

Abstract
Single molecule experimental methods have provided new insights into biomolecular function, dynamic disorder, and transient states that are all invisible to conventional measurements. A novel, non-fluorescent single molecule technique involves attaching single molecules to single-walled carbon nanotube field-effective transistors (SWNT FETs). These ultrasensitive electronic devices provide long-duration, label-free monitoring of biomolecules and their dynamic motions. However, generalization of the SWNT FET technique first requires design rules that can predict the success and applicability of these devices. Here, we report on the transduction mechanism linking enzymatic processivity to electrical signal generation by a SWNT FET. The interaction between SWNT FETs and the enzyme lysozyme was systematically dissected using eight different lysozyme variants synthesized by protein engineering. The data prove that effective signal generation can be accomplished using a single charged amino acid, when appropriately located, providing a foundation to widely apply SWNT FET sensitivity to other biomolecular systems.
Keywords: nanotube sensor, lysozyme, enzymology, single molecule, field-effect transistor
Nanoscale electronic devices like field-effect transistors (FETs) offer a sensitive, label-free method for the real-time detection of biomolecules. In these devices, semiconductor nanowires1–4 or carbon nanotubes5–9 provide the conductive channels for biosensing. Single-walled carbon nanotubes (SWNTs), in particular, have the requisite sensitivity and bandwidth to detect and monitor single molecule dynamics10–12. These characteristics make SWNT FETs an attractive tool to complement existing single molecule techniques. In particular, single molecule techniques like Förster resonance energy transfer (FRET)13 are inherently limited by the properties of the fluorophores employed as reporters. Fluorophores suffer from photobleaching14, intermittency15, and limited photon fluxes16, all of which limit the resolution of very short-lived states or the long duration recording of conformational variability of the same molecule. As a complementary new technique, SWNT FETs provide a platform for continuous, temporally-resolved single molecule monitoring independent of fluorophore limitations. This resolution may help expand single molecule research by, for example, revealing transient motions and intermediate states that are impossible to resolve by conventional, ensemble-averaged measurements13,17–19.
Label-free readout of single biomolecule functionality has many potential applications, and our past work has specifically focused on the kinetic variability of repetitive catalytic processing by single lysozyme enzymes11,12. Monitoring individual lysozymes tethered to SWNTs revealed a processive enzyme capable of hydrolyzing around one hundred glycosidic bonds without substrate dissociation. Furthermore, analysis of statistical variances uncovered hidden information about lysozyme dynamics11,12. In principle, any molecule of interest may be directly tethered to a SWNT FET, but further generalization of the SWNT FET technique first demands reliable design rules that can predict the success and applicability of these devices. This report addresses this need by focusing on the transduction mechanisms that link lysozyme processivity to electrical signal generation in a SWNT FET.
Ultimately, the sensitivity of SWNT FETs20–22 originates from their quasi one-dimensional electronic structure and low carrier concentration. Heller et al. clearly summarized the various possible mechanisms that can contribute to chemical sensing by SWNT FETs, and tried to distinguish among them23. In certain cases, charge transfer doping or chemisorption events were shown to be the main cause of slow, chemiresistive responses. Others have proven that interfacial effects at SWNT-electrode junctions can play important roles24,25. However, the observation of high bandwidth, dynamic electrical signals severely restricts the possible modes of transduction. The signals generated by a SWNT-bound lysozyme molecule exactly match the dynamics of enzyme processing, and therefore rule out mechanisms that are slow or irreversible, are caused by nonspecific binding, or are located at distant interfaces11,12.
Here, we investigate the hypothesis that signal transduction is driven by a simpler, electrostatic mechanism in which conformational motion of the lysozyme molecule electrostatically gates a short portion of the SWNT channel. Protein engineering was used to dissect the through-space interaction between eight different lysozyme variants and SWNT FET devices. The results prove the critical roles of two, charged, sidechain functionalities located close to the SWNT. Since all proteins have charged amino acids present on their surfaces, this transduction mechanism is straightforward to expand to a wide range of bioelectronic measurements.
Experimental Methods
Single molecule lysozyme measurements were accomplished here using an electronic technique reported previously11,26. Very briefly, single-SWNT FETs were fabricated and then decorated with pyrene-maleimide linker molecules, which strongly adhere to SWNT sidewalls by π-π stacking6,27. Next, the devices were incubated in a solution of the pseudo wild-type, single-cysteine variant of T4 lysozyme (C54T/C97A/S90C, hereafter referred to as S90C lysozyme)28,29. The thiol functionality of the lysozyme reacted with an SWNT-bound maleimide to produce a covalent bioconjugate. Reaction conditions were tailored to readily produce devices having only one lysozyme attachment, as verified by atomic force microscopy (AFM). Fig. 1a shows two example devices illustrating the high quality surfaces obtained and the ease of protein identification by AFM. Fig. 1b schematically depicts a likely orientation of lysozyme with respect to its C90 residue and the SWNT attachment point. Two moving domains surrounding a catalytically-active site are shown in light and dark grey. To date, over 100 devices using ten different lysozyme variants have been successfully fabricated and measured. Precise details on device fabrication and lysozyme synthesis and characterization are described more fully in the Supporting Information (SI).
Fig 1.

Single molecule SWNT FETs. (A) Example AFM images of SWNTs having single lysozyme molecules attached (arrows). Each SWNT is 1.2 to 2.0 nm in diameter, whereas the lysozyme stands 5 to 7 nm tall. Distant source and drain electrodes are protected by a PMMA coating shown across the top and bottom of each image. (B) Schematic representation of the device, highlighting the enzyme’s two active domains (light and dark grey), a peptidoglycan fragment (red) at the binding site, the C90 attachment site (green), and two charged residues K83 and R119 (blue) targeted for mutagenesis. (C) The SWNT FET current fluctuates between two levels in synchrony with enzyme opening and closing.
The electronic single molecule technique involved monitoring nanocircuit conductance. A small bias of 100 mV was applied between source and drain terminals and then the SWNT source-drain current I(t) was continuously recorded at a fixed gate bias, typically for no less than 600 s. In the case of lysozyme, binding of peptidoglycan substrate (Fig. 1b, red) at the active site occurs 3.1 nm distant from the C90 residue, but it also drives a large amplitude (0.8 nm), hinge-bending motion of the two domains. Previous work11,26 has shown that this substrate-driven mechanical motion dynamically modulates the conductance of the underlying SWNT FET to produce electronic fluctuations like those shown in Fig. 1c. The fluctuating portion of the signal, ΔI(t)=I(t)−<I(t)>, switches with statistics that closely match the signals from single molecule FRET studies30–33 and bulk, ensemble techniques34. Good agreement among the turnover rates from all three techniques indicated that the SWNT FET linkage at the C90 residue did not substantially interfere with the active site or the hinge motion.
Results
Debye screening in single-molecule SWNT FETs
One method of probing the interaction between lysozyme’s enzymatic activity and the SWNT electrical signal I(t) created by it is to vary the screening length of the surrounding electrolyte. I(t) from the S90C lysozyme variant was measured in phosphate buffer (10 mM, pH 7.5) with a variable level of NaCl from 10 to 300 mM (Fig. 2a) in order to examine the role of the electrolyte’s ionic strength in screening electrostatic effects35–37. The magnitude of single-molecule fluctuations |ΔI(t)| of the S90C lysozyme variant-containing devices peaked at a salt concentration of 50 mM and dropped sharply with increasing salt concentration.
Fig. 2.

Debye screening of signal transduction. (A) The signal amplitude ΔI of a typical lysozyme SWNT FET as a function of the buffer salt concentration (bottom axis) and corresponding Debye length λD (top axis). The solid line is a fit to the Debye-Huckel screening model described in the text. Note that the phosphate buffer includes an additional 10 mM sodium phosphate. (B) Ensemble measurements of lysozyme activity over the same concentration range show varying enzymatic activity. The drop in activity and thus signal transduction at the lowest salt concentrations is likely due to enzyme denaturation. All error bars depict ±1σ.
As the magnitude of ΔI decreased, the frequency distribution of the I(t) fluctuations was substantially unchanged. An independent, fluorescence-based ensemble assay verified enzymatic activity over the tested range of salt concentrations (Fig. 2b). The ensemble measurements suggested a concentration dependence, which was not observed in single molecule recordings. This disagreement is likely due to the combination of a broad, bi-exponential distribution of events and the relatively poor sampling of the full range of lysozyme’s dynamic disorder (even a 600 s recording observes fewer than 10,000 catalytic events). Presumably, infinite-duration single molecule measurements would reproduce ensemble rates. Thus, our investigation of signal transduction focused exclusively on the ΔI magnitude, which varied reproducibly and systematically between recordings as short as 10 s.
Except for measurement in 10 mM NaCl, the magnitude of ΔI fluctuations from the S90C variant of lysozyme can be described by the Debye-Hückel expression38
| (Eq. 1) |
where λD is the electrolyte Debye length39, and xo is the relevant screening distance for gating the SWNT FET. λD varies from 0.54 to 1.54 over the range of NaCl concentrations used here, and it is plotted on the top axis of Fig. 2a. Even though the experimental range of λD was narrow, the data fit Eq. 1 very well and fitting determines the value xo = 1.03 ± 0.10 nm. The fit implicates an electrostatic mechanism rather than a purely mechanical (e.g., strain-based) one. Experiments by other laboratories have drawn similar conclusions35–37; for example, Stern et al. have used linker molecules of differing lengths to show that the parameter xo accurately describes the distance between point charges and a sensitive, nanowire FET.
The small xo value is remarkable given that a lysozyme molecule has physical dimensions of 5 to 7 nm. The active site for peptidoglycan catalysis is located 3.1 nm from the C90 attachment site, a distance of 3xo. Thus, the small xo value suggests that the SWNT is responding to charge motions close to the C90 attachment site, rather than from events occurring exclusively at the active site, such as substrate binding. As predicted by theoretical modeling40,41, single molecule events at the enzyme’s active site should be very difficult to detect electronically under these screening conditions. More likely, large-scale, substrate-induced conformational motions alter the positions of amino acids near the SWNT C90 attachment site.
This conclusion is further supported by two observations. First, multilevel I(t) signals were observed when devices were fabricated using an alternate attachment site (SI, Fig. S5). Although both variants exhibited similar catalytic activities, the second attachment site (S36C instead of S90C) reproducibly generated more complex, multi-level I(t) signals. Secondly, the magnitude of the ΔI(t) signal did not distinguish between catalytic processing and non-productive binding of substrate11,12, indicating that sensitivity is to mechanical motions of the enzyme rather than substrate binding and hydrolysis. Thus, we conclude that the ΔI(t) electronic signal is generated by the enzyme’s mechanical conformational motions near the attachment site, and not its catalytic activity.
These results highlight the importance of the enzyme attachment site in lysozyme-functionalized SWNT FETs. The simple, two-level I(t) signal of the S90C variant of lysozyme provided an ideal system for more detailed investigations. Comparisons of the X-ray crystal structures of lysozyme in the open (unbound) and closed (substrate bound) states28,29 reveal many charged residues within ~xo of the C90 attachment site. Two of these, K83 and R119, are approximately 1.5 nm from the C90 attachment site and are particularly interesting as their charged sidechain functionalities move substantially when the enzyme transitions from an open to a closed state (Fig. 3). As the enzyme moves from an unbound to a substrate-bound state, positively charged K83 and R119 move further from the SWNT by approximately 0.15 nm each29,42. Unlike other surface charges, which remain relatively fixed in place, net movements of K83 and R119 could potentially affect the SWNT FET electrostatically. Consequently, we decided to directly measure the electrostatic contribution of each sidechain functionality.
Fig. 3.

Detailed view of the lysozyme-SWNT interface, showing X-ray structures of a small portion of the protein in its open (light gray) and closed (dark gray) conformations (Protein Data Base 1QTV and 148L, respectively). The C90 attachment to the pyrene-maleimide linker molecule can provide a fixed reference point, and the dashed line depicts the distance xo = 1.0 nm described in the text. In the vicinity of C90, only the amino acids at positions 83 and 119 have charged sidechains that move appreciably (highlighted). The pyrene and SWNT are independently free to rotate around the C90 site, but this illustration depicts an energetically likely orientation.
Electrostatic effects of specific amino acids
Site-specific engineering of proteins offers a powerful tool to probe the relationship between protein structure and function43. In this work, mutagenesis allowed the role of specific amino acids at the SWNT-lysozyme interface to be dissected. Seven variants of the S90C lysozyme were designed and synthesized; for each variant, the wild-type K83 and R119 residues were substituted with either neutral alanines, negatively charged glutamate residues, or a combination of the two amino acids. In the remainder of this report, the variants are frequently described in terms of a charge N, which here is defined as the sum of the bare elementary charges for only the two residues of interest, independent of the rest of the protein, electrolyte, or counterions. The variants were designed to have the net charge at positions 83 and 119 spanning from N = +2 to −2, with the initial S90C lysozyme variant accounting for the highest, N = +2 value.
To demonstrate the role of charged functionalities in signal transduction, Fig. 4 shows representative signals from two of these SWNT FET devices. In the presence of substrate, both devices exhibited two-level switching with overall statistics that match our previous results. The main difference between the two examples in Fig. 4 is a change in sign of the short-duration, transient events that correspond to the closed, substrate-bound conformation. The R119A variant has a single positive charge (N = +1), and produces positive I(t) fluctuations above a baseline current (Fig. 4a). The K83A/R119E variant has a single negative charge (N = −1), and produces I(t) fluctuations with the opposite sign (Fig. 4b).
Fig. 4.
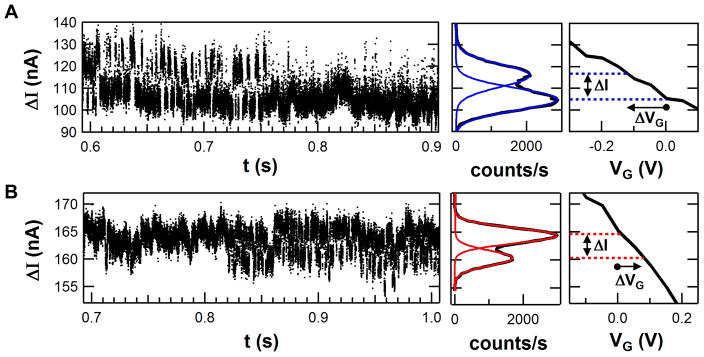
Dynamic I(t) signals during catalytic processing and inactive, substrate-free moments. (A) A positively charged variant (R119A, N = +1) produces short duration I(t) increases each time lysozyme closes upon the substrate. (B) A negatively charged variant (K83A/R119E, N = −1) produces I(t) decreases, with the opposite sign but otherwise similar timing and statistics. Next to each segment of raw data, a histogram is shown for one full second of filtered signal. Each histogram is fit to two Gaussians, the peaks of which determine a transduction magnitude ΔI for the device. The unique electrolyte gating response I(VG) of each device (right) can be used to convert the closing action of the lysozyme enzyme into an effective change of gating ΔVG from the operating point at VG = 0 V.
The left portions of Fig. 4 show 0.3 seconds of raw I(t) data from each, particularly selected to show a transition between active substrate-bound and the inactive, substrate-free conformation (Fig. 4a,b). Because the inactive enzymatic periods do not exhibit two level fluctuations, these transitional moments unambiguously identify the direction of the I(t) signal upon hinge closure, and therefore confirm a difference in sign for ΔI(t) produced by the two variants. The middle portions of Fig. 4 show histograms from longer, 1-second data sets obtained by first applying a 10-Hz, highpass filter to each I(t) signal. Both histograms are composed of a dominant peak for the hinge-open conformation and a smaller peak for the hinge-closed conformation, again revealing the difference in ΔI(t).
Quantitative comparisons among the variants were complicated by device-to-device variations of the SWNT FETs. For example, every SWNT had a unique resistance and gate sensitivity, and these factors caused the mean value <I(t)> to be completely independent of the lysozyme attachment. Nevertheless, we developed a robust and illuminating way of comparing the lysozyme component of ΔI(t) from dissimilar devices. First, every lysozyme-functionalized SWNT was electrolytically gated in a substrate-free buffer in order to record its baseline response to gate voltages I(VG). Second, I(t) histograms like those shown in Fig. 4 were fit to two Gaussian peaks, the positions and widths of which depended solely on the SWNT properties. Third, the separation ΔI in peak position was converted to an effective change in gate voltage ΔVG, as determined by each SWNT’s I(VG) curve. This step accounted for the fact that some SWNT FETs were semiconducting and very sensitive to electrostatic gating, while others were semi-metallic and much less so. The conversion from ΔI to ΔVG removed all of the device-specific differences to produce device-independent ΔVG values. In past work, this process for calculating ΔVG was applied to the S90C lysozyme variant, and a consistent ΔVG value of −0.19 ± 0.02 V was obtained, even when measured across 18 different SWNT devices varying from semiconducting to metallic11. Thus, the ΔVG should serve as a good variable to compare signal transduction by different lysozyme variants.
This normalization process is depicted graphically for the R119A (N = +1) and K83A/R119E (N = −1) lysozyme variants (Fig. 4, right). The Gaussian fits are overlaid on each I(t) histogram in color, and the peak positions are extended by dashed lines onto the I(VG) curves shown immediately to their right. The dominant peak of the R119A (N = +1) lysozyme variant at 105 nA corresponds to a device operating point around the applied gate bias of VG = 0 V. The smaller peak at 117 nA shows that enzyme closure provides an effective change in gate of ΔVG = −119 ± 52 mV (Fig. 4a, right). The operating point for the K83A/R119E (N = −1) lysozyme variant at 165 nA was shifted to a lower current by an effective gating ΔVG = +77 ± 23 mV (Fig. 4b, right). In both cases, the error in ΔVG is estimated from the width of the Gaussian fits. As demonstrated by Fig. S6, this width is primarily due to the large background noise typical of SWNT devices44–46 and biosensors37,47,48. Thus, the error does not necessarily indicate variations in the dynamic disorder or signal transduction by the enzyme.
The ΔVG from 30 different SWNT FETs, incorporating one of the seven charged lysozyme variants revealed the role of charged functionalities in signal transduction (Fig. 5). The ΔVG of each charged variant proved to be characteristic, and ranged from −205 to +135 mV (supporting data showing every device is provided in the SI, Fig. S7). The positive variants had ΔVG < 0 V whereas the negative variants had ΔVG > 0 V. The two variants with N = ± 2 exhibited the strongest signals, being approximately twice as large as from N = ± 1 variants. Furthermore, a single charged functionality could effectively transduce signal from enzyme to SWNT with a large ΔVG value.
Fig. 5.

Average transduction by seven charged variants. (A) Lysozyme positions 83 and 119 were mutated to have positive (blue), neutral (yellow), or negative (red) charged sidechains. The effective gating ΔVG by each variant varied from 116 to −205 mV, with a value of −34 mV for the neutral N = 0 variant. (B) For all variants, ΔVG is nearly proportional to N, with a slope of 87 ± 2 mV per unit charge. Raw data (open squares) shifted up +34 mV (solid squares) results in a response symmetric around zero. Error bars indicate three standard deviations as determined from n (indicated in parentheses) different devices fabricated with each particular variant.
Direct, quantitative comparisons of each lysozyme variant’s ability to transduce signal were generated by plotting ΔVG as a function of N. The observation of two-level switching by the neutral, K83A/R119A lysozyme variant (N = 0) identifies the likely contributions of all other charged residues at positions near residues 83 and 119. The observed gating of ΔVG = −34 mV in the neutral case agrees with previous calculations40 based on the distal domain’s net charge of +3, and its substantial hinge motion relative to C90. Fig. 5b shows the effective gating ΔVG of each variant, with (open squares) and without (solid squares) this contribution. After adjustment, the entire set of values became nearly symmetric about zero and linear to N for all six charged variants. The average slope dΔVG/dN = −91 mV per unit charge is a sensitivity nearly three times greater than from the rest of the protein combined. A more detailed consideration of signal transduction by these two positions follows.
Discussion
Taken together, results from Debye screening and site-directed mutagenesis demonstrate an electrostatic mechanism in which charged functionalities at S90C lysozyme positions 83 and 119 play a dominant role in signal transduction by gating the SWNT FET. Unique to these positions are key factors that, we believe, predict good quality signals. First, these positions have charged sidechains that move substantially as a result of catalytic processing. Second, these residues are much closer to the SWNT than other, similarly charged residues, making them less shielded than distant charges. The Debye screening experiments also suggest that salt concentration can be optimized within a biologically relevant range to maximize the signal from these two positions. For example, decreasing the salt concentration improves the signal-to-noise ratio, until low salt concentration prevents the enzyme from functioning normally.
To analyze the electrostatic mechanism further, we have calculated the theoretical electric fields created by charges at lysozyme positions 83 and 119 (Supplementary Table S1). Comparisons of the X-ray structures of lysozyme in its open and closed configurations provided the vector movement of each charge relative to C90 and to the attached pyrene-maleimide linker. The principal qualitative feature of the calculations is that both residues move away from the attachment site as lysozyme binds its substrate. Thus, the net electric field during closure becomes less positive in the N = +2 case. In the N = −2 case, the field becomes more positive. For both variants, the sign of the change agrees with the ΔVG observed experimentally.
Since the orientations of both lysozyme and the SWNT with respect to the pyrene-maleimide linker are not easily defined, there is uncertainty in the electric fields at the SWNT sidewall. Nevertheless, we have modeled an energetically likely orientation of the lysozyme with respect to pyrene-maleimide and the SWNT (Fig. 3). In this model, the dihedral angle of pyrene with respect to maleimide is fixed at its lowest energy conformation at an angle of −50°. This consideration restricted the orientation of lysozyme with respect to the SWNT to two possible lowest energy thioether rotamers43. One cysteine rotamer maintains the 83 and 119 residues at similar distances from the SWNT, resulting in nearly equal electrostatic effects from movements at either position. The second rotamer would cause a charged residue at 119 to have three to five times the gating effect as one at position 83, which is an asymmetric effect that has not been observed for any device to date. The data in Fig. 5 show both positions to have nearly equal gating effects on the SWNT, leading us to depict the first rotamer in Fig. 3.
The calculated fields predict that positions 83 and 119 should be somewhat distinguishable, as expected for any two charges located at slightly different positions. In fact, the main experimental difference between the two positions in Fig. 5 is not a difference in sensitivity dΔVG/dN, such as would arise from differing distances, but rather a reproducible offset that is systematically higher than the ΔVG = −34 mV value of the neutral variant. Specifically, the two N = ±1 variants with charge at position 119 result in a fit to the expression ΔVG = −92N − 11 ± 8 mV, whereas charges at position 83 give ΔVG = −90N − 26 ± 6 mV. The charge-symmetric response from both positions has a nearly identical slope dΔVG/dN, but with position-dependent offsets that are up to 2σ more positive than from the neutral variant.
The offset described above indicates that assigning a single, fixed ΔVG to all the non-targeted charges in the enzyme is too simplistic. Structural differences amongst the variants are expected to be minor, as the variants remain functional and fold consistently into similar circular dichroism-measured structures. Furthermore, protein structure typically remains indifferent to the substitution of small numbers of surface-exposed residues, as shown for many proteins; for example, T4 lysozyme has been mutated at position 119 without affecting its thermal stability44. An alternative and more attractive interpretation reconsiders the complexity of the charge substitutions themselves. The sidechain functional groups are not true point charges, but rather charge distributions resulting from different chemical functionalities in a complex, screening environment. Pursuing this hypothesis, we observe that small shifts of ΔN = −0.1 and −0.2 at the positions 83 and 119, respectively, can align the data to a single, constant offset ΔVG = −34 mV. More detailed analysis requires precise knowledge of the linker’s orientation, the SWNT’s response to the resultant fields, and a more accurate model of electrolyte screening in the immediate vicinity of the linker, all of which are formidable characterization challenges.
Another detail apparent in Supplementary Table S1 but not in Fig. 5 is the joint effect of the N = ±2 variants. Placing charges at both positions 83 and 119 approximately doubles the effective gating and suggests an additive effect. In fact, ΔVG is not quite twice as large as measured when N = ± 1, rather being greater by only a factor of 1.87. Experimentally, dΔVG/dN = 85 mV per unit charge, whereas the sum of the individual charges would predict dΔVG/dN = 91 mV. Inspection of Fig. 3 reveals the cause: the two sidechains do not move in parallel to each other; thus, the vector sum of their fields must be less than the sum of the two magnitudes. The vector calculation in Supplementary Table S1 accounts for this geometry.
Finally, we conclude with a short summary of factors that impact the effectiveness of these devices, since understanding their operation at the molecular level could generalize the approach to other single-enzyme experiments. Remarkably, the role of the particular SWNT is quite minimal, as both metallic and semiconducting SWNTs produce comparable signal-to-noise ratios. This ratio is not substantially improved by seeking the steeper I(VG) curves of semiconducting SWNTs, since their enhanced sensitivity is accompanied by greater noise from background fluctuations. These observations prove that transduction and sensitivity of the lysozyme-functionalized SWNT FETs is not merely due to carrier accumulation or depletion, as in more traditional semiconductor-based sensors. Instead, we note that the electrostatic I(VG) response in the functionalized devices is different from that of pristine SWNTs. In practice, the DC resistance of every SWNT FET is raised by the biofunctionalization11, with the added scattering located at the point of attachment. Thus, sensitivity is not controlled by electrostatic doping nor by selecting a SWNT with a particular bandstructure; rather, it results from a local scattering barrier induced by the attachment itself. This co-localization of the chemical sensitivity and electrical transduction, which has been studied in detail for covalent SWNT defects45–48, leads to signal amplification. We hypothesize that the enzyme’s motion modulates the height and/or width of this barrier, leading to an electostatic field effect that is distinct from carrier accumulation. Additional theoretical work in this area would be very beneficial, since past attempts to model SWNT sensitivity have pursued more traditional bandstructure or charge transfer models.
In addition to the proximity of charged functionalities to the enzyme attachment site described above, the location of this site within the overall protein structure is also critical to successful transduction. Firstly, the attachment site does not need to be located near the enzyme’s active site. In fact, attachments at such positions might interfere with activity, and could perturb enzymatic function. Secondly, we have selected an attachment site that is relatively rigid with respect to the molecule’s center of mass. C90 moves by less than 0.15 nm during catalytic processing. We suspect that similar positions of minimal motion could be key aspects of good attachment sites, since they can minimize perturbations to the enzymatic activity and mechanical stresses on the SWNT linkage. Thirdly, the presence of one or more charged groups moving in concert near the attachment site is critically enabling. The measurements here prove that the immediate environment of the attachment site is most important for transduction.
In conclusion, these results support a very simple, electrostatic gating mechanism for signal transduction by single-enzymes in SWNT FETs. By varying the surrounding electrolyte and the enzyme’s surface charges, we have built a precise understanding of signal generation in these devices. The data show that mechanical displacements of charged functionalities resulting from distal motions are the primary sources of transduction. Furthermore, through selection or introduction of such functionalities, the signal strength can be optimized. The findings suggest design rules by which structural data can guide the creation of similarly effective nanocircuits for single-molecule studies of other enzymes, binding proteins, aptamers, and ribozymes. In addition, single-mutation sensitivity has been demonstrated, providing a promising platform for the further study of molecular function with single molecule resolution.
Supplementary Material
Acknowledgments
We gratefully acknowledge support by the NCI of the NIH (R01 CA133592-01) and NSF (DMR-1104629 and ECCS-0802077).
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Materials, methods, control measurements, and tabular data is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cui Y, Wei QQ, Park HK, Lieber CM. Science. 2001;293:1289. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 2.Patolsky F, Zheng GF, Lieber CM. Analytical Chemistry. 2006;78:4260. doi: 10.1021/ac069419j. [DOI] [PubMed] [Google Scholar]
- 3.Patolsky F, Zheng GF, Lieber CM. Nat Protoc. 2006;1:1711. doi: 10.1038/nprot.2006.227. [DOI] [PubMed] [Google Scholar]
- 4.Gao ZQ, Agarwal A, Trigg AD, Singh N, Fang C, Tung CH, Fan Y, Buddharaju KD, Kong JM. Analytical Chemistry. 2007;79:3291. doi: 10.1021/ac061808q. [DOI] [PubMed] [Google Scholar]
- 5.Star A, Gabriel JCP, Bradley K, Gruner G. Nano Lett. 2003;3:459. [Google Scholar]
- 6.Besteman K, Lee JO, Wiertz FGM, Heering HA, Dekker C. Nano Lett. 2003;3:727. [Google Scholar]
- 7.So H-M, Won K, Kim YH, Kim B-K, Ryu BH, Na PS, Kim H, Lee J-O. J Am Chem Soc. 2005;127:11906. doi: 10.1021/ja053094r. [DOI] [PubMed] [Google Scholar]
- 8.Star A, Tu E, Niemann J, Gabriel JCP, Joiner CS, Valcke C. Proc Natl Acad Sci USA. 2006;103:921. doi: 10.1073/pnas.0504146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong X, Fu D, Xu Y, Wei J, Shi Y, Chen P, Li L-J. J Phys Chem C. 2008;112:9891. [Google Scholar]
- 10.Sorgenfrei S, Chiu C-y, Gonzalez RL, Yu Y-J, Kim P, Nuckolls C, Shepard KL. Nat Nano. 2011;6:126. doi: 10.1038/nnano.2010.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi Y, Moody IS, Sims PC, Hunt SR, Corso BL, Weiss GA, Collins PG. Science. 2012;335:319. doi: 10.1126/science.1214824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi Y, Moody IS, Sims PC, Hunt SR, Corso BL, Seitz DE, Blaszcazk LC, Collins PG, Weiss GA. J Am Chem Soc. 2012;134:2032. doi: 10.1021/ja211540z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy R, Hohng S, Ha T. Nat Methods. 2008;5:507. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong X, Nir E, Hamadani K, Weiss S. J Am Chem Soc. 2007;129:4643. doi: 10.1021/ja068002s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabanayagam CR, Eid JS, Meller A. J Chem Phys. 2005;123:224708. doi: 10.1063/1.2136157. [DOI] [PubMed] [Google Scholar]
- 16.Johnson S, Cain S. Appl Optics. 2008;47:5147. doi: 10.1364/ao.47.005147. [DOI] [PubMed] [Google Scholar]
- 17.Xie SN. Single Molecules. 2001;2:229. [Google Scholar]
- 18.Steiner M, Karunatilaka KS, Sigel RKO, Rueda D. Proc Natl Acad Sci USA. 2008;105:13853. doi: 10.1073/pnas.0804034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claridge SA, Schwartz JJ, Weiss PS. ACS Nano. 2011;5:693. doi: 10.1021/nn103298x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins PG, Bradley K, Ishigami M, Zettl A. Science. 2000;287:1801. doi: 10.1126/science.287.5459.1801. [DOI] [PubMed] [Google Scholar]
- 21.Kong J, Franklin NR, Zhou CW, Chapline MG, Peng S, Cho KJ, Dai HJ. Science. 2000;287:622. doi: 10.1126/science.287.5453.622. [DOI] [PubMed] [Google Scholar]
- 22.Lee K-J, Kong J. Chemical Sensing with SWNT-FETs. In: Javey A, Kong J, editors. Carbon Nanotube Electronics. Springer; New York: 2009. [Google Scholar]
- 23.Heller I, Janssens AM, Mannik J, Minot ED, Lemay SG, Dekker C. Nano Lett. 2008;8:591. doi: 10.1021/nl072996i. [DOI] [PubMed] [Google Scholar]
- 24.Bradley K, Gabriel JCP, Star A, Gruner G. App Phys Lett. 2003;83:3821. [Google Scholar]
- 25.Zhang J, Boyd A, Tselev A, Paranjape M, Barbara P. App Phys Lett. 2006;88:123112. [Google Scholar]
- 26.Choi Y, Moody IS, Sims PC, Hunt SR, Corso BL, Seitz DE, Blaszcazk LC, Collins PG, Weiss GA. J of the Am Chem Soc. 2012;134:2032. doi: 10.1021/ja211540z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen RJ, Zhan YG, Wang DW, Dai HJ. J Am Chem Soc. 2001;123:3838. doi: 10.1021/ja010172b. [DOI] [PubMed] [Google Scholar]
- 28.Kuroki R, Weaver LH, Matthews BW. Nature Structural Biology. 1995;2:1007. doi: 10.1038/nsb1195-1007. [DOI] [PubMed] [Google Scholar]
- 29.Kuroki R, Weaver LH, Matthews BW. Science. 1993;262:2030. doi: 10.1126/science.8266098. [DOI] [PubMed] [Google Scholar]
- 30.Lu HP, Xun LY, Xie XS. Science. 1998;282:1877. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 31.Hu D, Lu HP. Biophysical Journal. 2004;87:656. doi: 10.1529/biophysj.104.042101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu HP. Current Pharmaceutical Biotechnology. 2004;5:261. doi: 10.2174/1389201043376887. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Lu HP. J Phys Chem B. 2010;114:6669. doi: 10.1021/jp1004506. [DOI] [PubMed] [Google Scholar]
- 34.McHaourab HS, Oh KJ, Fang CJ, Hubbell WL. Biochemistry. 1997;36:307. doi: 10.1021/bi962114m. [DOI] [PubMed] [Google Scholar]
- 35.Stern E, Wagner R, Sigworth FJ, Breaker R, Fahmy TM, Reed MA. Nano Lett. 2007;7:3405. doi: 10.1021/nl071792z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heller I, Chatoor S, Mannik J, Zevenbergen MAG, Dekker C, Lemay SG. J Am Chem Soc. 2010;132:17149. doi: 10.1021/ja104850n. [DOI] [PubMed] [Google Scholar]
- 37.Sorgenfrei S, Chiu C-y, Johnston M, Nuckolls C, Shepard KL. Nano Lett. 2011;11:3739. doi: 10.1021/nl201781q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Debye P, Huckel E. Phys Z. 1923;24:185. [Google Scholar]
- 39.Israelachvili JN. Intermolecular and surface forces. Academic Press; London: 1991. [Google Scholar]
- 40.Prisbrey L, Schneider G, Minot E. J Phys Chem B. 2010;114:3330. doi: 10.1021/jp910946v. [DOI] [PubMed] [Google Scholar]
- 41.Abadir GB, Walus K, Pulfrey DL. Nanotechnology. 2010;21:015202. doi: 10.1088/0957-4484/21/1/015202. [DOI] [PubMed] [Google Scholar]
- 42.Kuroki R, Weaver LH, Matthews BW. Proc Natl Acad Sci USA. 1999;96:8949. doi: 10.1073/pnas.96.16.8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin L, Cohen FE, Wells JA. Proc Natl Acad Sci USA. 1994;91:113. doi: 10.1073/pnas.91.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins PG, Fuhrer MS, Zettl A. Appl Phys Lett. 2000;76:894. [Google Scholar]
- 45.Lin Y-M, Appenzeller J, Knoch J, Zhihong C, Avouris P. Nano Lett. 2006;6:930. doi: 10.1021/nl052528d. [DOI] [PubMed] [Google Scholar]
- 46.Tobias D, Ishigami M, Tselev A, Barbara P, Williams ED, Lobb CJ, Fuhrer MS. Phys Rev B. 2008;77:033407. [Google Scholar]
- 47.Mannik J, Heller I, Janssens AM, Lemay SG, Dekker C. Nano Lett. 2008;8:685. doi: 10.1021/nl073271h. [DOI] [PubMed] [Google Scholar]
- 48.Sorgenfrei S, Chiu C-y, Gonzalez RL, Yu Y-J, Kim P, Nuckolls C, Shepard KL. Nat Nano. 2011;6:126. doi: 10.1038/nnano.2010.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.