

Abstract
Background
Horizontal gene transfer (HGT) is considered to be a major force driving the evolutionary history of prokaryotes. HGT is widespread in prokaryotes, contributing to the genomic repertoire of prokaryotic organisms, and is particularly apparent in Rickettsiales genomes. Gene gains from both distantly and closely related organisms play crucial roles in the evolution of bacterial genomes. In this work, we focus on genes transferred from distantly related species into Rickettsiales species.
Results
We developed an automated approach for the detection of HGT from other organisms (excluding alphaproteobacteria) into Rickettsiales genomes. Our systematic approach consisted of several specialized features including the application of a parsimony method for inferring phyletic patterns followed by blast filter, automated phylogenetic reconstruction and the application of patterns for HGT detection. We identified 42 instances of HGT in 31 complete Rickettsiales genomes, of which 38 were previously unidentified instances of HGT from Anaplasma, Wolbachia, Candidatus Pelagibacter ubique and Rickettsia genomes. Additionally, putative cases with no phylogenetic support were assigned gene ontology terms. Overall, these transfers could be characterized as “rhizome-like”.
Conclusions
Our analysis provides a comprehensive, systematic approach for the automated detection of HGTs from several complete proteome sequences that can be applied to detect instances of HGT within other genomes of interest.
Keywords: Horizontal gene transfer, Rickettsiales, Candidatus Pelagibacter ubique, Sympatry
Background
Horizontal gene transfer (HGT) has played a significant role in bacterial evolution [1]. HGT is widespread in prokaryotes [2] and is considered to be a driving force in the innovation and evolution of bacterial genomes [3,4]. HGTs have been shown to contribute to the genomic repertoires of a number of prokaryotes, including Rickettsiales species.
The order Rickettsiales has been classified into two families, Rickettsiaceae and Anaplasmataceae. Members of the Rickettsiales order are primarily intracellular bacteria and can encompass both parasitic genera, such as Rickettsia and Orientia, or symbiotic genera, such as Wolbachia. Over the course of their evolution, the intracellular lifestyle of these members has led to the irreversible loss of genes [5,6]. Orientia species cause scrub typhus in human and are classified in a separate clade from Rickettsiae species [7-9]. Rickettsiae species are associated with a diverse host range that include vertebrates, arthropods, annelids, amoebae and plants and are known as arthropod-vector pathogens of vertebrate hosts [10,11]. The family Anaplasmataceae encompasses the three genera Anaplasma, Ehrlichia and Wolbachia[12]. A. phagocytophilum causes human granulocytic ehrlichiosis [13], E. chaffensis causes human monocytic ehrlichiosis [14,15] and Neorickettsia sennetsu causes sennetsu ehrlichiosis, an infectious mononucleosis-like disease [16]. The Wolbachia species live as symbionts in arthropods and annelids. The genomes of these species demonstrate both genome reduction and gene integration events between symbionts and host nuclear genomes [17]. Candidatus Pelagibacter ubique is a marine, free-living bacterium with a small, AT-rich genome that belongs to the SAR11 clade [18]. Candidatus Pelagibacter ubique was included under the sister group of the Rickettsiales clade in 2007 [19], but the placement of this free-living bacterium in an obligate intracellular phylum remains a subject of debate [20-24].
HGTs have occurred at high frequencies within alphaproteobacteria species [25] and within the Rickettsia[26]; therefore, we limited our analysis to the study of genes transferred from distantly related species into Rickettsiales species genomes. Previously, we have reported the existence of novel HGTs from a number of other genomes into Rickettsiales species [27-29]. In the current study, we have detected these new instances of transfer by developing an automated approach for the detection of HGT from complete proteomes.
Currently, a number of methods exist for detecting HGTs [30-40]. The two main approaches depend on the analysis of sequence composition and the use of phylogenetic methods. The first examines atypical GC content, codon usage bias, oligonucleotide frequencies, and genomic signatures [30-34]. The second enables the detection of inconsistencies in gene and genome evolutionary history either (i) by reconstructing the gene tree and comparing it with the reference species tree, i.e., via the test of topologies, bipartition, quartet bipartitions [35-37]; or (ii) by examining gene history, i.e., phyletic profiles [38,39]. We herein describe a new strategy for systematically searching for instances of HGT that depends on the use of stringent filters. Using this method, a cluster of orthologous protein sequences were first retrieved and examinated for sequence similarity with the organisms of interest. This was followed by the application of phyletic pattern detection for the identification of gain events. The results were then analyzed by Blast searches to retrieve sequences that were homologous to Rickettsiales genomes from other organisms, excluding alphaproteobacteria. These results were then examinated by automated phylogenetic analysis, followed by the detection of instances of HGT. Using this method, we were also able to verify the HGT events reported by previous studies. Here, we provide a detailed description of the transferred proteins, including their predicted function and ecological niches, as well as an analysis of the function of putative transfers with no phylogenetic support.
Results
Clustered Ortholog Groups (COGs)
We analyzed 38,895 proteins from 31 complete proteomes using OrthoMCL, of which 1,594 (4%) proteins were found to be less than 50 amino acids in length and therefore discarded. From the remaining 37,301 (96%) proteins, 4,240 (11%) belonged to non-orthologous groups that did not form clusters, and 33,061 (85%) were clustered into 3,537 orthologous groups. We found that 267 (7.6%) such Clustered Ortholog Groups (COGs) were conserved between Caulobacter cresentus CB15 and Rickettsiales species that were among the 31 species included in our study (Additional file 1, Table S3; Additional file 2, Figure S10). An additional 277 COGs were conserved among Rickettsia, Orientia, Wolbachia, Anaplasma, Ehrlichia, Neorickettsia, and Candidatus Pelagibacter ubique genome (Additional file 1, Table S4; Additional file 2, Figure S10), and 328 COGs were conserved among Rickettsia, Orientia, Wolbachia, Anaplasma, Ehrlichia, Neorickettsia genomes (Additional file 1, Table S5). We found 549 conserved COGs among Wolbachia, Anaplasma, Ehrlichia, and Neorickettsia genomes, and 497 conserved COGs were found only in Rickettsia and Orientia genomes. We also carried out functional analyses for these orthologous groups using BlastP against the COG database [41,42] with an E-value cutoff of less than 10-10 and annotated 1,709 (48.5%) orthologous groups. The results of these analyses are provided in Additional file 1, Table S6 and Additional file 2, Figure S10.
Detection of gain and loss events from 3,537 COGs
To characterize gains and losses, the presence and absence of genes from the species trees was mapped onto orthologous groups, and phyletic patterns of gene gain and gene loss events were reconstructed. The results thus obtained from pattern detection were logically classified into four types in the following manner: 0 gain, 0 loss; 0 gain, 1 loss; 1 gain, 0 loss; and at least 1 gain 1 loss (Figure 1). Accordingly, the results from each orthologous group that fell under each type were analyzed for gain and loss events. In this way, we obtained 267 COGs with 0 gain and 0 loss events, 752 COGs with 0 gain and 1 loss, and 2,518 COGs with at least 1 gain and 1 loss event (Additional file 2, Figure S5). As mentioned previously, we evaluated only instances of gain events due to HGT, which formed the main objective of our study.
Figure 1.

Flowchart representing the systematic pipeline for the automated detection of horizontal gene transfer in complete genomes. Step 1. Clustering of orthologous groups by OrthoMCL. Step 2. Detection of the presence or absence of genetic events on the species tree and the reconstruction of the phyletic pattern of gene gain and gene loss using PhyloPattern. Step 3. BlastP filter to retrieve the homologous sequences. Step 4. Construction of phylogenetic trees using FIGENIX. Step 5. Detection of HGT events from HGT agent using a transfer filter. Step 6. Validation of HGT cases using Bayesian method. Steps 1 through 5 are included in the pipeline.
Analysis of HGT cases from only gain event
The 2,518 COGs with at least 1 gain event were automatically identified using Blast filter, phylogenetic analysis, and HGT detection and were validated by a human expert using a Bayesian approach as described in the methods. We found that 838 (33.3%) proteins had hits outside of alphaproteobacterial species, while the remaining 1,680 (66.7%) proteins had hits within alphaproteobacterial species (Additional file 2, Figure S6a). The 838 proteins were subjected to automated phylogenetic reconstruction and HGT detection. Phylogenetic trees were successfully reconstructed for 56.9% of the cases, out of which 4.3% (36 cases) were associated with strong bootstrap support for HGT, while the remaining 52.2% were weakly supported by bootstrap values. The remaining 43.5% of cases failed to produce phylogenetic trees based on the application of stringenet FIGENIX parameters, which are listed as follows: gap sizes that exceeded the threshold by more than 30%; the presence of a greater number of repeats; an insufficient number of Blast hits that did not allow for phylogenetic reconstruction; and detection by puzzle filter of quickly evolving sites resulting from the duplication of paralogous groups that hindered phylogenetic reconstruction. The corresponding genes may be considered to be putative cases of HGT but with weak or no phylogenetic support. We will further describe the distribution of these genes based on Gene Ontology (GO) database [43] in order to classify their putative functions.
Finally, we found 4.3% of proteins (36 cases) with phylogenetic support for HGT with at least 1 gain event, of which 12 cases were detected from uncultured bacteria to Candidatus Pelagibacter ubique (data not shown) and were therefore excluded from further analysis. The remaining 24 cases were found to be good candidates for HGT from different organisms with well-supported bootstrap values. Of these 24 cases, two have been reported as HGTs in previous studies, gi:42520378 was reported as to be an instance of HGT from a Wolbachia strain to Aedes aegypti by Klasson et al., 2009 [44] and gi:190570783 was reported to be an instance of HGT from Wolbachia phage WO into Wolbachia species by Wu et al., 2004 [45] (Additional file 1, Table S8). The remaining 22 instances of HGTs were present in Anaplasma, Wolbachia and Rickettsia species genomes. These proteins were acquired from different origins, including bacterial species from Bacteroidetes, Firmicutes, Chlamydia, Spirochaetales, Nitrospirales, and Proteobacteria clades (Beta, Gamma and Delta). The functions of these transferred proteins are diverse, including ATPases, aldolases, transporter activities, cystathionine beta-lyases, sugar phosphate permeases, growth inhibitors and antitoxin activities, while the others were found to be hypothetical with no known function.
To determine the possible origin of these hypothetical proteins, we analyzed them using the ACLAME database. We report here only the best match, which was for gi:238651161 with plasmid:126745 from Rickettsia felis URRWXCal2, with an E-value of 3.5e-27 and 69.5% identity. The transfer of this gene (gi:238651161) was from Actinobacillus minor NB305 to R. felis and R. peacockii str. Rustic (Additional file 1, Table S8).
HGT cases from non-orthologous groups
We applied a similar procedure to the 4,240 non-orthologous proteins to detect transfer events. Of these, 1,795 proteins shared with Caulobacter cresentus CB15 and were therefore discarded from further analysis. The remaining 2,445 proteins were analyzed by BlastP against the NR database with a cutoff of E-value of 10-10. Of these, 680 (27.8%) proteins had hits outside of the alphaproteobacteria species, and 1,765 (72.2%) proteins had hits within alphaproteobacterial species (Additional file 2, Figure S6b). The 680 proteins were further analyzed for HGT in a similar manner to that discussed above. Phylogenetic trees were reconstructed for 61.6% of the cases, of which 53% were weakly supported and the remaining 8.6% (59 cases) were strongly supported by bootstrap values. The remaining 38.4% of cases did not result in phylogeny due to the reasons previously discussed. Thus, we identified 59 instances of HGT from non-orthologous proteins, of which 41 cases were from uncultured bacteria and were excluded from our analysis. The remaining 18 instances were found to be good candidates for HGT from different organisms with strong supported bootstrap values.
Of these 18 cases, we detected two instances of HGT that had been previously reported. The transfer of gi:67458540 from Yersinia species of gammaproteobacteria to Rickettsia felis URRWX Cal2 was reported by Merhej et al., 2011 [26], and the transfer of gi:71084007 from the CyanobacteriaSynechococcus and Prochlorococcus to two Candidatus Pelagibacter ubique strains was reported by Viklund et al., 2011 [22] (Additional file 1, Table S9). Of the remaining 16 cases, 50% of the transfers were observed in Candidatus Pelagibacter ubique, and the remaining were present in Wolbachia and Rickettsia species (Additional file 1, Table S9). These proteins were acquired from different origins, including bacterial species from the Firmicutes, Chlorobi, Cyanobacteria, Bacteroidetes, Actinobacteria and Proteobacteria clades (Epsilon, Delta, Beta and Gamma) as well as from Archaea and Metazoa (Hydra magnipapillata). Most of the transferred genes encoded unnamed proteins for which a function has not yet been determined, while others were involved in translocation and hydrolase and synthase activities. To determine the possible origins of these unnamed proteins, we compared them against the ACLAME database; however we were unable to identify genes with similarity above our cutoff value.
Functional annotation for putative HGT cases (without phylogenetic support) by Gene Ontology (GO)
Out of a total of 1,518 proteins from two groups (838 proteins having at least 1 gain event and 680 proteins from the non-orthologous group), 1,476 remained with no functional annotation. This group was therefore analyzed using the the GO ontology database. No GO terms were identified for 190 of the 1,476 proteins, while the remaining 1,286 proteins were assigned a GO term related to biological processes, molecular functions or cellular components. The results obtained from the GO analysis are described in Additional file 2, Figure S8 and Additional file 1, Table S10. We focused our analysis on the 9th and the 12th DAG levels of the GO hierarchy. At the 9th level, 25 proteins were associated with molecular functions. Of these, 5 were associated with DNA helicase activity and 4 were assigned ATP-dependent helicase activity. The remaining are showed in Additional file 1, Table S11. At the 12th level of the GO hierarchy, 33 proteins were associated with biological process, of which 18 were ATP catabolic and 4 were GTP catabolic. An analysis of the remaining 11 is provided in Additional file 1, Table S12. Thus, only 58 of 1,476 proteins (3.9%) demonstrated significant specificity for a particular GO term.
Discussion
Although previous studies have detected instances of horizontal gene transfer in Rickettsiae species [27-29]. HGTs have not been reported in other genomes such as Wolbachia, Anaplasma and Candidatus Pelagibacter ubique. In the current study, we carried out an exhaustive analysis to investigate additional instances of HGT in 31 complete Rickettsiales genomes and detected 38 new instances of HGT as well as 4 cases identified in previously published studies. Among these, several new cases were identified in Anaplasma and Candidatus Pelagibacter ubique as well as in Rickettsia species. Addionally, we carried out a functional analysis of the 42 HGTs identified using our automated pipeline (Figure 1). We considered the remaining cases (1,476 cases) with no phylogenetic support to be putative instances of transfer, and these were analyzed further by searching for GO terms associated with their potential functions.
HGTs between species from the same environment
We analyzed 42 instances of HGT with phylogenetic support and identified new instances of transfer into the genome of R. bellii from different bacterial phyla, including Firmicutes, Nitrospirales, Bacteroidetes, and Proteobacteria (Beta and Gamma). R. bellii has been previously reported to exchange genes related to amoebal symbionts [26], and there have been no reported cases of gene loss [27]. The newly introduced genes may be maintained as a consequence of the large size of the R. bellii genome. Our phylogenetic analyses also identified four instances of horizontal gene transfer among amoeba-resistant microorganisms (Additional file 1, Table S7, gi:157964915, gi:157825906, gi:157827021, gi:229586611). Three out of these four instances were among Amoebophilus asiasticus 5a2 and Rickettsia species, and one case was among Legionella species and Rickettsia species. Recently, genes were found to have been transferred between M. avium and L. pneumophila, which have sympatric lifestyles in free-living protozoa [46]. Additionally, empirical observations of transferred genes have been described in sympatric population [46,47]. Here, our phylogenetic reconstructions identified possible instances of HGT between the ancestors of Rickettsia and the amoeba-parasites. Our results support a model in which free-living amoeba act as a “melting pot” for genetic exchange, particularly HGT [47-49]. In addition to amoeba-resistant bacteria, fungi, giant virus and virophages have been detected in free-living protozoa [50-52], which suggests that bacteria living in sympatric populations facilitate more genetic exchange than those living in allopatric populations and that protists can serve as a hot spot for horizontal gene transfer.
We also analyzed instances of HGT from arthropods and their endosymbionts (Francisella tularensis). For example, the gene gi:225630595 was transferred from Francisella tularensis species to Wolbachia endosymbionts of the Culex species (Additional file 1, Table S7). This gene encodes for dihydroneopterin aldolase and participates in folate biosynthesis, which is absent from Wolbachia species [45]. Other cases include transfers from Francisella species to Orientia species and to the Wolbachia endosymbionts of Culex quinquefasciatus Pel (Additional file 1, Table S8, gi:189182975). Francisella-like bacterial endosymbionts were discovered in ticks of the Dermacentor family [53], and this transfer may have been the result of the co-habitation of Francisella and other species within the same host. In this work, we identified an instance of HGT from Aedes species to two Wolbachia strains (Additional file 1, Table S8, gi:42520378). This transfer was distinct from the case identified by Klasson et al., [44], we identified Wolbachia as the recipient species and Aedes as the donor species, whereas Klasson et al. reported a transfer in the opposite direction. Our interpretation of this transfer was based on several observations. First, our Bayesian phylogenetic tree for gene gi:4250378 had strong bootstrap support (pp=0.99) (Additional file 2, Figure S7), and the two sequences of Wolbachia were affiliated to form a monophyletic group not observed by Klasson et al. Second, we verified our Blast search results for the following 3 sequences: gi:4250378 from the Wolbachia endosymbiont of Drosophila melanogaster; gi:190571717 from the Wolbachia endosymbiont of Culex quinquefasciatus Pel; and gi:157104824 from Aedes aegypti. Moreover, the two protein sequences from Wolbachia demonstrated 62% amino acid identity to each other, but had only 51% amino acid identity to the protein from the Aedes species. Third, orthologous genes were not found in any other Wolbachia species. Taken together, we believe that this gene transfers occurred from the insect to the bacterium and that this first Wolbachia species may have subsequently transferred the gene to other species. Alternatively, the absence of this gene in other Wolbachia species may be due to subsequent gene loss. To date, the acquisition of eukaryotic genes by bacteria has rarely been documented. However, a typically eukaryotic glycoside-hydrolase that is necessary for starch breakdown in plants was recently found to have been transferred to bacteria via two successive transfers [54]. Moreover, observations supporting large-scale HGT from bacteria, fungi and plants into Bdelloid rotifers have been reported [55]. Horizontal gene transfers between distantly related organisms, i.e., bacteria and multicellular eukaryotes was thought to occur rarely, but our results demonstrate that these transfers may occur more frequently if they are between species that share the same arthropod host, i.e., ticks or beetles. The evolutionary history of these transfers remains a mystery and required further study. Here, we demonstrated two instances of HGT from the Wolbachia phage to the Wolbachia genome (Additional file 1, Table S8 gi:190570783, Table S9 gi:225630935). Wolbachia species habor virus-like particles of the phage WO [56,57], and in cases of Wolbachia-phage transfer, the phage is considered to be the vehicle of transfer via transduction mechanisms rather than the donor species [58].
In addition, we observed many instances of HGT in Candidatus Pelagibacter ubique. For example, the gene gi:71083892, which is involved in translocation activity, was transferred from several different species of gammaproteobacteria, such as Shewanella, Aeromonas, Vibrio, Moritella, and Photobacterium strains, as well as from Arcobacter butzleri RM4018 of the epsilonproteobacteria. Shewanella species are ubiquitous and are present on the North Atlantic coast as well as deep in the Baltic Sea [59]. Together with Shewanella species, Vibrio species occupy various aquatic habitats ranging from the deep sea to shallow coastal marine environments [60]. A phylogenetic tree based on the complete sequences of 16S rDNA demonstrated that Shewanella species are closely related to marine gammaproteobacteria such as Aeromonas, Vibrio, Photobacterium, Pseudoalteromonas, Colwellia and Psychromonas species [59]. Like Candidatus Pelagibacter ubique, these latter bacteria are marine organisms. Evidence of HGT has been reported recently in Shewanella species in warm aquatic environments [61]. We also analyzed other cases of HGT in Candidatus Nitrospira defluvii (Nitrospirales) (Additional file 1, Table S8, gi:157826324) and Hydra magnipapillata (Additional file 1, Table S9, gi:71083916) in aquatic environments, although the mechanism responsible for these transfers is not clearly understood. It has been shown that HGTs mediated by gene transfer agents frequently found in marine ecosystems facilitate the adaptation of these microorganisms to new environments [62]. Moreover, mobile genetic elements (MGEs) have been shown to play a crucial role in the expansion of the environmental niches of pathogenic and environmental Vibrio species [63]. Taken together, we believe that the aquatic environment may play a crucial role in facilitating such transfers in these organisms.
Overall, we analyzed a number of instances of HGT from different niches, including free-living protozoa, arthropod hosts and aquatic environments. In addition to the characteristic transfers between different ecological niches, HGTs were also analyzed from other origins, including the transfers of genes involved in IS5 family transposase activity from Archaeal species to Wolbachia species(Additional file 1, Table S9, gi:42520763). The acquisition of this gene may have been the result of massive gene exchange occurring between bacterial and archaeal species [64]. This horizontal gene transfer from different origins revealed a rhizome for Rickettsiales genomes [65] (Figure 2). We also analyzed the functions of novel genes, the origins of which remaine unknown, although previous studies have suggested that these new genes may stem from plasmidic or viral origin [66].
Figure 2.
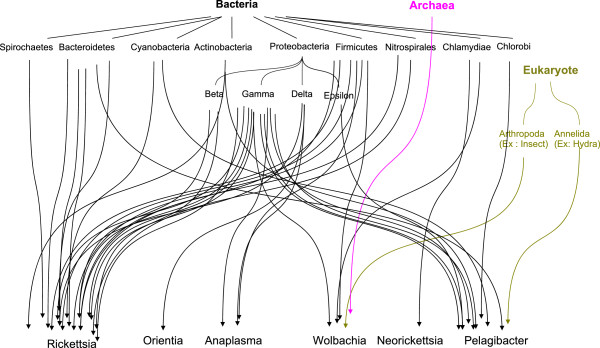
Rhizome of Rickettsiales genomes. Bacteria (in black), Archaea (in pink) and Eukaryotes (in yellow).
Functional assignment for putative transfers
Here, we analyzed putative instances of transfer (i.e., those with no phylogenetic support). Only 3.9% of a total of 1,476 proteins were assigned specific terms from the gene ontology database. The majority (18 cases) were assigned terms related to ATP catabolic processes, which are used in nucleotide metabolism, and genes involved in the synthesis for nucleotides are completely lacking in Rickettsia species [67]. Moreover, these translocase enzymes were identified in Rickettsia species more closely related to amoeba-symbionts than other Rickettsia[68-70]. The deficiency of these genes is likely to be linked to the activity of ATP/ADP translocase (tlc) which is used to import ATP from the host [71]. This parasitic enzyme may have been duplicated in the Chlamydiales ancestor, which may have been transferred to plant and Rickettsiae species approximately 1.16 billion years ago. Plant and algae plastids acquired tlc at approximately the same time as Parachlamydiaceae and Chlamydiaceae diverged [72], which suggests that such a transfer may have occurred at a time when Rickettsiales and Chlamydiaceae shared a common protist ancestral host. Furthermore, these bacteria may have been facultative intracellular bacteria that shared a common ancestral cell host, such as a free-living amoeba [72]. Likewise, it is likely that an ancestor of Rickettsia infected an amoeba-like protozoan ancestor and thereby exchanged genes with other symbionts [11]. Furthermore, it has been shown recently that R. bellii is able to survive within phagocytic amoeba and can multiply in the nucleus of eukaryotic cells [28]. It has also been reported that Rickettsiae species are deficient in tlc, which is a parasite enzyme involved in energy metabolism [72]. Rickettsiae acquired tlc from the Rickettsiales ancestor and are therefore able to import ATP from the host cell, whereas members of Anaplasmataceae and Orientia subsequently lost this gene [72] (Additional file 2, Figure S9). Lacking strong evidence in support of this loss, we therefore can only predict that these proteins may be the result of putative HGT.
Conclusion
We developed an automated approach for the detection of horizontal gene transfer events that depends on the use of stringent filters. This approach consists of a number of specialized features, including the application of a parsimony method for inferring phyletic patterns followed by a blast filter, automated phylogenetic reconstruction and horizontal gene transfer detection. Using this approach, we identified many new instances of transfer into genomes, including those of Wolbachia species, Anaplasma species, Candidatus Pelagibacter ubique and other species of Rickettsia. The current study provides a systematic pipeline for the automated detection of horizontal gene transfer events from several complete proteomic sequences and therefore can be applied to detect the instances of gens transfer for other genomes of interest. This study also provides i) a global analysis of HGT cases in different ecological niches, including protist and arthropods hosts and aquatic environments, and from diverse origins, such as prokaryotic, eukaryotic, archaeal and viral domains, and ii) gene functions resulting from putative instances of gene transfer. Our identification of HGT cases indicative of transfer from different origins allowed us to develop a rhizome for Rickettsiales genomes.
Methods
Complete Proteomes
The protein sets for 31 completely sequenced species (13 Rickettsia strains, 2 Orientia strains, 4 Anaplasma strains, 4 Ehrlichia strains, 4 Wolbachia strains, 2 Neorickettsia strains, Candidatus Pelagibacter ubique HTCC1602 and Caulobacter crescentus CB15) within the Rickettsiales order were downloaded from NCBI (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/). The details regarding the NCBI accession numbers for these genomes are provided in Additional file 1, Table S1.
Detection of orthologous groups
We employed OrthoMCL [73] to retrieve orthologous proteins. Only protein sequences longer than 50 amino acid residues were considered for further analysis. Homologous sequences were selected using the all-against-all BlastP algorithm [74] with an E value of less than 0.00001. Then, clustering of the orthologous sequences was analyzed using the Markov Cluster algorithm [75] which was based on probability and graph flow theory that allows the simultaneous classification of global relationships in a similarity space. The inflation index of 1.5 was used to regulate cluster tightness (granularity), and the resulting clustered ortholog groups were analyzed further.
16S rRNA tree for Rickettsiales
We reconstructed a phylogenetic tree using 16S rRNA sequences to form a species tree. The 16S RNA sequences were downloaded from NCBI (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/), and the alignments were carried out using MUSCLE 3.6 [76]. Next, the phylogenetic tree was constructed with Mega5 software [77] and the following three methods: Neighbor Joining (NJ), Maximum Parsimony(MP) and Maximum Likelihood (ML). The 16S rRNA species tree is provided as Additional file 2, Figure S1.
Detection of gene gain and loss by phyletic pattern
The ortholog groups identified by OrthoMCL were submitted to PhyloPattern [78], a software library based on the Prolog language [79], for the automated analysis and manipulation of phylogenetic trees (within the DAGOBAH framework). PhyloPattern contains three modules: (1) a tree annotation module that incorporates a traversal algorithm, infers phylogenetic trees composed of hierarchical nodes as binary structures, and functionally annotates each node; (2) a pattern-matching module to define patterns that can be used to search phylogenetic trees; and (3) a tree comparison module for the global comparison of tree topologies and the identification of matching nodes.
The 16S rRNA species tree was used as a reference for inferring topologies for further analysis. We mapped the characteristics “presence” and “absence” for genes on the nodes of the species tree using the tree annotation module. The presence and absence characteristics corresponded to the presence and absence of the gene in a species from a group of orthologs at a particular node. Next, we defined the distribution of ancestral characteristics by implementing the Sankoff parsimony algorithm [80]. Further analyses were carried out to search for gene gain and loss events in phylogenetic subtrees using the pattern-matching module in PhyloPattern [61]. A phylogenetic subtree consisted of an ancestral node and two descendant nodes. The pattern was defined as a loss event if the gene was present in the ancestral node and one descendant node but absent in another node. Conversely, the pattern wass defined as a gain event if the gene was absent in the ancestral node and one descendant node but was present in another descendant node (Additional file 2, Figure S2). This pattern for each local subtree was then used with the tree comparison module to infer the topologies of global trees. The total number of gain and loss events was subsequently calculated as the sum of all events of the subtrees. The gain and loss events inferred from the PhyloPattern are shown in Additional file 2, Figure S2 and Figure S3. Finally, one representative protein sequence from each ortholog group was analyzed using the Blast program. The selection of sequences was carried out in the following order: Rickettsia species, Orientia species, Wolbachia species, Anaplasma species, Ehrlichia species, Neorickettsia species and Candidatus Pelagibacter ubique.
Analysis of homologous sequences using the Blast filter
Based on the number of gene gain and loss events identified using PhyloPattern, one protein sequence from each representative event was analyzed by Blast search. We speculated that gain events may have been the result of HGT and that loss events may have been the result of gene loss during the course of a species’ evolution. As the main objective of the current study was to analyze HGT, we evaluated only instances of gene gain unless otherwise stated.
We carried out the BlastP search using cutoff E-values less than 10-10 against the NR NCBI database using the remote method. The first hundred homologous protein sequences from each query were retrieved and classified into two groups: (a). hits within the group of interest (within alphaproteobacteria) and (b). hits outside of the group of interest (out of the alphaproteobacteria). The protein sequences outside of the group of interest were retrieved and subjected to further phylogenetic analysis. We chose only those Blast results that returned at least one hit outside of alphaproteobacteria for further analysis. Because the results of the Blast search that provided hits only within the alphaproteobacteria were not considered for further analysis, these cases were not analyzed in the phylogenetic reconstructions. Interestingly, we also detected several protein sequences in the studied group that were lineage-specific, and we presumed that these represented new genes or novel genes. The origin of new genes can result from many types of genetic events, such as gene duplication, gene fission and fusion, lateral gene transfer, transposable elements (TEs) and de novo gene formation [81].
Phylogeny construction
The results obtained from the Blast analysis were submitted to FIGENIX [82,83] for phylogenetic reconstruction within the DAGOBAH framework. FIGENIX is an automated pipeline for structural and functional annotation in which the structural part represents the integration of abinitio and homology-based approaches and the functional annotation consists of fully automated modules for complex phylogenomic inference. Homologous sequences were retrieved from the NR NCBI database by BlastP search and were aligned using MUSCLE v3.6 [59]. Functional domains were detected for all aligned sequences using the HMMPFAM [84] database. Ambiguously aligned regions, including data that produced bias or noise (such as short sequences), were automatically excluded from the analysis, and a new alignment was built by merging the preserved parts of the domain alignments. Based on the new alignment, phylogenetic trees were constructed using three different methods: NJ with ClustalW software [85], MP using the PAUP package [86] (p-distance) and ML using the TREE-PUZZLE package with the default parameters [87]. The topologies of these trees were compared using the PSCORE command (“Templeton winning sites” test) from the PAUP package and the KISHINO-HASEGAWA test [88] from the TREE-PUZZLE package. All three trees were then merged into a final consensus tree. The consensus tree was compared to a reference species tree (Tree of Life) from NCBI, and phylogenies were inferred for orthologous protein sequences against the query sequence. The topology of the 16S rRNA sequence and the tree from NCBI were similar with the exception that the NCBI tree included nodes that were not bifurcated.
Detection of gene transfer events by HGT agent
The output generated by FIGENIX was submitted to the multi-agent system DAGOBAH [89], in which HGT events were detected using an in-house-built transfer filter called HGT agent. HGT agent uses PhyloPattern [61] to annotate each internal duplication node of the tree with three tags, including: recipient species, donor species and external species. Then, it applies a special phyletic pattern and searches the gene tree to find recipient species that are closer to donor species than to other external species that would otherwise be placed between the recipient and donor species in the species tree. In other words, a “donor” subtree must contain only species of a specific group and not those from the “recipient” group and vice versa, and there should be no common species between the donor and external groups. Using this agent, one can specify the names of the donor species and the recipient species according to their usage. In the current study, we defined the recipient species as the Rickettsiales group, the donor species as the non-alphaproteobacterial species and external species as different than donor species. We considered the donor species to be the most likely donors. The pattern used to detect HGT is shown in Additional file 2, Figure S4. In this way, the HGT agent used phyletic patterns to detect instances of HGT with strongly supported bootstrap values (greater than 50%), which were than confirmed by Bayesian methods to validate the results. The results of the Bayesian analysis are presented in Additional file 1, Table S2 and Additional file 2, Figure S7.
Confirmation of HGT events by Bayesian analysis
We used Phylobayes 3.2 [90] for the Bayesian analysis to detect HGT cases. Phylobayes was run using the CAT model and a gamma correction with 4 rate categories [91]. Four chains were run for at least two million generations. The first 250 trees were discarded as “burn-in”, and the remaining trees from each chain were used to test for convergence (less than 0.1) and computed for consensus trees. The trees were visualized using FigTree 3.1, a program for the graphical viewing of phylogenetic trees (http://tree.bio.ed.ac.uk/software/figtree/).
Detection of transfer from mobile genetic elements using the ACLAME database
To determine the possible origins of the hypothetical transferred proteins, we verified these against the ACLAME database [92], which is a collection of known mobile genetic elements (MGEs) from phage genomes, plasmids and transposons. BlastP searches were performed for sequences with an E-value of less than 10-3 and a percent of identity greater than 50%.
Functional annotation by Gene Ontology
Gene ontology can be represented as a direct acyclic graph (DAG) with a hierarchical structure encompassing numerous levels and terms (Gene Ontology project 2006) [93]. Genes associated with GO terms are more likely to be functionally enriched at each DAG level. General biological roles are identified at the higher levels, and specific roles are identified at the lower levels. Deeper DAG levels indicate high specificity for the gene [94]. As functions for the 42 HGT cases had been previously discussed, they were not included in the GO analysis. Best hit BlastP (E-values less than 0.0001) for each of the 1,476 proteins against the GO database were retrieved and analyzed.
Data Access
For cases of horizontal gene transfer, the Genbank IDs for the phylogenetic trees resulting from at least 1 gain and 1 loss event and the non-orthologous proteins are stored and can be obtained from the I.O.D.A. database [95] (http://ioda.univ-provence.fr).
Abbreviations
HGT: Horizontal gene transfer; NR: Non-redundant; COG: Cluster of Orthologous Groups.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PP and DR conceived of and designed the study. PG supervised the software development. LG and PTL developed the bioinformatic tool to adapt DAGOBAH to bacteria and the ortholog filter for the OrthoMCL groups. JP developed a preliminary version of the Blast filter, the HGT detection agent and the graphical interface package that visualized the results deployed on the I.O.D.A web site. OC installed OrthoMCL and performed the GO annotation. PTL performed the data production and the analysis. PTL and HGR analyzed the phylogenies. PTL, HGR, PP and DR wrote the manuscript. PTL, HGR, DR and PP analyzed the data. All authors read and approved the final manuscript.
Supplementary Material
Table S1 shows the NCBI accession numbers for the studied species.Table S2 shows the Bayesian analysis for the horizontal gene transfer cases from other species to Rickettsiales excluding alphaproteobacteria. Violet rows represent transfer cases from at least 1 gain event, and the brown rows represent transfer cases from a non-orthologous group. PP indicates the posterior probability value from the Bayesian analysis, aa indicates amino acids, and BV indicates the bootstrap values obtained with the NJ, MP, and ML methods. Table S3 is a list of annotations for the conserved genes from 267 Ortholog clusters of Rickettsiales and Caulobacter cresentus CB15 from the COG database. Table S4 is a list of annotations for the conserved genes from 277 Ortholog clusters of Rickettsia, Orientia, Wolbachia, Anaplasma, Ehrlichia, Neorickettsia and Candidatus Pelagibacter ubique from the COG database. Table S5 is a list of annotation for the conserved genes from 328 Ortholog clusters of Rickettsia, Orientia, Wolbachia, Anaplasma, Ehrlichia and Neorickettsia from the COG database. Table S6 is a list of annotation sfor the 1,709 orthologous groups based on the COG database. Table S7 is a table of the horizontal gene transfer cases identified from at least 1 gain and 1 loss event (part 1). Table S8 is a table of the horizontal gene transfer cases identified from at least 1 gain and 1 loss event (part 2). Table S9 is a table of the horizontal gene transfer cases identified from non-orthologous groups. Table S10 is a table showing the distribution of proteins annotated from the Gene Ontology database. Table S11 is a table showing the assignment of proteins annotated by Gene Ontology at the 9th level. Table S12 is a table showing the assignment of proteins annotated by Gene Ontology at the 12th level. Table S13 is a table showing the non-synonymous/synonymous substitution analysis for de novo genes.
Figure S1 shows the 16S rRNA species tree.Figure S2 shows the two principle patterns for the detection of gene gain and gene loss using PhyloPattern: a. Loss event and b. Gain event. Red circles represent the “present” characteristic, blue circles represent the “absent” characteristic. The pattern was defined as a loss event if the gene was present in the ancestral node and one descendant node but absent in another node (violet rhomboid). Conversely, the pattern was defined as a gain event if the gene was absent in the ancestral node and one descendant node but was present in another descendant node (green rhomboid). Figure S3 maps the gain and loss events from the phyletic patterns onto the species tree. Red circles represent the “present” characteristic, and blue circles represent the “absent” characteristic. Violet and green rhomboids represent loss and gain events, respectively. Figure S4 shows the transfer pattern detected by HGT agent from the multi-agent system DAGOBAH. HGT agent applied PhyloPattern to annotate the subtree (see Methods). Yellow circle with a D indicates that a duplication of the gene occurred at that node. Red, green and cyan circles represent recipient species, donor species and external species, respectively. Tax id: taxonomy identity. Figure S5 shows the results of the number of orthologous groups obtained from the gene gain and gene loss analysis by PhyloPattern in each respective event.
Figure S6a is a flowchart representing the detailed description of the systematic analysis of horizontal gene transfer detection with at least one gain event. Figure S6b is a flowchart representing the detailed description of the systematic analysis of horizontal gene transfer detection for non-orthologous groups. Figure S7 shows Bayesian phylogenetic trees for a subset of the detected instances of horizontal gene transfer from the group with at least 1 gain and 1 loss (No. 1 to 10) and the non-orthologous groups (No. 11 to 15). The scale bar represents the average number of substitutions per site. Numbers at nodes correspond to the posterior probabilities calculated by Phylobayes. The donor species are indicated in blue, and the recipient species are indicated in red. Figure S8 represents the distribution of proteins assignments using the Gene Ontology database. Figure S9 illustrates the hypothesis for the acquisition of the ADP/ATP translocase-encoding gene by Rickettsia species. The evolutionary time was estimated from the data provided by Greub and Raoult, 2003. Figure S10 is a histogram of COG size.
Contributor Information
Phuong Thi Le, Email: phuongle@hotmail.fr.
Hemalatha Golaconda Ramulu, Email: hema1752002@gmail.com.
Laurent Guijarro, Email: l.guij@orange.fr.
Julien Paganini, Email: julien.paganini@univ-provence.fr.
Philippe Gouret, Email: philippe.gouret@univ-provence.fr.
Olivier Chabrol, Email: olivier.chabrol@univ-provence.fr.
Dider Raoult, Email: didier.raoult@gmail.com.
Pierre Pontarotti, Email: Pierre.Pontarotti@univ-provence.fr.
Acknowledgements
PTL and HGR are funded by Infectiopole Sud. JP is funded by MIE from CNRS. We thank both anonymous reviewers for improving the manuscript.
References
- Doolittle WF. Lateral genomics. Trends in Cell Biol. 1999;9(12):M5—8. [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405(6784):299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer accelerates genome innovation and evolution. Mol biol evol. 2003;20(10):1598–1602. doi: 10.1093/molbev/msg154. [DOI] [PubMed] [Google Scholar]
- Merhej V, Royer-Carenzi M, Pontarotti P, Raoult D. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol Direct. 2009;4:13. doi: 10.1186/1745-6150-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussau B, Karlberg EO, Frank AC, Legault BA, Andersson SGE. Computational inference of scenarios for alpha-proteobacterial genome evolution. Proc National Acad Sci USA. 2004;101(26):9722–9727. doi: 10.1073/pnas.0400975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHILIP CB. Tsutsugamushi disease in World War II. J Parasitology. 1948;34(3):169–191. doi: 10.2307/3273264. [DOI] [PubMed] [Google Scholar]
- Seong SY, Choi MS, Kim IS. Orientia tsutsugamushi infection: overview and immune responses. Microbes and Infection / Institut Pasteur. 2001;3:11–21. doi: 10.1016/S1286-4579(00)01352-6. [DOI] [PubMed] [Google Scholar]
- Tamura A, Ohashi N, Urakami H, Miyamura S. Classification of Rickettsia tsutsugamushi in a new genus, Orientia gen. nov., as Orientia tsutsugamushi comb. nov. Int J Syst Bacteriol. 1995;45(3):589–591. doi: 10.1099/00207713-45-3-589. [DOI] [PubMed] [Google Scholar]
- Raoult D, Roux V. Rickettsioses as paradigms of new or emerging infectious diseases. Clin Microbio Rev. 1997;10(4):694–719. doi: 10.1128/cmr.10.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhej V, Raoult D. Rickettsial evolution in the light of comparative genomics. Biol Rev Cambridge Philos Soc. 2011;86(2):379–405. doi: 10.1111/j.1469-185X.2010.00151.x. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Karr TL. Wolbachia: evolutionary novelty in a rickettsial bacteria. BMC Evol Biol. 2001;1:10. doi: 10.1186/1471-2148-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SM, Dumler JS, Bakken JS, Walker DH. Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J Clin Microbiol. 1994;32(3):589–595. doi: 10.1128/jcm.32.3.589-595.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson BE, Dawson JE, Jones DC, Wilson KH. Ehrlichia chaffeensis, a new species associated with human ehrlichiosis. J Clin Microbiol. 1991;29(12):2838–2842. doi: 10.1128/jcm.29.12.2838-2842.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson JE, Anderson BE, Fishbein DB, Sanchez JL, Goldsmith CS, Wilson KH, Duntley CW. Isolation and characterization of an Ehrlichia sp. from a patient diagnosed with human ehrlichiosis. J Clin Microbiol. 1991;29(12):2741–2745. doi: 10.1128/jcm.29.12.2741-2745.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikihisa Y. The tribe Ehrlichieae and ehrlichial diseases. Clin microbiol rev. 1991;4(3):286–308. doi: 10.1128/cmr.4.3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoh N, Tanaka K, Shibata F, Kondo N, Hizume M, Shimada M, Fukatsu T. Wolbachia genome integrated in an insect chromosome: evolution and fate of laterally transferred endosymbiont genes. Genome Res. 2008;18(2):272–280. doi: 10.1101/gr.7144908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappé MS, Short JM, Carrington JC, Mathur EJ. Genome streamlining in a cosmopolitan oceanic bacterium. Sci (New York, N.Y.) 2005;309(5738):1242–1245. doi: 10.1126/science.1114057. [DOI] [PubMed] [Google Scholar]
- Williams KP, Sobral BW, Dickerman AW. A robust species tree for the alphaproteobacteria. J bacteriol. 2007;189(13):4578–4586. doi: 10.1128/JB.00269-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades K, Madoui MA, Le P, Robert C, Raoult D. Phylogenomic analysis of Odyssella thessalonicensis fortifies the common origin of Rickettsiales, Pelagibacter ubique and Reclimonas americana mitochondrion. PloS one. 2011;6(9):e24857. doi: 10.1371/journal.pone.0024857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash JC, Boyd A, Huggett MJ, Grote J, Carini P, Yoder RJ, Robbertse B, Spatafora JW, Rappé MS, Giovannoni SJ. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Sci rep. 2011;1:13. doi: 10.1038/srep00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viklund J, Ettema TJG, Andersson SGE. Independent Genome Reduction and Phylogenetic Reclassification of the Oceanic SAR11 Clade. Mol Biol Evol. 2011;29(2):599–615. doi: 10.1093/molbev/msr203. [DOI] [PubMed] [Google Scholar]
- Brindefalk B, Ettema TJG, Viklund J, Thollesson M, Andersson SGE. A Phylometagenomic Exploration of Oceanic Alphaproteobacteria Reveals Mitochondrial Relatives Unrelated to the SAR11 Clade. PloS One. 2011;6(9):e24457. doi: 10.1371/journal.pone.0024457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodráguez-Ezpeleta N, Embley TM. The SAR11 Group of Alpha-Proteobacteria Is Not Related to the Origin of Mitochondria. PloS One. 2012;7:e30520. doi: 10.1371/journal.pone.0030520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloesges T, Popa O, Martin W, Dagan T. Networks of gene sharing among 329 proteobacterial genomes reveal differences in lateral gene transfer frequency at different phylogenetic depths. Mol biol evol. 2011;28(2):1057–1074. doi: 10.1093/molbev/msq297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhej V, Notredame C, Royer-Carenzi M, Pontarotti P, Raoult D. The rhizome of life: the sympatric Rickettsia felis paradigm demonstrates the random transfer of DNA sequences. Mol Biol Evol. 2011;28(11):3213–3223. doi: 10.1093/molbev/msr239. [DOI] [PubMed] [Google Scholar]
- Georgiades K, Merhej V, El Karkouri K, Raoult D, Pontarotti P. Gene gain and loss events in Rickettsia and Orientia species. Biol Direct. 2011;6:6. doi: 10.1186/1745-6150-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, La Scola B, Audic S, Renesto P, Blanc G, Robert C, Fournier PE, Claverie JM, Raoult D. Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet. 2006;2(5):e76. doi: 10.1371/journal.pgen.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Ogata H, Robert C, Audic S, Claverie JM, Raoult D. Lateral gene transfer between obligate intracellular bacteria: evidence from the Rickettsia massiliae genome. Genome Res. 2007;17(11):1657–1664. doi: 10.1101/gr.6742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubin V, Lerat E, Perrière G. The source of laterally transferred genes in bacterial genomes. Genome Biol. 2003;4(9):R57. doi: 10.1186/gb-2003-4-9-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc National Acad Sci USA. 1998;95(16):9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SD, Berg OG. Detection of genes with atypical nucleotide sequence in microbial genomes. J mol evol. 2002;54(3):365–375. doi: 10.1007/s00239-001-0051-8. [DOI] [PubMed] [Google Scholar]
- Deschavanne P, Giron A, Vilain J, Dufraigne C, Fertil B. Genomic Signature is Preserved in Short DNA Fragments. IEEE Int Symp Bio-Informatics and Biomed Eng. 2000;33(1):e6. [Google Scholar]
- Dufraigne C, Fertil B, Lespinats S, Giron A, Deschavanne P. Detection and characterization of horizontal transfers in prokaryotes using genomic signature. Nucleic acids res. 2005;33:e6. doi: 10.1093/nar/gni004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst biol. 2002;51(3):492–508. doi: 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- Zhaxybayeva O, Hamel L, Raymond J, Gogarten JP. Visualization of the phylogenetic content of five genomes using dekapentagonal maps. Genome biol. 2004;5(3):R20. doi: 10.1186/gb-2004-5-3-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O, Gogarten JP, Charlebois RL, Doolittle WF, Papke RT. Phylogenetic analyses of cyanobacterial genomes: quantification of horizontal gene transfer events. Genome res. 2006;16(9):1099–1108. doi: 10.1101/gr.5322306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999;21:108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 2002;12:17–25. doi: 10.1101/gr.176501. [DOI] [PubMed] [Google Scholar]
- David LA, Alm EJ. Rapid evolutionary innovation during an Archaean genetic expansion. Nature. 2011;469(7328):93–96. doi: 10.1038/nature09649. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson L, Kambris Z, Cook PE, Walker T, Sinkins SP. Horizontal gene transfer between Wolbachia and the mosquito Aedes aegypti. BMC Genomics. 2009;10:33. doi: 10.1186/1471-2164-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC, McGraw EA, Martin W, Esser C, Ahmadinejad N, Wiegand C, Madupu R, Beanan MJ, Brinkac LM, Daugherty SC, Durkin AS, Kolonay JF, Nelson WC, Mohamoud Y, Lee P, Berry K, Young MB, Utterback T, Weidman J, Nierman WC, Paulsen IT, Nelson KE, Tettelin H, O’Neill SL, Eisen JA. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2(3):E69. doi: 10.1371/journal.pbio.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamrabet O, Merhej V, Pontarotti P, Raoult D, Drancourt M. The genealogic tree of mycobacteria reveals a long-standing sympatric life into free-living protozoa. PloS One. 2012;7(4):e34754. doi: 10.1371/journal.pone.0034754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer M, Yutin N, Pagnier I, Barrassi L, Fournous G, Espinosa L, Robert C, Azza S, Sun S, Rossmann MG, Suzan-Monti M, La Scola B, Koonin EV, Raoult D. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc National Acad Sci USA. 2009;106(51):21848–21853. doi: 10.1073/pnas.0911354106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moliner C, Fournier PE, Raoult D. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol Rev. 2010;34(3):281–94. doi: 10.1111/j.1574-6976.2009.00209.x. [DOI] [PubMed] [Google Scholar]
- Thomas V, Greub G. Amoeba/amoebal symbiont genetic transfers: lessons from giant virus neighbours. Intervirology. 2010;53(5):254–67. doi: 10.1159/000312910. [DOI] [PubMed] [Google Scholar]
- Chrisman CJ, Alvarez M, Casadevall A. Phagocytosis of Cryptococcus neoformans by, and nonlytic exocytosis from, Acanthamoeba castellanii. App environ microbiol. 2010;76(18):6056–6062. doi: 10.1128/AEM.00812-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoult D, Boyer M. Amoebae as genitors and reservoirs of giant viruses. Intervirology. 2010;53(5):321–9. doi: 10.1159/000312917. [DOI] [PubMed] [Google Scholar]
- La Scola B, Audic S, Robert C, Jungang L, de Lamballerie X, Drancourt M, Birtles R, Claverie JM, Raoult D. A giant virus in amoebae. Sci (New York, N.Y.) 2033;299(5615):2003. doi: 10.1126/science.1081867. [DOI] [PubMed] [Google Scholar]
- Scoles GA. Phylogenetic analysis of the Francisella-like endosymbionts of Dermacentor ticks. J Med Entomology. 2004;41(3):277–286. doi: 10.1603/0022-2585-41.3.277. [DOI] [PubMed] [Google Scholar]
- Arias MC, Danchin EGJ, Coutinho P, Henrissat B, Ball S. Eukaryote to gut bacteria transfer of a glycoside hydrolase gene essential for starch breakdown in plants. Mobile genet elem. 2012;2(2):81–87. doi: 10.4161/mge.20375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev EA, Meselson M, Arkhipova IR. Massive horizontal gene transfer in bdelloid rotifers. Sci (New York, N.Y.) 2008;320(5880):1210–1213. doi: 10.1126/science.1156407. [DOI] [PubMed] [Google Scholar]
- Masui S, Kamoda S, Sasaki T, Ishikawa H. Distribution and evolution of bacteriophage WO in Wolbachia, the endosymbiont causing sexual alterations in arthropods. J Mol Evol. 2000;51(5):491–497. doi: 10.1007/s002390010112. [DOI] [PubMed] [Google Scholar]
- Masui S, Kuroiwa H, Sasaki T, Inui M, Kuroiwa T, Ishikawa H. Bacteriophage WO and virus-like particles in Wolbachia, an endosymbiont of arthropods. Biochem Biophys Res Commun. 2001;283(5):1099–1104. doi: 10.1006/bbrc.2001.4906. [DOI] [PubMed] [Google Scholar]
- Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3(9):722–32. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- Zhao JS, Deng Y, Manno D, Hawari J. Shewanella spp. genomic evolution for a cold marine lifestyle and in-situ explosive biodegradation. PloS one. 2010;5(2):e9109. doi: 10.1371/journal.pone.0009109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reen FJ, Almagro-Moreno S, Ussery D, Boyd EF. The genomic code: inferring Vibrionaceae niche specialization. Nat rev Microbiol. 2006;4(9):697–704. doi: 10.1038/nrmicro1476. [DOI] [PubMed] [Google Scholar]
- Caro-Quintero A, Deng J, Auchtung J, Brettar I, Höfle MG, Klappenbach J, Konstantinidis KT. Unprecedented levels of horizontal gene transfer among spatially co-occurring Shewanella bacteria from the Baltic Sea. ISME J. 2011;5:131–140. doi: 10.1038/ismej.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel LD, Young E, Delaney J, Ruhnau F, Ritchie KB, Paul JH. High frequency of horizontal gene transfer in the oceans. (New York, N.Y.) 2010;330(6000):50. doi: 10.1126/science.1192243. [DOI] [PubMed] [Google Scholar]
- Hazen TH, Pan L, Gu JD, Sobecky PA. The contribution of mobile genetic elements to the evolution and ecology of Vibrios. FEMS Microbiol Ecol. 2010;74(3):485–499. doi: 10.1111/j.1574-6941.2010.00937.x. [DOI] [PubMed] [Google Scholar]
- Aravind L, Tatusov RL, Wolf YI, Walker DR, Koonin EV. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends in Genet: TIG. 1998;14(11):442–444. doi: 10.1016/S0168-9525(98)01553-4. [DOI] [PubMed] [Google Scholar]
- Raoult D. The post-Darwinist rhizome of life. Lancet. 2010;375(9709):104–105. doi: 10.1016/S0140-6736(09)61958-9. [DOI] [PubMed] [Google Scholar]
- Cortez D, Forterre P, Gribaldo S. A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol. 2009;10(6):R65. doi: 10.1186/gb-2009-10-6-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renesto P, Ogata H, Audic S, Claverie JM, Raoult D. Some lessons from Rickettsia genomics. FEMS, journal=Microbiol Rev. 2005. pp. 99–117. [DOI] [PubMed]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Sci (New York, N.Y.) 1998;282(5389):754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- Zomorodipour A, Andersson SG. FEBS lett. 1999. pp. 11–15. [DOI] [PubMed]
- Wolf YI, Aravind L, Koonin EV. Rickettsiae and Chlamydiae: evidence of horizontal gene transfer and gene exchange. Trends in Genet: TIG. 1999;15(5):173–175. doi: 10.1016/S0168-9525(99)01704-7. [DOI] [PubMed] [Google Scholar]
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Pontén T, Alsmark UC, Podowski RM, Näslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396(6707):133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- Greub G, Raoult D. History of the ADP/ATP-translocase-encoding gene, a parasitism gene transferred from a Chlamydiales ancestor to plants 1 billion years ago. Appl Environ Microbiol. 2003;69(9):5530–5535. doi: 10.1128/AEM.69.9.5530-5535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert J, Christian J, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen S. Graph clustering by flow simulation. PhD thesis. University of Utrecht, The Netherlands 2000.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouret P, Thompson JD, Pontarotti P. PhyloPattern: regular expressions to identify complex patterns in phylogenetic trees. BMC Bioinf. 2009;10:298. doi: 10.1186/1471-2105-10-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren D, Pereira L, Pereira F. Symposium on Artifical Intelligence and Programming Languages. New York USA: SIGPLAN Notices; 1997. Prolog: the language and its implementation compared with Lisp; pp. 109–15. [Google Scholar]
- Sankoff D. Minimal mutation trees of sequences. SIAM J App Methamatics, Volume 28. pp. 35–42. Available at http://ebookbrowse.com/1975-sankoff-pdf-d293771529.
- Chandrasekaran C, Betran E. Origins of new genes and pseudogenes. Nature Education. 2008. Available at, http://www.nature.com/scitable/topicpage/origins-of-new-genes-and-pseudogenes-835.
- Gouret P, Vitiello V, Balandraud N, Gilles A, Pontarotti P, Danchin EGJ. FIGENIX: intelligent automation of genomic annotation: expertise integration in a new software platform. BMC Bioinf. 2005;6:198. doi: 10.1186/1471-2105-6-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paganini J, Gouret P. Reliable Phylogenetic Trees Building: A New Web Interface for FIGENIX. Evolutionary bioinf online. 2012;8:417–421. doi: 10.4137/EBO.S9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ, CLUSTAL W. improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics (Oxford, England) 1998;14(9):755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Swofford D. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Method) Sunderland, Massachusetts: Sinauer Associates; 2003. [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics (Oxford, England) 2002;18(3):502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J mol evol. 1989;29(2):170–179. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- Gouret P, Paganini J, Dainat J, Louati D, Darbo E, Pontarotti P, Levasseur A. In: Evolutionary Biology - Concepts, Biodiversity, Marcroevolution and Genome Evolution. Pontarotti P, editor. Berlin Heidelberg: Springer - Verlag; 2011. Integration of Evolutionary Biology Concepts for Functional Annotation and Automation of Complex Research in Evolution: The Multi-Agent Software System DAGOBAH; pp. 71–76. [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics (Oxford, England) 2009;25(17):2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol biol evol. 2004;21(6):1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Leplae R, Lima-Mendez G, Toussaint A. ACLAME: a CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010;38(Database issue):D57—61. doi: 10.1093/nar/gkp938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology (GO) project in 2006. Nucleic acids res. 2006;34(Database issue):D322—326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Cui Y. GeneInfoViz: constructing and visualizing gene relation networks. In silico biol. 2004;4(3):323–333. [PubMed] [Google Scholar]
- Levasseur A, Paganini J, Dainat J, Thompson JD, Poch O, Pontarotti P, Gouret P. The chordate proteome history database. Evolutionary bioinf online. 2012;8:437–447. doi: 10.4137/EBO.S9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 shows the NCBI accession numbers for the studied species.Table S2 shows the Bayesian analysis for the horizontal gene transfer cases from other species to Rickettsiales excluding alphaproteobacteria. Violet rows represent transfer cases from at least 1 gain event, and the brown rows represent transfer cases from a non-orthologous group. PP indicates the posterior probability value from the Bayesian analysis, aa indicates amino acids, and BV indicates the bootstrap values obtained with the NJ, MP, and ML methods. Table S3 is a list of annotations for the conserved genes from 267 Ortholog clusters of Rickettsiales and Caulobacter cresentus CB15 from the COG database. Table S4 is a list of annotations for the conserved genes from 277 Ortholog clusters of Rickettsia, Orientia, Wolbachia, Anaplasma, Ehrlichia, Neorickettsia and Candidatus Pelagibacter ubique from the COG database. Table S5 is a list of annotation for the conserved genes from 328 Ortholog clusters of Rickettsia, Orientia, Wolbachia, Anaplasma, Ehrlichia and Neorickettsia from the COG database. Table S6 is a list of annotation sfor the 1,709 orthologous groups based on the COG database. Table S7 is a table of the horizontal gene transfer cases identified from at least 1 gain and 1 loss event (part 1). Table S8 is a table of the horizontal gene transfer cases identified from at least 1 gain and 1 loss event (part 2). Table S9 is a table of the horizontal gene transfer cases identified from non-orthologous groups. Table S10 is a table showing the distribution of proteins annotated from the Gene Ontology database. Table S11 is a table showing the assignment of proteins annotated by Gene Ontology at the 9th level. Table S12 is a table showing the assignment of proteins annotated by Gene Ontology at the 12th level. Table S13 is a table showing the non-synonymous/synonymous substitution analysis for de novo genes.
Figure S1 shows the 16S rRNA species tree.Figure S2 shows the two principle patterns for the detection of gene gain and gene loss using PhyloPattern: a. Loss event and b. Gain event. Red circles represent the “present” characteristic, blue circles represent the “absent” characteristic. The pattern was defined as a loss event if the gene was present in the ancestral node and one descendant node but absent in another node (violet rhomboid). Conversely, the pattern was defined as a gain event if the gene was absent in the ancestral node and one descendant node but was present in another descendant node (green rhomboid). Figure S3 maps the gain and loss events from the phyletic patterns onto the species tree. Red circles represent the “present” characteristic, and blue circles represent the “absent” characteristic. Violet and green rhomboids represent loss and gain events, respectively. Figure S4 shows the transfer pattern detected by HGT agent from the multi-agent system DAGOBAH. HGT agent applied PhyloPattern to annotate the subtree (see Methods). Yellow circle with a D indicates that a duplication of the gene occurred at that node. Red, green and cyan circles represent recipient species, donor species and external species, respectively. Tax id: taxonomy identity. Figure S5 shows the results of the number of orthologous groups obtained from the gene gain and gene loss analysis by PhyloPattern in each respective event.
Figure S6a is a flowchart representing the detailed description of the systematic analysis of horizontal gene transfer detection with at least one gain event. Figure S6b is a flowchart representing the detailed description of the systematic analysis of horizontal gene transfer detection for non-orthologous groups. Figure S7 shows Bayesian phylogenetic trees for a subset of the detected instances of horizontal gene transfer from the group with at least 1 gain and 1 loss (No. 1 to 10) and the non-orthologous groups (No. 11 to 15). The scale bar represents the average number of substitutions per site. Numbers at nodes correspond to the posterior probabilities calculated by Phylobayes. The donor species are indicated in blue, and the recipient species are indicated in red. Figure S8 represents the distribution of proteins assignments using the Gene Ontology database. Figure S9 illustrates the hypothesis for the acquisition of the ADP/ATP translocase-encoding gene by Rickettsia species. The evolutionary time was estimated from the data provided by Greub and Raoult, 2003. Figure S10 is a histogram of COG size.