

Abstract
Phenotypes for a gene deletion are often revealed only when the mutation is tested in a particular genetic background or environmental condition1,2. There are examples where many genes need to be deleted to unmask hidden gene functions3,4. Despite the potential for important discoveries, genetic interactions involving three or more genes are largely unexplored. Exhaustive searches of multi-mutant interactions would be impractical due to the sheer number of possible combinations of deletions. However, studies of selected sets of genes, such as sets of paralogs with a greater a priori chance of sharing a common function, would be informative.
In the yeast Saccharomyces cerevisiae, gene knockout is accomplished by replacing a gene with a selectable marker via homologous recombination. Because the number of markers is limited, methods have been developed for removing and reusing the same marker5,6,7,8,9,10. However, sequentially engineering multiple mutations using these methods is time-consuming because the time required scales linearly with the number of deletions to be generated.
Here we describe the Green Monster method for routinely engineering multiple deletions in yeast11. In this method, a green fluorescent protein (GFP) reporter integrated into deletions is used to quantitatively label strains according to the number of deletions contained in each strain (Figure 1). Repeated rounds of assortment of GFP-marked deletions via yeast mating and meiosis coupled with flow-cytometric enrichment of strains carrying more of these deletions lead to the accumulation of deletions in strains (Figure 2). Performing multiple processes in parallel, with each process incorporating one or more deletions per round, reduces the time required for strain construction.
The first step is to prepare haploid single-mutants termed 'ProMonsters,' each of which carries a GFP reporter in a deleted locus and one of the 'toolkit' loci—either Green Monster GMToolkit-a or GMToolkit-α at the can1Δ locus (Figure 3). Using strains from the yeast deletion collection12, GFP-marked deletions can be conveniently generated by replacing the common KanMX4 cassette existing in these strains with a universal GFP-URA3 fragment. Each GMToolkit contains: either the a- or α-mating-type-specific haploid selection marker1 and exactly one of the two markers that, when both GMToolkits are present, collectively allow for selection of diploids.
The second step is to carry out the sexual cycling through which deletion loci can be combined within a single cell by the random assortment and/or meiotic recombination that accompanies each cycle of mating and sporulation.
Keywords: Microbiology, Issue 70, Genetics, Synthetic Biology, Environmental Genomics, Genomics, Bioengineering, Biomedical Engineering, Cellular Biology, Multi-site genomic engineering, genetic interaction, green fluorescent protein, GFP, flow cytometry, Saccharomyces cerevisiae, yeast, Green Monster
Protocol
1. Generation of ProMonsters
Prepare the universal GFP replacement cassette by amplifying a tetO2-GFP marker and the URA3 marker from the plasmid pYOGM012 (ampicillin resistance) using primers with sequences: GGATCCCCGGGTTAATTAAGGCGCGCCAGATCTGTTTAGCTTGCCCAAGCTCCTCGAGTAATTCG and GGCGTTAGTATCGAATCGACAGCAGTATAGCGACCAGCATTCACGTACCGGGTAATAACTGATATAAT (bolded regions provide homology to the flanking regions of the KanMX4 cassette allowing targeted replacement). The PCR reaction should be carried out with Phusion polymerase, HF buffer, and 3% dimethyl sulfoxide (New England BioLabs) starting with the template DNA concentration of 0.5 ng/μl. The PCR program is 98 °C for 30 sec and 35 cycles of 98 °C for 10 sec, 49 °C for 30 sec, and 72 °C for 1.5 min. The reaction volume should be > 50 μl to provide enough DNA for each transformation experiment. We purify the product using a QIAquick PCR purification kit (Qiagen) with ~50 μl of the PCR reaction processed per column, but skipping the purification step may increase the yield of transformants. Alternatively, release a fragment containing the tetO2-GFP and URA3 markers, as well as homologous regions for KanMX4 targeting, by digesting the plasmid pYOGM057 (ampicillin resistance) using the restriction enzyme EcoRI (New England BioLabs). The DNA concentration in this reaction should be ~30 ng/μl. The reaction volume should be > 34 μl. For integrating the GFP cassette into a region other than KanMX4, adjust the homology region of the PCR primers above to the appropriate homologous sequences.
Transform an a-haploid strain from the KanMX4 yeast knockout collection12 with the universal GFP deletion cassette. Following the protocol by Woods and Gietz13 .34 μl of the DNA sample (purified PCR product or non-purified restriction digest) should be used for each transformation experiment. Plate one fifth of the transformation mixture onto a CAA-Ura plate (0.67% yeast nitrogen base, 2% glucose, 0.6% casamino acid, 0.0025% adenine hemisulfate, 0.005% tryptophan, and 2% agar).
Streak out the transformants on a fresh CAA-Ura plate to obtain isolated colonies.
Transfer cells from a colony onto a YPDA plate containing 200 μg/ml G418 to confirm the lack of growth. To confirm successful GFP integration, follow the steps (1.5) - (1.9) to perform colony PCR.
Add cells taken from a colony into 2 μl of lysis buffer (0.1 M sodium phosphate, pH 7.4, 1 unit zymolyase from ZymoResearch) in a 0.2-ml PCR tube or a well of a 96-well plate. Mix by pipetting the solution in and out of a pipette tip.
Overlay with one drop (roughly 20 μl) of mineral oil using a P200 Pipetman (Gilson).
Incubate the mixture at 37 °C for 20 min and then at 95 °C for 10 min.
Dilute the lysate with 50 μl water. Mix by pipetting the solution in and out ten times.
Analyze 1 μl of the lysate using 10-μl PCR reactions with (A) a forward primer that anneals to the upstream flanking region paired with a primer of the sequence CCTGAGAAAGCAACCTGACC (285 bp from the beginning of the cassette), (B) a reverse primer that anneals to the downstream region paired with a primer of the sequence GCATTGGGTCAACAGTATAG (375 bp from the end), (C) two primers that anneal to KanMX4 with the sequences CGTACTCCTGATGATGCATG and GACGAAATACGCGATCGCTG (181 bp), and (D) two primers that anneal to the wild-type sequence of the targeted gene (Figure 4). (D) is important because about 8% of the strains in the deletion collections are known to contain aneuploidy14. A correct transformant should be positive with (A) and (B), and negative with (C) and (D).
To introduce a GMToolkit into the transformant, add roughly 250,000 cells from the transformed strain and 250,000 cells of either RY0146 (containing GMToolkit-a) or RY0148 (containing GMToolkit-α) into a 1.6-ml Eppendorf tube. Vortex. The genotypes of RY0146 and RY0148 are MATα lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 can1Δ::GMToolkit-a [CMVpr-rtTA KanMX4 STE2pr-Sp-his5] and MATα lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 can1Δ::GMToolkit-α [CMVpr-rtTA NatMX4 STE3pr-LEU2], respectively. pr denotes promoter. Cell number should be estimated from optical density of the preceding culture.
Centrifuge at 800 x g for 2 min.
Remove the supernatant.
To allow for air exchange, make a hole on the lid using a pushpin treated with 70% ethanol.
Add 50 μl of YPDA medium (1% yeast extract, 2% peptone, 2% glucose, and 0.003% adenine hemisulfate). Vortex.
Centrifuge at 800 x g for 5 min.
Place the tube in a damp box. This can be created with an empty tip box, a small tip stand, and a wet paper towel.
Incubate the box overnight at 30 °C.
Select mated diploids by first streaking out the cells on a CAA-Ura plate. Incubate the plate at 30 °C for one day.
Taking cells from the bulk area, streak them out to make isolated colonies on YPDA plates (1% yeast extract, 2% peptone, 2% glucose, 0.003% adenine hemisulfate, and 2% agar) containing 200 μg/ml G418 for selecting diploids containing GMToolkit-a or 100 μg/ml nourseothricin (Nat)15 for selecting diploids containing GMToolkit-α.
Incubate the plate at 30 °C for 3 days.
Starting from an isolated colony, transfer most of the cells to 500 μl of GNA medium without agar (1% yeast extract, 3% Difco nutrient broth, and 5% glucose; autoclave glucose and the rest of the components separately as 2× solutions to avoid caramelization) in a 5-ml culture tube with the lid loosened.
Rotate the tube horizontally for five hours at 30 °C. The axis of the rotation needs to be parallel to the longitudinal axis of the tube.
Confirm that colonies used in (1.21) are Ura+ by streaking them out on a CAA-Ura plate.
Centrifuge the tube at 800 x g for 2 min. Discard the supernatant.
Add 500 μl of minimal sporulation medium (1% potassium acetate, 0.005% zinc acetate; filter-sterilize). Vortex.
Centrifuge the tube at 800 x g for 2 min. Discard the supernatant.
Repeat (1.25) - (1.26) twice.
Add 1 ml of minimal sporulation medium supplemented with 7.5 μg/ml lysine, 7.5 μg/ml leucine, 5 μg/ml histidine, 5 μg/ml methionine, and 1.25 μg/ml uracil (to enhance the sporulation rate of the auxotrophic strains). Vortex.
Rotate the tube at room temperature for one day.
Rotate the tube at 30 °C for 2 days.
Centrifuge 125 μl of this mixture in a 1.6-ml Eppendorf tube at 800 x g for 2 min. Discard the supernatant.
Add 50 μl of zymolyase solution (100 mM sodium phosphate buffer, pH 7.4, 1 M sorbitol, and 2 units zymolyase from ZymoResearch). Mix by pipetting the solution in and out of a pipette tip.
Incubate at 30 °C for 1 hr.
Add 50 μl of 0.02% NP-40. Mix by pipetting the solution in and out of a pipette tip.
Leave the tube at room temperature for five minutes.
Add 500 μl of water.
Place the tube on ice.
Sonicate this mixture twice at the output setting of 1 (weakest setting) for 15 sec using a 3.2-mm microtip with Sonifier 450 (Branson). Between the two rounds of sonication, put the tube on ice to return the temperature to ~0 °C.
Centrifuge the tubes at 800 x g for 5 min. Discard the supernatant.
Add 200 μl of SC-Ura-His (a) or SC-Ura-Leu (α) medium, depending on whether the cross is for incorporating GMToolkit-a or GMToolkit-α. To prepare the media, make SC-Ura-His-Leu medium (0.67% yeast nitrogen base, 2% glucose, 0.01% arginine, 0.01% aspartic acid, 0.01% glutamic acid, 0.01% isoleucine, 0.01% lysine, 0.01% methionine, 0.01% phenylalanine, 0.08% serine, 0.04% threonine, 0.002% tyrosine, 0.03% valine, 0.0025% adenine, 0.005% tryptophan, 0.003% adenine, and 0.005% tryptophan) and supplement it with sterile histidine or leucine before use to the final concentration of 0.01%.
Make a hole on the lid using a pushpin treated with 70% ethanol.
Rotate the tube horizontally at 30 °C for 2 days. The axis of the rotation needs to be parallel to the longitudinal axis of the tube.
Streak out the cells on a SC-Ura-His or SC-Ura-Leu agar plate (the same ingredients as above plus 2% agar; threonine and aspartic acid must be added after autoclaving; add histidine or leucine before pouring) to make isolated colonies for the ProMonster strains.
2. Sexual Cycling
Rotate a culture of an established GFP deletion strain in 100 μl of SC-His (a) or SC-Leu (α) depending on mating type at 30 °C overnight using a 1.6-ml Eppendorf tube. For air exchange, make a hole using a pushpin treated with 70% ethanol. Rotate the tube horizontally. The axis of the rotation needs to be parallel to the longitudinal axis of the tube. It is possible to cross multiple strains via en masse mating. When a sorted sample is directly used for this purpose, culture the cells in 200 μl at 30 °C for 2 days. To prepare the medium, add uracil (final concentration: 0.0025%) and histidine (0.01%) or leucine (0.01%) to SC-Ura-His-Leu medium.
Mix 250,000 a-haploid cells and 250,000 α-haploid cells of GFP deletion strains in a 1.6-ml Eppendorf tube. Vortex.
Mate these cells by following steps (1.11) - (1.17).
Transfer 10 μl of the mating mixture to 500 μl GNA medium containing 200 μg/ml G418 and 100 μg/ml Nat in a 5-ml culture tube with a loosened lid. The starting OD600 is roughly 0.1.
Rotate the culture at 30 °C for 24 hr. This allows for the selection of diploids because only diploids containing both KanMX4 in GMToolkit-a and NatMX4 in GMToolkit-α can grow in the presence of G418 and Nat (Figure 3).
Transfer 20 μl of the one-day culture to 500 μl of fresh GNA containing G418 and Nat. The starting OD600 is roughly 0.2.
Rotate the tube at 30 °C for 5 hr to bring cells to the log phase (OD600 of ~1).
Follow the steps (1.24)-(1.38) to sporulate the diploids and isolate spores.
Split the suspension of sporulated cells into two 300-μl parts and put each into a separate 1.5-ml Eppendorf tube.
Centrifuge the tubes at 800 x g for 5 min. Discard the supernatant.
To the pellet of one tube, add 100 μl of SC-His medium to select a-haploids. To the pellet of the other tube, add 100 μl of SC-Leu medium to select α-haploids.
Rotate the tubes at 30 °C overnight. The final OD600 should be less than 0.5. If OD600 tends to be too high, try culturing at room temperature.
To induce GFP, add 100 μl of fresh SC-His (a) or SC-Leu (α) medium containing 20 μg/ml doxycycline. The final concentration of doxycycline is 10 μg/ml. Vortex.
Rotate the tubes at 30 °C for 2 days.
3. Flow Cytometry
Add 900 μl of pre-filtered TE buffer (pH 7.5) containing 10 μg/ml doxycycline to a 5-ml polypropylene tube (BD Falcon #352063) with the cap swapped with a cell-strainer cap from a 5-ml polystyrene tube (BD Falcon #352235). Polypropylene tubes are suitable for flow cytometry. The strainer cap is desired for removing large particles.
Gently place 75 μl of TE buffer with doxycycline on the cell-strainer cap.
Add 25 μl of the induced cells to the solution on the cap.
While pressing the tip of Pipetman against the mesh, shoot all of the combined solution through the strainer.
Press down the strainer cap and vortex the tube.
Prepare collection tubes (1.6-ml Eppendorf tubes) filled with 200 μl of SC-His or SC-Leu.
Start the cell sorter and adjust settings according to the manufacturer manual.
Vortex both the collection tube (to coat the wall with the medium) and the tube containing cells before loading them onto the cell sorter.
Acquire flow cytometry data of the sample. A strain not containing GFP can be a negative control.
Draw gates for sorting. (A) To avoid cell aggregates, which could exhibit high GFP intensity, discard outliers with disproportionately large pulse width for forward scatter (FSC) using a plot of pulse width and pulse height for MoFlo sorter or disproportionately small FSC height using a plot of FSC height and FSC area for FACSAria sorter (Figure 5, top-left panel). (B) Since large FSC (area) is expected for cell aggregates, only take cells in the lower 20% in FSC, while avoiding regions with the lowest FSC and side scatter (SSC) values where cell debris is often found (Figure 5, top-right panel). (C) SSC (area) and GFP signal (area) are often positively correlated. Because simply taking the brightest cells given the gates above would also enrich for cells with a high SSC value, draw a gate along the periphery of the population using a plot of GFP intensity and SSC to take a similar percentage of the brightest cells from each point of the SSC axis (Figure 5, bottom-left diagram). The population selected in (C) should be 0.1-1% of the population selected in (B).
Sort 200 cells into the collection tube given the criteria in (3.10). For an en masse process or for other projects requiring a more complex population, sort more cells as needed.
Vortex the collection tube to cover the sorted cells with medium.
Plate a subset of cells (for example, 50 cells) on a YPDA plate for genotyping.
Incubate the plate at 30 °C for 2 days.
Make a lysate for ~12 colonies following the steps (1.5) - (1.8).
Transfer the residual cells on the tip used in (1.5) to a spot on a YPDA plate containing G418 and Nat by gently touching the plate with the tip. Selected haploids should not grow on this plate. Diploids may have more GFP copies and may therefore be brighter. This test can show the efficiency of haploid selection.
For genotyping, analyze 1 μl of the lysate using a 10-μl PCR reaction with a forward primer that anneals to the upstream flanking region of each deleted gene paired with a primer of the sequence CCTGAGAAAGCAACCTGACC. Multiplex PCR reactions if possible (conditions can be determined using lysates of single mutants). PCR can be done in a 96- or 384-well format. The latter format is suitable for the reaction volume of as little as 5 μl. Arraying of PCR master mixes differing in primers and addition of cell lysates to these mixes can be carried out using a multi-channel pipette or a liquid handling robot. Tip changes are optional when adding the same lysate to different PCR mixes.
Analyze PCR products. The Gel XL Ultra V-2 electrophoresis system (Labnet) is compatible with sample loading using multi-channel pipettes.
Based on information from (3.16) and (3.18), identify likely haploids that have desired genotypes.
Establish these strains on a fresh plate (YPDA, CAA-Ura, or SC-Ura-His/Leu). Also, freeze them in 25% glycerol at -80 °C. Additional tests such as confirmation of the absence of wild-type sequences for deleted genes and pulsed-field gel electrophoresis are recommended.
Repeat sexual cycling from (2.1) with useful strains.
Representative Results
When an a-haploid strain carrying four GFP-marked deletions (ycl033cΔ yer042wΔ ykl069wΔ yol118cΔ) was crossed with an α-haploid strain carrying four deletions (ycl033cΔ ydl242wΔ ydl227cΔ yer042wΔ), with two deletions (ycl033cΔ yer042wΔ) shared by two strains, a representative result was obtained. The mating mixture was cultured in YPDA medium containing G418 and Nat to select diploids. The resulting diploids were cultured in the sporulation medium. The spores were dispersed by zymolyase treatment and sonication, and germinated in haploid selection media. The cells were then induced to express GFP. Using gates with a flow cytometer, a population of cells was selected that is unlikely to contain debris or cell aggregates (Figure 5). Within this population, the brightest 1% of the cells were sorted. Sorted cells and unsorted cells were genotyped after they formed colonies on a plate. When randomly selected colonies were streaked out on a YPDA plate containing G418 and Nat, cells from 2 out of 16 colonies grew for the unsorted sample, and cells from 18 out of 26 colonies grew for the sorted sample, suggesting that some diploids gained the ability to express a haploid-selection marker and propagated during the haploid-selection and GFP-induction phases in this experiment. Diploids were further enriched in the sorted sample presumably due to the larger number of GFP copies they contained. When haploids were analyzed with PCR, the average number of deletions in the sorted cells was 5.1±0.4 (n = 8; SEM shown). Four of these cells had six deletions, the maximally possible number of deletions for this cross. Haploids of the unsorted sample had 3.4±0.2 deletions on average (n = 14).
In a separate experiment, an a-haploid strain carrying seven GFP-marked deletions (yer042wΔ yer108cΔ yfr057wΔ ykl069wΔ ykr011cΔ ylr123cΔ yol118cΔ) was crossed with an α-haploid strain carrying eight deletions (ycl033cΔ ydl227cΔ ydl242wΔ yer042wΔ yer108cΔ yfr057wΔ ygl109wΔ ykr011cΔ), with four common deletions (yer042wΔ yer108cΔ yfr057wΔ ykr011cΔ) contained in the two strains. When diploids were selected and subsequently cultured in the sporulation medium, roughly 20% of the cells sporulated based on microscopic observation (n > 100). After the spores were dispersed and germinated, the resulting haploids were induced to express GFP and sorted based on fluorescence as above. Only one of 61 randomly selected cells grew on a YPDA plate containing G418 and Nat, suggesting that sorted cells were predominantly haploid. When analyzed with PCR (Figure 6), the sorted cells had the average deletion number of 8.8±0.3 (n = 12; SEM shown), including five cells containing ten deletions. The expected number of deletions for an unsorted cell resulting from random segregation was 7.5.
 Figure 1. Green Monsters under the microscope. Fluorescence micrographs show a non-mutant strain (left), a ProMonster strain (middle), and a 16-GFP monster strain (right). Identical exposure, brightness, and contrast settings were used for images.
Figure 1. Green Monsters under the microscope. Fluorescence micrographs show a non-mutant strain (left), a ProMonster strain (middle), and a 16-GFP monster strain (right). Identical exposure, brightness, and contrast settings were used for images.
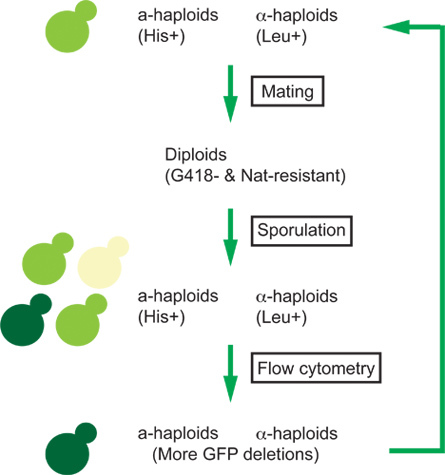 Figure 2. The scheme of the Green Monster process. Single-mutant haploids (light green) are mated. Meiotic recombination during sporulation of the mated diploids generates a mixture of 0-GFP cells (off-white), 1-GFP cells (light green), and 2-GFP cells (dark green). Flow cytometry is used to select the greenest cells enriched for the 2-GFP cells. Integrated molecular tools enable the selection of a-haploids (His+), α-haploids (Leu+), and diploids (G418- and Nat-resistance). This cycle is repeated to enrich for strains bearing an ever-increasing number of alterations.
Figure 2. The scheme of the Green Monster process. Single-mutant haploids (light green) are mated. Meiotic recombination during sporulation of the mated diploids generates a mixture of 0-GFP cells (off-white), 1-GFP cells (light green), and 2-GFP cells (dark green). Flow cytometry is used to select the greenest cells enriched for the 2-GFP cells. Integrated molecular tools enable the selection of a-haploids (His+), α-haploids (Leu+), and diploids (G418- and Nat-resistance). This cycle is repeated to enrich for strains bearing an ever-increasing number of alterations.
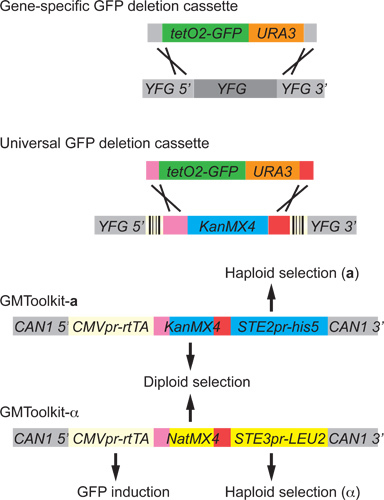 Figure 3. GFP deletion cassettes and GMToolkits. GFP deletion cassettes contain a GFP reporter gene and a yeast transformation marker (URA3). The tetO2 promoter is inducible by the addition of doxycycline in the medium through the action of the rtTA transcription factor. If the GFP level reaches the maximal capacity of cells to make the protein or to tolerate any toxic effect from it, the expression can be dialed down by lowering the doxycycline concentration (note however that we did not observe such saturation or toxicity effects even with 16 copies of GFP). This cassette can be targeted to any gene or region by attaching specific homologous sequences using PCR. Alternatively, the universal GFP deletion cassette with identical homologous sequences can be conveniently targeted to KanMX4 'landing pads' in strains of the yeast knockout collection. YFG denotes your favorite gene. Box with vertical lines denotes DNA barcode. GMToolkits are integrated into the CAN1 locus. The STE2-his5 marker and the STE3-LEU2 marker are expressed in a mating type specific manner in haploids to allow only a-haploids to grow in the absence of histidine and only α-haploids to grow in the absence of leucine, respectively. Selecting for resistance to G418 and Nat simultaneously selects for the KanMX4 locus in one GMToolkit and the NatMX4 locus in the other and thus selects for diploids.
Figure 3. GFP deletion cassettes and GMToolkits. GFP deletion cassettes contain a GFP reporter gene and a yeast transformation marker (URA3). The tetO2 promoter is inducible by the addition of doxycycline in the medium through the action of the rtTA transcription factor. If the GFP level reaches the maximal capacity of cells to make the protein or to tolerate any toxic effect from it, the expression can be dialed down by lowering the doxycycline concentration (note however that we did not observe such saturation or toxicity effects even with 16 copies of GFP). This cassette can be targeted to any gene or region by attaching specific homologous sequences using PCR. Alternatively, the universal GFP deletion cassette with identical homologous sequences can be conveniently targeted to KanMX4 'landing pads' in strains of the yeast knockout collection. YFG denotes your favorite gene. Box with vertical lines denotes DNA barcode. GMToolkits are integrated into the CAN1 locus. The STE2-his5 marker and the STE3-LEU2 marker are expressed in a mating type specific manner in haploids to allow only a-haploids to grow in the absence of histidine and only α-haploids to grow in the absence of leucine, respectively. Selecting for resistance to G418 and Nat simultaneously selects for the KanMX4 locus in one GMToolkit and the NatMX4 locus in the other and thus selects for diploids.
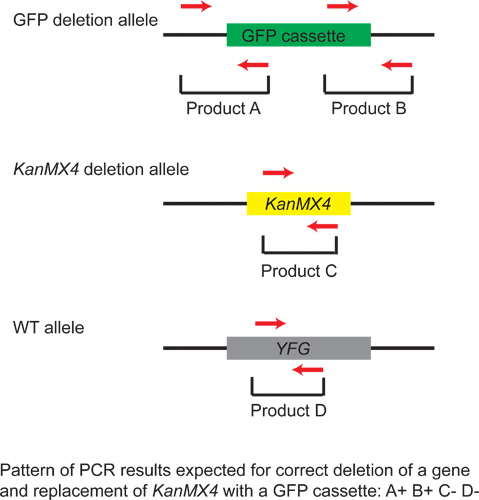 Figure 4. Confirming the correct integration of the GFP cassette. A universal GFP cassette can be used to convert any KanMX4-marked deletion available in the yeast deletion collection to a GFP-marked deletion. With a correctly generated GFP deletion allele, PCR reaction with a forward primer that anneals to the upstream flanking region paired with a reverse primer that anneals to the beginning of the cassette (PCR A of Step 1.9) should make a band. PCR with a reverse primer that anneals to the downstream flanking region paired with a forward primer that anneals to the end of the cassette (PCR B) should make a product. PCR with two primers that anneal within the KanMX4 cassette (PCR C) or with two primers that anneal to the wild-type sequence of the targeted gene (PCR D) should not produce a band. Red arrow denotes a PCR primer. Bracket shows a region amplified with a PCR reaction. Box indicates the GFP cassette (green), the KanMX4 cassette (yellow), or your favorite gene (YFG, gray). Black line indicates a flanking sequence in a yeast chromosome.
Figure 4. Confirming the correct integration of the GFP cassette. A universal GFP cassette can be used to convert any KanMX4-marked deletion available in the yeast deletion collection to a GFP-marked deletion. With a correctly generated GFP deletion allele, PCR reaction with a forward primer that anneals to the upstream flanking region paired with a reverse primer that anneals to the beginning of the cassette (PCR A of Step 1.9) should make a band. PCR with a reverse primer that anneals to the downstream flanking region paired with a forward primer that anneals to the end of the cassette (PCR B) should make a product. PCR with two primers that anneal within the KanMX4 cassette (PCR C) or with two primers that anneal to the wild-type sequence of the targeted gene (PCR D) should not produce a band. Red arrow denotes a PCR primer. Bracket shows a region amplified with a PCR reaction. Box indicates the GFP cassette (green), the KanMX4 cassette (yellow), or your favorite gene (YFG, gray). Black line indicates a flanking sequence in a yeast chromosome.
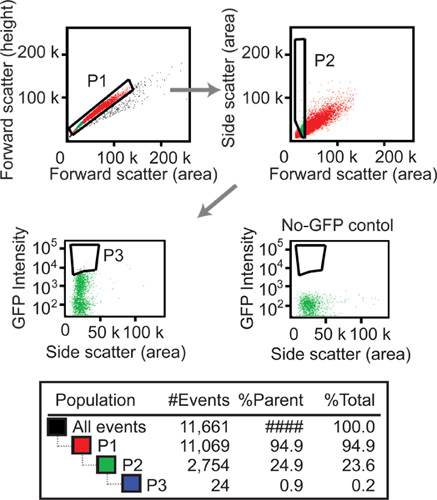 Figure 5. Flow cytometry. In this experiment, haploid progeny from a cross of two strains, one with the genotype ycl033cΔ yer042wΔ ykl069wΔ yol118cΔ and the other with the genotype ycl033cΔ ydl242wΔ ydl227cΔ yer042wΔ were induced to express GFP. Each gene deletion had a GFP cassette. By gating cells on the basis of forward scatter area and forward scatter height (P1), cell aggregates (which tend to have disproportionately large forward scatter area were excluded. By gating cells on the basis of forward scatter and side scatter (P2), cells in the lower 20% in forward scatter were accepted while excluding debris with the lowest forward scatter and side scatter. By gating cells on the basis of side scatter and GFP signal (P3), the more fluorescent cells among those in a given side scatter range were taken. To allow comparison of the meiotic mix and the negative control sample that does not express GFP, a gate identical to the one used for selecting the P3 population is shown in a diagram for the similarly selected cells of the negative control.
Figure 5. Flow cytometry. In this experiment, haploid progeny from a cross of two strains, one with the genotype ycl033cΔ yer042wΔ ykl069wΔ yol118cΔ and the other with the genotype ycl033cΔ ydl242wΔ ydl227cΔ yer042wΔ were induced to express GFP. Each gene deletion had a GFP cassette. By gating cells on the basis of forward scatter area and forward scatter height (P1), cell aggregates (which tend to have disproportionately large forward scatter area were excluded. By gating cells on the basis of forward scatter and side scatter (P2), cells in the lower 20% in forward scatter were accepted while excluding debris with the lowest forward scatter and side scatter. By gating cells on the basis of side scatter and GFP signal (P3), the more fluorescent cells among those in a given side scatter range were taken. To allow comparison of the meiotic mix and the negative control sample that does not express GFP, a gate identical to the one used for selecting the P3 population is shown in a diagram for the similarly selected cells of the negative control.
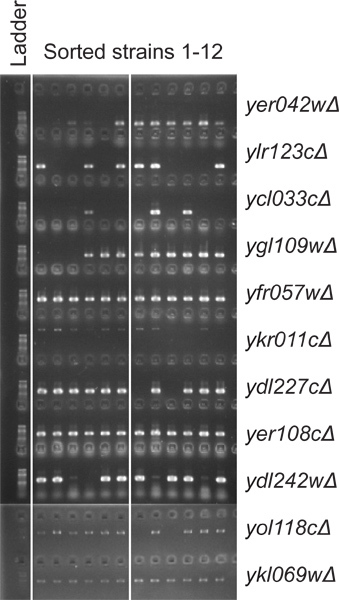 Figure 6. Genotyping the sorted strains. PCR was performed using a reverse primer that anneals to the downstream flanking region paired with a forward primer that anneals to the end of the cassette. The presence of band indicates the presence of the GFP cassette. A separate experiment showed that all of the twelve strains contain the yer042wΔ deletion and the ykr011cΔ deletion.
Figure 6. Genotyping the sorted strains. PCR was performed using a reverse primer that anneals to the downstream flanking region paired with a forward primer that anneals to the end of the cassette. The presence of band indicates the presence of the GFP cassette. A separate experiment showed that all of the twelve strains contain the yer042wΔ deletion and the ykr011cΔ deletion.
Discussion
As we developed the Green Monster approach, we were concerned with the possibility of recombination between different GFP replacement cassettes, leading to genome rearrangement. Mitigating against this possibility is our selection for cells that have successfully undergone multiple rounds of mating and meiosis. Cells bearing rearranged genomes are expected to be less fit after mating to cells without an identical rearrangement. Indeed, we did not observe any genome instability resulting from recombination between GFP cassettes11. However, we cannot entirely exclude this possibility, so it is recommended that users test the generated strains for rearrangement. Pulsed-field gel electrophoresis can be used to detect gross abnormality. PCR with primers annealing to sequences within the deletion can be used to confirm the absence of wild-type sequences.
Based on the fluorescence levels of established strains, GFP intensity had a near-linear relationship with the number of deletions, suggesting that saturation is not a problem within the tested range of one to 16 deletions11. However, even in later rounds with strains with many non-overlapping deletions, we were able to only increase the average number of deletions in the population by roughly two in each round, whereas theoretically a cell bearing the union of all deletions in the two parental strains could be combined at once if perfect stringency for GFP selection were achieved. One limitation is the cell-to-cell variation of GFP intensity amongst isogenic cells. In the second example in Representative Results, only 0.8% of cells in the population would be expected to have all 11 deletions that were possible in this cross (four of the 11 deletions were shared by both parental haploids). However, the more abundant ten-deletion strains will have a distribution of GFP intensity that overlaps that of the 11-deletion strains. Therefore, an even smaller portion of the population must be selected to preferentially select 11-deletion strains from the higher tail of the GFP intensity distribution of 11-deletion strains. One solution may be to simply raise the threshold to sort rarer cells with higher GFP intensity. Another solution may be to simultaneously measure an internal standard, a fluorescent protein reporter of a different color that is present in a single copy, expressed via the same tetO2 promoter. This strategy would be expected to cancel out extrinsic noise to reduce the cell-to-cell variation of thus-normalized GFP intensity measurements16,17.
The difficulty of obtaining rare multi-mutants can be exacerbated by the potentially slow growth rate of these strains. Multi-mutants are enriched using flow cytometry, but they may be outcompeted during the rest of the process by fitter siblings. To minimize the number of generations yeast strains undergo, we recommend small culture volumes that provide just-sufficient amplification of cells of the desired ploidy that are selected at each step.
Because multiple deletions can be acquired per cycle, the Green Monster method can be used to assemble multi-mutants more quickly than sequential methods11. If specific subsets of deletions have synthetic lethal relationships, 'dead ends' may be encountered with sequential approaches. This limitation can be circumvented by the Green Monster approach, which samples from the space of possible paths toward accumulating the full set of deletions. The more parallel en masse version of the Green Monster process should prove useful in finding a path to reach the largest tolerable number of deletions.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by the US Defense Advanced Research Projects Agency contract N66001-12-C-4039 to Y.S., a grant from the Alfred P. Sloan Foundation to R.S.L., and US National Institutes of Health grants R01 HG003224 and R21 CA130266 to F.P.R. F.P.R. was also supported by a fellowship from the Canadian Institute for Advanced Research and by the Canada Excellence Research Chairs program.
References
- Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science's STKE. 2001;294:2364–23. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beh CT, Cool L, Phillips J, Rine J. Overlapping functions of the yeast oxysterol-binding protein homologues. Genetics. 2001;157:1117. doi: 10.1093/genetics/157.3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorke R, et al. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS letters. 1999;464:123–128. doi: 10.1016/s0014-5793(99)01698-1. [DOI] [PubMed] [Google Scholar]
- Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akada R, et al. PCR-mediated seamless gene deletion and marker recycling in Saccharomyces cerevisiae. Yeast. 2006;23:399–405. doi: 10.1002/yea.1365. [DOI] [PubMed] [Google Scholar]
- Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic acids research. 1996;24:2519. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delneri D, et al. Exploring redundancy in the yeast genome: an improved strategy for use of the cre-loxP system. Gene. 2000;252:127–135. doi: 10.1016/s0378-1119(00)00217-1. [DOI] [PubMed] [Google Scholar]
- Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nat. Biotech. 2001;19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- Noskov VN, Segall-Shapiro TH, Chuang R. Tandem repeat coupled with endonuclease cleavage (TREC): a seamless modification tool for genome engineering in yeast. Nucl. Acids Res. 2010;38:2570–2576. doi: 10.1093/nar/gkq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, et al. Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat. Methods. 2011;8:159–164. doi: 10.1038/nmeth.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA. Functional Characterization of the S. Genome by Gene Deletion and Parallel Analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Woods RA, Gietz RD. High-efficiency transformation of plasmid DNA into yeast. Methods Mol. Biol. 2001;177:85–97. doi: 10.1385/1-59259-210-4:085. [DOI] [PubMed] [Google Scholar]
- Hughes TR, et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nature. 2000;25:333–337. doi: 10.1038/77116. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Newman JRS, et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Gene regulation at the single-cell level. Science. 2005;307:1962. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]