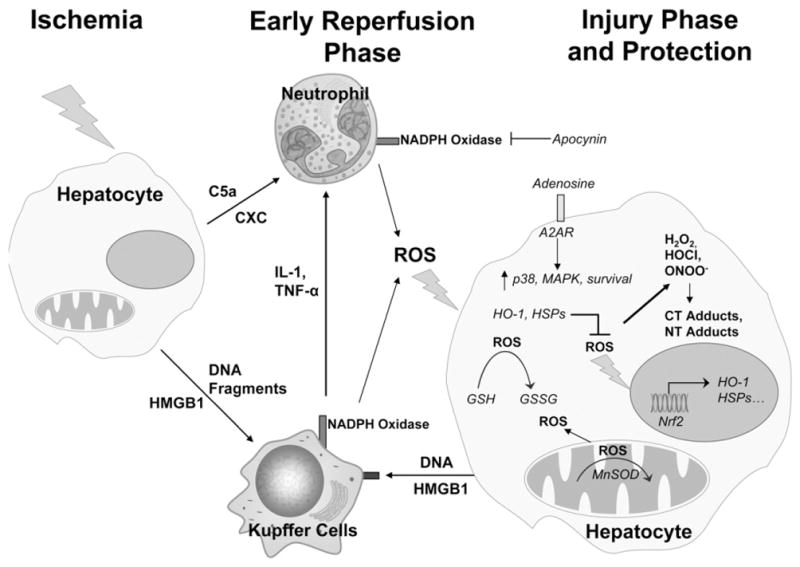
Mechanisms of ischemic and inflammatory injury and some potential antioxidant intervention strategies for hepatic ischemia reperfusion injury. Abbreviations: c5a, complement factor 5a; CXC, chemokines; HMGB1, high-mobility group box 1 protein; IL-1, interleukin 1; TNF-α, tumor necrosis factor-α; HO-1, heme oxygenase-1; MAPK, mitogen-activated protein kinase; A2AR, adenosine receptor 2a; HSPs, heat shock proteins; MnSOD, manganese superoxide dismutase; Nrf2, nuclear factor-erythroid 2 p45-related factor 2; GSH, glutathione; GSSG, glutathione disulfide; ROS, reactive oxygen species; H2O2, hydrogen peroxide; ONOO−, peroxynitrite; HOCl, hypochlorous acid; CT adducts, chlorotyrosine protein adducts; NT adducts, nitrotyrosine protein adducts.
4.1.1. Glutathione
The tripeptide glutathione (GSH) is a highly effective antioxidant present in high concentrations in hepatocytes [81]. GSH levels are regulated by the Nrf2-dependent gene glutamate-cysteine ligase [82]. Administration of the cysteine derivative N-acetylcysteine (NAC) increases intracellular glutathione levels, which can be used for the detoxification of hydrogen peroxide by glutathione peroxidase. However, hydrogen peroxide and hypochlorous acid as well as peroxynitrite can also react spontaneously with GSH [83,84]. Thus, GSH can detoxify these ROS in the presence (intracellularly) or absence (extracellularly) of the peroxidase. Because GSH is continuously released from hepatocytes into the vascular space, GSH can detoxify ROS generated by Kupffer cells [20]. Consequently, intravenous infusion of GSH has been shown to effectively protect against the vascular oxidant stress during reperfusion after warm or cold ischemia [83,85–87]. On the other hand, treatment with NAC can increase intracellular GSH levels and, through enhanced release into the vasculature, also strengthen the vascular defense. When given before [88,89] or continuously during ischemia/reperfusion injury [90], NAC is protective experimentally through maintenance of GSH levels and reduction of reactive oxygen species [91]. However, high doses of NAC may not only protect by scavenging ROS, but the excess can be used to support the mitochondrial energy metabolism [92]. Gene transfer of glutathione synthesis components glutamine cysteine ligase catalytic subunit (gclc), glutamine cysteine regulatory subunit (gclm), and glutathione synthase were protective against I/R injury by increasing intracellular glutathione levels [78,79]. Gene transfer is an especially attractive option for glutathione management, as glutathione has a very short half-life in humans in vivo [93]. Furthermore, clinical trials have reached phase IV for the use of NAC as a protective agent against both warm and cold ischemia, during surgical methods such as liver procurement, and during partial hepatectomy. Some of these trials have reported a reduction in biochemical markers of liver injury; however, there is no strong evidence in support of improved clinical outcome for these patients [94]. Thus, glutathione production and maintenance during ischemia/reperfusion may be an attractive target.
4.1.2. Superoxide dismutases
One of the first antioxidant interventions investigated was the use of exogenous catalase or superoxide dismutase (SOD) as a pretreatment before ischemic injury [2]. Consequentially, induction or maintenance of SOD levels has become another common explanation for mechanism of protection by numerous substances [95–98]. However, even intravenous administration of high doses resulted in only partial protection [2], largely due to poor bioavailability of the enzyme. Conjugation of these enzymes to carbohydrate structures improved bioavailability and protection [99]. Thus, potential antioxidant therapies using modified SODs, or catalase, may be a means for ameliorating injury. Indeed, gene delivery of cupric SOD1 and manganese SOD2 both reduced ischemia/reperfusion pathology, as did gene delivery of SOD3 when given intramuscularly [100]. Transgenic mice overexpressing SOD1 were also protected against ischemia–reperfusion injury [101]. Gene transfer as a therapeutic option is still in the beginning phase of successful implementation in humans; however, the capacity of SOD1, SOD2, and SOD3 to scavenge reactive oxygen and prevent formation of stronger radical species such as peroxynitrite makes them options for antioxidant therapeutic interventions. A caveat of some of the experimental studies is that alternate explanation for the results, that is, induction of other genes than SOD that may be responsible for the protection, is rarely considered. Thus, more detailed mechanistic studies are needed before these therapeutic approaches can be applied to the clinic.
4.1.3. Heat shock proteins
Heme oxygenase-1 (HSP32) is a stress response gene that can be induced by multiple different cellular insults, including ischemic injury [102–104]. Amersi et al. [105] showed that up-regulation of HO-1 using cobalt protoporphyrin or adenovirus HO-1 protected against I/R injury in fatty livers of the Zucker rat. Subsequent work has shown that many protective interventions discovered since also contain a component of HO-1 induction [106–108], commonly through the master stress response gene Nrf2. The mechanism by which HO-1 protects is thought to involve the formation of bilirubin and biliverdin, which both have antioxidant activity. However, carbon monoxide, another byproduct of HO-1 enzymatic activity, can induce p38, a key component of ischemic protection [109]. Induction of other HSPs, for example, HSP70, another Nrf2-inducible gene, increased survival and protected against I/R injury in the liver [110]. HO-1 induction appears to be currently one of the most promising therapeutic approaches in hepatic warm and cold ischemia models [111].
4.2. Preconditioning
Possibly the most common investigational methods of reducing ischemia–reperfusion injury is traditional ischemic preconditioning, done by preexposing the liver to a brief period of ischemia then reperfusion before the actual period of hepatic ischemia [112]. It has been postulated that low levels of oxidant stress are responsible for the preconditioning effect [113]. Ischemic preconditioning has been shown to lead to a reduced inflammatory response as well as reduced oxidant stress [113–115]. There are a number of common mechanisms involved in preconditioning therapies, including activation of the prosurvival p38/MAPK cascade by cAMP-activated protein kinase [116], induction of antioxidant survival genes such as HO-1 [117], and increased proliferation postinjury [118]. There is evidence that ischemic preconditioning is beneficial in the clinic [119–121]. Although, the protective effect may include improved antioxidant defenses, it is clearly multifactorial [122]. Ischemic postconditioning has also been shown to be protective against ischemic insult and has similar mechanisms as to preconditioning, such as activation of the prosurvival PI3K/Akt pathway [123]) and induction of antioxidant SODs and the vasodilator nitric oxide (NO) [124]). While NO can combine with superoxide to form peroxynitrite, a potent oxidant and nitrating species, NO also serves as a vasodilator during ischemic injury [125,126]. Because of the increased vasoconstriction during ischemia–reperfusion, NO is mainly beneficial by facilitating hepatic blood flow during reperfusion [125,126]. The presence of GSH limits detrimental effects of peroxynitrite [125].
Many attempts have been made to reproduce the benefits of ischemic preconditioning pharmacologically in order to avoid invasive measures and provide more persistent treatment postsurgery. By modulating intracellular or extracellular components known to be part of the protective pathway, it is possible to simulate the preconditioning effect. Adenosine 2A receptor (A2AR) activation (Fig. 2) is prevalent during ischemia–reperfusion injury physiologically [114,127,128] and protective when stimulated pharmacologically before ischemia–reperfusion with an A2A agonist [129] but not an A1A agonist [130]. Accordingly, antagonism of A2AR sensitizes hepatocytes to ischemia–reperfusion injury [131]. Other chemical agents such as atrial natriuretic peptide work through similar mechanisms and also precondition cells pharmacologically [132,133]. Some of the pharmacological preconditioning effects involve improved antioxidant capacity [134], but it has to be kept in mind that both surgical and pharmacological preconditioning involves likely a substantial number of additional mechanisms that promote cellular resistance to stress. However, this likely enhances the therapeutic potential in the clinic.
4.2.1. Anesthetic preconditioning
Volatile anesthetics as pharmacological preconditioning agents have received some investigation into their capacity to protect against hepatic ischemia reperfusion injury. Isoflurane can protect against ischemia–reperfusion injury at clinically relevant levels [135], and sevoflurane has been shown to reduce transaminase levels and improve outcome in clinical patients postsurgically, with an even better outcome in steatotic patients, although a mechanism has not been proposed [136]. It appears that anesthesia also induces both HO-1 [135] and HIF-1 [137], so the effect may be mediated through these enzymes’ antioxidant activity.
4.3. NADPH oxidase
Phagocytes including Kupffer cells and neutrophils generate superoxide through NADPH oxidase (NOX-2) [138]. As there is clear evidence for ROS formation by both cell types during hepatic ischemia–reperfusion [139], this enzyme could be an excellent therapeutic target [140]. In fact, NOX-2 inhibitors have been shown to protect against inflammatory injury including hepatic ischemia–reperfusion injury [38,47,48,141]. In addition, numerous strategies that indirectly reduce the inflammatory oxidant stress, for example, blocking adhesion molecules, depletion of Kupffer cells or neutrophils, and others, are highly effective against ischemia–reperfusion injury [139]. However, despite the efficacy of some of these interventions in preclinical models, for example, CD18 antibodies, clinical results were disappointing [142]. Although the antibody did not increase the risk for infection in these patients [143], no protection was observed in multiple ischemia–reperfusion diseases such as myocardial infarction and stroke [142]. This clearly argues for more efforts to understand the human pathophysiology of a disease rather than just rely on preclinical models.
4.4. Mitochondrial permeability transition (MPT) inhibition
The MPT is a central feature of cell death during hepatic ischemia–reperfusion [59]. ROS can induce the MPT, and the MPT can further promote the oxidant stress [40]. Thus, inhibitors of the MPT have been effective against hepatic ischemia–reperfusion injury [61,62], and a number of strategies to reduce the mitochondrial and extracellular ROS formation are an effective treatment against MPT formation and subsequent injury [40,144,145]. Administration of the antioxidants melatonin or edaravone both showed some preservation of mitochondrial respiration potential and a reduction in mitochondrial swelling when given before ischemia–reperfusion surgery; however, the authors failed to measure mitochondrial membrane potential in those papers [146,147]. Peroxiredoxin-6 knockout mice were found to be more susceptible to ischemia–reperfusion injury due to increased mitochondrial hydrogen peroxide [148]. More interestingly, the ischemia stress resulted in translocation of Prx-6 from the cytoplasm to the mitochondria, suggesting not only mitochondrial hydrogen peroxide production during injury but also a strong possibility that an enhanced native antioxidant capacity specifically in the mitochondria could protect against ischemia–reperfusion injury, which correlates with the beneficial effect of MnSOD induction [98]. The mitogen-activated protein (MAP) kinase c-jun-N-terminal kinase (JNK) can be activated by oxidant stress and translocate to the mitochondria, where it can further promote a mitochondrial oxidant stress [149,150]. Thus, inhibition of JNK may protect, in part, by reducing the postschemic oxidant stress and preventing the MPT [145]. In addition, targeting antioxidants directly to mitochondria may be most effective in preventing the MPT and cell death [151].
Edaravone is one example of a drug specifically designed as antioxidant to combat the effects of ischemia reperfusion injury [152]. Although the intent was for use in ischemic stroke, the drug is also highly protective in both warm and cold hepatic ischemia reperfusion injury in rodents [153–156]. Edaravone was also protective in larger mammals [157]. Edaravone may work through inhibition of mitochondrial MPT channel formation, as edaravone sustained ATP levels during ischemia–reperfusion injury in isolated hepatocytes, indicating mitochondrial protection [154]. This suggests that antioxidants targeting mitochondrial stress may be a successful drug class for the prevention of hepatic ischemia–reperfusion injury.
4.5. α-Tocopherol
RRR α-tocopherol is a vitamin E analogue that is preferentially absorbed in humans [158]. Oral pretreatment with α-tocopherol attenuated reperfusion injury and lipid peroxidation and increased synthesis of ATP postinjury when compared to control mice [159]. The effect on cellular ATP levels suggests that this treatment improved mitochondrial function. However, clinical trials using a full racemic mixture of α-tocopherol showed no reduction in clinical chemistry, but reduced length of intensive care unit stay in a double-blind randomized placebo controlled trial [160]. A recent derivative of vitamin E composed of vitamin E, taurine, and glutathione (ETS-GS) showed a reduction in hepatic I/R injury in rats [161]. This suggests that a combination of various antioxidants may be more effective than a single one.
4.6. Herbal antioxidants
Herbal antioxidants, for example, the grape skin component resveratrol and green tea extracts, have been tested in models of liver injury. Trans-resveratrol protected against ischemia–reperfusion injury when given both before ischemia [162] and before reperfusion [163] at low doses, largely through maintenance of GSH levels, and induction of SOD and catalase enzymes. Interestingly, when given at a high dose, resveratrol becomes a pro-oxidant and can actually damage the liver [162]. Green tea catechins (GTCs) are natural antioxidants found in green tea and have been shown to be protective in a dose-specific manner via maintenance of MnSOD and suppression of inflammation [164,165]. However, GTCs may suffer from poor bioavailability in humans, reducing their effectiveness [166]. Tetrandine, a traditional Chinese medicine with known anti-inflammatory and calcium-channel blocking capacity, also showed reduced oxidant stress, maintained SOD activity and reduced the inflammatory response [167]. Quercetin is a naturally occurring flavonoid molecule that may be protective against ischemia–reperfusion injury [168]. However, large-scale trials of quercetin have shown that it is likely to be more effective in cerebrovascular disorders [169]. Although many of these herbal compounds may be effective in reducing hepatic ischemia–reperfusion injury in various animal models, the limitations are that they generally require larger doses and prolonged pretreatment to be effective. In addition, the actual mechanism of protection is, in most cases, not clearly established. They could act as direct antioxidants, indirectly through activation of Nrf2 or even unrecognized mechanisms. Clearly, more mechanistic work needs to be done before these types of treatment could be tested in the clinic.
4.7. Future perspectives of antioxidant therapy
As outlined in this review, there is substantial evidence for the formation and critical involvement of ROS in various animal models of hepatic ischemia–reperfusion injury (Fig. 2). ROS are generated early during reperfusion, where the initial cell death triggers an inflammatory response with activation of tissue macrophages and recruitment of neutrophils, both of which cause cell damage by further ROS formation. Although a large number of intervention strategies that presumably target ROS have been shown to be beneficial in different animal models, the caveat is that most studies show more correlation between the intervention and the protection but do not clearly establish preventing ROS formation or scavenging ROS as the cause of the reduced injury. Without an established mechanism of action, it is difficult to predict potential efficacy in a different animal model or in humans. Furthermore, despite the progress in understanding disease mechanisms in animal models, our knowledge of the human pathophysiology in warm or cold ischemia–reperfusion injury is poor. Thus, the limited mechanistic understanding of an intervention strategy and the limited insight into the various human pathomechanisms are a combination that makes success in the clinic unlikely. Therefore, more translational studies are needed to improve our understanding of the mechanisms of liver cell injury, inflammation, and regeneration particularly in humans but also in animal models. In addition, more and improved antioxidant strategies need to be developed and mechanistically tested. Overall, it appears that therapeutic interventions that target multiple antioxidants pathways such as ischemic or chemical preconditioning including Nrf2 activation may be more promising strategies than attempt to improve the antioxidant capacity of cells by a single compound acting as free radical scavenger.
Acknowledgments
Work in the authors’ laboratory was supported, in part, by the National Institutes of Health grants R01 DK070195 and R01 AA12916 to H.J. and by grants P20 RR016475 and P20 RR021940 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health. B. Woolbright was supported by the “Training Program in Environmental Toxicology” (T32 ES007079-26A2) from the National Institute of Environmental Health Sciences.
Footnotes
The authors have no conflict of interest to report.
References
- 1.Nordstrom G, Seeman T, Hasselgren PO. Beneficial effect of allopurinol in liver ischemia. Surgery. 1985;97:679–84. [PubMed] [Google Scholar]
- 2.Atalla SL, Toledo-Pereyra LH, MacKenzie GH, et al. Influence of oxygen-derived free radical scavengers on ischemic livers. Transplantation. 1985;40:584–90. doi: 10.1097/00007890-198512000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez L, Heredia N, Grande L, et al. Preconditioning protects liver and lung damage in rat liver transplantation: role of xanthine/ xanthine oxidase. Hepatology. 2002;36:562–72. doi: 10.1053/jhep.2002.34616. [DOI] [PubMed] [Google Scholar]
- 4.Metzger J, Dore SP, Lauterburg BH. Oxidant stress during reperfusion of ischemic liver: No evidence for a role of xanthine oxidase. Hepatology. 1988;8:580–4. doi: 10.1002/hep.1840080324. [DOI] [PubMed] [Google Scholar]
- 5.Jaeschke H, Smith CV, Mitchell JR. Reactive oxygen species during ischemia–reflow injury in isolated perfused rat liver. J Clin Invest. 1988;81:1240–6. doi: 10.1172/JCI113441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaeschke H, Smith CV, Mitchell JR. Hypoxic damage generates reactive oxygen species in isolated perfused rat liver. Biochem Biophys Res Commun. 1988;150:568–74. doi: 10.1016/0006-291x(88)90431-7. [DOI] [PubMed] [Google Scholar]
- 7.Jaeschke H, Mitchell JR. Mitochondria and xanthine oxidase both generate reactive oxygen species in isolated perfused rat liver after hypoxic injury. Biochem Biophys Res Commun. 1989;160:140–7. doi: 10.1016/0006-291x(89)91632-x. [DOI] [PubMed] [Google Scholar]
- 8.Engerson TD, McKelvey TG, Rhyne DB, et al. Conversion of xanthine dehydrogenase to oxidase in ischemic rat tissues. J Clin Invest. 1987;79:1564–70. doi: 10.1172/JCI112990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong JY, Lebofsky M, Farhood A, et al. Oxidant stress-induced liver injury in vivo: role of apoptosis, oncotic necrosis and JNK activation. Am J Physiol Gastrointest Liver Physiol. 2009;296:G572–81. doi: 10.1152/ajpgi.90435.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeon BR, Yeom DH, Lee SM, et al. Protective effect of allopurinol on hepatic energy metabolism in ischemic and reperfused rat liver. Shock. 2001;15:112–7. doi: 10.1097/00024382-200115020-00006. [DOI] [PubMed] [Google Scholar]
- 11.Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–41. [PubMed] [Google Scholar]
- 12.Knight TR, Jaeschke H. Acetaminophen-induced inhibition of Fas receptor-mediated liver cell apoptosis: mitochondrial dysfunction versus glutathione depletion. Toxicol Appl Pharmacol. 2002;181:133–41. doi: 10.1006/taap.2002.9407. [DOI] [PubMed] [Google Scholar]
- 13.Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;36:976–80. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 14.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaeschke H. Antioxidant defense mechanisms. In: McQueen CA, editor. Comprehensive Toxicology. Vol. 9. Oxford: Academic Press; 2010. pp. 319–37. [Google Scholar]
- 16.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JS, Nitta T, Mohuczy D, et al. Impaired autophagy: a mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;5:1725–36. doi: 10.1002/hep.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang JH, Ahn IS, Fischer TD, et al. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141:2188–99. doi: 10.1053/j.gastro.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaeschke H. Vascular oxidant stress and hepatic ischemia/reperfusion injury. Free Radic Res Commun. 1991;12–13:737–43. doi: 10.3109/10715769109145853. [DOI] [PubMed] [Google Scholar]
- 20.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia–reperfusion injury in rat liver. Am J Physiol. 1991;260:G355–62. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 21.Caldwell-Kenkel JC, Currin RT, Tanaka Y, et al. Kupffer cell activation and endothelial cell damage after storage of rat livers: effects of reperfusion. Hepatology. 1991;13:83–95. [PubMed] [Google Scholar]
- 22.Jaeschke H, Bautista AP, Spolarics Z, et al. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun. 1991;15:277–84. doi: 10.3109/10715769109105223. [DOI] [PubMed] [Google Scholar]
- 23.Shibuya H, Ohkohchi N, Seya K, et al. Kupffer cells generate superoxide anions and modulate reperfusion injury in rat livers after cold preservation. Hepatology. 1997;25:356–60. doi: 10.1053/jhep.1997.v25.pm0009021947. [DOI] [PubMed] [Google Scholar]
- 24.Rymsa B, Wang JF, de Groot H. O2 release by activated Kupffer cells upon hypoxia-reoxygenation. Am J Physiol. 1991;261:G602–7. doi: 10.1152/ajpgi.1991.261.4.G602. [DOI] [PubMed] [Google Scholar]
- 25.Jaeschke H, Farhood A, Bautista AP, et al. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am J Physiol. 1993;264:G801–9. doi: 10.1152/ajpgi.1993.264.4.G801. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, McGuire GM, Fisher MA, et al. Activation of Kupffer cells and neutrophils for reactive oxygen formation is responsible for endotoxin-enhanced liver injury after hepatic ischemia. Shock. 1995;3:56–62. [PubMed] [Google Scholar]
- 27.Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia–reperfusion. J Exp Med. 2005;201:1135–43. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Decker K. Biologically active products of stimulated liver macrophages (Kupffer cells) Eur J Biochem. 1990;192:245–61. doi: 10.1111/j.1432-1033.1990.tb19222.x. [DOI] [PubMed] [Google Scholar]
- 29.Jaeschke H. Enhanced sinusoidal glutathione efflux during endotoxin-induced oxidant stress in vivo. Am J Physiol. 1992;263:G60–8. doi: 10.1152/ajpgi.1992.263.1.G60. [DOI] [PubMed] [Google Scholar]
- 30.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. 2008;30:279–89. doi: 10.1007/s00281-008-0118-3. [DOI] [PubMed] [Google Scholar]
- 31.Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 1990;4:3355–9. [PubMed] [Google Scholar]
- 32.Jaeschke H, Bautista AP, Spolarics Z, et al. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. J Leukocyte Biol. 1992;52:377–82. doi: 10.1002/jlb.52.4.377. [DOI] [PubMed] [Google Scholar]
- 33.Hasegawa T, Malle E, Farhood A, et al. Generation of hypochlorite-modified proteins by neutrophils during ischemia–reperfusion injury in rat liver: attenuation by ischemic preconditioning. Am J Physiol Gastrointest Liver Physiol. 2005;289:G760–7. doi: 10.1152/ajpgi.00141.2005. [DOI] [PubMed] [Google Scholar]
- 34.Mavier P, Preaux AM, Guigui B, et al. In vitro toxicity of polymorphonuclear neutrophils to rat hepatocytes: evidence for a proteinase-mediated mechanism. Hepatology. 1988;8:254–8. doi: 10.1002/hep.1840080211. [DOI] [PubMed] [Google Scholar]
- 35.Harbrecht BG, Billiar TR, Curran RD, et al. Hepatocyte injury by activated neutrophils in vitro is mediated by proteases. Ann Surg. 1993;218:120–8. doi: 10.1097/00000658-199308000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaeschke H. Mechanisms of neutrophil-mediated liver cell injury during ischemia–reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083–8. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 37.Jaeschke H, Ho YS, Fisher MA, et al. Glutathione peroxidase deficient mice are more susceptible to neutrophil-mediated hepatic parenchymal cell injury during endotoxemia: Importance of an intracellular oxidant stress. Hepatology. 1999;29:443–50. doi: 10.1002/hep.510290222. [DOI] [PubMed] [Google Scholar]
- 38.Gujral JS, Hinson JA, Farhood A, et al. NADPH oxidase-derived oxidant stress is critical for neutrophil-induced cytotoxicity during endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2004;287:G243–52. doi: 10.1152/ajpgi.00287.2003. [DOI] [PubMed] [Google Scholar]
- 39.Gujral RS, Farhood A, Bajt ML, et al. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology. 2003;38:355–63. doi: 10.1053/jhep.2003.50341. [DOI] [PubMed] [Google Scholar]
- 40.Nieminen AL, Saylor AK, Tesfai SA, et al. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochem J. 1995;307:99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nieminen AL, Byrne AM, Herman B, et al. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am J Physiol. 1997;272:C1286–94. doi: 10.1152/ajpcell.1997.272.4.C1286. [DOI] [PubMed] [Google Scholar]
- 42.Uchiyama A, Kim JS, Kon K, et al. Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology. 2008;48:1644–54. doi: 10.1002/hep.22498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shappell SB, Toman C, Anderson DC, et al. Mac-1 (CD11b/CD18) mediates adherence-dependent hydrogen peroxide production by human and canine neutrophils. J Immunol. 1990;144:2702–11. [PubMed] [Google Scholar]
- 44.Entman ML, Youker K, Shoji T, et al. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J Clin Invest. 1992;90:1335–45. doi: 10.1172/JCI115999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaeschke H, Farhood A, Smith CW. Neutrophil-induced liver cell injury in endotoxin shock is a CD11b/CD18—dependent mechanism. Am J Physiol. 1991;261:G1051–6. doi: 10.1152/ajpgi.1991.261.6.G1051. [DOI] [PubMed] [Google Scholar]
- 46.Jaeschke H, Farhood A, Bautista AP, et al. Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia–reperfusion injury in rat liver. Hepatology. 1993;17:915–23. [PubMed] [Google Scholar]
- 47.Lehnert M, Arteel GE, Smutney OM, et al. Dependence of liver injury after hemorrhage/resuscitation in mice on NADPH oxidase-derived superoxide. Shock. 2003;19:345–51. doi: 10.1097/00024382-200304000-00009. [DOI] [PubMed] [Google Scholar]
- 48.Dorman RB, Wunder C, Saba H, et al. NAD(P)H oxidase contributes to the progression of remote hepatic parenchymal injury and endothelial dysfunction, but not microvascular perfusion deficits. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1025–32. doi: 10.1152/ajpgi.00246.2005. [DOI] [PubMed] [Google Scholar]
- 49.Bamboat ZM, Balachandran VP, Ocuin LM, et al. Toll-like receptor 9 inhibition confers protection from liver ischemia–reperfusion injury. Hepatology. 2010;51:621–32. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan J, Li Y, Levy RM, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol. 2007;178:6573–80. doi: 10.4049/jimmunol.178.10.6573. [DOI] [PubMed] [Google Scholar]
- 51.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: toll-like receptors. Free Radic Biol Med. 2010;48:1121–32. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Negre-Salvayre A, Auge N, Ayala V, et al. Pathological aspects of lipid peroxidation. Free Radic Res. 2010;44:1125–71. doi: 10.3109/10715762.2010.498478. [DOI] [PubMed] [Google Scholar]
- 53.Mathews WR, Guido DM, Fisher MA, et al. Lipid peroxidation as molecular mechanism of liver cell injury during reperfusion after ischemia. Free Radic Biol Med. 1994;16:763–70. doi: 10.1016/0891-5849(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 54.Galley HF, Richardson N, Howdle PD, et al. Total antioxidant capacity and lipid peroxidation during liver transplantation. Clin Sci (Lond) 1995;89:329–32. doi: 10.1042/cs0890329. [DOI] [PubMed] [Google Scholar]
- 55.Serrano E, Diaz J, Acosta F, et al. Relationship between cold ischemia time and lipid peroxidation in liver transplantation. Transplant Proc. 2000;32:2648. doi: 10.1016/s0041-1345(00)01822-4. [DOI] [PubMed] [Google Scholar]
- 56.Risby TH, Maley W, Scott RP, et al. Evidence for free radical-mediated lipid peroxidation at reperfusion of human orthotopic liver transplants. Surgery. 1994;115:94–101. [PubMed] [Google Scholar]
- 57.Parola M, Bellomo G, Robino G, et al. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal. 1999;1:255–84. doi: 10.1089/ars.1999.1.3-255. [DOI] [PubMed] [Google Scholar]
- 58.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J Gastroenterol Hepatol. 2011;26(Suppl 1):173–9. doi: 10.1111/j.1440-1746.2010.06592.x. [DOI] [PubMed] [Google Scholar]
- 59.Lemasters JJ, Qian T, He L, et al. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–81. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 60.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- 61.Theruvath TP, Zhong Z, Pediaditakis P, et al. Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/ reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology. 2008;1:236–46. doi: 10.1002/hep.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhong Z, Ramshesh VK, Rehman H, et al. Activation of the oxygen-sensing signal cascade prevents mitochondrial injury after mouse liver ischemia–reperfusion. Am J Physiol Gastrointest Liver Physiol. 2008;95:G823–32. doi: 10.1152/ajpgi.90287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Molchanova LV, Nickulina SE, Ivanova TN, et al. Role of cAMP in regulation of activity of acid hydrolases of rat heart and liver during ischemia and after recirculation. Resuscitation. 1991;22:261–74. doi: 10.1016/0300-9572(91)90034-v. [DOI] [PubMed] [Google Scholar]
- 64.Grek OR, Pupyshev AB, Tikhonova EV. Effect of transitory ischemia on liver lysosomal apparatus in rats with different resistance to hypoxia. Bull Exp Biol Med. 2003;136:11–3. doi: 10.1023/a:1026016224694. [DOI] [PubMed] [Google Scholar]
- 65.Rüdiger HA, Graf R, Clavien PA. Liver ischemia: apoptosis as a central mechanism of injury. J Invest Surg. 2003;16:149–59. [PubMed] [Google Scholar]
- 66.Jones BE, Lo CR, Liu H, et al. Role of caspases and NF-kappaB signaling in hydrogen peroxide- and superoxide-induced hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol. 2000;278:G693–9. doi: 10.1152/ajpgi.2000.278.5.G693. [DOI] [PubMed] [Google Scholar]
- 67.Conde de la Rosa L, Schoemaker MH, Vrenken TE, et al. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–29. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 68.Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol. 2007;22(Suppl 1):S45–8. doi: 10.1111/j.1440-1746.2006.04646.x. [DOI] [PubMed] [Google Scholar]
- 69.Yan HM, Ramachandran A, Bajt ML, et al. The oxygen tension modulates acetaminophen-induced mitochondrial oxidant stress and cell injury in cultured hepatocytes. Toxicol Sci. 2010;117:515–23. doi: 10.1093/toxsci/kfq208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–57. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 71.Gujral JS, Bucci TJ, Farhood A, et al. Mechanism of cell death during warm hepatic ischemia–reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33:397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 72.Gerwig T, Meissner H, Bilzer M, et al. Atrial natriuretic peptide preconditioning protects against hepatic preservation injury by attenuating necrotic and apoptotic cell death. J Hepatol. 2003;39:341–8. doi: 10.1016/s0168-8278(03)00240-x. [DOI] [PubMed] [Google Scholar]
- 73.Jaeschke H, Gujral JS, Bajt ML. Apoptosis and necrosis in liver disease. Liver Int. 2004;24:85–9. doi: 10.1111/j.1478-3231.2004.0906.x. [DOI] [PubMed] [Google Scholar]
- 74.Baskin-Bey ES, Washburn K, Feng S, et al. Clinical trial of the pan-caspase inhibitor, IDN-6556, in human liver preservation injury. Am J Transplant. 2007;7:218–25. doi: 10.1111/j.1600-6143.2006.01595.x. [DOI] [PubMed] [Google Scholar]
- 75.Schotte P, Declercq W, Van Huffel S, et al. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 1999;442:117–21. doi: 10.1016/s0014-5793(98)01640-8. [DOI] [PubMed] [Google Scholar]
- 76.Kohli V, Gao W, Camargo CA, Jr, et al. Calpain is a mediator of preservation-reperfusion injury in rat liver transplantation. Proc Natl Acad Sci U S A. 1997;94:9354–9. doi: 10.1073/pnas.94.17.9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baskin-Bey ES, Canbay A, Bronk SF, et al. Cathepsin B inactivation attenuates hepatocyte apoptosis and liver damage in steatotic livers after cold ischemia-warm reperfusion injury. Am J Physiol Gastro-intest Liver Physiol. 2005;288:G396–402. doi: 10.1152/ajpgi.00316.2004. [DOI] [PubMed] [Google Scholar]
- 78.Klaassen CD, Reisman SA. Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicol Appl Pharmacol. 2010;244:57–65. doi: 10.1016/j.taap.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1–Nrf2–ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 80.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1–Nrf2 pathway. Arch Toxicol. 2011;85:241–72. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 81.Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. 2009;30:29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 82.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu P, Fisher MA, Farhood A, et al. Beneficial effects of extracellular glutathione against endotoxin-induced liver injury during ischemia and reperfusion. Circ Shock. 1994;43:64–70. [PubMed] [Google Scholar]
- 84.Knight TR, Ho YS, Farhood A, et al. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–75. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- 85.Schauer RJ, Kalmuk S, Gerbes AL, et al. Intravenous administration of glutathione protects parenchymal and non-parenchymal liver cells against reperfusion injury following rat liver transplantation. World J Gastroenterol. 2004;10:864–70. doi: 10.3748/wjg.v10.i6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schauer RJ, Gerbes AL, Vonier D, et al. Glutathione protects the rat liver against reperfusion injury after prolonged warm ischemia. Ann Surg. 2004;239:220–31. doi: 10.1097/01.sla.0000110321.64275.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bilzer M, Baron A, Schauer R, et al. Glutathione treatment protects the rat liver against injury after warm ischemia and Kupffer cell activation. Digestion. 2002;66:49–57. doi: 10.1159/000064415. [DOI] [PubMed] [Google Scholar]
- 88.Smyrniotis V, Arkadopoulos N, Kostopanagiotou G, et al. Attenuation of ischemic injury by N-acetylcysteine preconditioning of the liver. J Surg Res. 2005;129:31–7. doi: 10.1016/j.jss.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 89.Nagasaki H, Nakano H, Boudjema K, et al. Efficacy of preconditioning with N-acetylcysteine against reperfusion injury after prolonged cold ischaemia in rats liver in which glutathione had been reduced by buthionine sulphoximine. Eur J Surg. 1998;164:139–46. doi: 10.1080/110241598750004805. [DOI] [PubMed] [Google Scholar]
- 90.Fusai G, Glantzounis GK, Hafez T, et al. N-acetylcysteine ameliorates the late phase of liver ischaemia/reperfusion injury in the rabbit with hepatic steatosis. Clin Sci (Lond) 2005;109:465–73. doi: 10.1042/CS20050081. [DOI] [PubMed] [Google Scholar]
- 91.Sener G, Tosun O, Sehirli AO, et al. Melatonin and N-acetylcysteine have beneficial effects during hepatic ischemia and reperfusion. Life Sci. 2003;72:2707–18. doi: 10.1016/s0024-3205(03)00187-5. [DOI] [PubMed] [Google Scholar]
- 92.Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51:246–54. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wendel A, Cikryt P. The level and half-life of glutathione in human plasma. FEBS Lett. 1980;120:209–11. doi: 10.1016/0014-5793(80)80299-7. [DOI] [PubMed] [Google Scholar]
- 94.Jegatheeswaran S, Siriwardena AK. Experimental and clinical evidence for modification of hepatic ischaemia–reperfusion injury by N-acetylcysteine during major liver surgery. HPB (Oxford) 2011;13:71–8. doi: 10.1111/j.1477-2574.2010.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peralta C, León OS, Xaus C, et al. Protective effect of ozone treatment on the injury associated with hepatic ischemia–reperfusion: antioxidant–prooxidant balance. Free Radic Res. 1999;31:191–6. doi: 10.1080/10715769900300741. [DOI] [PubMed] [Google Scholar]
- 96.Shen SQ, Zhang Y, Xiang JJ, et al. Protective effect of curcumin against liver warm ischemia/reperfusion injury in rat model is associated with regulation of heat shock protein and antioxidant enzymes. World J Gastroenterol. 2007;13:1953–61. doi: 10.3748/wjg.v13.i13.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rao J, Zhang C, Wang P, et al. All-trans retinoic acid alleviates hepatic ischemia/reperfusion injury by enhancing manganese super-oxide dismutase in rats. Biol Pharm Bull. 2010;33:869–75. doi: 10.1248/bpb.33.869. [DOI] [PubMed] [Google Scholar]
- 98.Pardo M, Budick-Harmelin N, Tirosh B, et al. Antioxidant defense in hepatic ischemia–reperfusion injury is regulated by damage-associated molecular pattern signal molecules. Free Radic Biol Med. 2008;45:1073–83. doi: 10.1016/j.freeradbiomed.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 99.Yabe Y, Kobayashi N, Nishihashi T, et al. Prevention of neutrophil-mediated hepatic ischemia/reperfusion injury by super-oxide dismutase and catalase derivatives. J Pharmacol Exp Ther. 2001;298:894–9. [PubMed] [Google Scholar]
- 100.Wheeler MD, Katuna M, Smutney OM, et al. Comparison of the effect of adenoviral delivery of three superoxide dismutase genes against hepatic ischemia–reperfusion injury. Hum Gene Ther. 2001;12:2167–77. doi: 10.1089/10430340152710513. [DOI] [PubMed] [Google Scholar]
- 101.Suzuki M, Takeuchi H, Kakita T, et al. The involvement of the intracellular superoxide production system in hepatic ischemia–reperfusion injury. In vivo and in vitro experiments using transgenic mice manifesting excessive CuZn–SOD activity. Free Radic Biol Med. 2000;29:756–63. doi: 10.1016/s0891-5849(00)00369-5. [DOI] [PubMed] [Google Scholar]
- 102.Takeda A, Onodera H, Sugimoto A, et al. Increased expression of heme oxygenase mRNA in rat brain following transient forebrain ischemia. Brain Res. 1994;666:120–4. doi: 10.1016/0006-8993(94)90292-5. [DOI] [PubMed] [Google Scholar]
- 103.Maines MD, Mayer RD, Ewing JF, et al. Induction of kidney heme oxygenase-1 (HSP32) mRNA and protein by ischemia/reperfusion: possible role of heme as both promotor of tissue damage and regulator of HSP32. J Pharmacol Exp Ther. 1993;264:457–62. [PubMed] [Google Scholar]
- 104.Yamaguchi T, Terakado M, Horio F, et al. Role of bilirubin as an antioxidant in an ischemia–reperfusion of rat liver and induction of heme oxygenase. Biochem Biophys Res Commun. 1996;223:129–35. doi: 10.1006/bbrc.1996.0857. [DOI] [PubMed] [Google Scholar]
- 105.Amersi F, Buelow R, Kato H, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest. 1999;104:1631–9. doi: 10.1172/JCI7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmidt R, Tritschler E, Hoetzel A, et al. Heme oxygenase-1 induction by the clinically used anesthetic isoflurane protects rat livers from ischemia/reperfusion injury. Ann Surg. 2007;245:931–42. doi: 10.1097/01.sla.0000256891.45790.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Man K, Ng KT, Lee TK, et al. FTY720 attenuates hepatic ischemia–reperfusion injury in normal and cirrhotic livers. Am J Transplant. 2005;5:40–9. doi: 10.1111/j.1600-6143.2004.00642.x. [DOI] [PubMed] [Google Scholar]
- 108.Ke B, Shen XD, Lassman CR, et al. Cytoprotective and antiapoptotic effects of IL-13 in hepatic cold ischemia/reperfusion injury are heme oxygenase-1 dependent. Am J Transplant. 2003;3:1076–82. doi: 10.1034/j.1600-6143.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 109.Amersi F, Shen XD, Anselmo D, et al. Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology. 2002;35:815–23. doi: 10.1053/jhep.2002.32467. [DOI] [PubMed] [Google Scholar]
- 110.Kume M, Yamamoto Y, Saad S, et al. Ischemic preconditioning of the liver in rats: implications of heat shock protein induction to increase tolerance of ischemia–reperfusion injury. J Lab Clin Med. 1996;128:251–8. doi: 10.1016/s0022-2143(96)90026-8. [DOI] [PubMed] [Google Scholar]
- 111.Tsuchihashi S, Fondevila C, Kupiec-Weglinski JW. Heme oxygenase system in ischemia and reperfusion injury. Ann Transplant. 2004;9:84–7. [PubMed] [Google Scholar]
- 112.de Rougemont O, Lehmann K, Clavien PA. Preconditioning, organ preservation, and postconditioning to prevent ischemia–reperfusion injury to the liver. Liver Transpl. 2009;15:1172–82. doi: 10.1002/lt.21876. [DOI] [PubMed] [Google Scholar]
- 113.Sindram D, Rüdiger HA, Upadhya AG, et al. Ischemic preconditioning protects against cold ischemic injury through an oxidative stress dependent mechanism. J Hepatol. 2002;36:78–84. doi: 10.1016/s0168-8278(01)00229-x. [DOI] [PubMed] [Google Scholar]
- 114.Carini R, De Cesaris MG, Splendore R, et al. Signal pathway involved in the development of hypoxic preconditioning in rat hepatocytes. Hepatology. 2001;33:131–9. doi: 10.1053/jhep.2001.21050. [DOI] [PubMed] [Google Scholar]
- 115.Peralta C, Bulbena O, Xaus C, et al. Ischemic preconditioning: a defense mechanism against the reactive oxygen species generated after hepatic ischemia reperfusion. Transplantation. 2002;73:1203–11. doi: 10.1097/00007890-200204270-00004. [DOI] [PubMed] [Google Scholar]
- 116.Serafín A, Fernández-Zabalegui L, Prats N, et al. Ischemic preconditioning: tolerance to hepatic ischemia–reperfusion injury. Histol Histopathol. 2004;19:281–9. doi: 10.14670/HH-19.281. [DOI] [PubMed] [Google Scholar]
- 117.Schauer RJ, Gerbes AL, Vonier D, et al. Induction of cellular resistance against Kupffer cell-derived oxidant stress: a novel concept of hepatoprotection by ischemic preconditioning. Hepatology. 2003;37:286–95. doi: 10.1053/jhep.2003.50064. [DOI] [PubMed] [Google Scholar]
- 118.Teoh N, Dela Pena A, Farrell G. Hepatic ischemic preconditioning in mice is associated with activation of NF-kappaB, p38 kinase, and cell cycle entry. Hepatology. 2002;36:94–102. doi: 10.1053/jhep.2002.33134. [DOI] [PubMed] [Google Scholar]
- 119.Clavien PA, Yadav S, Sindram D, et al. Protective effects of ischemic preconditioning for liver resection performed under inflow occlusion in humans. Ann Surg. 2000;232:155–62. doi: 10.1097/00000658-200008000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Clavien PA, Selzner M, Rüdiger HA, et al. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann Surg. 2003;238:843–50. doi: 10.1097/01.sla.0000098620.27623.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jassem W, Fuggle SV, Cerundolo L, et al. Ischemic preconditioning of cadaver donor livers protects allografts following transplantation. Transplantation. 2006;81:169–74. doi: 10.1097/01.tp.0000188640.05459.37. [DOI] [PubMed] [Google Scholar]
- 122.Jassem W, Fuggle S, Thompson R, et al. Effect of ischemic preconditioning on the genomic response to reperfusion injury in deceased donor liver transplantation. Liver Transpl. 2009;15:1750–65. doi: 10.1002/lt.21936. [DOI] [PubMed] [Google Scholar]
- 123.Dal Ponte C, Alchera E, Follenzi A, et al. Pharmacological postconditioning protects against hepatic ischemia/reperfusion injury. Liver Transpl. 2011;17:474–82. doi: 10.1002/lt.22256. [DOI] [PubMed] [Google Scholar]
- 124.Guo Y, Yang T, Lu J, et al. Rb1 postconditioning attenuates liver warm ischemia–reperfusion injury through ROS–NO–HIF pathway. Life Sci. 2011;88:598–605. doi: 10.1016/j.lfs.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 125.Wang Y, Mathews WR, Guido DM, et al. Inhibition of nitric oxide synthesis aggravates reperfusion injury after hepatic ischemia and endotoxemia. Shock. 1995;4:282–8. doi: 10.1097/00024382-199510000-00009. [DOI] [PubMed] [Google Scholar]
- 126.Wang Y, Lawson JA, Jaeschke H. Differential effect of 2-aminoethyl-isothiourea, an inhibitor of the inducible nitric oxide synthase, on microvascular blood flow and organ injury in models of hepatic ischemia–reperfusion and endotoxemia. Shock. 1998;10:20–5. doi: 10.1097/00024382-199807000-00004. [DOI] [PubMed] [Google Scholar]
- 127.Carini R, De Cesaris MG, Splendore R, et al. Ischemic preconditioning reduces Na(+) accumulation and cell killing in isolated rat hepatocytes exposed to hypoxia. Hepatology. 2000;31:166–72. doi: 10.1002/hep.510310125. [DOI] [PubMed] [Google Scholar]
- 128.Hart ML, Much C, Gorzolla IC, et al. Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology. 2008;135:1739–50. doi: 10.1053/j.gastro.2008.07.064. [DOI] [PubMed] [Google Scholar]
- 129.Nakayama H, Yamamoto Y, Kume M, et al. Pharmacologic stimulation of adenosine A2 receptor supplants ischemic preconditioning in providing ischemic tolerance in rat livers. Surgery. 1999;126:945–54. doi: 10.1016/s0039-6060(99)70037-1. [DOI] [PubMed] [Google Scholar]
- 130.Arai M, Thurman RG, Lemasters JJ. Contribution of adenosine A(2) receptors and cyclic adenosine monophosphate to protective ischemic preconditioning of sinusoidal endothelial cells against storage/ reperfusion injury in rat livers. Hepatology. 2000;32:297–302. doi: 10.1053/jhep.2000.8896. [DOI] [PubMed] [Google Scholar]
- 131.Arai M, Tejima K, Ikeda H, Tomiya T, Yanase M, Inoue Y, Nagashima K, Nishikawa T, Watanabe N, Omata M, Fujiwara K. Ischemic preconditioning in liver pathophysiology. J Gastroenterol Hepatol. 2007;(Suppl 1):S65–7. doi: 10.1111/j.1440-1746.2006.04656.x. [DOI] [PubMed] [Google Scholar]
- 132.Gerbes AL, Vollmar AM, Kiemer AK, et al. The guanylate cyclase-coupled natriuretic peptide receptor: a new target for prevention of cold ischemia–reperfusion damage of the rat liver. Hepatology. 1998;28:1309–17. doi: 10.1002/hep.510280520. [DOI] [PubMed] [Google Scholar]
- 133.Kiemer AK, Gerbes AL, Bilzer M, et al. The atrial natriuretic peptide and cGMP: novel activators of the heat shock response in rat livers. Hepatology. 2002;35:88–94. doi: 10.1053/jhep.2002.30080. [DOI] [PubMed] [Google Scholar]
- 134.Bilzer M, Jaeschke H, Vollmar AM, et al. Prevention of Kupffer cell-induced oxidant injury in rat liver by atrial natriuretic peptide. Am J Physiol. 1999;276:G1137–44. doi: 10.1152/ajpgi.1999.276.5.G1137. [DOI] [PubMed] [Google Scholar]
- 135.Lv X, Yang L, Tao K, et al. Isoflurane preconditioning at clinically relevant doses induce protective effects of heme oxygenase-1 on hepatic ischemia reperfusion in rats. BMC Gastroenterol. 2011;11:31. doi: 10.1186/1471-230X-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Beck-Schimmer B, Breitenstein S, Urech S, et al. A randomized controlled trial on pharmacological preconditioning in liver surgery using a volatile anesthetic. Ann Surg. 2008;248:909–18. doi: 10.1097/SLA.0b013e31818f3dda. [DOI] [PubMed] [Google Scholar]
- 137.Zhang L, Huang H, Cheng J, et al. Pre-treatment with isoflurane ameliorates renal ischemic-reperfusion injury in mice. Life Sci. 2011;88:1102–7. doi: 10.1016/j.lfs.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 138.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–4. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 139.Jaeschke H. Molecular mechanisms of hepatic ischemia–reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 140.Jaquet V, Scapozza L, Clark RA, et al. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11:2535–52. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 141.Liu PG, He SQ, Zhang YH, et al. Protective effects of apocynin and allopurinol on ischemia/reperfusion-induced liver injury in mice. World J Gastroenterol. 2008;14:2832–7. doi: 10.3748/wjg.14.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dove A. CD18 trials disappoint again. Nat Biotechnol. 2000;18:817–8. doi: 10.1038/78412. [DOI] [PubMed] [Google Scholar]
- 143.Rhee P, Morris J, Durham R, et al. Recombinant humanized monoclonal antibody against CD18 (rhuMAb CD18) in traumatic hemorrhagic shock: results of a phase II clinical trial. Traumatic Shock Group J Trauma. 2000;49:611–9. doi: 10.1097/00005373-200010000-00007. [DOI] [PubMed] [Google Scholar]
- 144.Kantrow SP, Tatro LG, Piantadosi CA. Oxidative stress and adenine nucleotide control of mitochondrial permeability transition. Free Radic Biol Med. 2000;28:251–60. doi: 10.1016/s0891-5849(99)00238-5. [DOI] [PubMed] [Google Scholar]
- 145.Theruvath TP, Snoddy MC, Zhong Z, et al. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 2008;85:1500–4. doi: 10.1097/TP.0b013e31816fefb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Okatani Y, Wakatsuki A, Reiter RJ, et al. Protective effect of melatonin against mitochondrial injury induced by ischemia and reperfusion of rat liver. Eur J Pharmacol. 2003;469:145–52. doi: 10.1016/s0014-2999(03)01643-1. [DOI] [PubMed] [Google Scholar]
- 147.Okatani Y, Wakatsuki A, Enzan H, et al. Edaravone protects against ischemia/reperfusion-induced oxidative damage to mitochondria in rat liver. Eur J Pharmacol. 2003;465:163–70. doi: 10.1016/s0014-2999(03)01463-8. [DOI] [PubMed] [Google Scholar]
- 148.Eismann T, Huber N, Shin T, et al. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia–reperfusion in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G266–74. doi: 10.1152/ajpgi.90583.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hanawa N, Shinohara M, Saberi B, et al. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–77. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2010;246:8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Edeas M. Strategies to target mitochondria and oxidative stress by antioxidants: key points and perspectives. Pharm Res. 2011;28:2771–9. doi: 10.1007/s11095-011-0587-2. [DOI] [PubMed] [Google Scholar]
- 152.Watanabe T, Tahara M, Todo S. The novel antioxidant edaravone: from bench to bedside. Cardiovasc Ther. 2008;26:101–14. doi: 10.1111/j.1527-3466.2008.00041.x. [DOI] [PubMed] [Google Scholar]
- 153.Ninomiya M, Shimada M, Harada N, et al. Beneficial effect of MCI-186 on hepatic warm ischemia–reperfusion in the rat. Transplantation. 2002;74:1470–2. doi: 10.1097/00007890-200211270-00021. [DOI] [PubMed] [Google Scholar]
- 154.Abe T, Unno M, Takeuchi H, et al. A new free radical scavenger, edaravone, ameliorates oxidative liver damage due to ischemia–reperfusion in vitro and in vivo. J Gastrointest Surg. 2004;8:604–15. doi: 10.1016/j.gassur.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 155.Suzuki F, Hashikura Y, Ise H, et al. MCI-186 (edaravone), a free radical scavenger, attenuates hepatic warm ischemia–reperfusion injury in rats. Transpl Int. 2005;18:844–53. doi: 10.1111/j.1432-2277.2005.00094.x. [DOI] [PubMed] [Google Scholar]
- 156.Ninomiya M, Shimada M, Harada N, et al. The hydroxyl radical scavenger MCI-186 protects the liver from experimental cold ischaemia–reperfusion injury. Br J Surg. 2004;91:184–90. doi: 10.1002/bjs.4401. [DOI] [PubMed] [Google Scholar]
- 157.Totsuka O, Takeyoshi I, Tsutsumi H, et al. Effects of a free radical scavenger, MCI-186, on ischemia–reperfusion injury during extended liver resection in dogs. Hepatogastroenterology. 2005;52:1545–8. [PubMed] [Google Scholar]
- 158.Rigotti A. Absorption, transport, and tissue delivery of vitamin. E Mol Aspects Med. 2007;28:423–36. doi: 10.1016/j.mam.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 159.Marubayashi S, Dohi K, Ochi K, et al. Role of free radicals in ischemic rat liver cell injury: prevention of damage by alpha-tocopherol administration. Surgery. 1986;99:184–92. [PubMed] [Google Scholar]
- 160.Bartels M, Biesalski HK, Engelhart K, et al. Pilot study on the effect of parenteral vitamin E on ischemia and reperfusion induced liver injury: a double blind, randomized, placebo-controlled trial. Clin Nutr. 2004;23:1360–70. doi: 10.1016/j.clnu.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 161.Koga H, Hagiwara S, Inomata M, et al. Vitamin E derivative ETS-GS reduces liver ischemia–reperfusion injury in rats. J Surg Res. 2011 doi: 10.1016/j.jss.2011.02.045. in press. [DOI] [PubMed] [Google Scholar]
- 162.Hassan-Khabbar S, Cottart CH, Wendum D, et al. Postischemic treatment by trans-resveratrol in rat liver ischemia–reperfusion: a possible strategy in liver surgery. Liver Transpl. 2008;14:451–9. doi: 10.1002/lt.21405. [DOI] [PubMed] [Google Scholar]
- 163.Gedik E, Girgin S, Ozturk H, et al. Resveratrol attenuates oxidative stress and histological alterations induced by liver ischemia/ reperfusion in rats. World J Gastroenterol. 2008;14:7101–6. doi: 10.3748/wjg.14.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sehirli O, Ozel Y, Dulundu E, et al. Grape seed extract treatment reduces hepatic ischemia–reperfusion injury in rats. Phytother Res. 2008;22:43–8. doi: 10.1002/ptr.2256. [DOI] [PubMed] [Google Scholar]
- 165.Liang R, Nickkholgh A, Kern M, et al. Green tea extract ameliorates reperfusion injury to rat livers after warm ischemia in a dose-dependent manner. Mol Nutr Food Res. 2011;55:855–63. doi: 10.1002/mnfr.201000643. [DOI] [PubMed] [Google Scholar]
- 166.Peters CM, Green RJ, Janle EM, et al. Formulation with ascorbic acid and sucrose modulates catechin bioavailability from green tea. Food Res Int. 2010;43:95–102. doi: 10.1016/j.foodres.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cheng F, Li Y, Feng L, Li S. Effects of tetrandine on ischemia/ reperfusion injury in mouse liver. Transplant Proc. 2008;40:2163–6. doi: 10.1016/j.transproceed.2008.07.082. [DOI] [PubMed] [Google Scholar]
- 168.Su JF, Guo CJ, Wei JY, et al. Protection against hepatic ischemia–reperfusion injury in rats by oral pretreatment with quercetin. Biomed Environ Sci. 2003;16:1–8. [PubMed] [Google Scholar]
- 169.Firuzi O, Miri R, Tavakkoli M, et al. Antioxidant therapy: current status and future prospects. Curr Med Chem. 2011;18:3871–88. doi: 10.2174/092986711803414368. [DOI] [PubMed] [Google Scholar]