

Abstract
Background and Purpose
S-nitrosylated hemoglobin (S-nitrosohemoglobin) has been implicated in the delivery of O2 to tissues through the regulation of microvascular blood flow. This study tested the hypothesis that enhancement of S-nitrosylated hemoglobin by ethyl nitrite inhalation improves outcome after experimental subarachnoid hemorrhage (SAH).
Methods
A preliminary dosing study identified 20 ppm ethyl nitrite as a concentration that produced a 4-fold increase in S-nitrosylated hemoglobin concentration with no increase in methemoglobin. Mice were subjected to endovascular perforation of the right anterior cerebral artery and were treated with 20 ppm ethyl nitrite in air, or air alone for 72 hours, after which neurologic function, cerebral vessel diameter, brain water content, cortical tissue PO2, and parenchymal red blood cell flow velocity were measured.
Results
At 72 hours after hemorrhage, air- and ethyl nitrite– exposed mice had similarly sized blood clots. Ethyl nitrite improved neurologic score and rotarod performance; abated SAH-induced constrictions in the ipsilateral anterior, middle cerebral, and internal carotid arteries; and prevented an increase in ipsilateral brain water content. Ethyl nitrite inhalation increased red blood cell flow velocity and cortical tissue PO2 in the ipsilateral cortex with no effect on systemic blood pressure.
Conclusions
Targeted S-nitrosylation of hemoglobin improved outcome parameters, including vessel diameter, tissue blood flow, cortical tissue PO2, and neurologic function in a murine SAH model. Augmenting endogenous PO2-dependent delivery of NO bioactivity to selectively dilate the compromised cerebral vasculature has significant clinical potential in the treatment of SAH.
Keywords: brain, mouse, subarachnoid hemorrhage, S-nitrosylated hemoglobin, ethyl nitrite
Red blood cells (RBCs) regulate tissue O2 delivery by using hemoglobin (Hb) as both an O2 sensor and a transducer of NO vasodilator activity to match local tissue blood flow to that region’s O2 requirements.1 Impairment of this microcirculatory interrelationship may occur in pathophysiologic conditions, including subarachnoid hemorrhage (SAH).
After SAH, delayed narrowing of vessels impairs delivery of O2 and nutrients to brain tissue. This delayed arteriopathy is due, at least in part, to local disruption of NO bioactivity.2 Addressing this disruption is problematic. Systemic administration of nonspecific vasoactive agents has shown limited efficacy due to dose-limiting arterial hypotension.3,4 Central (directed) administration of NO donors has been reported to be beneficial in some,5,6 but not other,7 applications and is complicated by the need for invasive access. Nonetheless, affected cerebral vessels appear to maintain vasoreactivity,8 so a different course of action may be augmentation of the body’s innate ability to selectively increase blood flow to areas of low O2 tension.
Increasing the circulating pool of physiologic NO bioactivity (that is, S-nitrosothiols, including S-nitrosylated hemoglobin, SNO-Hb) could selectively improve flow to focal ischemic brain tissue without altering flow to other tissue beds. This does not involve the generation of free NO, which is rapidly metabolized by Hb. Instead, hypoxic vasodilation results from a series of transnitrosylation reactions when NO bioactivity is released by the RBC. Accumulating evidence suggests that a small S-nitrosothiol,4 S-nitrosoglutathione, which is derived from RBC SNO-Hb, subserves hypoxic regulation of O2 delivery.9–11
The goal of the present study was to test the hypothesis that augmentation of SNO-Hb improves outcome after experimental SAH. We reasoned that increased vessel diameter and enhancement of cortical tissue PO2 (tPO2) in the affected cortex would be reflected in improved neurologic outcome. The experiments were conducted in mice and utilized ethyl nitrite (ENO), a selective nitrosylating agent that preferentially forms SNO-Hb12,13 and other nitrosylated thiols on exposure to blood (the other reaction product is ethanol). ENO has not previously been tested as a therapy for a focal pathologic condition such as SAH, but it has shown benefit in a disparate collection of disorders characterized by disruptions in O2 delivery, including pulmonary hypertension13 and laparoscopic surgery.14
Materials and Methods
The Duke University institutional animal care and use committee approved all aspects of the study design. Experiments were conducted on male C57Bl/6J mice (20 to 25 g; The Jackson Laboratory, Bar Harbor, ME). Gas exposures occurred within a 5.5-L acrylic box kept at room temperature and normal atmospheric pressure. ENO (Custom Gas Solutions, Durham, NC) was blended with N2 to the desired concentration at the time of delivery. In this SAH model, cerebral vasospasm has been reported to peak 72 hours after SAH.15 We therefore examined mice at this recovery interval in the following studies.
Dose Finding and NO Measurements
Mice (n=5 per group) were exposed to ENO (0, 20, 40, or 80 ppm) in air for 72 hours. Mice were then anesthetized, and arterial blood (0.5 mL) was sampled via cardiac puncture for determination of total Hb and methemoglobin with a Gem Premier 3000 co-oximeter (Instrumentation Laboratory, Lexington, MA). In a separate set of mice, the 72-hour 20-ppm ENO exposure was repeated to measure final blood SNO-Hb and RBC NO concentrations (that is, FeNO and SNO) by Hg-coupled photolysis/chemiluminescence.16,17 On the basis of the methemoglobin and SNO-Hb values obtained from these experiments and evidence that 10 ppm ENO attenuates lipopolysaccharide-induced lung inflammation,18 we elected to further study 20 ppm (0.002% atm) ENO.
Post-SAH Neurologic Function, Vessel Diameter, and Brain Edema
The following experiments were conducted with animals randomly assigned to experimental groups. SAH was induced in isoflurane-anesthetized, mechanically ventilated mice by endovascular perforation of the anterior cerebral artery (ACA) with a 5-0 nylon mono-filament suture, according to previously reported procedures.19,20 Pericranial temperature was maintained at 37.0±0.2°C. After ACA perforation, the mice were awakened. At 60 minutes after SAH, mice were moved to the exposure chamber for 72 hours. Fresh gas inflow was 21% O2, with (n=20) or without (n=23) 20 ppm ENO, balanced with N2. In a separate experiment, sham mice were subjected to all procedures (except ACA perforation), including the 72-hour exposure to air (n=5) or ENO (n=5) to determine ENO effects on normal vessel diameter.
After the exposure/treatment period, mice underwent neurologic examination by an observer blinded to treatment group (0=no observable neurologic deficit; 1=failure to extend the left forepaw; 2=circling to the left; 3=falling to the left; and 4=unable to walk spontaneously)21 and rotarod testing.22 Rotarod performance (performed in triplicate with the best score analyzed) was compared against pre-SAH baseline values.
Mice were then anesthetized, and standardized casting of the cerebral vasculature was performed with an India ink/gelatin solution.19,20,22 Hemorrhage size was graded by an observer blinded to experimental group. Hemorrhage area was scored as follows: 1=SAH extends anteriorly <1.0 mm from the middle cerebral artery (MCA)-ACA bifurcation; 2=SAH extends >1.0 mm anteriorly from the bifurcation; and 3=SAH extends >1.0 mm anteriorly from the bifurcation with posterior extension across the internal carotid artery (ICA).19 Hemorrhage density was scored as follows: 1=underlying brain parenchyma visualized through the clot and 2=underlying brain parenchyma not visualized through the clot. Hemorrhage grade (2 to 5) was determined by the sum of the size and density scores. Absence of hemorrhage was scored as 0.19
Blood vessels were imaged with a video-linked dissecting microscope controlled by a calibrated image analyzer.19 Two regions of the ipsilateral MCA were analyzed: a 1.0-mm segment proximal to the ACA-MCA bifurcation and a 1.0-mm segment distal to the bifurcation. The ipsilateral ICA and ACA were divided into proximal and distal 0.8-mm segments. The smallest lumen diameter within each segment was measured by an observer blinded to experimental group, with the smaller diameter in each vessel chosen for further analysis. We also measured optical density of the ventral cortex surface as a qualitative measure of tissue perfusion with the casting solution.23
A separate cohort of treated and untreated mice (n=14 per group) was reanesthetized and decapitated at 72 hours after SAH. The forebrains were removed and divided into left and right hemispheres. The tissue was weighed, dried at 105°C for 24 hours, and reweighed by an observer blinded to treatment group. Percent water content was calculated as (wet weight−dry weight)×100.
Time-Dependent ENO Effects on Cortical Blood Flow Velocity and Systemic Blood Pressure
Mice were subjected to SAH and exposed to air for 24 or 72 hours or to sham surgery (all procedures except ACA perforation). Mice (n=3 per group) were then anesthetized with orotracheal isoflurane and mechanically ventilated with 30% O2, balance N2. A femoral artery was cannulated. A transcranial laser Doppler flow (LDF) probe was positioned over the right cerebral cortex. Mean arterial pressure (MAP) and LDF were continuously measured for a 30-minute baseline interval and for 60 minutes after onset of ENO (20 ppm) treatment.
Acute Effects of ENO on Brain tPO2 72 Hours After SAH
Brain tPO2 was measured according to a previously described polarographic method.24 Mice were exposed to SAH or sham surgery (n=6 per group) and treated with air during 72 hours of recovery. Mice were then anesthetized with 1.0% isoflurane in 30% O2/balance N2. A femoral artery was cannulated. A burr hole was made just caudal to bregma and 3.5 mm right of the midline. A transcranial LDF probe was positioned adjacent to the burr hole. An NaCl reference electrode was clipped to the tail, and a 10-μm-diameter Pt microelectrode was inserted 1 mm into the cortex. The system was calibrated in artificial cerebrospinal fluid at 37°C equilibrated with 0, 8, or 21% O2. MAP, LDF, and tPO2 were recorded every 5 minutes. After 30 minutes of stabilization, 20 ppm ENO was added to the inspired gas mixture. Measurements continued for 60 minutes. Arterial blood gas/glucose values were then measured.
Statistical Analysis
Power calculations with this model indicated that 20 mice per experimental group would have an 80% power to detect a <30% difference in MCA diameter.20 Nonparametric values (neurologic score, hemorrhage size, and tissue optical density) were compared by the Mann–Whitney U statistic and are reported as median ± interquartile range. Physiologic values, rotarod latency, vessel diameter, and brain water content were compared with the unpaired Student’s t test. The paired Student’s t test was used to compare baseline values of MAP, LDF, and tPO2 versus those at 60 minutes after ENO exposure onset. Methemoglobin and Hb concentrations were compared by 1-way ANOVA. Parametric values are reported as mean ± SD.
Results
Blood Parameters
There were no differences among groups for total Hb (0 ppm=14±1, 20 ppm=15±1, 40 ppm=14±1, 80 ppm=14±1 g/dL; P=0.08). Methemoglobin was increased by 40 and 80 ppm ENO (0 ppm=0.4±0.2%, 20 ppm=0.2±0.1%, 40 ppm=2.0±0.3%, 80 ppm=5.2±1.2%; main effect P<0.001). ENO (20 ppm) increased SNO-Hb and total RBC NO (Figure 1).
Figure 1.
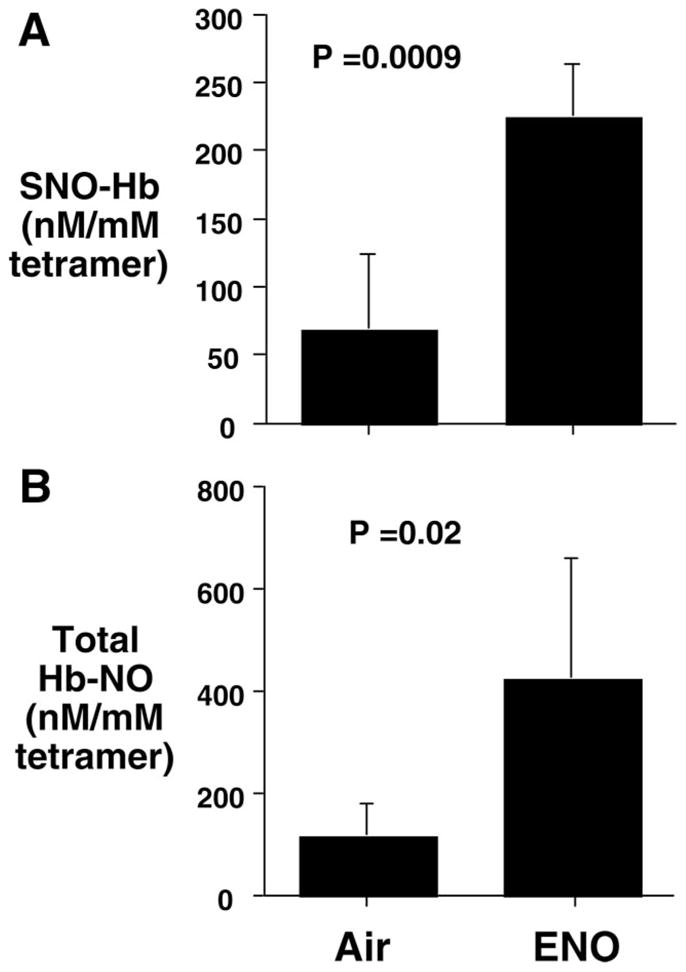
Preferential S-nitrosylation of Hb. RBC NO bioactivity after 72 hours of air or 20 ppm ENO inhalation. A, SNO-Hb, and B, total Hb NO. Values are mean±SD (n=5 per group).
Post-SAH Neurologic Function and Vessel Diameter
Three SAH mice died during the 72-hour recovery interval (1 from the ENO group and 2 from the air-only group). This was likely due to intracranial hypertension.25 There was no intergroup difference in body weight change from baseline (SAH-air=−3±4g, SAH-ENO=−2±2 g; P=0.25). SAH grades were similar (air=4±0.25, ENO=4±0; P=0.27). ENO improved 72-hour post-SAH neurologic scores (P=0.009, Figure 2A) and rotarod performance (P=0.003, Figure 2B). Right ACA, MCA, and ICA diameters were greater in the ENO group (Figure 3). There was no effect of ENO on vessel diameters in shams. Basilar artery diameters were similar among groups (sham-air=179±12 μm, sham-ENO=172±12 μm, SAH-air=183±2 μm, SAH-ENO=187±11 μm; P=0.61). This indicates that the effects of ENO were injury-specific, because there was no clot in this location. Relative tissue optical density was greater with ENO (SAH-air=1.17±0.04 vs SAH-ENO=1.23±0.02, P<0.0001), with values similar to shams (sham-air=1.23±0.01, sham-ENO=1.23±0.02).
Figure 2.

Post-SAH neurologic function. Mice were treated with either air (n=21) or 20 ppm ENO (n=19) for 72 hours after SAH. ENO improved neurologic score (A) (0=no deficit, P=0.009) and rotarod latency to fall (B) (P=0.003). Open circles indicate individual mouse values. Horizontal bars indicate median values for neurologic score and mean values for rotarod. There was no effect of ENO on sham (n=5) performance (P>0.99, P=0.25, respectively).
Figure 3.

Cerebral vessel diameters. Diameters of the ACA (A), MCA (B), and ICA (C) ipsilateral to the right ACA perforation were measured after 72 hours of treatment with air (n=21) or 20 ppm ENO (n=19). ENO increased post-SAH diameter in the ACA (P=0.003), MCA (P=0.04), and ICA (P<0.0001). Open circles indicate individual mouse values. Horizontal bars indicate mean values. There was no effect of ENO on sham (n=5) vessel diameters (P≥0.85).
Edema, LDF, MAP, and Cortical tPO2
ENO decreased cerebral edema in the hemisphere ipsilateral to the hemorrhage (Figure 4). In anesthetized shams, 20 ppm ENO inhalation did not alter MAP or LDF (Figure 5A). Mice were allowed to survive 24 or 72 hours after SAH in air. At 24 hours, the ENO LDF response was episodic, indicating an unstable effect of ENO at this stage of disease progression (Figure 5B). Although MAP remained constant, acute ENO administration rapidly and consistently improved LDF at 72 hours (Figure 5C), indicating that the postinjury vasculature can still respond to an increase in SNO-Hb. Another set of SAH and sham mice recovered for 72 hours in air. Acute ENO inhalation increased both LDF and cortical tPO2 (Figure 6), with no effect on MAP. At completion of the experiment, pH=7.36±0.07, PaCO2 = 35±7 mm Hg, PaO2 =163±34 mm Hg, and glucose=169±49 mg/dL.
Figure 4.
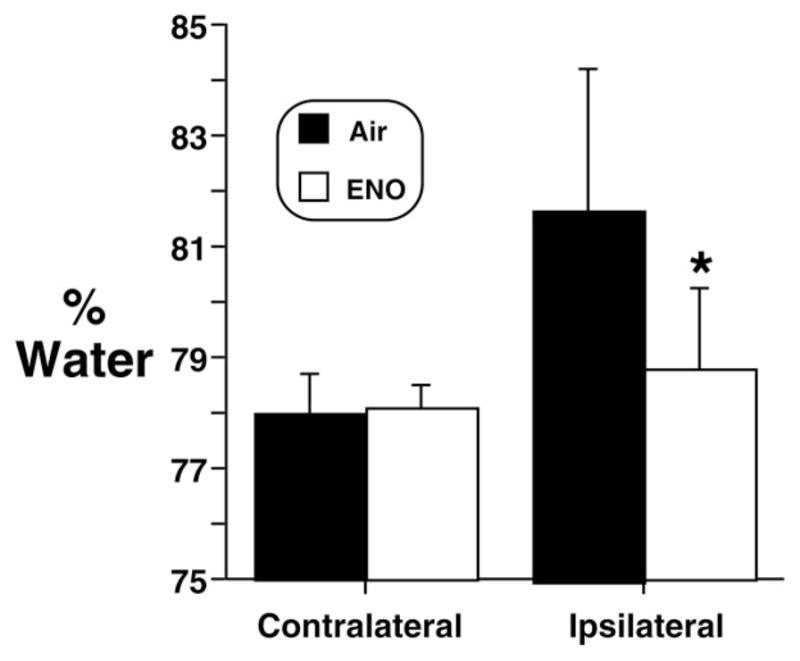
Brain edema. Brain water content (contralateral and ipsilateral to the ACA perforation) was measured after SAH in mice treated with 20 ppm ENO (n=14) in air vs air alone (n=14) for 72 hours (P=0.002). Values are mean±SD.
Figure 5.

Effect of acute ENO exposure on brain blood flow in sham and SAH mice. A, Sham-operated mice (n=3), without SAH, were monitored for cortical RBC velocity (by LDF; values are normalized to percent of baseline measured immediately before ENO treatment) and MAP 30 minutes before and for 60 minutes after 20 ppm ENO was introduced (vertical dashed lines) into the inspiratory gas mixture. SAH mice were allowed to survive for 24 (B, n=3) or 72 (C, n=3) hours after SAH in air and then exposed to 20 ppm ENO for 60 minutes.
Figure 6.

Effect of ENO on brain parenchymal PO2 in sham and SAH mice. Mice underwent SAH (n=6) or sham (n=6) surgery and recovered in air for 72 hours. MAP (in mm Hg), cortical RBC flow velocity (by LDF; values are normalized to percent of baseline measured immediately prior to ENO treatment onset), and tPO2 were then measured for 30 minutes. ENO (20ppm, vertical dashed line) was maintained within the inspiratory gas mixture for the remainder of the experiment. Values are mean±SD.
*P<0.05 vs 30-minute baseline.
Discussion
Inhaled ENO improved neurologic deficits attributable to experimental SAH. This was associated with greater vessel diameter, decreased brain edema, and improved LDF and tPO2, but ENO had no effect on MAP. ENO increases tissue oxygenation selectively in the ischemic brain and suggests that SNO-Hb provides a route to regulate microvascular blood flow.
The release of NO bioactivity from SNO-Hb is regulated allosterically by O2 saturation: NO bioactivity is liberated preferentially in environments that favor O2 offloading.26 Decreased tPO2 occurs in clinical SAH.27 After SAH, tPO2 values observed in this experiment were increased to a viable range by ENO (for example, 32±10 mm Hg). A likely basis for this response was an ENO-induced increase in circulating SNO-Hb concentration. MAP remained unaffected by ENO, as did LDF in the unaffected cortex, consistent with the normal tPO2 values in sham-operated animals. This hypoxic selectivity provides perhaps the most clinically relevant attribute of pharmacologically increased SNO-Hb concentrations.
A target for S-nitrosylation is the β93 cysteine thiol in Hb. The extent to which Hb is S-nitrosylated is dependent on the Hb oxygenation state.1,28 Oxyhemoglobin is readily nitrosylated and causes the S-nitrosothiol to face inward, protecting the NO moiety from solvent.1 In the deoxy state, the cysteine residue is allosterically rotated outward into the blood phase, thereby enabling SNO-Hb to transnitrosylate other moieties. Thus, SNO-Hb provides a hypoxia-activated source of NO bioactivity that constitutes a basis for increased delivery of O2 to a hypoxic region.
There have been brief human exposures to ENO. Treatment of chronic pulmonary hypertension with ENO (0 to 70 ppm) by facemask increased SNO-Hb, decreased pulmonary artery pressure and resistance, and increased arterial PO2 without affecting systemic vascular resistance or MAP.13 Critically ill newborns with persistent pulmonary hypertension were exposed to ENO concentrations of up to 80 ppm, which dose-dependently increased arterial O2 saturation.12 Methemoglobin levels remained in a clinically acceptable range.
Humans have not undergone sustained ENO exposure. Although we saw no adverse effects in mice exposed to ENO for 72 hours, the clinical course of delayed arteriopathy after SAH is many days. It remains to be determined whether sustained ENO exposure is required to provide benefit. It is noteworthy that prolonged ENO exposure served to abate vasospasm onset and that acute ENO exposure 72 hours after SAH provided beneficial effects on tPO2 and LDF once vasospasm had become established.
S-nitrosylation of proteins may play other roles in SAH.29,30 S-nitrosylation inhibits both caspase-3 and nuclear factor-κB activation, thereby attenuating inflammation and cell death.31,32 Treatment with ENO blocks nuclear factor-κB activation in acute lung injury.18 The N-methyl-D-aspartate receptor undergoes S-nitrosylation during hyperexcitation associated with hypoxia, with S-nitrosylation inhibiting N-methyl-D-aspartate– evoked currents.33 Apoptosis,34 nuclear factor-κB activation,35 and elevated glutamate36 are known SAH consequences. Although we have provided direct evidence of improved tissue perfusion and tPO2 from ENO, favorable effects on cellular events may have also contributed to improved neurologic function.
ENO improved acute recovery from murine SAH. Adverse effects were not observed. Pharmacologic enhancement of SNO-Hb provides a novel strategy for SAH therapeutic intervention. Optimal ENO dosing strategies, the presence of sustained efficacy in long-term outcome analyses, direct comparison with established treatments, and confirmation of purported molecular mechanisms of action require continued investigation.
Acknowledgments
Sources of Funding
This study was supported by US Public Health Service grants 5R21N3063108, 5R21NS058321, 1R01HL091876, and 1R01HL095463.
Footnotes
Disclosures
Duke University has filed a use patent application for ENO in SAH. Individual authors have no conflicts of interest to disclose.
References
- 1.Stamler J, Jia L, Eu J, McMahon T, Demchenko I, Bonaventura J, Gernert K, Piantadosi C. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- 2.Hatake K, Wakabayashi I, Kakishita E, Hishida S. Impairment of endothelium-dependent relaxation in human basilar artery after subarachnoid hemorrhage. Stroke. 1992;23:1111–1116. doi: 10.1161/01.str.23.8.1111. [DOI] [PubMed] [Google Scholar]
- 3.Otten ML, Mocco J, Connolly ES, Jr, Solomon RA. A review of medical treatments of cerebral vasospasm. Neurol Res. 2008;30:444–449. doi: 10.1179/174313208X284089. [DOI] [PubMed] [Google Scholar]
- 4.Pluta RM, Oldfield EH. Analysis of nitric oxide (NO) in cerebral vasospasm after aneurysmal bleeding. Rev Recent Clin Trials. 2007;2:59– 67. doi: 10.2174/157488707779318062. [DOI] [PubMed] [Google Scholar]
- 5.Afshar JK, Pluta RM, Boock RJ, Thompson BG, Oldfield EH. Effect of intracarotid nitric oxide on primate cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 1995;83:118–122. doi: 10.3171/jns.1995.83.1.0118. [DOI] [PubMed] [Google Scholar]
- 6.Clatterbuck RE, Gailloud P, Tierney T, Clatterbuck VM, Murphy KJ, Tamargo RJ. Controlled release of a nitric oxide donor for the prevention of delayed cerebral vasospasm following experimental subarachnoid hemorrhage in nonhuman primates. J Neurosurg. 2005;103:745–751. doi: 10.3171/jns.2005.103.4.0745. [DOI] [PubMed] [Google Scholar]
- 7.Aihara Y, Jahromi BS, Yassari R, Sayama T, Macdonald RL. Effects of a nitric oxide donor on and correlation of changes in cyclic nucleotide levels with experimental vasospasm. Neurosurgery. 2003;52:661– 667. doi: 10.1227/01.neu.0000048188.88980.86. [DOI] [PubMed] [Google Scholar]
- 8.Vatter H, Weidauer S, Dias S, Preibisch C, Ngone S, Raabe A, Zimmermann M, Seifert V. Persistence of the nitric oxide-dependent vasodilator pathway of cerebral vessels after experimental subarachnoid hemorrhage. Neurosurgery. 2007;60:179–187. doi: 10.1227/01.NEU.0000249212.96719.95. [DOI] [PubMed] [Google Scholar]
- 9.Lipton AJ, Johnson MA, Macdonald T, Lieberman MW, Gozal D, Gaston B. S-nitrosothiols signal the ventilatory response to hypoxia. Nature. 2001;413:171–174. doi: 10.1038/35093117. [DOI] [PubMed] [Google Scholar]
- 10.McMahon TJ, Moon RE, Luschinger BP, Carraway MS, Stone AE, Stolp BW, Gow AJ, Pawloski JR, Watke P, Singel DJ, Piantadosi CA, Stamler JS. Nitric oxide in the human respiratory cycle. Nat Med. 2002;8:711–717. doi: 10.1038/nm718. [DOI] [PubMed] [Google Scholar]
- 11.Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moya MP, Gow AJ, Califf RM, Goldberg RN, Stamler JS. Inhaled ethyl nitrite gas for persistent pulmonary hypertension of the newborn. Lancet. 2002;360:141–143. doi: 10.1016/S0140-6736(02)09385-6. [DOI] [PubMed] [Google Scholar]
- 13.McMahon TJ, Ahearn GS, Moya MP, Gow AJ, Huang YC, Luchsinger BP, Nudelman R, Yan Y, Krichman AD, Bashore TM, Califf RM, Singel DJ, Piantadosi CA, Tapson VF, Stamler JS. A nitric oxide processing defect of red blood cells created by hypoxia: deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proc Natl Acad Sci U S A. 2005;102:14801–14806. doi: 10.1073/pnas.0506957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimazutsu K, Uemura K, Auten KM, Baldwin MF, Belknap SW, La Banca F, Jones MC, McClaine DJ, McClaine RJ, Eubanks WS, Stamler JS, Reynolds JD. Inclusion of a nitric oxide congener in the insufflation gas repletes S-nitrosohemoglobin and stabilizes physiologic status during prolonged carbon dioxide pneumoperitoneum. Clin Transl Sci. 2009;2:405– 412. doi: 10.1111/j.1752-8062.2009.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamii H, Kato I, Kinouchi H, Chan PH, Epstein CJ, Akabane A, Okamoto H, Yoshimoto T. Amelioration of vasospasm after subarachnoid hemorrhage in transgenic mice overexpressing CuZn-superoxide dismutase. Stroke. 1999;30:867– 871. doi: 10.1161/01.str.30.4.867. [DOI] [PubMed] [Google Scholar]
- 16.Hausladen A, Rafikov R, Angelo M, Singel DJ, Nudler E, Stamler JS. Assessment of nitric oxide signals by triiodide chemiluminescence. Proc Natl Acad Sci U S A. 2007;104:2157–2162. doi: 10.1073/pnas.0611191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMahon TJ, Stamler JS. Concerted nitric oxide/oxygen delivery by hemoglobin. Methods Enzymol. 1999;301:99–114. doi: 10.1016/s0076-6879(99)01073-3. [DOI] [PubMed] [Google Scholar]
- 18.Marshall HE, Potts EN, Kelleher ZT, Stamler JS, Foster WM, Auten RL. Protection from lipopolysaccharide-induced lung injury by augmentation of airway S-nitrosothiols. Am J Respir Crit Care Med. 2009;180:11–18. doi: 10.1164/rccm.200807-1186OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 19.Parra A, McGirt MJ, Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Mouse model of subarachnoid hemorrhage associated cerebral vasospasm: methodological analysis. Neurol Res. 2002;24:510–516. doi: 10.1179/016164102101200276. [DOI] [PubMed] [Google Scholar]
- 20.McGirt MJ, Lynch JR, Parra A, Sheng H, Pearlstein RD, Laskowitz DT, Pelligrino DA, Warner DS. Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke. 2002;33:2950–2956. doi: 10.1161/01.str.0000038986.68044.39. [DOI] [PubMed] [Google Scholar]
- 21.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 22.Mesis RG, Wang H, Lombard FW, Yates R, Vitek MP, Borel CO, Warner DS, Laskowitz DT. Dissociation between vasospasm and functional improvement in a murine model of subarachnoid hemorrhage. Neurosurg Focus. 2006;21:E4. doi: 10.3171/foc.2006.21.3.4. [DOI] [PubMed] [Google Scholar]
- 23.Takata K, Sheng H, Borel CO, Laskowitz DT, Warner DS, Lombard FW. Long-term cognitive dysfunction following experimental subarachnoid hemorrhage: new perspectives. Exp Neurol. 2008;213:336–344. doi: 10.1016/j.expneurol.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, Piantadosi CA. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci. 2008;28:2015–2024. doi: 10.1523/JNEUROSCI.5654-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26:1086–1091. doi: 10.1161/01.str.26.6.1086. discussion 1091–1082. [DOI] [PubMed] [Google Scholar]
- 26.Sonveaux P, Kaz AM, Snyder SA, Richardson RA, Cardenas-Navia LI, Braun RD, Pawloski JR, Tozer GM, Bonaventura J, McMahon TJ, Stamler JS, Dewhirst MW. Oxygen regulation of tumor perfusion by S-nitrosohemoglobin reveals a pressor activity of nitric oxide. Circ Res. 2005;96:1119–1126. doi: 10.1161/01.RES.0000168740.04986.a7. [DOI] [PubMed] [Google Scholar]
- 27.Vath A, Kunze E, Roosen K, Meixensberger J. Therapeutic aspects of brain tissue PO2 monitoring after subarachnoid hemorrhage. Acta Neurochir Suppl. 2002;81:307–309. doi: 10.1007/978-3-7091-6738-0_78. [DOI] [PubMed] [Google Scholar]
- 28.Allen BW, Stamler JS, Piantadosi CA. Hemoglobin, nitric oxide and molecular mechanisms of hypoxic vasodilation. Trends Mol Med. 2009;15:452– 460. doi: 10.1016/j.molmed.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawaguchi AT, Nakai K, Fukumoto D, Yamano M, Haida M, Tsukada H. S-nitrosylated pegylated hemoglobin reduces the size of cerebral infarction in rats. Artif Organs. 2009;33:183–188. doi: 10.1111/j.1525-1594.2008.00705.x. [DOI] [PubMed] [Google Scholar]
- 30.Pei DS, Song YJ, Yu HM, Hu WW, Du Y, Zhang GY. Exogenous nitric oxide negatively regulates c-Jun N-terminal kinase activation via inhibiting endogenous NO-induced S-nitrosylation during cerebral ischemia and reperfusion in rat hippocampus. J Neurochem. 2008;106:1952–1963. doi: 10.1111/j.1471-4159.2008.05531.x. [DOI] [PubMed] [Google Scholar]
- 31.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall HE, Stamler JS. Nitrosative stress-induced apoptosis through inhibition of NF-κB. J Biol Chem. 2002;277:34223–34228. doi: 10.1074/jbc.M201638200. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi H, Shin Y, Cho SJ, Zago WM, Nakamura T, Gu Z, Ma Y, Furukawa H, Liddington R, Zhang D, Tong G, Chen HS, Lipton SA. Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron. 2007;53:53–64. doi: 10.1016/j.neuron.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabri M, Kawashima A, Ai J, Macdonald RL. Neuronal and astrocytic apoptosis after subarachnoid hemorrhage: a possible cause for poor prognosis. Brain Res. 2008;1238:163–171. doi: 10.1016/j.brainres.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou ML, Shi JX, Hang CH, Cheng HL, Qi XP, Mao L, Chen KF, Yin HX. Potential contribution of nuclear factor-κB to cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. J Cereb Blood Flow Metab. 2007;27:1583–1592. doi: 10.1038/sj.jcbfm.9600456. [DOI] [PubMed] [Google Scholar]
- 36.Kett-White R, Hutchinson PJ, Al-Rawi PG, Gupta AK, Pickard JD, Kirkpatrick PJ. Adverse cerebral events detected after subarachnoid hemorrhage using brain oxygen and microdialysis probes. Neurosurgery. 2002;50:1213–1221. doi: 10.1097/00006123-200206000-00008. discussion 1221–1222. [DOI] [PubMed] [Google Scholar]