

Abstract
Background
Targeted therapies in renal cell carcinoma can have different effects on primary and metastatic tumors. To pave the way for predictive biomarker development, we assessed differences in expression of targets of currently approved drugs in matched primary and metastatic specimens from 34 patients.
Methods
Four cores from each site were embedded in tissue microarray blocks. Expression of B-Raf, C-Raf, cKIT, FGF-R1, HIF-2α, mTOR, PDGF-Rβ, VEGF-R1, VEGF-R2, VEGF-R3, VEGF, VEGF-B, VEGF-C, VEGF-D, MEK1, and ERK1/2 was studied using a quantitative immunofluorescence method.
Results
No significant differences were observed in global expression levels in primary and metastatic renal cell carcinoma tumors, with the exception of MEK, which had higher expression in metastatic than primary specimens. Similarly, more ki67 positive cells were seen in metastatic specimens. Correlations between marker expression in primary and metastatic specimens were variable, with the lowest correlation seen for FGF-R1 and VEGF-D. There were no significant differences in the degree of heterogeneity in primary versus metastatic tumors.
Conclusions
Expression of most of the studied markers was similar in primary and metastatic renal cell carcinoma tumors, suggesting that predictive biomarker testing for these markers can be conducted on either the primary or metastatic tumors for most markers.
Keywords: Renal cell carcinoma, Targeted therapy, Predictive biomarkers, VEGF
Background
In recent years, the incidence of renal cell carcinoma (RCC) has increased from 38,000 new cases a year in 2006 to over 64,000 estimated for 2012 [1,2]. This increase is largely due to incidental radiographic identification of renal masses; within this expanding population, RCC diagnoses are shifting towards earlier stage, smaller tumors [3,4]. Despite early detection, the RCC mortality rate remains fairly stable at 13,570 estimated annual deaths [2]. The five-year survival rates for patients with organ-confined disease is >85%, and >50% for patients with regional spread [5], suggesting that tumor biology is variable within the different disease stages.
Surgery followed by surveillance imaging is the standard of care for RCC patients with localized disease. Fine needle aspiration or core needle biopsies are commonly employed for diagnosis of metastatic disease in the 10-50% of these patients with recurring disease. More than 20% of RCC patients present with metastatic disease without having a previously known localized primary tumor [6,7].
RCC is very resistant to standard chemotherapy. Despite advances in biological and immune-based therapies, treatment options for patients with unresectable or metastatic RCC (mRCC) are limited; response rates remain at about 15–44%, and five-year survival under 10% for those with distant metastases [6,8-10]. Immunotherapy once represented the standard treatment for mRCC; interferon-alpha (IFN-α) produces objective responses in 10-15% of patients with a median survival of 12 months, while high-dose Interleukin-2 (IL-2) induces durable remissions in approximately 10% of patients [7,11]. Both are associated with substantial toxicity [12]. Alternative approaches have thus been developed in recent years.
A growing understanding of the pathogenesis of clear cell RCC, the most common histologic subtype, has facilitated development of RCC-targeting therapies. The discovery of Von Hippel-Lindau (VHL) tumor-suppressor gene inactivation and subsequent hypoxia-induced factor (HIF) activation of genes and downstream pathways important to tumor progression have provided the impetus for development of new agents that target angiogenesis and proliferation pathways. A number of drugs that target the vascular endothelial growth factor (VEGF) pathway and downstream signaling molecules have been approved for mRCC. These include the small molecule tyrosine kinase inhibitors (TKIs) sunitinib, sorafenib, pazopanib, and axitinb [10,13-15], the anti-VEGF antibody bevacizumab [16-18], and the mTOR inhibitors temsirolimus and everolimus [19,20]. Other histologic subtypes have different underlying molecular abnormalities, although responses to VEGF and mTOR targeting therapies have been seen in subsets of non-clear cell tumors as well. Although these new agents improve progression free survival and, in some cases, overall survival, none are curative and duration of response is often limited.
Response of primary RCC (as opposed to metastatic deposits) to targeted therapies has not been well studied, however highly sensitive cases are thought to be relatively uncommon. Several groups have reported that pre-surgical targeted therapy can be effective in mRCC patients who have not had their primary tumors removed [21-24]. However, a recent comprehensive study showed minimal to no decrease in the primary tumor diameter in mRCC patients. Patients with a >10% reduction in size of their primary tumors are 2.25 times more likely to have a partial response or stable disease with systemic therapy, indicating that there can be discordance in tumor shrinkage between primary and metastatic tumors in mRCC patients who have not had a nephrectomy [25]. Ongoing clinical trials are assessing the benefits of targeted therapy prior to cytoreductive nephrectomy (NCT00930033, NCT01099423).
At present no predictive biomarkers have been developed to identify patients whose tumors are more likely to respond to any of the currently available therapies. In addition, biomarkers predictive of discordant response between primary and metastatic tumors are lacking. It is therefore necessary to establish patterns of expression of drug targets in tumors in order to attempt to develop predictive tissue based biomarker assays. Given that these drugs exert their effects on the proteome, protein-level predictive biomarkers are a logical place to start. Patterns of expression of drug targets in primary and metastatic RCC tumors have not been previously well demonstrated. Youssif et al. studied 25 matched primary and metastatic samples for correlations between mTOR pathway targets (PTEN, PI3K, p-Akt, phospho-mTOR and p70S6) and found a strong correlation for phospho-mTOR [26]. Here we assess levels of a number of proteins, focusing on targets of currently approved drugs, in four cores from primary tumors and corresponding metastatic deposits. Given the limitations of immunohistochemistry in terms of subjectivity and qualitative assessment of protein expression, we employed a method of quantitative immunofluorescence to measure protein levels. We found that while levels of most markers were not significantly different between primary and metastatic tumors, some markers showed less concordance.
Methods
Cohort details and tissue microarray (TMA) construction
Thirty-four patients with archived matched primary and metastatic RCC tumors were identified. Resections were done between 1972 and 2011. TMAs were constructed using cores measuring 0.6 mm in diameter, spaced 0.8 mm apart. Tumors from each of the primary and metastatic tumors were represented by four cores from different areas of the specimen (eight cores in total from each patient) and placed in two TMA blocks. Specimens and clinical information were collected with approval of a Yale University Institutional Review Board.
Immunofluorescence
Pairs of slides (containing two cores from different areas of each matching primary and metastatic tumor per patient) were stained individually for the target markers; B-Raf, C-Raf, cKIT, FGF-R1 (fibroblast growth factor receptor-1), HIF-2α (hypoxia induced factor-2α), mTOR (mammalian target of rapamycin), PDGF-Rβ (platelet-derived growth factor receptor-β), VEGF-R1, VEGF-R2, VEGF-R3, VEGF, VEGF-B, VEGF-C, VEGF-D, MEK1 (mitogen activated protein kinase-1), and ERK1/2 (extracellular signal related kinase 1/2). TMAs were also stained for ki67 as a marker of proliferation. Fluorescent staining for our Automated, Quantitative Analysis (AQUA) method was performed as previously described [27,28]. Briefly, slides were incubated with mouse monoclonal anti-human VEGF-D (R&D Systems, Minneapolis, MN, USA), FGF-R1 (QED Bioscience, San Diego, CA, USA), HIF-2α (Novus Biologicals, Littleton, CO, USA), ERK1/2 (Cell Signaling Technologies, Danvers, MA, USA), PDGF-Rβ (Cell Signaling Technologies, Danvers, MA, USA), ki67 (BD Biosciences, San Jose, California, USA), rabbit monoclonal anti-human C-Raf(Upstate/EMD Millipore, Billerica, MA, USA) mTOR (Cell Signaling Technologies, Danvers, MA, USA), MEK1 (Upstate/EMD Millipore, Billerica, MA, USA) and rabbit polyclonal B-Raf, VEGF-R1, VEGF-R2, VEGF-R3 (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), c-Kit (DAKO, Carpinteria, CA, USA) VEGF, VEGF-B (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), VEGF-C (Invitrogen, Carlsbad, CA, USA) overnight at 4°C. Goat anti-mouse (or anti-rabbit) HRP-decorated polymer backbone (Envision, Dako, Carpinteria, CA, USA) was used as a secondary reagent. Slides were incubated with Cyanine5-tyramide (Perkin Elmer, Waltham, MA) in the supplied amplification buffer for 10 min at room temperature. Slides were incubated twice for 7 minutes with 100 mM benzoic hydrazide (B13071, Sigma, St. Louis, MO) and 50 mM hydrogen peroxide in PBS to quench the HRP. To create a tumor mask, slides were incubated with a cocktail of rabbit (for VEGF-D, FGF-R1, HIF-2α, ERK1/2, PDGF-Rβ, ki67) or mouse (for B-Raf, C-Raf, VEGF-R1, VEGF-R2, VEGF-R3, c-Kit, VEGF, VEGF-B, VEGF-C, mTOR and MEK1) anti-cytokeratin (Dako, Carpinteria, CA, USA) 1:100 and HRP-streptavidin(Sigma, St. Louis, MO, USA) for 1 hour at room temperature. HRP-streptavidin binds endogenous biotin which is present is high amounts in RCC and renal tubules. Goat anti-mouse (or anti-rabbit) HRP-decorated polymer backbone (Envision, Dako, Carpinteria, CA, USA) was used as a secondary reagent. Slides were incubated with Cyanine2-tyramide (Perkin Elmer, Waltham, MA, USA) in the supplied amplification buffer for 10 min at room temperature. To create a nuclear mask, TMAs were incubated with 4, 6-diamidine-2-phenylindole (DAPI) at a concentration of 1:500 in 0.3% BSA in TBS. Coverslips were mounted with ProLong Gold antifade reagent with 4, 6-diamidine-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA).
Automated image acquisition and analysis
Images were acquired and analyzed using previously described algorithms [29]. Briefly, monochromatic, high-resolution (1024 × 1024 pixel) images were obtained of each histospot. Tumor was distinguished from stroma by the cytokeratin/streptavidin signal. Cell surface coalescence of cytokeratin was used to localize membranes and DAPI to identify nuclei. The target signal (B-Raf, C-Raf, cKIT, FGF-R1, HIF-2α, mTOR, PDGF-Rβ, VEGF-R1, VEGF-R2, VEGF-R3, VEGF, VEGF-B, VEGF-C, VEGF-D, MEK1, and ERK1/2) from the pixels within the cytoplasm was normalized to area of tumor mask and scored on a scale of 0–255 (the AQUA score). Ki-67 positivity was calculated as a percentage of tumor cells. Histospots were excluded if the tumor mask represented <5% of the histospot area or if there was anomalous staining (lacking DAPI or cytokeratin, or necrotic tissue).
Statistical analysis
Statview and JMP 5.0 software were used (SAS Institute, Cary, NC). AQUA scores for replicate tumor cores were averaged. Associations between continuous AQUA scores of the target and clinical and pathological parameters were assessed using ANOVA. Correlations between the AQUA scores of matched primary and metastatic histospots were calculated by the log rank method. Intratumor heterogeneity was assessed by Pearson linear regression.
Results
Patient characteristics are summarized in Table 1. Histological subtypes included clear cell (ccRCC) (91.2%) and mixed histology (8.8%). Age at diagnosis was 17–72 years (median-56). Performance status, LDH, hemoglobin and calcium levels were not available.
Table 1.
Patient characteristics
| Patient | Progression time: time to metastasis (months) | Age at primary | Tumor size (cm) | Histology | Fuhrman grade | Gender | Metastasis location |
|---|---|---|---|---|---|---|---|
| 1 |
6 |
51 |
5 |
Clear Cell |
3 |
M |
Lung |
| 2 |
72 |
64 |
3.2 |
Clear Cell |
3 |
M |
Lung |
| 3 |
6 |
50 |
10 |
Clear Cell |
3 |
M |
Abdominal Wall |
| 4 |
12 |
45 |
7 |
Clear Cell |
3 |
M |
Skin |
| 5 |
12 |
70 |
4 |
Clear Cell |
2 |
M |
Lung |
| 6 |
6 |
58 |
3 |
Clear Cell |
2 |
F |
Colon |
| 7 |
24 |
17 |
13 |
Clear Cell |
3 |
F |
Lung |
| 8 |
36 |
66 |
4 |
Clear Cell |
2 |
M |
Lung |
| 9 |
24 |
64 |
10.7 |
Clear Cell |
3 |
M |
Liver |
| 10 |
48 |
69 |
6 |
Clear Cell |
3 |
F |
Skin |
| 11 |
36 |
61 |
8 |
Clear Cell |
3 |
F |
Lung |
| 12 |
24 |
61 |
8 |
Clear Cell |
3 |
F |
Liver |
| 13 |
36 |
68 |
4.5 |
Clear Cell |
1 |
M |
Bone |
| 14 |
6 |
56 |
11 |
Clear Cell |
3 |
F |
Pituitary |
| 15 |
120 |
47 |
8 |
Clear Cell |
3 |
M |
Testes |
| 16 |
24 |
54 |
6.5 |
Clear Cell |
2 |
F |
Soft Tissue |
| 17 |
48 |
62 |
10.5 |
Clear Cell |
3 |
M |
Bone |
| 18 |
156 |
56 |
3.5 |
Clear Cell |
3 |
M |
Lung |
| 19 |
108 |
59 |
8.5 |
Clear Cell |
2 |
M |
Lung |
| 20 |
12 |
28 |
4 |
Mixed |
3 |
M |
Lymph Node |
| 21 |
6 |
54 |
Unknown |
Clear Cell |
2 |
M |
Lung |
| 22 |
12 |
40 |
3.7 |
Mixed |
4 |
M |
Lung |
| 23 |
6 |
64 |
3.5 |
Clear Cell |
2 |
M |
Bone |
| 24 |
84 |
46 |
14.5 |
Clear Cell |
2 |
F |
Lung |
| 25 |
36 |
55 |
8.5 |
Clear Cell |
2 |
M |
Lung |
| 26 |
36 |
66 |
6.5 |
Clear Cell |
2 |
M |
Adrenal |
| 27 |
132 |
72 |
5 |
Clear Cell |
2 |
F |
Adrenal |
| 28 |
12 |
72 |
6 |
Mixed |
3 |
F |
Soft Tissue |
| 29 |
6 |
52 |
3.5 |
Clear Cell |
3 |
F |
Bone |
| 30 |
12 |
50 |
12 |
Clear Cell |
2 |
F |
Skin |
| 31 |
6 |
33 |
3.5 |
Clear Cell |
2 |
M |
Bone |
| 32 |
36 |
69 |
10.5 |
Clear Cell |
3 |
F |
Bone |
| 33 |
12 |
62 |
6 |
Clear Cell |
3 |
M |
Bone |
| 34 | 36 | 47 | 8 | Clear Cell | 3 | F | Lung |
Each lot of antibodies was subjected to immunoblotting to verify presence of a single dominant band of the appropriate size (not shown). We note that a number of antibodies to VEGF-R2 are commercially available, and a recent publication demonstrated higher specificity of the 55B11 antibody (Cell Signaling Technologies, Danvers, MA) than the A-3 antibody (Santa Cruz Biotechnologies, Inc, Santa Cruz, CA) [30]. However, in our hands, using quantitative immunofluorescence, we found superior correlations between redundant spots, membrane-specific staining and better reproducibility of results with the A-3 antibody. Western blotting for the A-3 antibody showed a single band at the associated protein size. The discrepancy between our findings and those of Molhoek et al. [30] might be due to batch-to-batch variability and the quantitative staining method used here. Information on antibodies used is given in the Additional file 1: Table S1.
Figure 1a-d shows an example of C-Raf expression in corresponding primary and metastatic tissues of one patient. AQUA scores for the primary and metastatic tumors for this patient were 66.28 and 64.14, respectively. To assess intratumor heterogeneity, four distinct cores from both the primary and metastatic sites were used to evaluate expression of all the markers. Subsequently, scores from corresponding cores were averaged to obtain a single concatenated score for each tumor for each marker.
Figure 1.

Example of Automated Quantitative Analysis (AQUA) staining for CRAF in matched primary (upper panels) and metastatic (lower panels) specimens from a single patient: We used anti-cytokeratin antibodies to create a cytoplasmic compartment (two upper right quadrants). A tumor mask was made by filling in holes (upper left quadrants). 4’, 6-diamidino-2-phenylindole (DAPI) defines the nuclear compartment within the tumor mask (left lower quadrants). CRAF expression is measured within the cytoplasmic compartments, within the tumor mask (lower right quadrants), and each clinical case is assigned a score based on pixel intensity per unit area within the tumor mask. The upper panel shows an example of a histospot from a primary specimen and the lower shows the corresponding metastatic tumor. CRAF staining was strong and similar in both specimens.
Global distribution of scores in primary and metastatic spots was not significantly different for any of the markers with the exception of MEK1. The mean AQUA scores and the differences between primary and metastatic tumors by paired t-tests are shown in Table 2. No significant differences were found between expression in primary and metastatic specimens for B-Raf, C-Raf, cKIT, FGF-R1, HIF-2α, mTOR, PDGF-Rβ, VEGF-R1, VEGF-R2, VEGF-R3, VEGF, VEGF-B, VEGF-C, VEGF-D, and ERK1/2. Expression of MEK1 was somewhat higher in metastatic than primary tumors (P = 0.002). The p-value of 0.002 for MEK is below the Bonferroni adjusted p-value (0.003) for the 16 markers analyzed at an alpha of 0.05. As MEK is an important component of the major intracellular proliferation signaling pathway, we looked at percentage of cells in tumors with ki67 staining, and found that ki67 positivity was significantly higher (P = 0.0006) in metastatic than primary tumors.
Table 2.
Tyrosine kinase and mTOR inhibitor target expression in primary and metastatic RCC samples
| Target | AQUA scores, primary specimens (mean ± SD) | AQUA scores, metastatic specimens (mean ± SD) | t-statistic | p-value |
|---|---|---|---|---|
| B-Raf |
32.1 ± 8.9 |
31.2 ± 10.2 |
−0.417 |
0.678 |
| C-Raf |
26.7 ± 2.1 |
28.4 ± 2.5 |
0.519 |
0.605 |
| cKit |
23.3 ± 1.3 |
23.4 ± 1.3 |
0.079 |
0.937 |
| FGF-R1 |
29.1 ± 1.1 |
29.9 ± 0.9 |
0.579 |
0.564 |
| HIF-2α |
58.2 ± 1.9 |
58.2 ± 2.0 |
0.014 |
0.989 |
| mTOR |
18.8 ± 1.4 |
22.4 ± 1.8 |
1.637 |
0.106 |
| PDGF-Rβ |
24.7 ± 0.9 |
27.9 ± 1.5 |
1.879 |
0.064 |
| VEGF-R1 |
22.6 ± 0.9 |
23.9 ± 1.2 |
0.34 |
0.735 |
| VEGF-R2 |
36.5 ± 2.4 |
33.5 ± 1.7 |
−1.294 |
0.200 |
| VEGF-R3 |
45.6 ± 2.0 |
46.5 ± 1.6 |
0.371 |
0.712 |
| VEGF |
24.7 ± 1.0 |
26.0 ± 1.1 |
0.907 |
0.367 |
| VEGF-B |
11.2 ± 0.6 |
11.8 ± 0.9 |
0.486 |
0.628 |
| VEGF-C |
17.0 ± 1.3 |
14.3 ± 1.2 |
−1.544 |
0.127 |
| VEGF-D |
35.0 ± 1.2 |
36.8 ± 1.3 |
1.018 |
0.312 |
| MEK1 |
37.5 ± 2.5 |
50.3 ± 3.1 |
3.183 |
0.002 |
| ERK1/2 | 17.0 ± 1.1 | 18.6 ± 1.7 | 0.802 | 0.425 |
Given that archival tissue is often available from either the primary or the metastatic site, but not both, we determined the associations between marker expression in paired primary and metastatic samples using the Pearson correlation test, as displayed in Table 3. Levels correlated well between primary and metastatic specimens for most markers, with the poorest correlation seen for FGF-R1 and VEGF-D (0.15 and 0.28, respectively).
Table 3.
Correlations between paired primary and metastatic samples
| Marker | R value |
|---|---|
| B-Raf |
0.63 |
| c-Kit |
0.59 |
| C-Raf |
0.83 |
| MEK1 |
0.53 |
| FGF-R1 |
0.15 |
| HIF-2α |
0.77 |
| mTOR |
0.66 |
| PDGF-Rβ |
0.65 |
| VEGFR-1 |
0.69 |
| VEGFR-2 |
0.84 |
| VEGFR-3 |
0.53 |
| VEGF |
0.49 |
| VEGF-B |
0.66 |
| VEGF-C |
0.81 |
| VEGF-D |
0.28 |
| ERK 1/2 | 0.36 |
To determine whether there were differences in intra-tumor heterogeneity in primary and metastatic specimens, we used the four measurements from each tumor. Each core is represented by a vector of measurements of all markers, denoted as the core vector. For each patient we computed the median core vector and measured its L1 distances from the corresponding four core vectors. We defined the composite median absolute deviation (MAD) as the median of these four L1 distances, and used it as a proxy for estimating intra-tumor heterogeneity. For each patient, the composite MAD is computed separately for his/her primary and metastatic tumors. Using the Wilcoxon paired, two-sided signed rank test, we found no significant differences in heterogeneity between primary and metastatic tumors (P = 0.38), as shown in Figure 2.
Figure 2.
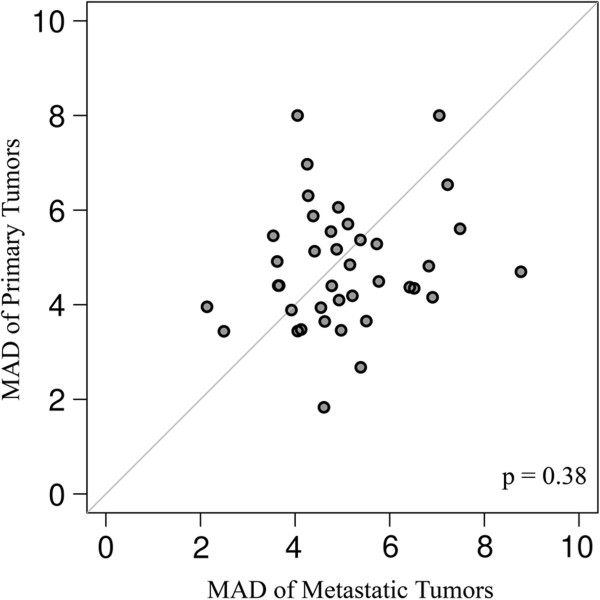
Comparison between heterogeneity within primary and metastatic specimens, estimated using a composite median absolute deviation (MAD) across all the markers: Each patient is represented by a dot. Dots above the diagonal represent patients with larger heterogeneity in the primary tumors, while dots below the diagonal represent greater heterogeneity in the corresponding metastatic tumors. The central diagonal grey line represents identical heterogeneity in primary and metastatic tumors. The Wilcoxon paired, two-sided signed rank test shows no significant difference between the heterogeneities of the primary and matched metastatic tumors (p = 0.38).
Discussion
Molecular targeted therapies that inhibit members of the VEGF pathway and mTOR are now widely used for the treatment of metastatic RCC. At present, no predictive biomarkers have been established for this class of drugs. Given that these agents inhibit this pathway at the protein level, target protein expression might be associated with response to therapy. Many metastatic RCC patients have either primary or metastatic tumor tissue (but not both) available for analysis, and our purpose was therefore to determine differences in expression of these drug targets in matched primary and metastatic specimens.
Target expression levels were not globally different between primary and metastatic tumors, with the exception of MEK1, which was higher in metastatic specimens. Given that MEK1 is a key component of the major intracellular proliferation signal transduction pathway, we studied ki67 expression in primary and metastatic samples and found that the percentage of ki67 positive cells was also significantly higher in the metastases. This is consistent with the commonly seen clinical picture in which the primary tumor grows over years, yet the course for metastatic disease patients is often much shorter due to rapid metastatic tumor growth.
Our data indicate that our ability to predict expression in primary specimens based on measurements from a corresponding metastasis and vice-versa is marker dependent. The intra-patient correlations were variable across markers, with the worst correlation shown for VEGF-D and FGF-R1, while other makers such as C-Raf, VEGF-R2 and cKIT demonstrated excellent correlations between levels in primary and metastatic specimens. These findings are consistent with the only other similar published study of which we are aware in which mTOR pathway members were assessed for concordance between primary and metastatic sites using standard immunohistochemistry (IHC) [26]. Levels of phosphorylated mTOR were similar in primary and metastatic sites, while levels of PI3K, p-Akt, PTEN and p70S6 had much weaker intra-patient correlations. We elected not to study levels of phosphorylated proteins in our study, as many of these specimens were old and fixation times were not uniform. Phosphatase activity, therefore, cannot be accounted for in our samples. Use of nominal IHC scores might make similarities in expression less apparent than use of continuous AQUA scores.
To date, none of the approved drugs for mRCC has an associated companion diagnostic biomarker test. A number of initial attempts have been made at developing predictive biomarkers that are primarily centered around VHL pathway markers, such as VHL mutations, HIF levels, VEGF isoforms and VEGF receptor levels. Phase II trials of axitinib or temsirolimus revealed no association between VHL mutational status and response to therapy [10,31]. Higher levels of pS6 and p-AKT in pre-treatment tissues were associated with response to temsirolimus, but no significant difference was found between primary and metastatic tissues. In addition, a number of studies have looked at VHL loss and response to immunotherapy [31,32]. A study of 123 patients with clear cell RCC showed that patients with wild-type VHL had a lower likelihood of responding to VEGF-pathway targeted therapies than patients with VHL mutations or VHL loss by hypermethylation [33]. This finding, however, requires further validation.
Small studies have suggested a potential role for VEGF and soluble forms of the VEGF receptors as predictors of response to VEGF pathway targeting therapies and cytokine therapies. Sabatino et al. [34] measured serum protein levels using multiplex protein arrays, and showed that high pretreatment levels of VEGF and fibronectin were negative predictors of response to IL-2. Trials using bevacizumab with interferon or sorafenib showed no predictive value of baseline VEGF levels in patients [13,17]. In a phase II trial, Hutson et al. found that decreased expression of soluble VEGFR-2 correlated with tumor response to pazopanib [35]. Similarly, in a phase II trial of sunitinib, increases in soluble VEGFR-2, VEGFR-3, and VEGF at day 28 were associated with a greater likelihood of response [36].
Clinical observations of discordance in response of primary and metastatic tumors suggest possible differences in biology. Alternatively, differences in response could be due to variable tumor microenvironment in the primary and metastatic sites. Primary RCC tumors do not seem to respond as well as RCC metastatic sites to VEGF pathway targeted therapies [25]. Studies evaluating targeted therapies in RCC for their preoperative potential to reduce the size of primary tumors with the hope of making them more resectable are ongoing. Prior studies evaluating sunitinib and/or sorafenib in patients with localized and metastatic RCC disease concluded that these agents might be useful in reducing primary tumor burden [21-24,37]. A phase II study of presurgical sunitinib resulted in only 1 partial primary tumor response, while another study concluded that preoperative sunitinib can be effective for cytoreduction. Eighty percent of patients demonstrated variable primary tumor shrinkage, with a median of 1.6 cm (range 0.4- 5.1) [38,39]. A larger study of 168 mRCC patients who received targeted therapy with their primary tumors in situ found noted negligible decreases in the size of the primary tumors [25]. Although contradictory, these studies do suggest discordant responses to drugs in primary and metastatic tumors. Additional trials are needed to determine whether any of the biomarkers studied here is predictive of response to VEGF pathway targeting therapies, and when discordant tumor shrinkage is observed, whether it can be explained by differences in biomarker expression in primary and metastatic samples.
Biomarker studies related to evolving biomarkers and experimental drugs are being done by our group and others. The clinical relevance of HIF-1α and HIF-2α are being studied as the hypoxia induced pathway is regularly aberrant in RCC. HIF-1α has been shown to be expressed in most RCC tumors while HIF-2α is relatively absent in early tumors, but is highly expressed in metastatic tumors [40,41]. B7-H1 is another target that is being heavily explored, with multiple clinical trials of B7-H1 targeting ongoing. A study by Thompson et. al in primary and metastatic RCC showed high B7-H1 expression is associated with a poor prognosis. Although only 1 patient was represented in both cohorts, more metastatic specimens had high B7-H1 expression than primary specimens (54.3% versus 44.4%, respectively) [42]. Tumor suppressor gene p53 was significantly higher in primary tumors versus metastatic tumors in a study by Zigeuner et. al (22.8% versus 51.8%), however the specimens were not matched [43]. In a study of mTOR and hypoxia-induced pathway members including 135 primary RCC and 41 unrelated metastasis, differential global patterns of expression were measured. Levels of p-AKT, p-S6, 4EBP1, and c-myc were higher in metastatic lesions compared to both primary and benign tissues [44].
The tumors studied here exhibited variable intratumor heterogeneity in the four tumor cores. The degree of heterogeneity is not significantly different in primary and metastatic samples. Although our study evaluates protein expression, recent DNA sequencing studies have shown intratumor heterogeneity in primary renal cell carcinoma. The majority of somatic mutations (over 60%) were not present throughout the tumor in the 4 samples examined. Moreover, DNA signatures of both good and poor prognosis were detected in different regions of the same tumor. The authors suggest that intratumor heterogeneity is the cause of lack of reproducible predictive biomarkers [45]. Utilizing single-cell exome sequencing in a single patient, Xu et al. demonstrated that there was no dominant clone throughout the tumor, and similarly demonstrated heterogeneity at the DNA level. This may provide insight into the observed heterogeneity in this study [46].
Conclusion
Our studies showed good concordance between primary and metastatic samples for most of the markers studied. The biomarkers with the least concordance were FGF-R1 and VEGF-D. The discordance in levels of VEGF-D might be due to the fact that this is a secreted protein, and levels of FGF-R1 might be more influenced by the tumor microenvironment than the other markers studied. Conversely, other biomarkers showed excellent concordance between primary and metastatic samples. As predictive biomarkers are developed, careful studies are needed to define concordance versus discordance for individual biomarkers in order to determine whether primary specimen measurements can be used as surrogates for metastatic specimens and vice-versa.
Competing interests
R.L. Camp is a co-founder, stockholder and consultant for a company called HistoRx that has licensed the technology for automated tissue analysis used in this study.
Authors’ contributions
SA drafted the manuscript, acquired the data, and participated in the analysis of data. JS helped with the acquisition of data and the analysis. AA performed the pathology review of specimens. FP and YK conducted statistical and computational analysis for this study. RC designed the tissue microarrays and performed pathology review. HK designed and coordinated the study and participated in the drafting of this manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
List of Antibodies.
Contributor Information
Saadia A Aziz, Email: saadia.aziz@yale.edu.
Joshua A Sznol, Email: joshua.sznol@yale.edu.
Adebowale Adeniran, Email: adebowale.adeniran@yale.edu.
Fabio Parisi, Email: fabio.parisi@yale.edu.
Yuval Kluger, Email: yuval.kluger@yale.edu.
Robert L Camp, Email: robert.camp@yale.edu.
Harriet M Kluger, Email: harriet.kluger@yale.edu.
Acknowledgements
The authors would like to thank Dr. Lucia Jilaveanu for her assistance in designing the tissue microarrays and for critical review of components of the manuscript. This work was supported by NIH grants R0-1 CA158167 (to H. Kluger), R0-1 CA129034 (to P. Febbo) and by American Cancer Society Award M130572 (to H. Kluger).
References
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Kane CJ, Mallin K, Ritchey J, Cooperberg MR, Carroll PR. Renal cell cancer stage migration: analysis of the National Cancer Data Base. Cancer. 2008;113:78–83. doi: 10.1002/cncr.23518. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin FM, Millan-Rodriguez F, Urdaneta-Pignalosa G, Rubio-Briones J, Villavicencio-Mavrich H. Small renal masses: incidental diagnosis, clinical symptoms, and prognostic factors. Adv Urol. 2008. p. 310694. [DOI] [PMC free article] [PubMed]
- Oudard S, George D, Medioni J, Motzer R. Treatment options in renal cell carcinoma: past, present and future. Ann Oncol. 2007;18(Suppl 10):x25–x31. doi: 10.1093/annonc/mdm411. [DOI] [PubMed] [Google Scholar]
- Lam JS, Leppert JT, Belldegrun AS, Figlin RA. Novel approaches in the therapy of metastatic renal cell carcinoma. World J Urol. 2005;23:202–212. doi: 10.1007/s00345-004-0466-0. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865–875. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- Figlin RA. Renal cell carcinoma: management of advanced disease. J Urol. 1999;161:381–386. doi: 10.1016/S0022-5347(01)61897-4. discussion 386–387. [DOI] [PubMed] [Google Scholar]
- Bukowski RM. Prognostic factors for survival in metastatic renal cell carcinoma: update 2008. Cancer. 2009;115:2273–2281. doi: 10.1002/cncr.24226. [DOI] [PubMed] [Google Scholar]
- Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, Michaelson MD, Gorbunova VA, Gore ME, Rusakov IG. et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995;13:688–696. doi: 10.1200/JCO.1995.13.3.688. [DOI] [PubMed] [Google Scholar]
- Facchini G, Perri F, Caraglia M, Pisano C, Striano S, Marra L, Fiore F, Aprea P, Pignata S, Iaffaioli RV. New treatment approaches in renal cell carcinoma. Anticancer Drugs. 2009;20:893–900. doi: 10.1097/CAD.0b013e32833123d4. [DOI] [PubMed] [Google Scholar]
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F. et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27:3312–3318. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST. et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A. et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S, Ravaud A, Golding S, Jethwa S, Sneller V. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi: 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
- Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Archer L, Atkins JN, Picus J, Czaykowski P, Dutcher J, Small EJ. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28:2137–2143. doi: 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I. et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- Amin C, Wallen E, Pruthi RS, Calvo BF, Godley PA, Rathmell WK. Preoperative tyrosine kinase inhibition as an adjunct to debulking nephrectomy. Urology. 2008;72:864–868. doi: 10.1016/j.urology.2008.01.088. [DOI] [PubMed] [Google Scholar]
- Shuch B, Riggs SB, LaRochelle JC, Kabbinavar FF, Avakian R, Pantuck AJ, Patard JJ, Belldegrun AS. Neoadjuvant targeted therapy and advanced kidney cancer: observations and implications for a new treatment paradigm. BJU Int. 2008;102:692–696. doi: 10.1111/j.1464-410X.2008.07660.x. [DOI] [PubMed] [Google Scholar]
- Thomas AA, Rini BI, Lane BR, Garcia J, Dreicer R, Klein EA, Novick AC, Campbell SC. Response of the primary tumor to neoadjuvant sunitinib in patients with advanced renal cell carcinoma. J Urol. 2009;181:518–523. doi: 10.1016/j.juro.2008.10.001. discussion 523. [DOI] [PubMed] [Google Scholar]
- van der Veldt AA, Meijerink MR, van den Eertwegh AJ, Bex A, de Gast G, Haanen JB, Boven E. Sunitinib for treatment of advanced renal cell cancer: primary tumor response. Clin Cancer Res. 2008;14:2431–2436. doi: 10.1158/1078-0432.CCR-07-4089. [DOI] [PubMed] [Google Scholar]
- Abel EJ, Culp SH, Tannir NM, Matin SF, Tamboli P, Jonasch E, Wood CG. Primary tumor response to targeted agents in patients with metastatic renal cell carcinoma. Eur Urol. 2011;59:10–15. doi: 10.1016/j.eururo.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou Youssif T, Fahmy MA, Koumakpayi IH, Ayala F, Al Marzooqi S, Chen G, Tamboli P, Squire J, Tanguay S, Sircar K. The mammalian target of rapamycin pathway is widely activated without PTEN deletion in renal cell carcinoma metastases. Cancer. 2011;117:290–300. doi: 10.1002/cncr.25402. [DOI] [PubMed] [Google Scholar]
- Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med. 2002;8:1323–1327. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- Aziz SA, Davies M, Pick E, Zito C, Jilaveanu L, Camp RL, Rimm DL, Kluger Y, Kluger HM. Phosphatidylinositol-3-kinase as a therapeutic target in melanoma. Clin Cancer Res. 2009;15:3029–3036. doi: 10.1158/1078-0432.CCR-08-2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger HM, Siddiqui SF, Angeletti C, Sznol M, Kelly WK, Molinaro AM, Camp RL. Classification of renal cell carcinoma based on expression of VEGF and VEGF receptors in both tumor cells and endothelial cells. Lab Invest. 2008;88:962–972. doi: 10.1038/labinvest.2008.65. [DOI] [PubMed] [Google Scholar]
- Molhoek KR, Erdag G, Rasamny JK, Murphy C, Deacon D, Patterson JW, Slingluff CL Jr, Brautigan DL. VEGFR-2 expression in human melanoma: revised assessment. Int J Cancer. 2011;129:2807–2815. doi: 10.1002/ijc.25963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D, Signoretti S, Dabora S, Regan M, Seeley A, Mariotti M, Youmans A, Polivy A, Mandato L, McDermott D. et al. Potential histologic and molecular predictors of response to temsirolimus in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2007;5:379–385. doi: 10.3816/CGC.2007.n.020. [DOI] [PubMed] [Google Scholar]
- Lam JS, Leppert JT, Figlin RA, Belldegrun AS. Role of molecular markers in the diagnosis and therapy of renal cell carcinoma. Urology. 2005;66:1–9. doi: 10.1016/j.urology.2005.06.112. [DOI] [PubMed] [Google Scholar]
- Choueiri TK, Vaziri SA, Jaeger E, Elson P, Wood L, Bhalla IP, Small EJ, Weinberg V, Sein N, Simko J. et al. von Hippel-Lindau gene status and response to vascular endothelial growth factor targeted therapy for metastatic clear cell renal cell carcinoma. J Urol. 2008;180:860–865. doi: 10.1016/j.juro.2008.05.015. discussion 865–866. [DOI] [PubMed] [Google Scholar]
- Sabatino M, Kim-Schulze S, Panelli MC, Stroncek D, Wang E, Taback B, Kim DW, Deraffele G, Pos Z, Marincola FM, Kaufman HL. Serum vascular endothelial growth factor and fibronectin predict clinical response to high-dose interleukin-2 therapy. J Clin Oncol. 2009;27:2645–2652. doi: 10.1200/JCO.2008.19.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson TE, Davis ID, Machiels JP, De Souza PL, Rottey S, Hong BF, Epstein RJ, Baker KL, McCann L, Crofts T. et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2010;28:475–480. doi: 10.1200/JCO.2008.21.6994. [DOI] [PubMed] [Google Scholar]
- Hutson TE. Sunitinib (SUTENT) for the treatment of metastatic renal cell carcinoma. Expert Rev Anticancer Ther. 2008;8:1723–1731. doi: 10.1586/14737140.8.11.1723. [DOI] [PubMed] [Google Scholar]
- Cowey CL, Amin C, Pruthi RS, Wallen EM, Nielsen ME, Grigson G, Watkins C, Nance KV, Crane J, Jalkut M. et al. Neoadjuvant clinical trial with sorafenib for patients with stage II or higher renal cell carcinoma. J Clin Oncol. 2010;28:1502–1507. doi: 10.1200/JCO.2009.24.7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffioen AW, Mans LA, de Graaf AM, Nowak-Sliwinska P, de Hoog CL, de Jong TA, Vyth-Dreese FA, van Beijnum JR, Bex A, Jonasch E. Rapid angiogenesis onset after discontinuation of sunitinib treatment of renal cell carcinoma patients. Clin Cancer Res. 2012;18:3961–3971. doi: 10.1158/1078-0432.CCR-12-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini BI, Garcia J, Elson P, Wood L, Shah S, Stephenson A, Salem M, Gong M, Fergany A, Rabets J. et al. The effect of sunitinib on primary renal cell carcinoma and facilitation of subsequent surgery. J Urol. 2012;187:1548–1554. doi: 10.1016/j.juro.2011.12.075. [DOI] [PubMed] [Google Scholar]
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, Wykoff CC, Maher ER, Harris AL, Ratcliffe PJ, Maxwell PH. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–468. doi: 10.1016/S1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, Chen L, Zincke H, Blute ML, Leibovich BC, Kwon ED. Costimulatory molecule B7-H1 in primary and metastatic clear cell renal cell carcinoma. Cancer. 2005;104:2084–2091. doi: 10.1002/cncr.21470. [DOI] [PubMed] [Google Scholar]
- Zigeuner R, Ratschek M, Rehak P, Schips L, Langner C. Value of p53 as a prognostic marker in histologic subtypes of renal cell carcinoma: a systematic analysis of primary and metastatic tumor tissue. Urology. 2004;63:651–655. doi: 10.1016/j.urology.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Schultz L, Chaux A, Albadine R, Hicks J, Kim JJ, De Marzo AM, Allaf ME, Carducci MA, Rodriguez R, Hammers HJ. et al. Immunoexpression status and prognostic value of mTOR and hypoxia-induced pathway members in primary and metastatic clear cell renal cell carcinomas. Am J Surg Pathol. 2011;35:1549–1556. doi: 10.1097/PAS.0b013e31822895e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H. et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of Antibodies.