

Abstract
In eukaryotes, most of the messenger RNAs (mRNAs) that encode secreted and membrane proteins are localized to the surface of the endoplasmic reticulum (ER). However, the visualization of these mRNAs can be challenging. This is especially true when only a fraction of the mRNA is ER-associated and their distribution to this organelle is obstructed by non-targeted (i.e. "free") transcripts. In order to monitor ER-associated mRNAs, we have developed a method in which cells are treated with a short exposure to a digitonin extraction solution that selectively permeabilizes the plasma membrane, and thus removes the cytoplasmic contents, while simultaneously maintaining the integrity of the ER. When this method is coupled with fluorescent in situ hybridization (FISH), one can clearly visualize ER-bound mRNAs by fluorescent microscopy. Using this protocol the degree of ER-association for either bulk poly(A) transcripts or specific mRNAs can be assessed and even quantified. In the process, one can use this assay to investigate the nature of mRNA-ER interactions.
Keywords: Cellular Biology, Issue 70, Biochemistry, Genetics, Molecular Biology, Genomics, mRNA localization, RNA, digitonin extraction, cell fractionation, endoplasmic reticulum, secretion, microscopy, imaging, fluorescent in situ hybridization, FISH, cell biology
Introduction
In eukaryotes, mRNA encoding secreted and membrane proteins can be targeted to the ER co-translationally by the signal recognition particle1,2 and can be maintained on the ER via the direct interactions between ribosomes and translocons during the translation3,4. However, whether mRNAs can be targeted and maintained on the ER independent of either ribosomes or translation was unclear until very recently. Previous studies attempted to address whether there is translation-independent mRNA association with the ER using cellular fractionation techniques. Because harsh chemical conditions were required to disassociate ribosomes from ER derived vesicles, which might disrupt the potentially delicate mRNA-ER association, these studies were inconclusive, providing evidence for5-8 and against9,10 ribosomal-independent anchoring of mRNA to the ER.
To circumvent these problems we have developed a protocol to isolate and image ER-bound mRNAs. This procedure involves a mild extraction treatment, which effectively removes all the cytoplasmic content of the cell (including non ER-bound mRNAs) while simultaneously preserving the ER morphology and all of its associated molecules. Using this protocol we have demonstrated that a subset of mRNAs are targeted and then maintained on the ER independently of ribosomes or translation11.
Protocol
1. Preparation of Materials for Extraction
- Preparation of Cells
- Seed tissue culture cells on acid-treated coverslips for at least one day prior to the experiment. We use COS-7 or U2OS, as these two cell lines exhibit robust production of secreted protein and have a well defined ER. Note that to achieve high extraction efficiency, cells should not exceed 80% confluency on the coverslip on the day of the experiment.
- If a particular exogenous mRNA is being investigated, cells are transfected one day after seeding with plasmids containing the gene of interest using standard transfection protocols. The cells are then allowed to express the mRNA for at least 18 hr prior to extraction.
- Treating the Cells with Translation Inhibitors
- To test the effect of intact ribosomes on the maintenance of mRNAs association with the ER, cells can be treated with translation inhibitors that disrupt the association of ribosomes with the mRNA11.
- Inhibitors of translation initiation, such as homoharringtonin (HHT), pactamycin, or 2-(4-methyl-2,6-dinitroanilino)-N-methylpropionamide (MDMP), prevent new ribosomes from associating with the transcript while simultaneously allowing engaged ribosomes to complete translation12-14. Since the translation rate for most proteins in mammalian cells is 5 amino acids per second, regardless of codon usage or mRNA length15, a treatment of 30 min should clear ribosomes off of messages with open reading frames as long as 27,000 nucleotides (coding for proteins as long as 9,000 amino acids).
- Inhibitors such as puromycin promote the premature ejection of the nascent chain and thus disrupt polysomes12. However to completely disrupt the association of puromycin-treated ribosomes with the mRNA, magnesium chelators such as EDTA must be added to the digitonin extraction buffer11.
- In this experiment, cells are treated with 200 μM puromycin, 5 μM HHT, or control medium for 30 min prior to extraction.
- Preparation of Digitonin Extraction Buffer. To visualize ER-localized mRNAs without the obstruction of free (i.e. cytoplasmic, non-ER-associated) transcripts, we selectively permeabilize the cell with digitonin, a steroid glycoside that interacts preferentially with 3β-hydroxysterols. At low concentrations, digitonin selectively permeabilizes portions of the plasma membrane, causing 'leakiness'16. In contrast, the ER membrane and nuclear envelope, which are poor in cholesterol, are left intact16,17.
- Digitonin (Sigma Aldrich) powder is dissolved in distilled water at 5% weight/volume and this stock solution is aliquoted into small volumes and stored in -20 °C. In our experience, multiple freeze-thaw cycles decreases the efficiency of dissolved digitonin.
- A stock solution of 10x CHO buffer (1x working solution concentration: 115 mM KAc, 25 mM HEPES pH 7.4, 2.5 mM MgCl2, 2 mM EGTA and 150 mM Sucrose) is prepared with RNase-free reagents and stored at -20 °C. A working solution (1x CHO buffer) is prepared by diluting the stock solution with RNase-free water and can be stored at 4 °C.
2. Digitonin Extraction
- Preparing Extraction Solutions
- A heating block with a flat surface is pre-heated to 40 °C and maintained at this temperature during to the extraction.
- To form a flat working surface that is RNase free, the heating block is moistened with some water and overlaid with a fresh piece of parafilm M (Bemis Company, Inc). Air bubbles between the parafilm sheet and heating block are smoothed out using RNase-free gloves and kimwipes (VWR).
- 1x CHO buffer is warmed to 37 °C by incubating in a water bath.
- 6-well plates are used to wash and fix the cells. Each row of 3 wells is used for one cover-slip. To the first two wells in the row, 2 ml of warmed CHO buffer is added. To the last well, 2 ml of the fixation solution (PBS + 4% paraformaldehyde (Electron Microscope Sciences)) is added. This tray can be stored in the 37 °C incubator until the cells are ready for extraction.
- Digitonin extraction solution is prepared by diluting the stock digitonin solution to 0.025% with warm CHO buffer just prior to use. Again note that for puromycin-treated cells, the extraction buffer must additionally contain 20 mM EDTA to efficiently disrupt ribosomes11. Drops of 100 μl of the extraction solution are placed on the parafilm-covered heating block immediately before extraction.
- Digitonin Extraction and Cell Fixation
- Remove the cells from the 37 °C incubator.
- During the extraction process, coverslips are manipulated with the help of jewler's forceps. With the forceps pick up the coverslip and dip it into the first and then the second well containing CHO buffer to wash of the growth medium.
- Quickly blot off excess buffer off the back side of the coverslip with a kimwipe and immediately place the coverslip, cell side facing down, on the extraction solution.
- After 10 sec, pick up the coverslip with the forceps and immediately transfer it, cell side facing up, to the well containing the 4% paraformaldehyde fixing buffer.
- Let the sample incubate in fixative for at least 15 min at room temperature.
- Preparation of Unextracted Control Cells. To determine the total amount of mRNA in the cytoplasm it is a good idea to prepare an unextracted control sample.
- Cells grown on coverslips are prepared as above (see section 2.2), except that the digitonin extraction step (2.2.3) is skipped.
- After fixation, the un-extracted cells need to be permeabilized in PBS + 0.1% Triton X-100 for 15 min at room temperature.
3. FISH Staining
- Designing probes for fluorescence in situ hybridization
- The primary sequence of the mRNA of interested is folded using RNA secondary structure prediction software, such as RNAstructure 5.018. The mRNA folds are visually assessed to identify a region of about 50 nucleotides that is free of major secondary structures.
- The reverse complement of this region is synthesized with an Alexa546 fluorophore attached to the 5' end of the probe (purchased from Integrated DNA Technologies).
- The probe is diluted with RNase-free water to a stock concentration of 100 μM which can be stored at -20 °C.
- Staining with FISH probes
- Note that although the digitonin-extracted cells do not require further permeabilization, treating these fixed samples with 2 ml of 0.1% Triton X-100 diluted in PBS for 15 min at room temperature prior to FISH staining, helps to reduce the background signal.
- The slides are then washed twice with PBS to remove any residual paraformaldehyde or Triton X-100.
- The cells are then washed twice with 25% to 60% formamide in 1X Sodium Citrate Buffer (150 mM NaCl and 15 mM sodium citrate). Generally, for specific FISH probes that are 50-60 nucleotides in length, 60% formamide is used, but for poly(A) mRNA staining with oligo(dT) FISH probe, the level of formamide is reduced down to 25%.
- To prepare the staining chamber, the bottom of a 150 mm Petri dish is covered with water and a piece of parafilm is floated on the water. The water is removed by turning the dish over, thus allowing the parafilm to adhere to the bottom of the dish. Air bubbles are manually removed with kimwipes.
- For each coverslip to be stained, pipette a 100 μl drop of the hybridization solution containing FISH probe of interest onto the parafilm in the staining chamber. The stock FISH probe is diluted with 25% to 60% formamide in hybridization buffer (1x SSC, 100 mg/ml dextran sulphate, 1 mg/ml yeast tRNA, 5mM vanadyl riboside complex, 25-60% formamide) to a working concentration of 0.2 μM. Note that the amount of formamide should be optimized for the particular probe being used and is present at the same concentration as in the SSC wash buffer (see step 3.3.3).
- The coverslip is placed cell side face down onto the FISH solution in the staining chamber and incubated for 5-24 hr at 37 °C in a humidified chamber such as the tissue culture incubator.
- Washing and mounting the stained samples
- To prepare an RNase free washing area, lay down a strip of parafilm on a level flat surface, such as a lab bench. To ensure that the parafilm is flat, wet the level surface, place the parafilm on top and manually remove any air bubbles with kimwipes.
- The staining chamber is removed from the incubator and placed on a flat level surface. The coverslips are then floated by pipetting approximately 500 μl of FISH wash buffer near the edge of each coverslip. The solution will be drawn in-between the coverslip and the parafilm via capillary action.
- For each coverslip, 1 ml of wash buffer (1x SCC with 25-60% formamide, see step 3.3.3) is pipetted onto the washing area parafilm (see step 3.4.1).
- Using forceps, the coverslips are removed from the staining chamber and each is placed onto the drop of wash buffer and incubated at room temperature for 5 min. The washing process is repeated 3 times.
- Glass slides are washed with 70% ethanol, and dried with kimwipes.
- For each coverslip, 25 μl of mounting solution (DAPI Fluoromount G, Southern Biotech) is pipetted onto the slide. Note that this solution contains 4',6-diamidino-2-phenylindole (DAPI), which stains DNA and allows for the visualization of nuclei.
- Using forceps and kimwipes, the back of the coverslip is dried and excess FISH wash buffer is blotted off without drying the sample. Each coverslip is then placed cell side facing down, onto the drop of mounting solution.
- Mounted samples are then labeled and can be store at 4 °C.
4. Imaging and Quantification
- Imaging
- An epifluorescence microscope is used to image the cells following FISH staining. The transfected cell can be differentiated from untransfected cells by bright fluorescence signal in the appropriate channel. Ideally, each acquired field will also contain an untransfected cell to be used to determine the background fluorescence for quantification.
- For each field images of the FISH and DAPI channel are acquired. In addition, if the cells are co-stained with protein markers, an image of the immunofluorescence channel is also acquired. Note that digitonin extraction can also be used to visualize ER-bound ribosomes and proteins11.
- To ensure that the fluorescence intensities between fields can be compared, the exposure time for each channel should remain constant for each set of experiment.
- Again to ensure that the fluorescent intensities between cells on different coverslips can be compared, the day-to-day changes in epiluminescence intensity or decay in the FISH signal should be minimized by imaging all of the coverslips in a single sitting.
- Quantification. To quantify the amount of mRNA that is localized on the ER, we use Nikon NIS Element software. However other image analysis software such as ImageJ can be used.
- Using the Nikon NIS ELEMENT image analysis tool, the cell periphery and the nucleus are outlined as separate Region of Interests (ROIs).
- For each ROI, the fluorescent intensity (IT for the total cell intensity and IN for the nuclear intensity) and the area (AT and AN) are recorded and exported to an excel spreadsheet.
- For each image, the background fluorescent intensity (IB) is determined by recording the intensity of a non-transfected cell. If poly(A) mRNA levels are being analyzed, a cell-free area is selected.
- The total amount of ER (in extracted cells) or cytoplasmic (in unextracted cells) fluorescence is calculated by the formula: [AT][ IT - IB]- [AN][ IN - IB]
- The nuclear staining intensity can also be calculated and used as internal control between various treatments. Since nuclei are not affected by digitonin treatment17 or ribosome disruption11, the amount of nuclear mRNA should remain constant between each of these treatments. To calculate nuclear fluorescence the following formula is used: [AN][ IN - IB]
- The percent of cytoplasmic mRNA that is ER-targeted can be calculated by taking the average ER fluorescence of 30-40 extracted cells (see 4.2.4) divided by the average cytoplasmic fluorescence from 30-40 unextracted cells (again see 4.2.4). It is critical that the extracted and unextracted cells are prepared, stained and imaged in parallel.
Representative Results
To determine the percentage of mRNA that is ER-associated, COS-7 cells that were transfected with plasmids that contained the placental alkaline phosphatase (ALPP) or cytochrome P450-8B1 (CYP8B1) were either extracted with digitonin and then fixed, or directly fixed (see Figure 1, compare "Unextracted Ctrl" to "Extracted Ctrl"). The non-nuclear fluorescence was quantified in both samples and the fraction of ER-associated mRNA was calculated (Figure 2). Our data clearly shows that for both ALPP and CYP8B1, about 60% of the cytoplasmic mRNA is associated with the ER. Since both of these mRNAs encode proteins that are processed in the ER-lumen, our results are consistent with the idea that such mRNAs are translated on the surface of the ER.
To monitor the ER-association of these transcripts after ribosome dissociation, transfected COS-7 cells were treated with control media, puromycin or HHT for 30 min then extracted, fixed and stained using specific FISH probes directed against each mRNA (for representative images see Figure 1, for quantification of ER- and nuclear-associated mRNA see Figure 3). Note that only ALPP, and not CYP8B1 mRNA, is maintained on the ER after ribosomes are disassembled with puromycin or HHT (Figures 2-3). Importantly the level of nuclear mRNA did not change between the variously treated samples for either mRNA (Figure 3). This constant nuclear FISH signal indicates that the changes in fluorescence intensity in the ER fraction are not due to alterations in mRNA expression or variations in FISH staining.
From these results we conclude that a subset of ER-targeted mRNAs, such as the ALPP transcript, is maintained on the surface of this organelle after ribosome disassembly.
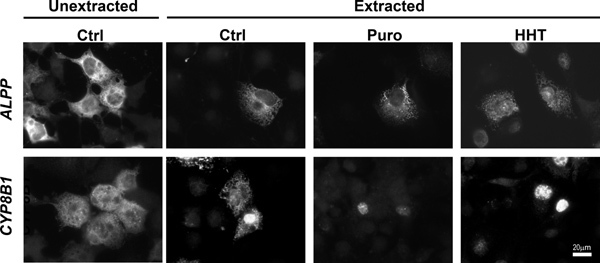 Figure 1. ALPP, but not CYP8B1 mRNA remains associated with the ER independently of ribosomes and translation. COS-7 cells were transfected with plasmids containing either the ALPP (top row), or CYP8B1 (bottom row) genes and allowed to express mRNA for 18-24 hr. The cells were then treated with DMSO control medium ("Ctrl"), puromycin ("Puro") or HHT for 30 min. Cells were either directly fixed ("unextracted") or first extracted then fixed. Note that while the control and HHT-treated cells were extracted with digitonin alone, Puro-treated cells were extracted with digitonin and EDTA. Cells were stained for mRNA using specific FISH probes, and imaged. Scale bar = 20 μm.
Figure 1. ALPP, but not CYP8B1 mRNA remains associated with the ER independently of ribosomes and translation. COS-7 cells were transfected with plasmids containing either the ALPP (top row), or CYP8B1 (bottom row) genes and allowed to express mRNA for 18-24 hr. The cells were then treated with DMSO control medium ("Ctrl"), puromycin ("Puro") or HHT for 30 min. Cells were either directly fixed ("unextracted") or first extracted then fixed. Note that while the control and HHT-treated cells were extracted with digitonin alone, Puro-treated cells were extracted with digitonin and EDTA. Cells were stained for mRNA using specific FISH probes, and imaged. Scale bar = 20 μm.
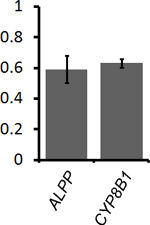 Figure 2. Quantification of the level of ER-association for ALPP and CYP8B1 mRNA. Transfected COS-7 cells were either directly fixed to determine the total level of mRNA in the cytoplasm (see Figure 1, "Unextracted Ctrl" cells) or first extracted and then fixed to determine the amount of ER-associated mRNA (see Figure 1, "Extracted Ctrl" cells). For each experiment the average ER-associated fluorescence of 30-40 extracted cells was divided by the average cytoplasmic fluorescence of 30-40 unextracted cells, to give the fraction of ER-bound mRNA (y-axis). Each bar represents the average value and standard error of three independent experiments.
Figure 2. Quantification of the level of ER-association for ALPP and CYP8B1 mRNA. Transfected COS-7 cells were either directly fixed to determine the total level of mRNA in the cytoplasm (see Figure 1, "Unextracted Ctrl" cells) or first extracted and then fixed to determine the amount of ER-associated mRNA (see Figure 1, "Extracted Ctrl" cells). For each experiment the average ER-associated fluorescence of 30-40 extracted cells was divided by the average cytoplasmic fluorescence of 30-40 unextracted cells, to give the fraction of ER-bound mRNA (y-axis). Each bar represents the average value and standard error of three independent experiments.
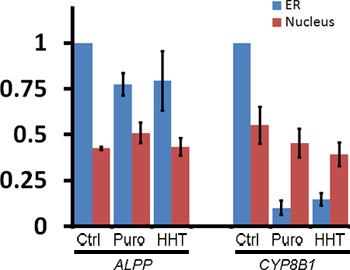 Figure 3. Quantification of the ER-association of ALPP and CYP8B1 mRNA after ribosome dissociation. Transfected COS-7 cells were treated with control medium or various translation inhibitors, extracted, fixed, stained for mRNA using specific FISH probes and imaged (see Figure 1, "Extracted" cells). The fluorescence intensities of mRNA in the ER and nucleus were quantified. Each bar represents the average and standard error of three independent experiments, each consisting of the average integrated intensity of 30 cells over background and normalized to the fluorescence in the ER of control-treated cells (y-axis). Note that although ribosome disruption caused CYP8B1 mRNA to dissociate from the ER, the levels of nuclear mRNA were unaffected.
Figure 3. Quantification of the ER-association of ALPP and CYP8B1 mRNA after ribosome dissociation. Transfected COS-7 cells were treated with control medium or various translation inhibitors, extracted, fixed, stained for mRNA using specific FISH probes and imaged (see Figure 1, "Extracted" cells). The fluorescence intensities of mRNA in the ER and nucleus were quantified. Each bar represents the average and standard error of three independent experiments, each consisting of the average integrated intensity of 30 cells over background and normalized to the fluorescence in the ER of control-treated cells (y-axis). Note that although ribosome disruption caused CYP8B1 mRNA to dissociate from the ER, the levels of nuclear mRNA were unaffected.
Discussion
The localization of mRNAs to various subcellular sites, through the interaction of transcripts with mRNA localization proteins, is a widespread phenomenon important for the proper sorting of proteins to their final destination, and for the fine tuning of gene expression to the local requirements of a subcellular region19,20.
Using the assay described here, we revisited the question of whether mRNAs that encode secretory proteins can be maintained on the ER in the presence or absence of ribosomes or translation. Indeed, we found that a subset of these mRNAs have this activity11. For example, using this extraction protocol, we determined that both ALPP and CYP8B1 mRNAs are localized on the surface of the ER in control treated COS-7 cells (Figures 1-2). However, after treating the cells with translation inhibitors puromycin or HHT, only ALPP, but not CYP8B1 mRNA, remained associated with the ER in the absence of functional ribosomes and translation (Figures 1, 3). In addition we previously found that ALPP mRNA remains ER-associated in COS-7 cells treated with pactamycin11. Since these various treatments act through divergent mechanisms to clear mRNA of associated ribosomes, it is unlikely that the ER-association of ALPP is the result of some indirect cellular response to any particular drug. To confirm that these translation inhibitors are effective, it is also helpful to test whether they inhibit the incorporation of 35S-methionine into newly synthesized proteins. For example, we have found that MDMP-treatment (100 μM, 15 min) can only inhibit 40-60% of all protein synthesis in COS-7 cells (X.A. Cui and A.F. Palazzo, unpublished observations). As a result, this drug does not efficiently disrupt the ER-localization of mRNAs that require translation for their targeting and maintenance (X.A. Cui and A.F. Palazzo, unpublished observations).
To further ensure that ER-targeting of the mRNA is not dependent on encoded signal sequence or transmembrane domains, one can alter the exogenous transcript so that it encodes a cytoplasmic protein. Indeed, we previously demonstrated that a version of the ALPP mRNA that lacks both signal sequence and transmembrane coding regions is still able to localize to the ER11.
It is also worth noting that the method presented here when combined with other assays used to analyze mRNA nuclear export21, can be used to define the kinetics and requirements of mRNA transport from one compartment to another (e.g. from the nucleus to the surface of the ER). Indeed, by microinjecting plasmids that contain the ALPP gene into the nuclei of COS-7 cell that were pretreated with translation inhibitors, we previously demonstrated that newly made ALPP mRNAs are targeted to the ER independently of translation or ribosome-association11.
Although we do not fully understand how these mRNAs are targeted and anchored to the ER, it is likely that this organelle contains mRNA receptors. Indeed we recently identified p180, a large positively-charged membrane-bound protein, as being required for the ability of ALPP mRNA to be maintained on the ER independently of translation11. However, it is likely that other mRNA receptors must be present on the surface of the ER11. With the help of the technique outlined here, we hope to identify and dissect many of the molecular mechanisms that help regulate mRNA localization in mammalian cells.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by grants from the National Science and Engineering Research Council of Canada to X.A.C. and A.F.P.
References
- Walter P, Ibrahimi I, Blobel G. Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 1981;91:545–550. doi: 10.1083/jcb.91.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Blobel G. Translocation of proteins across the endoplasmic reticulum. II. Signal recognition protein (SRP) mediates the selective binding to microsomal membranes of in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 1981;91:551–556. doi: 10.1083/jcb.91.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grlich D, Prehn S, Hartmann E, Kalies KU, Rapoport TA. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell. 1992;71:489–503. doi: 10.1016/0092-8674(92)90517-g. [DOI] [PubMed] [Google Scholar]
- Grlich D, Rapoport TA. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell. 1993;75:615–630. doi: 10.1016/0092-8674(93)90483-7. [DOI] [PubMed] [Google Scholar]
- Milcarek C, Penman S. Membrane-bound polyribosomes in HeLa cells: association of polyadenylic acid with membranes. J. Mol. Biol. 1974;89:327–338. doi: 10.1016/0022-2836(74)90522-1. [DOI] [PubMed] [Google Scholar]
- Lande MA, Adesnik M, Sumida M, Tashiro Y, Sabatini DD. Direct association of messenger RNA with microsomal membranes in human diploid fibroblasts. J. Cell Biol. 1975;65:513–528. doi: 10.1083/jcb.65.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardelli J, Long B, Pitot HC. Direct association of messenger RNA labeled in the presence of fluoroorotate with membranes of the endoplasmic reticulum in rat liver. J. Cell Biol. 1976;70:47–58. doi: 10.1083/jcb.70.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adesnik M, Lande M, Martin T, Sabatini DD. Retention of mRNA on the endoplasmic reticulum membranes after in vivo disassembly of polysomes by an inhibitor of initiation. J. Cell Biol. 1976;71:307–313. doi: 10.1083/jcb.71.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruppa J, Sabatini DD. Release of poly A(+) messenger RNA from rat liver rough microsomes upon disassembly of bound polysomes. J. Cell Biol. 1977;74:414–427. doi: 10.1083/jcb.74.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adesnik M, Maschio F. Segregation of specific classes of messenger RNA into free and membrane-bound polysomes. Eur. J. Biochem. 1981;114:271–284. doi: 10.1111/j.1432-1033.1981.tb05146.x. [DOI] [PubMed] [Google Scholar]
- Cui XA, Zhang H, Palazzo AF. p180 Promotes the Ribosome-Independent Localization of a Subset of mRNA to the Endoplasmic Reticulum. PLoS Biol. 2012;10:e1001336. doi: 10.1371/journal.pbio.1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S. Inhibitors of ribosome functions. Annu. Rev. Microbiol. 1971;25:487–562. doi: 10.1146/annurev.mi.25.100171.002415. [DOI] [PubMed] [Google Scholar]
- Fresno M, Jimnez A, Vzquez D. Inhibition of translation in eukaryotic systems by harringtonine. Eur. J. Biochem. 1977;72:323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- Weeks DP, Baxter R. Specific inhibition of peptide-chain initiation by 2-(4-methyl-2,6-dinitroanilino)-N-methylpropionamide. Biochemistry. 1972;11:3060–3064. doi: 10.1021/bi00766a018. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner RS, et al. Partitioning and translation of mRNAs encoding soluble proteins on membrane-bound ribosomes. RNA. 2003;9:1123–1137. doi: 10.1261/rna.5610403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam SA, Marr RS, Gerace L. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J. Cell Biol. 1990;111:807–816. doi: 10.1083/jcb.111.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lcuyer E, et al. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell. 2007;131:174–187. doi: 10.1016/j.cell.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Lcuyer E, Yoshida H, Krause HM. Global implications of mRNA localization pathways in cellular organization. Curr. Opin. Cell Biol. 2009;21:409–415. doi: 10.1016/j.ceb.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Gueroussov S, Tarnawsky SP, Cui XA, Mahadevan K, Palazzo AF. Analysis of mRNA nuclear export kinetics in mammalian cells by microinjection. J Vis Exp. 2010;46:e2387. doi: 10.3791/2387. [DOI] [PMC free article] [PubMed] [Google Scholar]