

Abstract
Insulin-, and contraction-induced GLUT4 and fatty acid (FA) transporter translocation may share common trafficking mechanisms. Our objective was to examine the effects of partial Munc18c ablation on muscle glucose and FA transport, FA oxidation, GLUT4 and FA transporter (FAT/CD36, FABPpm, FATP1, FATP4) trafficking to the sarcolemma, and FAT/CD36 to mitochondria. In Munc18c−/+ mice, insulin-stimulated glucose transport and GLUT4 sarcolemmal appearance were impaired, but were unaffected by contraction. Insulin- and contraction-stimulated FA transport, sarcolemmal FA transporter appearance, and contraction-mediated mitochondrial FAT/CD36 were increased normally in Munc18c−/+ mice. Hence, Munc18c provides stimulus-specific regulation of GLUT4 trafficking, but not FA transporter trafficking.
Keywords: FAT/CD36, FABPpm, FATP1, FATP4, Fatty acid transport, Glucose transport
1. Introduction
The clearance of glucose and long chain fatty acids (FA) from the circulation are important regulatory processes for maintaining metabolic homeostasis. Transport of glucose and FA into the cell occur via highly regulated protein-mediated mechanisms involving glucose transporter GLUT4 (cf. [1,2]) and selected FA transporters, including fatty acid translocase (FAT/CD36), plasma membrane associated fatty acid binding protein (FABPpm), and the family of fatty acid transporter proteins (FATP 1–6) (cf. [3]). During muscle contraction FAT/CD36 is also translocated to the mitochondria, contributing to upregulating mitochondrial FA oxidation (cf. [3]).
The dysregulation of both glucose and FA transport, and the trafficking of their transport proteins, are implicated in skeletal muscle insulin resistance (cf. [3]). However, the signaling pathways involved in the translocation of FA transporters to the plasma membrane (PM) are largely unknown, although in muscle FAT/CD36 may share some similarities to the insulin- and contraction-mediated signaling cascades of GLUT4 (cf. [3,4]). However, in cardiac cells, independent signaling mechanisms do exist for GLUT4 and FAT/CD36 [5,6]. Whether GLUT4 and FA transporter trafficking to the PM share similar mechanisms remains to be determined.
The subcellullar trafficking of GLUT4 to the PM is far better characterized than for FA transporters. GLUT4 trafficking is a vesicle-mediated process following the soluble N-ethylmaleimide-sensitive factor-attachment protein receptor (SNARE) hypothesis where vesicle associated SNARE (vSNARE) proteins associate with complimentary target SNAREs (tSNARE) at the PM (cf. [7]). Formation of the vSNARE-tSNARE complex enables docking, fusion, and integration of GLUT4 vesicles into the PM. A number of accessory proteins are also involved in the formation of the SNARE complex, including vesicle associated membrane proteins (VAMPs), SNARE-related protein (SNAPs), and syntaxins. These accessory proteins facilitate the complementary binding of vSNARE and tSNARE vesicles, ensuring proper PM insertion of GLUT4. In addition, three homologs (a, b, and c) of Munc18, from the Sec1 protein family are known to be present at the mammalian PM [8,9]. Of these, Munc18c is ubiquitously expressed and has binding specificity for the tSNARE protein syntaxin4 [10]. In skeletal muscle, partial ablation of Munc18c in heterozygous knockout mice (Munc18c−/+) induces insulin resistance due to impaired GLUT4 translocation [11].
Munc18c may however only be involved in insulin-stimulated GLUT4 trafficking, since GLUT4 trafficking in insulin resistant muscle is impaired while in the same muscles contraction-stimulated GLUT4 trafficking remains normal [12–14]. Conversely, in insulin resistant muscle, sarcolemmal FAT/CD36, but not FABPpm, is upregulated. However, insulin-, and contraction-stimulated FAT/CD36, but not FABPpm, translocations are impaired (cf. [3]). Collectively, it appears that there may be (i) different exocytosis mechanisms for GLUT4 and FA transporters, and (ii) depending on the stimulus provided, different proteins may be required for the trafficking of GLUT4 and/or FA transporters to the PM. Such a system would provide for selective regulation of GLUT4 and/or FA transporter trafficking to the PM.
Our aim was to examine the effects of Munc18c on insulin-, and contraction-stimulated glucose and FA transport. We hypothesized that Munc18c differentially affects (a) insulin- and contraction-stimulated glucose and FA transport and (b) the translocation of GLUT4 and FA transporters to the PM, as well as (c) contraction-induced mitochondrial FA oxidation and (d) FAT/CD36 trafficking to mitochondria [15,16].
2. Materials and methods
2.1. Animals
Due to an embryonically lethal homozygous genotype, we used male, 8–10 week old heterozygous Munc18c−/+ mice [11] (22.7 ± 3.0 g) bred on site with C57/BL6 wildtype (WT) mice (24.1 ± 3.1 g), and kept at 22 °C, 40% humidity, 12:12-h light-dark cycle, and given chow and water ad libitum. Experiments were performed on anesthetized mice (sodium pentobarbital 6 mg/100 g body wt ip; MTC Pharmaceuticals, Cambridge, ON, Canada) using principles of laboratory animal care (National Institutes of Health publication No. 85-23, revised 1985; http://grants1.nih.gov/grants/olaw/references/phspol.htm. The Munc18c−/+ genotype was confirmed using standard DNA isolation and PCR methods (Extract-N-Amp, Sigma-Aldrich, St. Louis, MO, USA), as we have reported previously [11].
2.2. Experimental treatments
Fasted (12 h) WT and Munc18c−/+ animals (N = 4–6/experiment) were designated as control, or treated with (a) insulin (Humulin, 1.0 U/kg body wt, ip, 15 min; Eli Lilly, Toronto, ON, Canada), or (b) muscle contraction (sciatic nerve stimulation: 3 × 5 min, 2 min rest between stimulations, 100 Hz/3 s, 5 V, train 200 ms, pulse 10 ms) [17,18]. Homogenates, giant sarcolemmal vesicles (GSV) and mitochondria were prepared from hindlimb muscles [17–19]. Intraperitoneal glucose (1.0 g/kg body wt) and insulin (1.0 U/kg body wt) tolerance tests were determined in separate animals. Tail vein glucose was determined using a glucose meter (Ascensia Elite XL, Bayer Inc., Toronto, ON, Canada).
2.3. Giant sarcolemmal vesicles and substrate transport
GSV from hindlimb muscles were used to determine FA and glucose transport and the presence of PM transport proteins, as described previously [18,19].
2.4. Mitochondrial isolation and palmitate oxidation
After muscle contraction, hindlimb muscles were harvested for isolation of subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria, for determination of FA oxidation, using standard procedures [15–17]. Mitochondrial recovery is 26% [20] and they are highly purified [21].
2.5. Western blotting
Protein levels were measured in GSV, muscle homogenate, and mitochondria using Western blotting [17,18]. Antibodies against Munc18c and syntaxin4 (each 1:1000) were generated in-house [11]. Antibodies against GLUT4 (1:4000) (Millipore, Temecula, CA), FAT/CD36, FATP1, FATP4 (each 1:500) (Santa Cruz Biotechnology, Santa Cruz, CA, USA [22]), AMPK and Akt2, and phosphorylated AMPKa Thr172, Akt Thr308, and Akt Ser473 (each 1:1000) (Cell Signaling (Danvers, MA, USA). FABPpm (1:30000, J. Calles-Escandon, Wake Forest University) and MCT1 (1:3000, H. Hatta, University of Tokyo) were gifts. Secondary antibodies, (Santa Cruz Biotechnology), were used as follows: Munc18c, syntaxin4, GLUT4, FABPpm and MCT1 (1:3000 anti-rabbit), FAT/CD36 (1:5000 anti-mouse), FATP1 (1:5000 anti-rabbit), FATP4 (1:5000 anti-goat), and all total and phosphorylated Akt and AMPK (1:1000 anti-rabbit). Blots were analyzed with the ChemiGenius2 Bioimaging and GeneTools software (SynGene, Cambridge, UK) [17,18]. Ponceau-S staining was used to ensure protein loading, as well as COXIV (Invitrogen, Burlington, ON; 1:30000 dilution) for mitochondria.
2.6. Statistics
Data were analyzed using analysis of variance and Fisher's LSD post hoc test when appropriate. All data are reported as mean ± sem.
3. Results
A 50% reduction in Munc18c protein (Fig. 1), along with a comparable reduction in mRNA (data not shown, [11,23]) occurred in skeletal muscle of Munc18c−/+ animals. Syntaxin4 and transport proteins (GLUT4, FAT/CD36, FABPpm, FATP1, FATP4) remained unaltered (Fig. 1A). Basal blood glucose concentrations were comparable in mice (WT (6.1 mM ± 1.1; Munc18c−/+ 6.3 ± 1.0 mM). Munc18c−/+ mice exhibited insulin resistance and glucose intolerance (Fig. 1 B and C).
Fig 1.
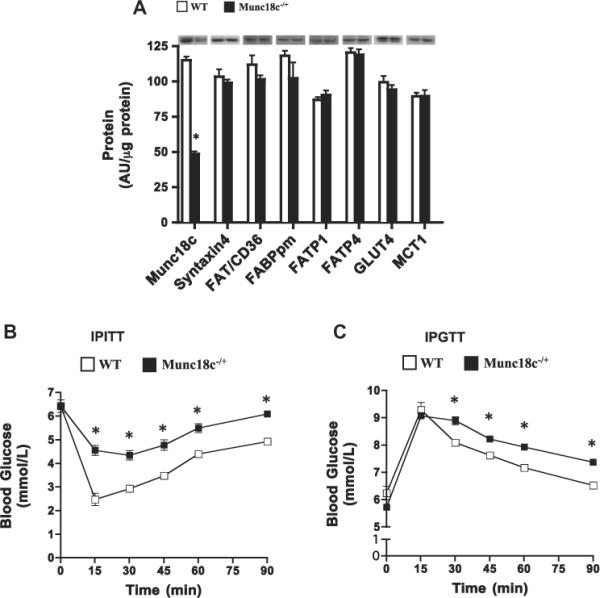
Muscle protein content of Munc18c, syntaxin4, and glucose and FA transport proteins (A), IPITT (B), and IPGTT (C) in WT and Munc18c−/+ mice. N = 4–6, mean ± sem. Error bars are smaller than symbols in B an C. As noted in Section 2, Ponceau staining was used to confirm equal loading of proteins. MCT1, a monocarboxylate transporter, served as a positive control. *p < 0.05, Munc18c−/+ vs WT mice.
3.1. Knockdown of Munc18c does not affect insulin-, or contraction-mediated signal transduction
Insulin increased the phosphorylation of Akt-Ser473 (+150%) and Akt-Thr308 (+220%) comparably in WT and Munc18c−/+ mice (Fig. 2A and B), as reported previously [11]. For unknown reasons, muscle contraction did not alter Akt phosphorylation (Fig. 2) as observed elsewhere [24], possibly owing to differences in the contractile stimulus used (Fig. 2). Total Akt2 was comparable in WT and Munc18c−/+ mice (Fig. 2C).
Fig 2.

Effects of insulin (INS) and muscle contraction (CT) on the phosphorylation of Akt Ser473 (A), Akt Thr308 (B), total Akt2 (C), phosphorylated AMPKα Thr172 (D), and total AMPKα (E) in WT and Munc18c−/+ mice. N = 4–6 mean ± sem. As noted in Section 2, Ponceau staining was used to confirm equal loading of proteins. *p < 0.05, insulin or contraction stimulation in WT and Munc18c−/+ compared to basal phosphorylation.
Muscle contraction, not insulin, increased AMPKα Thr172 phosphorylation (+400%) (Fig. 2D). Total AMPKα was comparable in WT and Munc18c−/+ mice (Fig. 2E).
3.2. Knockdown of Munc18c reduces insulin-, but not contraction-mediated glucose transport and GLUT4 translocation
Basal glucose transport and PM GLUT4 were comparable between WT and Munc18c−/+ mice (Fig. 3), whereas PM Munc18c was reduced 50% (Fig. 4A). In WT mice, insulin increased glucose transport (+133%) and PM GLUT4 (+60%) (Fig. 3), whereas in Munc18c−/+ mice, insulin-stimulated glucose transport and GLUT4 appearance at the PM were impaired (Fig. 3). In WT mice, but not in Munc18c−/+ mice, PM Munc18c was slightly increased (26%) by insulin. Syntaxin4 (Fig. 4) was not altered by insulin in either group (Fig. 4).
Fig 3.
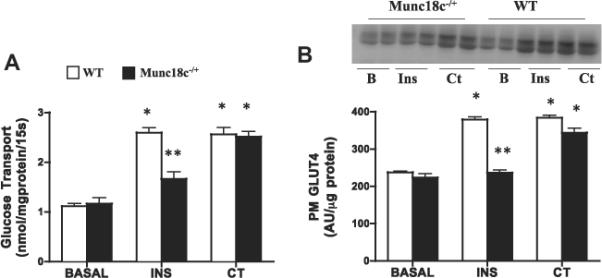
Effects of insulin (INS) and muscle contraction (CT) on glucose transport (A) and plasma membrane (PM) GLUT4 (B) in WT and Munc18c−/+ mice. N = 4–6 independent determinations, mean ± sem (3 mice were pooled for each independent experiment). Blots of two independent experiments are shown. MCT1 served as a positive control, as it is present only at the sarcolemma [41]. It was not altered by the experimental treatments in WT and in Munc18c−/+ mice (data not shown). As noted in Section 2, Ponceau staining was used to confirm equal loading of proteins. INS = Insulin; CT = Contraction. *p < 0.05, stimulation with insulin or contraction vs respective basal. **p < 0.05, Munc18c−/+ vs WT.
Fig 4.
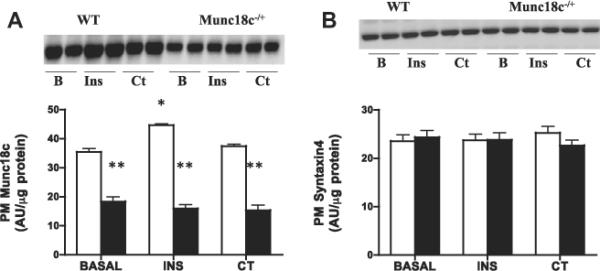
Effects of insulin (INS) and muscle contraction (CT) on plasma membrane (PM) Munc18c (A) and PM Syntaxin4 (B) in WT and Munc18c−/+ mice. N = 4–6 independent determinations, mean ± sem (3 mice were pooled for each independent experiment). Blots of two independent experiments are shown. MCT1 served as a positive control, as it is present only at the sarcolemma [41]. It was not altered by the experimental treatments in WT and in Munc18c−/+ mice (data not shown). As noted in Section 2, Ponceau staining was used to confirm equal loading of proteins. INS = Insulin; CT = Contraction. *p < 0.05, stimulation with insulin or contraction vs respective basal. **p < 0.05, Munc18c−/+ vs WT.
Contraction-induced increases in glucose transport (+130%) and PM GLUT4 (62%) were comparable in WT and Munc18c−/+ mice (Fig. 3). Muscle contraction did not alter the PM Munc18c or syntaxin4 in either group (Fig. 4).
3.3. Knockdown of Munc18c does not affect insulin- or contraction-mediated FA transport or FA transporters translocation
Basal FA transport and PM FA transport proteins were comparable in WT and Munc18c−/+ mice (Fig. 5). In both groups, insulin stimulated FA transport (+41% (Fig. 5A)) and FA transporters at the PM comparably (FAT/CD36 +82%; FATP1 +40%; FABPpm +39%; FATP4 +33%; (Fig. 5B–E)). Similar increases were also observed in both groups with muscle contraction (FA transport +40%; FAT/CD36 +84%; FATP1 +38%; FABPpm +43%; FATP4 +32%; (Fig. 5B–E)).
Fig 5.
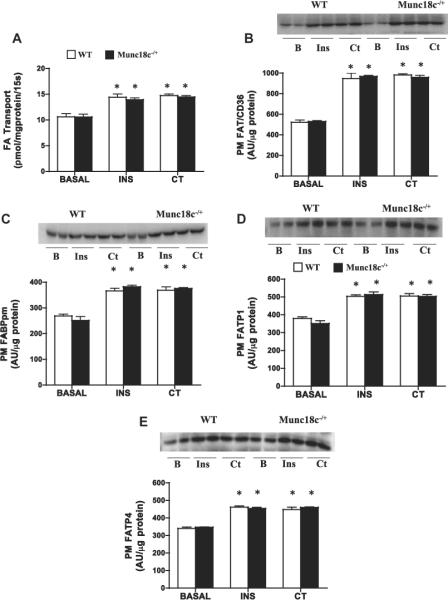
Effects of insulin (INS) and muscle contraction (CT) on FA transport (A) and PM FA transport proteins FAT/CD36 (B), FABPpm (C), FATP1 (D), FATP4 (E) in WT and Munc18c−/+ mice. N = 4–6 independent determinations mean ± sem (3 mice were pooled for each independent experiment). Blots of two independent experiments are shown. MCT1 served as a positive control, as it is present only at the sarcolemma [41]. It was not altered by the experimental treatments in WT and in Munc18c−/+ mice (data not shown). As noted in Section 2, Ponceau staining was used to confirm equal loading of proteins. INS = Insulin; CT = Contraction. *p < 0.05, stimulation with insulin or contraction vs respective basal.
3.4. Knockdown of Munc18c does not affect contraction-stimulated FA oxidation or FAT/CD36 translocation to mitochondria
Only contraction-stimulated FA oxidation was examined in mitochondria (SS, IMF), as insulin does not alter mitochondrial FA oxidation directly. Neither basal nor contraction-stimulated FA oxidation (+40%, Fig. 6A) and FAT/CD36 (+63%, Fig. 6B) differed between WT and Munc18c−/+ mice.
Fig 6.
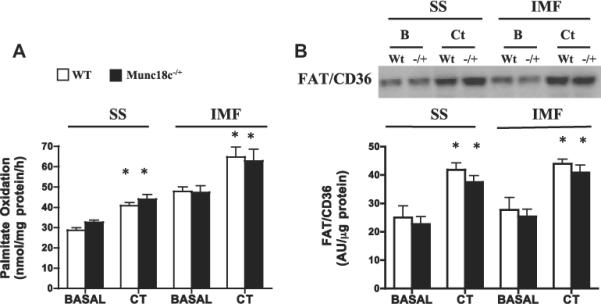
Effects of muscle contraction (CT) on FA oxidation (A) and FAT/CD36 protein (B) in SS and IMF mitochondria in WT and Munc18c−/+ mice. COXIV was used as a loading control. N = 4–6 independent determinations, mean ± sem (3 mice were pooled for each independent experiment). As noted in Section 2, CT = Contraction. *p < 0.05, stimulation with contraction vs respective basal.
4. Discussion
We examined the role of Munc18c, on contraction-, and insulin-stimulated (i) glucose and FA transport, (ii) appearance of GLUT4 and FA transporters at the PM, and (iii) contraction-stimulated FA oxidation and FAT/CD36 appearance at mitochondria. We show that Munc18c is involved in insulin-stimulated, but not contraction-stimulated GLUT4 trafficking to the PM. A novel observation is that Munc18c is not required for the trafficking of FA transporters to the PM, or FAT/CD36 to mitochondria. Thus, (i) insulin-stimulated, but not contraction-stimulated, GLUT4 trafficking to the PM is Munc18c-dependent, whereas (ii) FA transporter trafficking to the PM, induced by insulin or contraction, is Munc18c-independent.
4.1. Insulin- and contraction-stimulated glucose uptake and PM GLUT4
Munc18c binds with syntaxin4, thereby converting syntaxin4 to an open conformational state, allowing interactions with other VAMP and SNAP trafficking proteins [8,10]. The binding between Munc18c and syntaxin4 is required for the docking and fusion of GLUT4 vesicles into the PM [25]. Thus, it is likely that a 50% reduction in Munc18c protein in skeletal muscle, unlike in the heart [26], accounted for the impaired insulin-stimulated GLUT4 appearance at the PM, and glucose transport into muscle, as insulin-stimulated Akt phosphorylation and syntaxin4 protein levels were normal in the Munc18c−/+ mice, as shown previously [11].
Contraction-induced trafficking of GLUT4 to the sarcolemma did not require Munc18c. Apparently, only syntaxin4 is required for muscle GLUT4 translocation [27]. The stimulus-specific trafficking of GLUT4 is presumably due to the presence of separate insulin-, and contraction-sensitive endosomal GLUT4 storage vesicles [28,29].
4.2. Insulin- and contraction-stimulated PM Munc18c and syntaxin4
In muscle, Munc18c was also translocated to the PM by insulin, but not contraction. This parallels observations in insulin-stimulated adipocytes [30]. It remains unknown why insulin-stimulated translocation of Munc18c was not observed in the Munc18c−/+ mice.
4.3. Insulin- and contraction-stimulated FA uptake and PM FA transporters
Reductions in Munc18c did not affect basal FA transport or expression of any FA transporters Although Munc18c+/− mice were insulin resistant and glucose intolerant, we did not observe upregulation of plasmalemmal FAT/CD36 under basal conditions, as was previously observed in more severe models of insulin resistance (obese Zucker rats, human obesity and type 2 diabetes (cf. [3])). This suggests that in the Munc18c+/− model insulin resistance is not associated with a basal upregulation of plasmalemmal FAT/CD36.
Partial ablation of Munc18c did not affect FA transport, nor the FA transporter translocation to the PM, in either insulin-, or contraction-stimulated muscle. Thus, while some components of insulin- and contraction-induced GLUT4 and FA transporter translocation to the PM appear to share similar signal transduction pathways (cf. [3]), the insulin-stimulated trafficking of FA transporters differs from GLUT4. Indirect evidence suggests that contraction-induced FA transporter and GLUT4 translocation to the PM require different trafficking proteins. In insulin resistant muscle, contraction-stimulated GLUT4 translocation is normal whereas contraction-stimulated FAT/CD36 translocation is not increased further beyond its upregulated, basal sarcolemmal levels (cf. [3]).
Whether other components of the GLUT4 trafficking machinery are involved in directing FA transporters to the PM is unknown. In cultured cardiac myocytes, VAMP trafficking proteins are involved in the localization of FAT/CD36 to the PM, and distinct VAMP isoforms are specific for GLUT4 or FAT/CD36, and their translocation is stimulus dependent [31]. In addition, in cardiac muscle the Rab11 trafficking protein co-localizes in the same endosomal pools as FAT/CD36 [32] and may regulate endocytosis/recycling of this FA transporter in H9c2-hIR cells [33]. Thus, there is evidence suggesting the involvement of trafficking proteins other than Munc18c in the regulation of FA transporters. It is unlikely that GLUT4 and FA transporters reside in the same intracellular pools as shown by fractionation and immuno-isolation studies in cardiac tissue [32], and the presence of GLUT4-, and FAT/CD36-specific VAMP isoforms in cultured cardiac myocytes [31]. The negative correlation between PM levels of GLUT4 and FAT/CD36 in insulin resistant muscle suggests differential metabolic regulation between these transporters [34].
We did not examine the translocation of GLUT4, and possibly FA transporters, to the transverse tubules [35]. Preliminary studies indicate that this is more complex than it might appear, especially for the FA transporters (Bonen and Stefanyk, unpublished observations). The heterozygous knockout Munc18c model may also complicate our conclusion that Munc18c is not involved in FA transporter trafficking, as it is possible that less Munc18c is required for PM FA transporter translocation in comparison to GLUT4.
4.4. Mitochondrial FA oxidation and FA transporters
FAT/CD36 has been identified on mitochondria in human [16,20] and rodent muscle [15,36], and FAT/CD36 co-immunoprecipitates with CPT1 [37,38]. Muscle contraction is known to stimulate FA oxidation, but insulin does not directly affect muscle FA oxidation [39]. We confirm previous work [17,20] that contraction increases mitochondrial FA oxidation and induces FAT/CD36 trans-location to mitochondria. This trafficking process is Munc18c-independent. While the possible trafficking pathways of FAT/CD36 to the mitochondria are unknown, a novel isoform of VAMP1 has been associated with mitochondria in human epithelial cells suggesting the interesting possibility that mitochondria may also participate in vesicular trafficking [40].
4.5. Summary
Munc18c is required for insulin-stimulated, but not contraction-mediated trafficking of GLUT4 to the PM. Munc18c is not required for FA transporter trafficking to the PM or to mitochondria. It is possible that insulin-sensitive and/or contraction-sensitive pools of FA transporters are selectively regulated by specific, trafficking protein isoforms. In view of the known upregulation of plasmalemmal FAT/CD36 in insulin resistant muscle (cf. [3]), identification of their trafficking proteins is important, as these may be possible therapeutic targets.
Acknowledgements
Rported by the Canadian Institutes of Health Research (CIHR), and the Canada Research Chair program to A.B., and the National Institutes of Health (NIH), DK076614 and DK067912 to D.C.T.
S.J. held an NSERC Canada Graduate Scholarship.
Abbreviations
- FA
fatty acid
- FAT/CD36
fatty acid translocase
- GLUT4
glucose transporter
- FABPpm
plasma membrane associated fatty acid binding protein
- FATP
fatty acid transport protein
- PM
plasma membrane
- AMPK
5'AMP-activated protein kinase
- SNARE
soluble N-ethylmaleimide-sensitive factor-attachment protein receptor
- SS
subsarcolemmal mitochondria
- IMF
intermyofibrillar mitochondria
References
- [1].Jessen N, Goodyear LJ. Contraction signaling to glucose transport in skeletal muscle. J. Appl. Physiol. 2005;99:330–337. doi: 10.1152/japplphysiol.00175.2005. [DOI] [PubMed] [Google Scholar]
- [2].Klip A. The many ways to regulate glucose transporter 4. Appl. Physiol. Nutr. Metab. 2009;34:481–487. doi: 10.1139/H09-047. [DOI] [PubMed] [Google Scholar]
- [3].Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- [4].Samovski D, Su X, Xu Y, Abumrad NA, Stahl PD. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J. Lipid Res. 2012;53:709–717. doi: 10.1194/jlr.M023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Steinbusch LK, Wijnen W, Schwenk RW, Coumans WA, Hoebers NT, Ouwens DM, Coumans WA, Hoebers NT, Diamant M, Bonen A, Glatz JF, Luiken JJ. Differential regulation of cardiac glucose and fatty acid uptake by endosomal pH and actin filaments. Am. J. Physiol. Cell Physiol. 2010;298:C1549–C1559. doi: 10.1152/ajpcell.00334.2009. [DOI] [PubMed] [Google Scholar]
- [6].Dirkx E, Schwenk RW, Coumans WA, Hoebers N, Angin Y, Viollet B, Bonen A, van Eys GJ, Glatz JF, Luiken JJ. Protein kinase D1 is essential for contraction-induced glucose uptake but is not involved in fatty acid uptake into cardiomyocytes. J. Biol. Chem. 2012;287:5871–5881. doi: 10.1074/jbc.M111.281881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thurmond DC, Pessin JE. Molecular machinery involved in the insulin-regulated fusion of GLUT4-containing vesicles with the plasma membrane (review) Mol. Membr. Biol. 2001;18:237–245. doi: 10.1080/09687680110082400. [DOI] [PubMed] [Google Scholar]
- [8].Halachmi N, Lev Z. The Sec1 family: a novel family of proteins involved in synaptic transmission and general secretion. J. Neurochem. 1996;66:889–897. doi: 10.1046/j.1471-4159.1996.66030889.x. [DOI] [PubMed] [Google Scholar]
- [9].Tellam JT, McIntosh S, James DE. Molecular identification of two novel Munc-18 isoforms expressed in non-neuronal tissues. J. Biol. Chem. 1995;270:5857–5863. doi: 10.1074/jbc.270.11.5857. [DOI] [PubMed] [Google Scholar]
- [10].Tellam JT, Macaulay SL, McIntosh S, Hewish DR, Ward CW, James DE. Characterization of Munc-18c and syntaxin-4 in 3T3-L1 adipocytes. Putative role in insulin-dependent movement of GLUT-4. J. Biol. Chem. 1997;272:6179–6186. doi: 10.1074/jbc.272.10.6179. [DOI] [PubMed] [Google Scholar]
- [11].Oh E, Spurlin BA, Pessin JE, Thurmond DC. Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance. Diabetes. 2005;54:638–647. doi: 10.2337/diabetes.54.3.638. [DOI] [PubMed] [Google Scholar]
- [12].Zierath JR, Houseknecht KL, Gnudi L, Kahn BB. High-fat feeding impairs insulin-stimulated GLUT4 recruitment via an early insulin-signaling defect. Diabetes. 1997;46:215–223. doi: 10.2337/diab.46.2.215. [DOI] [PubMed] [Google Scholar]
- [13].Brozinick J, Jr., Etgen GJ, Jr., Yaspelkis BB, 3rd, Ivy JL. Contraction-activated glucose uptake is normal in insulin-resistant muscle of the obese Zucker rat. J. Appl. Physiol. 1992;73:382–387. doi: 10.1152/jappl.1992.73.1.382. [DOI] [PubMed] [Google Scholar]
- [14].Etgen GJ, Jr., Wilson CM, Jensen J, Cushman SW, Ivy JL. Glucose transport and cell surface GLUT-4 protein in skeletal muscle of the obese Zucker rat. Am. J. Physiol. 1996;271:E294–E301. doi: 10.1152/ajpendo.1996.271.2.E294. [DOI] [PubMed] [Google Scholar]
- [15].Campbell SE, Tandon NN, Woldegiorgis G, Luiken JJ, Glatz JF, Bonen A. A novel function for fatty acid translocase (FAT)/CD36: involvement in long chain fatty acid transfer into the mitochondria. J. Biol. Chem. 2004;279:36235–36241. doi: 10.1074/jbc.M400566200. [DOI] [PubMed] [Google Scholar]
- [16].Bezaire V, Bruce CR, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Identification of fatty acid translocase on human skeletal muscle mitochondrial membranes: essential role in fatty acid oxidation. Am. J. Physiol. Endocrinol. Metab. 2006;290:E509–E515. doi: 10.1152/ajpendo.00312.2005. [DOI] [PubMed] [Google Scholar]
- [17].Holloway GP, Jain SS, Bezaire V, Han XX, Glatz JF, Luiken JJ, Harper ME, Bonen A. FAT/CD36-null mice reveal that mitochondrial FAT/CD36 is required to upregulate mitochondrial fatty acid oxidation in contracting muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;297:R960–R967. doi: 10.1152/ajpregu.91021.2008. [DOI] [PubMed] [Google Scholar]
- [18].Jain SS, Chabowski A, Snook LA, Schwenk RW, Glatz JF, Luiken JJ, Bonen A. Additive effects of insulin and muscle contraction on fatty acid transport and fatty acid transporters, FAT/CD36, FABPpm, FATP1, 4 and 6. FEBS Lett. 2009;583:2294–2300. doi: 10.1016/j.febslet.2009.06.020. [DOI] [PubMed] [Google Scholar]
- [19].Koonen DP, Coumans WA, Arumugam Y, Bonen A, Glatz JF, Luiken JJ. Giant membrane vesicles as a model to study cellular substrate uptake dissected from metabolism. Mol. Cell. Biochem. 2002;239:121–130. [PubMed] [Google Scholar]
- [20].Holloway GP, Bezaire V, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Mitochondrial long chain fatty acid oxidation, fatty acid translocase/CD36 content and carnitine palmitoyltransferase I activity in human skeletal muscle during aerobic exercise. J. Physiol. 2006;571:201–210. doi: 10.1113/jphysiol.2005.102178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yoshida Y, Holloway GP, Ljubicic V, Hatta H, Spriet LL, Hood DA, Bonen A. Negligible direct lactate oxidation in subsarcolemmal and intermyofibrillar mitochondria obtained from red and white rat skeletal muscle. J. Physiol. 2007;582:1317–1335. doi: 10.1113/jphysiol.2007.135095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nickerson JG, Alkhateeb H, Benton CR, Lally J, Nickerson J, Han XX, Wilson MH, Jain SS, Snook LA, Glatz JF, Chabowski A, Luiken JJ, Bonen A. Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1 and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J. Biol. Chem. 2009;284:16522–16530. doi: 10.1074/jbc.M109.004788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Oh E, Thurmond DC. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes. 2009;58:1165–1174. doi: 10.2337/db08-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sakamoto K, Arnolds DE, Fujii N, Kramer HF, Hirshman MF, Goodyear LJ. Role of Akt2 in contraction-stimulated cell signaling and glucose uptake in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006;291:E1031–E1037. doi: 10.1152/ajpendo.00204.2006. [DOI] [PubMed] [Google Scholar]
- [25].Thurmond DC, Kanzaki M, Khan AH, Pessin JE. Munc18c function is required for insulin-stimulated plasma membrane fusion of GLUT4 and insulin-responsive amino peptidase storage vesicles. Mol. Cell. Biol. 2000;20:379–388. doi: 10.1128/mcb.20.1.379-388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Habets DD, Thurmond DC, Coumans WA, Bonen A, Glatz JF, Luiken JJ. Munc18c is not rate-limiting for glucose and long-chain fatty acid uptake in the heart. Mol. Cell. Biochem. 2009;322:81–86. doi: 10.1007/s11010-008-9942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang C, Coker KJ, Kim JK, Mora S, Thurmond DC, Davis AC, Yang B, Williamson RA, Shulman GI, Pessin JE. Syntaxin 4 heterozygous knockout mice develop muscle insulin resistance. J. Clin. Invest. 2001;107:1311–1318. doi: 10.1172/JCI12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aledo JC, Lavoie L, Volchuk A, Keller SR, Klip A, Hundal HS. Identification and characterization of two distinct intracellular GLUT4 pools in rat skeletal muscle: evidence for an endosomal and an insulin-sensitive GLUT4 compartment. Biochem. J. 1997;325(Pt 3):727–732. doi: 10.1042/bj3250727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lemieux K, Han XX, Dombrowski L, Bonen A, Marette A. The transferrin receptor defines two distinct contraction-responsive GLUT4 vesicle populations in skeletal muscle. Diabetes. 2000;49:183–189. doi: 10.2337/diabetes.49.2.183. [DOI] [PubMed] [Google Scholar]
- [30].Nelson BA, Robinson KA, Buse MG. Insulin acutely regulates Munc18-c subcellular trafficking: altered response in insulin-resistant 3T3-L1 adipocytes. J. Biol. Chem. 2002;277:3809–3812. doi: 10.1074/jbc.C100645200. [DOI] [PubMed] [Google Scholar]
- [31].Schwenk RW, Dirkx E, Coumans WA, Bonen A, Klip A, Glatz JF, Luiken JJ. Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia. 2010 doi: 10.1007/s00125-010-1832-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Muller H, Deckers K, Eckel J. The fatty acid translocase (FAT)/CD36 and the glucose transporter GLUT4 are localized in different cellular compartments in rat cardiac muscle. Biochem. Biophys. Res. Commun. 2002;293:665–669. doi: 10.1016/S0006-291X(02)00276-0. [DOI] [PubMed] [Google Scholar]
- [33].Schwenk RW, Luiken JJ, Eckel J. FIP2 and Rip11 specify Rab11a-mediated cellular distribution of GLUT4 and FAT/CD36 in H9c2-hIR cells. Biochem. Biophys. Res. Commun. 2007;363:119–125. doi: 10.1016/j.bbrc.2007.08.111. [DOI] [PubMed] [Google Scholar]
- [34].Chabowski A, Chatham JC, Tandon NN, Calles-Escandon J, Glatz JF, Luiken JJ, Bonen A. Fatty acid transport and FAT/CD36 are increased in red but not in white skeletal muscle of ZDF rats. Am. J. Physiol. Endocrinol. Metab. 2006;291:E675–E682. doi: 10.1152/ajpendo.00096.2006. [DOI] [PubMed] [Google Scholar]
- [35].Marette A, Burdett E, Douen A, Vranic M, Klip A. Insulin induces the translocation of GLUT4 from a unique intracellular organelle to transverse tubules in rat skeletal muscle. Diabetes. 1992;41:1562–1569. doi: 10.2337/diab.41.12.1562. [DOI] [PubMed] [Google Scholar]
- [36].Holloway GP, Benton CR, Mullen KL, Yoshida Y, Snook LA, Han XX, Glatz JF, Luiken JJ, Lally J, Dyck DJ, Bonen A. In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am. J. Physiol. Endocrinol. Metab. 2009;296:E738–E747. doi: 10.1152/ajpendo.90896.2008. [DOI] [PubMed] [Google Scholar]
- [37].Schenk S, Horowitz JF. Coimmunoprecipitation of FAT/CD36 and CPT I in skeletal muscle increases proportionally with fat oxidation after endurance exercise training. Am. J. Physiol. Endocrinol. Metab. 2006;291:E254–E260. doi: 10.1152/ajpendo.00051.2006. [DOI] [PubMed] [Google Scholar]
- [38].Smith BK, Jain SS, Rimbaud S, Dam A, Quadrilatero J, Ventura-Clapier R, Bonen A, Holloway GP. FAT/CD36 is located on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem. J. 2011;437:125–134. doi: 10.1042/BJ20101861. [DOI] [PubMed] [Google Scholar]
- [39].Turcotte LP, Swenberger JR, Yee AJ. High carbohydrate availability increases LCFA uptake and decreases LCFA oxidation in perfused muscle. Am. J. Physiol. Endocrinol. Metab. 2002;282:E177–E183. doi: 10.1152/ajpendo.00316.2001. [DOI] [PubMed] [Google Scholar]
- [40].Isenmann S, Khew-Goodall Y, Gamble J, Vadas M, Wattenberg BW. A splice-isoform of vesicle-associated membrane protein-1 (VAMP-1) contains a mitochondrial targeting signal. Mol. Biol. Cell. 1998;9:1649–1660. doi: 10.1091/mbc.9.7.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bonen A, Miskovic D, Tonouchi M, Lemieux K, Wilson MC, Marette A, Halestrap AP. Abundance and subcellular distribution of MCT1 and MCT4 in heart and fast-twitch skeletal muscles. Am. J. Physiol. Endocrinol. Metab. 2000;278:E1067–E1077. doi: 10.1152/ajpendo.2000.278.6.E1067. [DOI] [PubMed] [Google Scholar]