

Abstract
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a potent ovarian toxicant. Previously, we demonstrated that in vitro TCDD (1 nM) exposure decreases production/secretion of the sex steroid hormones progesterone (P4), androstenedione (A4), testosterone (T), and 17β-estradiol (E2) in mouse antral follicles. The purpose of this study was to determine the mechanism by which TCDD inhibits steroidogenesis. Specifically, we examined the effects of TCDD on the steroidogenic enzymes, atresia, and the aryl hydrocarbon receptor (AHR) protein. TCDD exposure for 48 h increased levels of A4, without changing HSD3B1 protein, HSD17B1 protein, estrone (E1), T or E2 levels. Further, TCDD did not alter atresia ratings compared to vehicle at 48 h. TCDD, however, did down regulate the AHR protein at 48 h. TCDD exposure for 96 h decreased transcript levels for Cyp11a1, Cyp17a1, Hsd17b1, and Cyp19a1, but increased Hsd3b1 transcript. TCDD exposure particularly lowered both Hsd17b1 transcript and HSD17B1 protein. However, TCDD exposure did not affect levels of E1 in the media nor atresia ratings at 96 h. TCDD, however, decreased levels of the proapoptotic factor Bax. Collectively, these data suggest that TCDD exposure causes a major block in the steroidogenic enzyme conversion of A4 to T and E1 to E2 and that it regulates apoptotic pathways, favoring survival over death in antral follicles. Finally, the down-regulation of the AHR protein in TCDD exposed follicles persisted at 96 h, indicating that the activation and proteasomal degradation of this receptor likely plays a central role in the impaired steroidogenic capacity and altered apoptotic pathway of exposed antral follicles.
Keywords: TCDD, Antral follicle, Atresia, Aryl hydrocarbon receptor, HSD17B, Bax
Introduction
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is the most toxic member of a class of chemicals called dioxins and is a persistent contaminant found in the human food chain, in the waterways, and in the air due to industrial waste (Birnbaum and DeVito, 1995; Frakes et al., 1993; Hites, 2011; Tuppurainen et al., 2003). TCDD has a long half-life in the body and can remain in humans for more than 10 years after a single exposure due to its highly lipophilic nature, slow metabolism, and slow excretion (Miniero et al., 2001; Sorg et al., 2009). Dioxins, like TCDD, have been detected in human fat tissue, serum, breast milk, and ovarian follicular fluid (Birnbaum and DeVito, 1995; Humblet et al., 2011; Schecter et al., 2006; Tsutsumi et al., 2011; Ulaszewska et al., 2011).
TCDD is an endocrine disruptor and potent ovarian toxicant. Numerous studies show that besides its actions on the hypothalamus-pituitary axis to disrupt reproductive processes such as ovulation in rats, TCDD acts directly on the ovary (Cao et al., 2011; Gao et al., 2001; Mizuyachi et al., 2002; Petroff et al., 2000, 2001, 2003).
Studies also indicate that TCDD acts directly on antral follicles and inhibits steroidogenesis (Karman et al., 2012). The antral follicle is the main functional unit of the ovary, and is the primary source of sex steroid hormones in females. The process of sex steroid hormone synthesis in the rodent antral follicle (see Diagram 1) begins in the thecal cells and involves mobilization of cholesterol into the inner mitochondrial membrane via steroidogenic acute regulatory protein (STAR) where it is cleaved by the enzyme CYP11A1 to pregnenolone (P5). The thecal smooth endoplasmic reticulum is the site for hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1 (HSD3B1) isozyme conversion of P5 to progesterone (P4) and dehydroepiandrosterone (DHEA) to androstenedione (A4); and for the conversion of P5 to DHEA under cytochrome P450, family 17, subfamily a, polypeptide 1 (CYP17A1). A4 then diffuses across the basement membrane to the granulosa cells where it is either aromatized by cytochrome P450, family 19, subfamily a, polypeptide 1 (CYP19A1) to estrone (E1) and converted via hydroxysteroid (17-beta) dehydrogenase 1 (HSD17B1) to estradiol-17β (E2), or metabolized to testosterone (T) via HSD17B1. T is then aromatized to E2 by CYP19A1 (for a more detailed review of ovarian steroidogenesis, see Edson et al., 2009).
Previously, we demonstrated that an environmentally relevant exposure to TCDD (1 nM) leads to reduced sex steroid hormone production/secretion by intact mouse antral follicles, without affecting their growth in vitro (Karman et al., 2012). Further, we were able to bypass the reduced hormone production/secretion by co-treatment with pregnenolone, an early substrate in the sex steroid hormone synthesis pathway, suggesting that TCDD may inhibit cholesterol mobilization or its metabolism to P5 by CYP11A1 in the mouse ovary. Thus, one goal of the current studies was to determine which enzymes in the steroidogenic pathway are affected by TCDD exposure in antral follicles. We focused on the effects of TCDD on CYP11A1 and HSD3B1 because they are critical enzymes present in the thecal cells necessary for the reduction of cholesterol to P5 and conversion of P5 to A4. We also focused on HSD17B1 and CYP19A1 because these enzymes are critical for interconverting A4 to T, E1, and E2 in the granulosa cell compartment of the follicle.
Another process in the ovary that can affect steroidogenesis is cell death. During the process of folliculogenesis in the ovary, most of the follicles (>95%) undergo a process of programmed cell death called atresia and are eventually eliminated from the ovary (Kaipia and Hsueh, 1997). Any alteration in the tight balance between follicular atresia and survival can lead to decreased sex steroid hormone levels and/or infertility. Also, E2 itself is an important survival factor that helps to protect follicles from cell death (Kaipia and Hsueh, 1997). Further, it has been demonstrated that atresia caused by other environmental contaminants is primarily through apoptosis and not via other forms of cell death such as necrosis (Matikainen et al., 2001; Pru et al., 2009; Springer et al., 2012; Takai et al., 2003).
Apoptosis is a normal process in eukaryotic cells and is important for eliminating damaged or unwanted cells (Kerr et al., 1972). Disruption of mitochondrial membrane integrity by altering expression of bcl-2 proto-oncogene gene family members such as B cell leukemia/lymphoma 2 (Bcl2) and BCL2-associated X protein (Bax) is one pathway known to initiate apoptosis. Bax is known to promote apoptosis by compromising mitochondrial membrane integrity, causing release of cytochrome c and activation of the caspases, while other factors such as Bcl2 help to maintain mitochondrial membrane integrity (Danial and Korsmeyer, 2004). An imbalance of either Bcl2 or Bax could be an indication of a cell under stress and altered expression of these factors has been identified in some cancers (Christensen et al., 1999). Notably, chronic TCDD exposures cause promotion of ovarian tumors in rats and acute exposure is associated with an increased incidence of ovarian cancer in young women (Davis et al., 2000; Pesatori et al., 1993).
Interestingly, TCDD has been identified as both an inducer and a suppressor of apoptosis depending on the tissue, the experimental model, or dose and length of TCDD exposure. Specifically, TCDD has been shown to induce apoptosis in thymocytes, but to inhibit apoptosis in liver cells (Chopra et al., 2009; Kamath et al., 1997; Worner and Schrenk, 1996; Wörner and Schrenk, 1998). Further, studies investigating the effects of TCDD exposure on human luteinized granulosa cells found that apoptosis was induced in a time and dose dependent manner, while ovaries from rats exposed to TCDD did not contain more atretic follicles compared to vehicle controls (Heimler et al., 1998a,b). Thus, currently it is unclear whether TCDD increases programmed cell death and atresia in the ovary.
TCDD exposure in mice has been shown to act through the AHR to increase levels of the proapoptotic gene Bax in the thymus, contributing to apoptosis (Camacho et al., 2005; Fernandez-Salguero et al., 1996; Zeytun et al., 2002). TCDD is considered a liver tumor promoter and acts primarily through the AHR, as AHR−/− mice are resistant to the effects of TCDD (Fernandez-Salguero et al., 1996; Pitot et al., 1980). In liver cells, TCDD exposure does not alter the expression of Bcl2 or Bax and inhibits apoptosis in liver cells undergoing programmed cell death due to ultraviolet light treatment (Chopra et al., 2009; Worner and Schrenk, 1996). Also, a functional aryl hydrocarbon receptor response element (AHRE) was identified in the Bax promoter (Matikainen et al., 2001). Specifically, it was demonstrated that a metabolite of dimethylbenz[a]anthracene (DMBA), and not TCDD, induced increased transcription of Bax through this AHRE in oocytes transfected with a Bax promoter-GFP reporter construct (Matikainen et al., 2001). Collectively, these experimental results suggest AHR ligand and tissue specific action on Bax promoter activities. Thus, because E2 production/secretion is dramatically reduced in TCDD exposed mouse antral follicles without affecting growth, we hypothesized that the intracellular signaling pathway for apoptosis is altered, resulting in an environment that favors survival over death. To test this hypothesis we examined the effects of TCDD on atresia ratings and on the expression of Bcl2 and Bax in antral follicles.
Finally, TCDD is a known ligand of the protein transcription factor, the AHR. The endogenous functions of the AHR and its role in toxicology are complex. The AHR has been identified as an important player in ovarian function, including steroidogenesis (Hernández-Ochoa et al., 2009). When the AHR is not bound to a ligand such as TCDD, it is primarily located in the cytoplasm of the cell where it associates with various heat shock proteins (Gu et al., 2000; Pocar et al., 2005; Whitlock, 1999). When it is bound to TCDD, it goes through a conformational change which allows it to enter the nucleus. Once in the nucleus, it can bind to proteins such as the aryl hydrocarbon receptor nuclear translocator (ARNT) and act as a transcription factor by binding to various regulatory elements within the promoters of responsive genes such as Bax (Matikainen et al., 2001). Further, it has been demonstrated in numerous cell and tissue types that TCDD can provoke AHR protein degradation via a ubiquitin-proteasome pathway without altering levels of AHR mRNA (Giannone et al., 1998; Pollenz, 2002; Prokipcak and Okey, 1991; Roberts and Whitelaw, 1999). Previously, we did not observe a change in AHR mRNA levels after TCDD exposure in mouse antral follicles (Karman et al., 2012). Thus, the final goal of these studies was to determine the effects of TCDD on AHR protein levels in antral follicles.
Materials and methods
Chemicals
TCDD dissolved in dimethyl sulphoxide (DMSO) at 50 μg/mL (#ED-901-B) was purchased from Cambridge Isotope Laboratories, Inc., Andover, MA. DMSO (D2650), 100× insulin, transferrin, and selenium (ITS), penicillin and streptomycin were purchased from Sigma-Aldrich (St. Louis, MO). Alpha minimal essential media (α-MEM) was purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals, Lawrenceville, GA. Human recombinant follicle-stimulating hormone (hFSH) was obtained from Dr. A. F. Parlow, National Hormone and Peptide Program, Harbor-UCLA Medical Center, Torrance, CA.
Animals
CD-1 mice were purchased from Charles River and maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the University of Illinois Animal Care and Use Committee. Upon arrival at the University of Illinois animal care facility, mice were allowed to acclimate for a minimum of 48 h prior to tissue collection. All mice were housed under strict 12L:12D lighting and temperature was maintained at 22±1 °C. Food and water were provided ad libitum.
In vitro mouse antral follicle culture
Antral follicles from recent cycling mice were cultured as previously described (Karman et al., 2012). Briefly, female mice were euthanized by CO2 asphyxiation followed by cervical dislocation on post-natal day (PND) 33 and their ovaries were removed. Small antral follicles were isolated mechanically from the ovaries of 2–4 mice for each experiment based on relative size (250–350 μm) and cleaned of interstitial tissue in unsupplemented α-MEM using fine #5 watchmaker forceps. Follicles were placed randomly one per individual well in a 96-well culture plate with α-MEM prior to treatment. For each experiment, a minimum of eight follicles were plated per treatment group. TCDD or vehicle (DMSO) alone was added to supplemented α-MEM at an equal volume (0.75 μL/mL media) to maintain the vehicle concentration at a constant of 0.075% for each treatment. Follicles were incubated for either 48 or 96 h at 37 °C in 95% air and 5% CO2. We elected to use 1 nM TCDD because this environmentally relevant dose significantly affected sex steroid hormone production/secretion in antral follicles in a previous study (Karman et al., 2012). Interestingly, lower and higher doses of TCDD do not always significantly affect hormone production/secretion (Karman et al., 2012).
Measurement of sex steroid hormones
Media were collected after 48 or 96 h culture and frozen at −80 °C until they were subjected to enzyme-linked immunosorbent assays (ELISAs) for A4, T, E1, and E2 levels. ELISA kits were purchased from DRG Diagnostics (USA) and procedures were followed using the manufacturer’s protocols. The media were randomly selected from 8 to 23 wells and were assayed individually for each treatment group from 3 to 6 experiments. The intra-assay and inter-assay variabilities were less than 10%.
Analysis of transcript levels
Transcript levels were compared as previously described (Karman et al., 2012). Briefly, at the end of the cultures, 8–16 follicles from each treatment group were pooled and immediately snap frozen in liquid nitrogen and stored at −80 °C until quantitative real-time PCR (qPCR) analysis. Total RNA (250 ng) was reverse transcribed using an iScript cDNA synthesis kit according to manufacturer’s instructions (Bio-Rad, Hercules, CA).
All qPCR reactions were performed in triplicate using the CFX96 Real-time System C1000 Thermal Cycler (Bio-Rad). A BLASTN search was performed in GenBank to ensure that all primers were unique to the gene of interest. To avoid amplification from genomic DNA contamination, all primer sets spanned a large exon–intron–exon junction. Primer sequences are shown in Table 1.
Table 1.
qPCR primer abbreviations, accession numbers, and sequences.
| Transcript | NCBI reference sequence |
Forward primer sequence 5′–3′ |
Reverse primer sequence 5′–3′ |
|---|---|---|---|
| Actb | NM_007393.3 | GGG CAC AGT GTG GGT GAC |
CTG GCA CCA CAC CTT CTA C |
| Star | NM_011485.4 | CAG GGA GAG GTG GCT ATG CA |
CCG TGT CTT TTC CAA TCC TCT G |
| Cyp11a1 | NM_008841.2 | AGA TCC CTT CCC CTG GTG ACA ATG |
CGC ATG AGA AGA GTA TCG ACG CAT C |
| Cyp17a1 | NM_007809.3 | CCA GGA CCC AAG TGT GTT CT |
CCT GAT ACG AAG CAC TTC TCG |
| Hsd3b1 | NM_008293.3 | CAG GAG AAA GAA CTG CAG GAG GTC |
GCA CAC TTG CTT GAA CAC AGG C |
| Hsd17b1 | NM_010475.1 | CTG CCA TTC CAC GAA GTG TAC TGT |
TTG CTC ATA ACC ACG CAG GTA GTG |
| Cyp19a1 | NM_007810.3 | CAT GGT CCC GGA AAC TGT GA |
GTA GTA GTT GCA GGC ACT TC |
| Bcl2 | NM_009741.3 | ATG CCT TTG TGG AAC TAT ATG GC |
GGT ATG CAC CCA GAG TGA TGC |
| Bax | NM_007527.3 | TGA AGA CAG GGG CCT TTT TG |
AAT TCG CCG GAG ACA CTC G |
Relative transcript amount was calculated by a mathematical model developed by Pfaffl (Pfaffl, 2001). Briefly, the method involves calculating the relative expression ratio of the target gene based on the amplification efficiency of each amplicon and the ΔCt of the treated samples versus the vehicle control. These ratios were then compared to the expression of the reference gene Actb. Actb was verified as a good internal control because its levels were unchanged with treatment (data not shown). The data were reported as mean transcript expression ratios relative to Actb from 3 to 4 separate follicle culture experiments.
Analysis of protein levels
Protein levels were compared as previously described (Karman et al., 2012). Briefly, at the end of each of the cultures, 12–16 follicles from each treatment group were pooled and immediately snap frozen in liquid nitrogen and stored at −80 °C until analysis by western blot. The pooled follicles then were lysed and homogenized in 20 μl T-Per (Thermo Fisher Scientific, Rockford, IL) containing protease inhibitors (Roche Diagnostics, Mannheim, Germany). For positive and negative controls, a PND 33 female mouse was euthanized as previously described and the ovaries, heart, lung, and skeletal muscle were removed and immediately snap frozen and stored at −80 °C until analysis by western blot. A bicinchoninic acid (BCA) assay kit was used to determine protein concentration (Thermo Fisher Scientific, Rockford, IL). Electrophoresis and immunoblotting were performed using XCell SureLock Mini-Cell Blot Module Kit and recommended reagents as per manufacturer’s protocol (Invitrogen, Carlsbad, CA). The protein lysate (2 μg) was loaded on precast 4–12% bis-tris sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE; Invitrogen, Carlsbad, CA), followed by transfer of the proteins to 0.45 μm Invitralon™ polyvinylidene difluoride membranes (Invitrogen, Carlsbad, CA) at 4 °C. The blots were blocked with 5% non-fat milk for 1 h at room temperature and then incubated overnight at 4 °C with primary antibodies. The primary antibodies and concentrations used were as follows: CYP11A1 (C-16) sc-18043, 1:100 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), 3β-HSD (P-18) sc-30820, 1:200 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), HSD17B1 NBP1-56295, 0.5 μg/mL (Novus Biologicals, Littleton, CO), CYP19 (C-16) sc-14245, 1:200 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and AHR BML-SA210, 1:5000 (Enzo Life Sciences, Plymouth Meeting, PA). The secondary antibodies and concentrations used were as follows goat anti-rabbit HRP ab6721, 1:5000 (Abcam Inc., Cambridge, MA) or donkey anti-goat HRP sc-2020, 1:5000 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). To ensure uniform loading of the proteins, the blots were then incubated with the anti-beta actin antibody (ACTB) ab8227, 1:5000 (Abcam Inc., Cambridge, MA). The immune complexes on the blots were visualized using an enhanced chemiluminescence detection kit (Cell Signaling Technologies, Danvers, MA) and captured on X-ray film. Densitometric units of the CYP11A1, HSD3B1, HSD17B1, CYP19A1, and AHR protein bands were normalized to the densitometric units of the corresponding ACTB protein bands on each blot for quantification using Image J software (http://rsb.info.nih.gov/ij/).
Analysis of atresia
At the end of each follicle culture experiment, supplemented α-MEM was removed from each well and Dietrich’s solution was immediately added to fix the follicles. The tissues were dehydrated, embedded in cold-curing Technovit 7100 plastic resin (EB Sciences, Germany), serially sectioned (2 μm), mounted on charged glass slides, and stained with Lee’s methylene blue-basic fuchsin. Each follicle section containing the oocyte was examined for the level of atresia as evidenced by the presence of pyknotic bodies. Data were reported as the average percentage of pyknotic bodies to total cell number for each follicle section examined from 8–10 follicles per treatment group from 4 separate follicle culture experiments. Follicles were rated on a scale of 1–4 for the percentage of pyknotic bodies present as previously described (Miller et al., 2006; Paulose et al., 2012): a rating of 1 is a healthy follicle, a rating of 2 is an early atretic follicle with <10% pyknotic bodies, a rating of 3 is a mid atretic follicle with 10–30% pyknotic bodies, and a rating of 4 is a late atretic follicle with >30% pyknotic bodies. All analyses were conducted without knowledge of treatment group. Ratings were averaged and plotted to compare the effect of TCDD on atresia levels.
Statistical analysis
Independent samples t-tests were used to analyze the data. If the data did not pass the assumption of homogeneity of variance, the non-parametric Mann–Whitney U test was employed. Statistical significance was assigned at p≤0.05 for all comparisons. All data were analyzed using SPSS 11.0 statistical software (SPSS Inc., Chicago, IL).
Results
Effect of TCDD exposure on antral follicle steroid secretion
Previously, we determined that in vitro TCDD (1 nM) exposure in antral follicles decreases levels of P4, A4, T and E2 in the media at 96 h (Karman et al., 2012). To determine when TCDD has an effect on antral follicle steroid hormone production/secretion, antral follicles were cultured for 48 h with vehicle or TCDD (1 nM). At the end of the 48 h culture, the media were collected and assayed for levels of A4, T, E1, and E2. TCDD exposure for 48 h increased levels of A4 in the media, but did not change T, E1, and E2 levels compared to vehicle control (Figs. 1A–D).
Diagram 1.
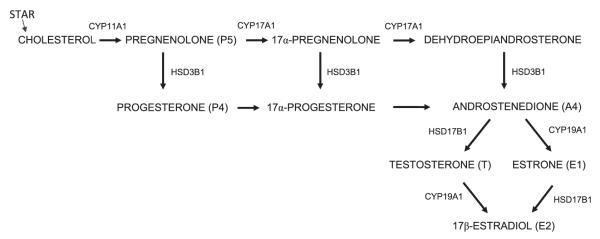
Steroidogenesis pathway in antral follicles of the ovary.
Previously, we measured hormone levels in the media for P4, A4, T, and E2 after 96 h TCDD exposure, but did not measure E1 levels (Karman et al., 2012). Thus, to test the hypothesis that TCDD alters E1 production/secretion after the longer exposure, antral follicles were cultured for 96 h with vehicle or TCDD (1 nM). At the end of the 96 h culture, the media were collected and assayed for levels of E1. TCDD did not significantly affect E1 levels in the media at 96 h (Fig. 2).
Fig. 1.

Effect of 48 h in vitro TCDD exposure on A4, E1, T, and E2 production/secretion by antral follicles. Antral follicles were mechanically isolated from young cycling CD1 mice and exposed for 48 h in culture to TCDD (1 nM) or vehicle alone. After the 48 h culture period, media were collected and subjected to hormone assays for (A) androstenedione, (B) estrone, (C) testosterone, and (D) estradiol levels. The graphs represent the mean hormone levels ±SEM from 8 to 16 culture wells from four separate follicle culture experiments. Column with an asterisk (*) is significantly different from the vehicle control (p≤0.05).
Effect of TCDD exposure on the expression of transcript levels for key steroidogenic enzymes in antral follicles
To determine whether the decrease in sex steroid hormone levels observed in the follicle culture media after 96 h TCDD exposure was due to altered/impaired regulation of genes responsible for cholesterol transport and/or genes for enzymes metabolizing cholesterol to sex steroid hormones, follicles were collected at the end of the culture period and subjected to measurements of transcripts for Star, Cyp11a1, Cyp17a1, Hsd3b1, Hsd17b1, and Cyp19a1. TCDD exposure did not alter the transcript levels for Star (Fig. 3A), but significantly decreased Cyp11a1, Cyp17a1, Hsd17b1, and Cyp19a1 transcript levels compared with controls (Figs. 3B, C, E and F). Additionally, TCDD exposure increased Hsd3b1 transcript levels in antral follicles compared to controls (Fig. 3D).
Fig. 2.
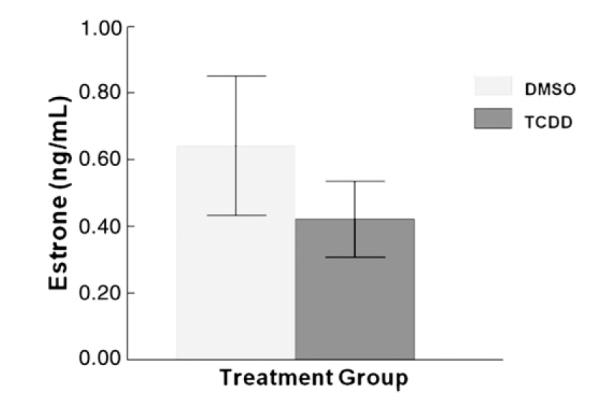
Effect of 96 h in vitro TCDD exposure on E1 production/secretion by antral follicles. Antral follicles were mechanically isolated from young cycling CD1 mice and exposed for 96 h in culture to TCDD (1 nM) or vehicle alone. After the 96 h culture period, media were collected and subjected to hormone assays for estrone. The graph represents the mean hormone levels ±SEM from 12 culture wells from four separate follicle culture experiments.
Effect of TCDD on protein levels for key steroidogenic enzymes in antral follicles
Since proteins are ultimately the enzymes that catalyze metabolism of the sex steroid hormones, we compared protein levels for key steroidogenic enzymes from the vehicle and TCDD exposed antral follicles after 48 h. We focused on CYP11A1 because previously we were able to restore sex steroid hormone production/secretion by antral follicles exposed to TCDD with P5 co-treatment (Karman et al., 2012), suggesting TCDD affects upstream steroidogenic enzymes. Thus, we tested the hypothesis that TCDD exposure alters the protein levels for CYP11A1 because this enzyme is upstream of P5 in the steroidogenic pathway. After 48 h, TCDD exposure did not change protein levels for CYP11A1 compared with vehicle (Figs. 4A and B).
Fig. 3.

Effect of 96 h in vitro TCDD exposure on the expression of steroidogenic enzyme transcripts in antral follicles. At the end of each of the 96 h cultures, 8–16 follicles from each treatment group were pooled and immediately snap frozen in liquid nitrogen and assayed by qPCR for levels of (A) Star (B) Cyp11a1, (C) Cyp17a1, (D) Hsd3b1, (E) Hsd17b1, and (F) Cyp19a1. Levels of these key steroidogeneic enzymes were normalized to β-actin (Actb). Data are expressed as mean relative expression ratios ±SEM calculated from 3–4 separate culture experiments. Columns with asterisks indicate *p≤0.05, **p≤0.01, ***p≤0.001.
Since 48 h TCDD exposure increased accumulation of the sex steroid hormone intermediate A4 in the media, we further hypothesized that TCDD exposure increases conversion of DHEA to A4 via increased expression of the HSD3B1 enzyme in antral follicles. TCDD exposure for 48 h did not change protein levels for HSD3B1 in antral follicles (Figs. 4E and F).
Another possibility for the increased A4 levels in the media could be due to accumulation of this sex steroid hormone intermediate because of inhibition of downstream enzymes such as HSD17B1 and CYP19A1. To test this hypothesis, we determined the effect of 48 h exposure to vehicle or TCDD on HSD17B1 and CYP19A1 protein levels. TCDD did not alter protein levels of HSD17B1 (Figs. 5A and B) or CYP19A1 (Figs. 5E and F) compared with vehicle exposed follicles.
Fig. 4.

Effect of 48 and 96 h in vitro TCDD exposure on CYP11A1 and HSD3B1 protein expression in antral follicles. At the end of each of the cultures, 12–16 follicles from each treatment group were pooled and immediately snap frozen in liquid nitrogen and analyzed by western blot for CYP11A1 (A–D) and HSD3B1 (E–H) protein levels. Antral follicles (250–350 μm) (F), ovary (O), heart (H), lung (L), and a biopsy of skeletal muscle (M) were removed from a PND 33 female mouse and analyzed as positive and negative controls. Densitometric units of the CYP11A1 and HSD3B1 protein bands were normalized to the densitometric units of the corresponding ACTB protein bands on each blot for quantification. Data are expressed as mean relative expression ratios ±SEM calculated from four separate culture experiments (graphs A and E represent the 48 h cultures and graphs C and G represent the 96 h cultures). B, D, F, and H are representative images of the protein bands from western blots for CYP11A1 and HSD3B1 with an image of the corresponding ACTB protein bands that were used as loading controls below. V = vehicle control, T=1 nM TCDD, and X = blank lane.
To determine whether the effects on these steroidogenic enzymes by TCDD were time dependent, we compared their protein levels from vehicle and TCDD exposed antral follicles after the longer 96 h exposure period. TCDD did not significantly change protein levels of CYP11A1 (Figs. 4C and D), HSD3B1 (Figs. 4G and H), and CYP19A1 (Figs. 5C and D) compared to controls. Interestingly, TCDD significantly decreased protein levels for HSD17B1 compared to vehicle controls at 96 h (Figs. 5G and H).
Effect of TCDD exposure on antral follicle atresia
To determine whether the effects of TCDD exposure on antral follicle steroidogenesis were due to an ability of TCDD to kill antral follicles, antral follicles were collected at the end of the 48 and 96 h cultures and evaluated for atresia. TCDD exposure did not alter the level of atresia in antral follicles compared with controls at any time point (Figs. 6A–D). Additionally, pyknotic bodies were only present in the antral space and not in the mural granulosa layer or in the thecal layer in control and TCDD treated follicles. When the oocyte nucleus was present for evaluation, the germinal vesicle was intact, evidence that oocytes remained in meiotic arrest in both control and TCDD exposed antral follicles.
Fig. 5.

Effect of 48 and 96 h in vitro TCDD exposure on HSD17B1 and CYP19A1 protein expression in antral follicles. At the end of each of the cultures, 12–16 follicles from each treatment group were pooled and immediately snap frozen in liquid nitrogen and analyzed by western blot for HSD17B1 (A–D) and CYP19A1 (E–H) protein levels. Antral follicles (250–350 μm) (F), ovary (O), heart (H), lung (L), and a biopsy of skeletal muscle (M) were removed from a PND 33 female mouse and analyzed as positive and negative controls. Densitometric units of the HSD17B1 and CYP19A1 protein bands were normalized to the densitometric units of the corresponding ACTB protein bands on each blot for quantification. Data are expressed as mean relative expression ratios±SEM calculated from four separate culture experiments (graphs A and E represent the 48 h cultures and graphs C and G represent the 96 h cultures). B, D, F, and H are representative images of the protein bands from western blots for HSD17B1 and CYP19A1, with an image of the corresponding ACTB protein bands that were used as loading controls below. V = vehicle control, T=1 nM TCDD, and X = blank lane.
Effect of TCDD exposure on the expression of Bcl2 and Bax transcript levels
To determine whether TCDD exposure alters expression of genes responsible for apoptosis, follicles were collected at the end of the culture period and subjected to measurements of transcripts for the anti-apoptotic factor Bcl2 and the proapoptotic factor Bax. TCDD exposure did not alter the transcript levels for Bcl2 (Fig. 7A), but significantly decreased Bax (Fig. 7B) transcript levels compared with controls.
Fig. 6.

Effect of 48 and 96 h in vitro TCDD exposure on atresia in antral follicles. At the end of each follicle culture experiment, supplemented α-MEM was removed from each well and Dietrich’s solution was immediately added to fix the follicles. The tissues were dehydrated, embedded in cold-curing plastic resin, serially sectioned (2 μm), mounted on charged glass slides, stained, and analyzed for atresia rating. All analyses were conducted without knowledge of treatment group. Ratings were averaged and plotted to compare the effect of TCDD on atresia levels. Graphs represent the mean atresia rating for each follicle section examined from 8–10 follicles per treatment group from 4 separate follicle culture experiments ±SEM. A = graph for atresia ratings and B = representative (10×) microscope images of antral follicle sections from follicles exposed for 48 h to DMSO or TCDD. C = graph for atresia ratings and D = representative (10×) light microscope images of antral follicle sections from follicles exposed for 96 h to DMSO or TCDD.
Effect of TCDD exposure on the expression of the AHR protein in antral follicles
Degradation of the AHR protein is considered a hallmark of its activation by ligands such as TCDD (Davarinos and Pollenz, 1999; Pollenz, 2002). Furthermore, the AHR is at the crossroads of toxicology and physiology because it has been identified as an important regulator of ovarian function and is activated by TCDD (Hernández-Ochoa et al., 2009). Thus, to determine if and when the AHR is activated in antral follicles by TCDD, antral follicles were cultured for either 48 or 96 h with vehicle or TCDD (1 nM). At the end of the culture period, follicles were assayed by western blot for levels of AHR protein. TCDD treatment resulted in down-regulation of the AHR protein in antral follicles after both 48 and 96 h in vitro exposure (Figs. 8A and B).
Fig. 7.

Effect of TCDD exposure on the expression of Bcl2 and Bax transcript levels. To determine whether TCDD exposure impairs expression of genes responsible for apoptosis, follicles were collected at the end of the culture period and subjected to measurements of transcripts for the anti-apoptotic factor Bcl2 (A) and the proapoptotic factor Bax (B). Levels of Bcl2 and Bax were normalized to β-actin (Actb). Data are expressed as mean relative expression ratios ±SEM calculated from 3 to 4 separate culture experiments. Column with asterisk indicates *p≤0.05.
Discussion
The experimental results reported here and by others overwhelmingly support the hypothesis that TCDD exerts much of its toxicity directly on the ovary through inhibiting steroidogenesis, particularly at the level of the steroidogenic enzymes. To date, the majority of studies aimed at unraveling the mechanism of TCDD’s actions on steroidogenesis utilized isolated ovarian cells in culture. Although the investigation of isolated ovarian cells has been a key to better understanding the mechanisms of TCDD toxicity in the ovary, isolated cells lack connections between the thecal and granulosa follicular cell types and are missing the oocyte compartment of the follicle necessary for proper functioning (Matzuk et al., 2002). Previously, we demonstrated that an intact mouse antral follicle culture system is a valuable model for studying TCDD toxicity in the ovary (Karman et al., 2012). The cultured follicles maintain much of their steroidogenic and proliferative capacity throughout the 96 h culture period; maintaining the presence of granulosa cells, thecal cells, and a basement membrane and oocytes in meiotic arrest (Cortvrindt and Smitz, 2002; Cortvrindt et al., 1997). Thus, we used this culture system to examine the mechanism by which TCDD inhibits steroidogenesis.
The first rate limiting step necessary for steroid synthesis in the antral follicle is the mobilization of cholesterol into the inner mitochondrial membrane via STAR. Previously, we were able to bypass the reduced steroidogenesis in TCDD exposed mouse antral follicles by co-culturing with P5, suggesting that TCDD could be inhibiting STAR (Karman et al., 2012). Thus, in this study, we hypothesized that TCDD exposure in vitro decreases levels of Star in antral follicles. We did not find decreased levels of Star mRNA after 96 h TCDD exposure in antral follicles, suggesting that the ability of TCDD to impair steroidogenesis is not through STAR. Our results are similar to studies reporting that TCDD exposure did not alter the levels of Star mRNA in TCDD exposed rat granulosa cells (Hirakawa et al., 2008). Additionally, TCDD did not change STAR protein levels even though it decreased Star mRNA in pubertal rat ovaries following embryonic and lactational TCDD exposures (Myllymäki et al., 2005; Pesonen et al., 2006).
Our results differ from studies investigating the adrenal and testes from rats exposed to TCDD and from studies examining the promoter activities of the human Star gene in adrenal tumor cells. Specifically, it has been demonstrated that reduced steroidogenesis in the rat adrenal and testes from animals exposed to TCDD is primarily due to impairment of cholesterol transport, possibly via STAR (DiBartolomeis et al., 1987; Moore et al., 1991). Further, it was demonstrated in an adrenal tumor cell line transfected with a human Star promoter construct that the Star gene is transcriptionally regulated by the ligand activated AHR (Sugawara et al., 2001). It is possible that while Star may be a target for TCDD toxicity in various steroidogenic organs in the rat and human, it may not be in the mouse ovary. Also, TCDD could affect cholesterol transport and STAR function through the benzodiazepine receptors as was demonstrated in adrenal mitochondria (Yanagibashi et al., 1989). Finally, the systemic effects from in vivo TCDD studies could explain some of the discrepancies between in vivo and in vitro studies.
The next step in the steroidogenic pathway is conversion of cholesterol to P5 via CYP11A1. To test the hypothesis that TCDD acts on the CYP11A1 enzyme in mouse antral follicles, we measured CYP11A1 protein levels after 48 and 96 h and transcript levels after 96 h in vitro TCDD exposure in antral follicles. TCDD significantly down regulated Cyp11a1 mRNA after 96 h, however, TCDD did not affect protein levels for CYP11A1 after both 48 and 96 h exposure. These results indicate that transcriptional AHR activation by TCDD ligand binding may regulate Cyp11a1 gene expression. Usually mRNA and protein levels for the P450 enzymes correlate (Conley et al., 1994; Havelock et al., 2006), but in this case, it could be that the western blot is not a sensitive enough method for detecting small changes in protein levels or the small, but significant changes observed in Cyp11a1 mRNA levels after 96 h TCDD exposure are not enough to significantly lower the protein levels to a measurable amount. It is possible that while TCDD exposure affects Cyp11a1 transcription, it may not affect protein levels. Also, these data do not eliminate the possibility that TCDD may have acute effects on the protein, leading to changes in post-translational modification and/or activity (Kojima et al., 2001; Li et al., 2012; Matocha and Waterman, 1985; Moore et al., 1991).
Similar to our findings, one study conducted using FSH–stimulated isolated rat granulosa cells found that Cyp11a1 mRNA levels were decreased after TCDD exposure (Dasmahapatra et al., 2000). Also, juvenile post-natal day 16 rat ovaries had lower Cyp11a1 mRNA levels after in utero and lactational TCDD exposure (Myllymäki et al., 2005). CYP11A1 is primarily expressed in the thecal cells of early antral follicles, but is also expressed in the granulosa cells of preovulatory follicles and is stimulated by FSH (Goldring et al., 1987). In the current study, it is difficult to distinguish whether the thecal cells or the granulosa cells are a more sensitive target for the actions of TCDD on Cyp11a1 transcription.
To determine whether TCDD exposure impacts the conversion of P5 to DHEA or P4 to A4 under CYP17A1, we measured transcript levels for Cyp17a1 in antral follicles following 96 h exposure. TCDD significantly reduced Cyp17a1 levels in antral follicles. This is consistent with studies conducted using human luteinized granulosa cells exposed to TCDD. In those studies, Morán et al. demonstrated that the reduced steroidogenesis in human luteinized granulosa cells was the result of an impairment in the supply of androgens due to a decrease in the lyase activity of the CYP17A1 enzyme as well as decreased levels of CYP17A1 protein (Morán et al., 2003a,b).
HSD3B1 is responsible for isozyme conversion of P5 to P4, 17α-pregnenolone to 17α-progesterone, and DHEA to A4. Interestingly, we observed an increase in A4 levels in the media after 48 h antral follicle culture with TCDD. This was followed by an increase in Hsd3b1 transcript levels and a major decrease in A4 levels by 96 h (Karman et al., 2012), indicating a possible rescue attempt by the TCDD exposed follicle to compensate for the reduced supply of hormone substrates (i.e. P4 or DHEA) due to decreased expression of the upstream enzymes CYP11A1 and CYP17A1. Supportive of this idea, though it has been suggested that human luteinized granulosa cells have a super active HSD3B1 enzyme, the addition of DHEA to TCDD exposed cultured human luteinized granulosa cells restored E2 levels in the media to control levels (Morán et al., 2003a). Interestingly, mouse antral follicles did not have altered levels of HSD3B1 protein after both 48 and 96 h TCDD exposure, suggesting that this enzyme is not a major target for TCDD induced toxicity.
The increase in A4 levels after 48 h TCDD exposure could also be due to accumulation of this hormone substrate due to inhibited downstream enzymes such as CYP19A1 and HSD17B1. HSD17B1 is an important enzyme because it is considered the major HSD17 enzyme involved in E2 synthesis in the rodent ovary and catalyzes the interconversion between the less active 17-oxosteroid and more active 17β-hydroxysteroid hormone forms. In the mouse follicle, HSD17B1 is expressed in the granulosa cells of growing antral follicles and catalyzes the reduction of A4 to T and E1 to E2 with equal efficiency (Ghersevich et al., 1994a; Nokelainen et al., 1996). Thus, we tested the hypothesis that TCDD exposure affects expression of the HSD17B1 enzyme in antral follicles. We found that TCDD exposure significantly reduced both transcript and protein levels for HSD17B1 in antral follicles after 96 h exposure, while there was no change in the protein levels after only 48 h exposure. These data suggest that actions of TCDD on the HSD17B1 enzyme are time dependent and may follow a reduction in A4, T and E2 as well as a reduction in cAMP. In rats, HSD17B1 is primarily regulated by FSH and modulated by sex steroid hormones and growth factors in the follicle (Ghersevich et al., 1994a,b). Specifically, FSH stimulates HSD17B1 expression through a protein kinase A-dependent pathway in isolated rat granulosa cells (Ghersevich et al., 1994b). Additionally, estrogens and androgens enhance the stimulatory effects of FSH on HSD17B1 expression and activity (Ghersevich et al., 1994a). Though we did not measure cAMP or growth factor levels in TCDD exposed antral follicles, we did observe that E2 and T levels in the media were not altered after 48 h TCDD exposure when HSD17B1 protein levels were unaffected. HSD17B1 transcript and protein levels were only affected after longer TCDD exposure when E2 and T levels were significantly down. Since both transcript and protein levels for HSD17B1 were significantly affected by TCDD exposure, it is likely that this enzyme plays a more important role in mouse follicle steroidogenesis and is a major target for ovarian toxicity when compared to CYP19A1. To our knowledge, this is the first report on the actions of TCDD on the HSD17B1 enzyme.
The effects of TCDD on the CYP19A1 enzyme in the ovary have been more extensively studied than the effects of TCDD on other steroidogenic enzymes. Like HSD17B1, CYP19A1 is located in the granulosa cells of antral follicles and is primarily regulated by FSH, sex steroid hormones, and growth factors (Stocco, 2008). It is the enzyme responsible for aromatizing A4 to E1 and T to E2 in the ovary. TCDD did not significantly alter the levels of E1 after 48 h exposure, but caused a non-significant decrease after 96 h. The slight decrease in E1 levels after 96 h suggests that TCDD could be targeting the CYP19A1 enzyme in the mouse antral follicle. Thus, we tested the hypothesis that TCDD exposure affects CYP19A1 transcript and protein levels in mouse antral follicles. Interestingly, TCDD exposure decreased Cyp19a1 transcript levels after 96 h, but there was no effect on protein levels after both 48 and 96 h exposure. Similarly, studies conducted by Dasmahapatra et al. using FSH-stimulated rat granulosa cells exposed to TCDD found that the addition of A4 partially restored E2 levels, suggesting actions on both CYP19A1 and HSD17B1 enzymes. Although they did not measure HSD17B1, upon further investigation, Dasmahapatra et al. found that TCDD inhibited the FSH-stimulated increase in CYP19A1 mRNA levels and enzyme activity usually exhibited by these cells, indicating possible actions on this enzyme as well as the follicle-stimulating hormone receptor (Dasmahapatra et al., 2000). Gregoraszczuk also found evidence of reduced CYP19A1 activity in porcine granulosa and thecal cell co-cultures after TCDD exposure (Gregoraszczuk, 2002). Further, Myllymäki et al. found decreased Cyp19a1 mRNA levels in pubertal ovaries and decreased CYP19A1 enzyme activity in isolated follicles from rats exposed in utero and lactationally to TCDD (Myllymäki et al., 2005). Although transcript levels were not assayed, similar to our findings for CYP19A1, protein and activity levels were unaffected in human luteinized granulosa cells (Morán et al., 2000). While another environmental toxicant (DMBA) has been shown to enhance the transcription of Cyp19a1 in the mouse ovary through AHR binding to an AHRE in the ovary specific Cyp19a1 gene promoter causing a rise in E2 production, it is possible that TCDD could have the opposite effect and repress the promoter activities of this gene leading to a decrease in E2 synthesis (Baba et al., 2005). Future studies could be directed at comparing the enzyme activity levels between vehicle and TCDD exposed follicles in vitro to ascertain the biological relevance of the decreased Cyp19a1 transcript levels in our culture system.
Atresia is a normal process in the ovary, is hormonally regulated, and is responsible for elimination of >95% of the follicles from the ovary by the time of reproductive senescence (Kaipia and Hsueh, 1997). Atresia is mainly controlled by apoptosis (Kaipia and Hsueh, 1997). Further, it has been demonstrated that atresia caused by other environmental contaminants is primarily through apoptosis and not via other forms of cell death, such as necrosis (Matikainen et al., 2001; Pru et al., 2009; Springer et al., 2012; Takai et al., 2003). TCDD has been identified as both an inducer and a suppressor of apoptosis depending on the tissue or the experimental model (Chopra and Schrenk, 2011; Puga et al., 2009). Thus, we hypothesized that TCDD exposure alters the amount of atresia in the antral follicle in vitro. TCDD exposure did not alter the amount of atresia in antral follicles compared to vehicle treated follicles after both 48 and 96 h exposure. These results suggest that TCDD does not increase apoptosis of antral follicles, which is consistent with the findings from in vivo studies in rats, but is contrary to the findings in human luteinized granulosa cells. Specifically, rats gestationally exposed to TCDD had fewer large preantral and early antral follicles with no difference in the level of apoptotic cell death or atresia ratings when compared with vehicle control treated rats (Heimler et al., 1998b). Another study found that TCDD induced apoptosis in human luteinized granulosa cells in a dose and time dependent manner (Heimler et al., 1998a). The effects seen in human luteinized granulosa cells could be due to the higher doses of TCDD that were employed or could be due to the endocrine status of the donor cells from infertility patients. The highest dose of TCDD (3.1 μM) also resulted in an increase in E2 levels in the media from exposed human luteinized granulosa cells, which was attributed to possible changes in membrane permeability due to the observation that the TCDD exposed human luteinized granulosa cells had extracellular DNA, not membrane enclosed, as examined via transmission electron microscopy techniques.
In other studies, both E2 and AHR have been implicated in modulating apoptosis through the Bcl2 family members in the ovary (Hu et al., 2001; Kaipia and Hsueh, 1997; Kee et al., 2010; Matikainen et al., 2001; Springer et al., 2012; Takai et al., 2003; Thompson et al., 2005). In the granulosa cells of healthy antral follicles, E2 acts with other factors to suppress transcription of proapoptotic factors such as Bax (Matsuda et al., 2012). Bcl2 acts to suppress the actions of Bax. Interestingly, TCDD significantly reduced Bax levels, but did not change Bcl2 levels in antral follicles compared to controls. These data suggest that even though TCDD does not increase atresia, it still may regulate apoptosis pathways in antral follicles. It is likely TCDD does so through the AHR. When the AHR protein is down regulated by TCDD, this could lead to a deregulation of the proapoptotic factor Bax, and an environment that favors survival over death in the follicle. This could explain why, despite the severe reduction in E2 production/secretion from TCDD exposed antral follicles, the follicles still grow and do not have increased atresia ratings when compared with vehicle controls (Karman et al., 2012). This is similar in liver cells, where TCDD inhibits apoptosis, possibly through the AHR (Chopra and Schrenk, 2011; Chopra et al., 2009).
The AHR has been identified as an important factor that mediates steroidogenesis and hormone signaling in the ovary. In pigs, primates and rodents, the AHR is expressed in all follicle cell types, but particularly in the granulosa cells (Baldridge and Hutz, 2007; Robles et al., 2000; Sakurada et al., 2011; Wojtowicz et al., 2005). Levels of the AHR change in the ovary with the reproductive cycle, and FSH and E2 are modulators of AHR expression (Bussmann and Baranao, 2006; Chaffin et al., 1999, 2000). Besides FSH and E2, multiple cellular factors and processes have been identified that alter AHR levels and activity in various tissues (Harper et al., 2006). It has been suggested that LH may be a regulator of AHR transcriptional activity in the ovary by inducing the aryl hydrocarbon receptor repressor (AHRR) (Baba et al., 2005). Furthermore, antral follicles from AHR −/− females produce less E2 and their ovaries express lower levels of Cyp19a1 mRNA without an increase in antral follicle atresia compared to wild type mice (Baba et al., 2005; Barnett et al., 2007; Benedict et al., 2003).
There is evidence supporting the hypothesis that the proteolytic degradation of the AHR protein could be one mechanism for attenuating AHR-mediated gene regulation (Pollenz et al., 2005). Thus, because we observed a rise in Cyp1b1 mRNA levels (Karman et al., 2012) and a decrease in Bax mRNA levels after 96 h TCDD exposure, it is possible that the AHR is recruited to the Cyp1b1 and Bax promoters in antral follicles. Concurrently, the down-regulation of the AHR protein after both 48 and 96 h TCDD exposure in antral follicles indicates that a ubiquitin-proteasome pathway is active in the ovary. Similar to our findings, Bussmann et al. demonstrated the rapid proteasomal degradation of the AHR after ligand exposure in rat granulosa cells (Bussmann and Baranao, 2006). Specifically, they found that the degradation occurred as early as 4 h after β-naphthoflavone exposure and that inhibition of proteasomal degradation with a chemical inhibitor caused a super-induction of the Cyp1a1 gene. They also observed a rise in AHR mRNA levels after 48 h of β-naphthoflavone exposure. TCDD exposure did not result in significant changes in AHR mRNA levels after 96 h in mouse antral follicles (Karman et al., 2012). The differences between the two studies could be due to the higher stability of TCDD when compared to other AHR ligands such as β-naphthoflavone, repressing the AHR protein and maintaining constant mRNA levels for longer periods (Pollenz, 2002).
In conclusion, the direct actions of TCDD on the ovary appear to be primarily on the steroidogenic enzymes, leading to reduced steroidogenic capacity of antral follicles without affecting atresia (please see Table 2 for a summary of findings). Evidence reported here and by others indicates that TCDD acts mainly on the granulosa cells, evidenced by its major effects on HSD17B1 and CYP19A1. This is the first report (to our knowledge) of the actions of TCDD on the HSD17B1 enzyme in the ovary. Transcriptional AHR activation by TCDD and other ligands may lead to proteasomal degradation and chronic down-regulation of the protein in the antral follicle. This chronic down-regulation may dampen the toxic response pathway, rescuing the follicle from death, but also could cause deregulation of the endogenous actions of the AHR protein in the ovary. These actions of TCDD on the ovary could collectively contribute to ovotoxicity, endocrine disruption, and cancer promotion. Future studies should be targeted at understanding the mechanisms of endogenous AHR function in the ovary and the consequences of exogenous ligand binding to this receptor.
Table 2.
TCDD-induced transcript and protien level changes in antral at 48 and 96 h.
| STAR | CYP11A1 | Cyp17A1 | HSD3B1 | Cyp19A1 | HSD17B1 | BCL2 | BAX | AHR | |
|---|---|---|---|---|---|---|---|---|---|
| 48 h transcript | ? | ? | ? | ? | ? | ? | ? | ? | ? |
| 48 h protein | ? |

|
? |

|

|

|
? | ? |

|
| 96 h transcript |

|

|

|

|

|

|

|

|

|
| 96 h protein | ? |

|

|

|

|

|
? | ? |

|
Table 2 key: ? = not measured in this study
 = no difference in levels between vehicle and TCDD exposed follicles
= no difference in levels between vehicle and TCDD exposed follicles
 = levels increased in TCDD exposed follicles compared with vehicle exposed follicles
= levels increased in TCDD exposed follicles compared with vehicle exposed follicles
 = levels decreased in TCDD exposed follicles compared with vehicle exposed follicles.
= levels decreased in TCDD exposed follicles compared with vehicle exposed follicles.
Note: the transcript levels for the AHR were measured in Karman et al., 2012.
Fig. 8.

Effect of 48 and 96 h in vitro TCDD exposure on AHR protein expression in antral follicles. At the end of each of the cultures, 12–16 follicles from each treatment group were pooled and immediately snap frozen in liquid nitrogen and analyzed by western blot for AHR protein levels. Antral follicles (250–350 μm) (F), ovary (O), heart (H), lung (L), and a biopsy of skeletal muscle (M) were removed from a PND 33 female mouse and analyzed as positive controls. Densitometric units of the AHR protein bands were normalized to the densitometric units of the corresponding ACTB protein bands on each blot for quantification. Data are expressed as mean relative expression ratios ±SEM calculated from four separate culture experiments. Graph A represents the 48 h cultures and graph C represents the 96 h cultures. B and D are representative images of the protein bands from western blots for AHR with an image of the corresponding ACTB protein bands that were used as loading controls below. V = vehicle control, T = 1 nM TCDD, and X = blank lane. Columns with asterisks indicate *p≤0.05 and ***p≤0.001.
Acknowledgments
This work was supported by NIH R01 HD 047275, NIH R01 ES 019178 (JAF), NIEHS ES07326 Research Training Program in Endocrine, Developmental and Reproductive Toxicology (BNK), and an Environmental Toxicology Predoctoral Fellowship (MSB). The authors would also like to thank Liying Gao, Dr. Zelieann R. Craig, Dr. Wei Wang, Dr. Tessie Palouse, and Ayelet Ziv Gal for their technical assistance in isolating antral follicles.
Footnotes
Conflict of interest statement The authors have no conflicts of interest.
References
- Baba T, Mimura J, Nakamura N, Harada N, Yamamoto M, Morohashi K, Fujii-Kuriyama Y. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol. Cell. Biol. 2005;25:10040–10051. doi: 10.1128/MCB.25.22.10040-10051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MG, Hutz RJ. Autoradiographic localization of aromatic hydrocarbon receptor (AHR) in rhesus monkey ovary. Am. J. Primatol. 2007;69:681–691. doi: 10.1002/ajp.20381. [DOI] [PubMed] [Google Scholar]
- Barnett KR, Tomic D, Gupta RK, Miller KP, Meachum S, Paulose T, Flaws JA. The aryl hydrocarbon receptor affects mouse ovarian follicle growth via mechanisms involving estradiol regulation and responsiveness. Biol. Reprod. 2007;76:1062–1070. doi: 10.1095/biolreprod.106.057687. [DOI] [PubMed] [Google Scholar]
- Benedict JC, Miller KP, Lin TM, Greenfeld C, Babus JK, Peterson RE, Flaws JA. Aryl hydrocarbon receptor regulates growth, but not atresia, of mouse preantral and antral follicles. Biol. Reprod. 2003;68:1511–1517. doi: 10.1095/biolreprod.102.007492. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS, DeVito MJ. Use of toxic equivalency factors for risk assessment for dioxins and related compounds. Toxicology. 1995;105:391–401. doi: 10.1016/0300-483x(95)03237-a. [DOI] [PubMed] [Google Scholar]
- Bussmann UA, Baranao JL. Regulation of aryl hydrocarbon receptor expression in rat granulosa cells. Biol. Reprod. 2006;75:360–369. doi: 10.1095/biolreprod.106.053017. [DOI] [PubMed] [Google Scholar]
- Camacho IA, Singh N, Hegde VL, Nagarkatti M, Nagarkatti PS. Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. J. Immunol. 2005;175:90–103. doi: 10.4049/jimmunol.175.1.90. [DOI] [PubMed] [Google Scholar]
- Cao J, Patisaul HB, Petersen SL. Aryl hydrocarbon receptor activation in lactotropes and gonadotropes interferes with estradiol-dependent and -independent preprolactin, glycoprotein alpha and luteinizing hormone beta gene expression. Mol. Cell. Endocrinol. 2011;333:151–159. doi: 10.1016/j.mce.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffin CL, Stouffer RL, Duffy DM. Gonadotropin and steroid regulation of steroid receptor and aryl hydrocarbon receptor messenger ribonucleic acid in macaque granulosa cells during the periovulatory interval. Endocrinology. 1999;140:4753–4760. doi: 10.1210/endo.140.10.7056. [DOI] [PubMed] [Google Scholar]
- Chaffin CL, Trewin AL, Hutz RJ. Estrous cycle-dependent changes in the expression of aromatic hydrocarbon receptor (AHR) and AHR-nuclear translocator (ARNT) mRNAs in the rat ovary and liver. Chem. Biol. Interact. 2000;124:205–216. doi: 10.1016/s0009-2797(99)00157-x. [DOI] [PubMed] [Google Scholar]
- Chopra M, Schrenk D. Dioxin toxicity, aryl hydrocarbon receptor signaling, and apoptosis-persistent pollutants affect programmed cell death. Crit. Rev. Toxicol. 2011;41:292–320. doi: 10.3109/10408444.2010.524635. [DOI] [PubMed] [Google Scholar]
- Chopra M, Dharmarajan AM, Meiss G, Schrenk D. Inhibition of UV-C light-induced apoptosis in liver cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2009;111:49–63. doi: 10.1093/toxsci/kfp128. [DOI] [PubMed] [Google Scholar]
- Christensen JG, Romach EH, Healy LN, Gonzales AJ, Anderson SP, Malarkey DE, Corton JC, Fox TR, Cattley RC, Goldsworthy TL. Altered bcl-2 family expression during non-genotoxic hepatocarcinogenesis in mice. Carcinogenesis. 1999;20:1583–1590. doi: 10.1093/carcin/20.8.1583. [DOI] [PubMed] [Google Scholar]
- Conley AJ, Howard HJ, Slanger WD, Ford JJ. Steroidogenesis in the preovulatory porcine follicle. Biol. Reprod. 1994;51:655–661. doi: 10.1095/biolreprod51.4.655. [DOI] [PubMed] [Google Scholar]
- Cortvrindt RG, Smitz JE. Follicle culture in reproductive toxicology: a tool for in-vitro testing of ovarian function? Hum. Reprod. Update. 2002;8:243–254. doi: 10.1093/humupd/8.3.243. [DOI] [PubMed] [Google Scholar]
- Cortvrindt R, Smitz J, Van Steirteghem AC. Assessment of the need for follicle stimulating hormone in early preantral mouse follicle culture in vitro. Hum. Reprod. 1997;12:759–768. doi: 10.1093/humrep/12.4.759. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Dasmahapatra AK, Wimpee BA, Trewin AL, Wimpee CF, Ghorai JK, Hutz RJ. Demonstration of 2,3,7,8-tetrachlorodibenzo-p-dioxin attenuation of P450 steroidogenic enzyme mRNAs in rat granulosa cell in vitro by competitive reverse transcriptase-polymerase chain reaction assay. Mol. Cell. Endocrinol. 2000;164:5–18. doi: 10.1016/s0303-7207(00)00245-8. [DOI] [PubMed] [Google Scholar]
- Davarinos NA, Pollenz RS. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytoplasmic proteasome following nuclear export. J. Biol. Chem. 1999;274:28708–28715. doi: 10.1074/jbc.274.40.28708. [DOI] [PubMed] [Google Scholar]
- Davis BJ, McCurdy EA, Miller BD, Lucier GW, Tritscher AM. Ovarian tumors in rats induced by chronic 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Cancer Res. 2000;60:5414–5419. [PubMed] [Google Scholar]
- DiBartolomeis MJ, Moore RW, Peterson RE, Christian BJ, Jefcoate CR. Altered regulation of adrenal steroidogenesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Biochem. Pharmacol. 1987;36:59–67. doi: 10.1016/0006-2952(87)90382-0. [DOI] [PubMed] [Google Scholar]
- Edson MA, Nagaraja AK, Matzuk MM. The Mammalian ovary from genesis to revelation. Endocr. Rev. 2009;30:624–712. doi: 10.1210/er.2009-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- Frakes RA, Zeeman CQ, Mower B. Bioaccumulation of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) by fish downstream of pulp and paper mills in Maine. Ecotoxicol. Environ. Saf. 1993;25:244–252. doi: 10.1006/eesa.1993.1023. [DOI] [PubMed] [Google Scholar]
- Gao X, Mizuyachi K, Terranova PF, Rozman KK. 2,3,7,8-Tetrachlorodibenzo-p-dioxin decreases responsiveness of the hypothalamus to estradiol as a feedback inducer of preovulatory gonadotropin secretion in the immature gonadotropin-primed rat. Toxicol. Appl. Pharmacol. 2001;170:181–190. doi: 10.1006/taap.2000.9099. [DOI] [PubMed] [Google Scholar]
- Ghersevich S, Nokelainen P, Poutanen M, Autio-Harmainen H, Rajaniemi H, Vihko R. Rat 17 beta-hydroxysteroid dehydrogenase type 1: primary structure and regulation of enzyme expression in rat ovary by diethylstilbestrol and gonadotropins in vivo. Endocrinology. 1994a;135:1477–1487. doi: 10.1210/endo.135.4.7925110. [DOI] [PubMed] [Google Scholar]
- Ghersevich S, Poutanen M, Tapanainen J, Vihko R. Hormonal regulation of rat 17 beta-hydroxysteroid dehydrogenase type 1 in cultured rat granulosa cells: effects of recombinant follicle-stimulating hormone, estrogens, androgens, and epidermal growth factor. Endocrinology. 1994b;135:1963–1971. doi: 10.1210/endo.135.5.7956918. [DOI] [PubMed] [Google Scholar]
- Giannone JV, Li W, Probst M, Okey AB. Prolonged depletion of AH receptor without alteration of receptor mRNA levels after treatment of cells in culture with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol. 1998;55:489–497. doi: 10.1016/s0006-2952(97)00493-0. [DOI] [PubMed] [Google Scholar]
- Goldring NB, Durica JM, Lifka J, Hedin L, Ratoosh SL, Miller WL, Orly J, Richards JS. Cholesterol side-chain cleavage P450 messenger ribonucleic acid: evidence for hormonal regulation in rat ovarian follicles and constitutive expression in corpora lutea. Endocrinology. 1987;120:1942–1950. doi: 10.1210/endo-120-5-1942. [DOI] [PubMed] [Google Scholar]
- Gregoraszczuk EL. Dioxin exposure and porcine reproductive hormonal activity. Cad. Saude Publica. 2002;18:453–462. doi: 10.1590/s0102-311x2002000200010. [DOI] [PubMed] [Google Scholar]
- Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- Harper PA, Riddick DS, Okey AB. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem. Pharmacol. 2006;72:267–279. doi: 10.1016/j.bcp.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Havelock JC, Rainey WE, Bradshaw KD, Carr BR. The post-menopausal ovary displays a unique pattern of steroidogenic enzyme expression. Hum. Reprod. 2006;21:309–317. doi: 10.1093/humrep/dei373. [DOI] [PubMed] [Google Scholar]
- Heimler I, Rawlins RG, Owen H, Hutz RJ. Dioxin perturbs, in a dose- and time-dependent fashion, steroid secretion, and induces apoptosis of human luteinized granulosa cells. Endocrinology. 1998a;139:4373–4379. doi: 10.1210/endo.139.10.6264. [DOI] [PubMed] [Google Scholar]
- Heimler I, Trewin AL, Chaffin CL, Rawlins RG, Hutz RJ. Modulation of ovarian follicle maturation and effects on apoptotic cell death in Holtzman rats exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin(TCDD) in utero and lactationally. Reprod. Toxicol. 1998b;12:69–73. doi: 10.1016/s0890-6238(97)00101-9. [DOI] [PubMed] [Google Scholar]
- Hernández-Ochoa I, Karman BN, Flaws JA. The role of the aryl hydrocarbon receptor in the female reproductive system. Biochem. Pharmacol. 2009;77:547–559. doi: 10.1016/j.bcp.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa T, Minegishi T, Abe K, Kishi H, Inoue K, Ibuki Y, Miyamoto K. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the expression of follicle-stimulating hormone receptors during cell differentiation in cultured granulosa cells. Endocrinology. 2008;141:1470–1476. doi: 10.1210/endo.141.4.7424. [DOI] [PubMed] [Google Scholar]
- Hites RA. Dioxins: an overview and history. Environ. Sci. Technol. 2011;45:16–20. doi: 10.1021/es1013664. [DOI] [PubMed] [Google Scholar]
- Hu X, Christian P, Sipes IG, Hoyer PB. Expression and redistribution of cellular Bad, Bax, and Bcl-X(L) protein is associated with VCD-induced ovotoxicity in rats. Biol. Reprod. 2001;65:1489–1495. doi: 10.1095/biolreprod65.5.1489. [DOI] [PubMed] [Google Scholar]
- Humblet O, Williams PL, Korrick SA, Sergeyev O, Emond C, Birnbaum LS, Burns JS, Altshul L, Patterson DG, Jr., Turner WE, Lee MM, Revich B, Hauser R. Dioxin and polychlorinated biphenyl concentrations in mother’s serum and the timing of pubertal onset in sons. Epidemiology. 2011;22:827–835. doi: 10.1097/EDE.0b013e318230b0d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaipia A, Hsueh AJ. Regulation of ovarian follicle atresia. Annu. Rev. Physiol. 1997;59:349–363. doi: 10.1146/annurev.physiol.59.1.349. [DOI] [PubMed] [Google Scholar]
- Kamath AB, Xu H, Nagarkatti PS, Nagarkatti M. Evidence for the induction of apoptosis in thymocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in vivo. Toxicol. Appl. Pharmacol. 1997;142:367–377. doi: 10.1006/taap.1996.8049. [DOI] [PubMed] [Google Scholar]
- Karman BN, Basavarajappa MS, Craig ZR, Flaws JA. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activates the aryl hydrocarbon receptor and alters sex steroid hormone secretion without affecting growth of mouse antral follicles in vitro. Toxicol. Appl. Pharmacol. 2012;261:88–96. doi: 10.1016/j.taap.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee K, Flores M, Cedars MI, Reijo Pera RA. Human primordial germ cell formation is diminished by exposure to environmental toxicants acting through the AHR signaling pathway. Toxicol. Sci. 2010;117:218–224. doi: 10.1093/toxsci/kfq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K, Yamasaki E, Kitada S, Ogishima T, Ito A. Recognition of mitochondrial protein precursor lacking arginine at position −2 by mitochondrial processing peptidase: processing of bovine cytochrome P450(SCC) precursor. J. Biochem. 2001;130:497–502. doi: 10.1093/oxfordjournals.jbchem.a003012. [DOI] [PubMed] [Google Scholar]
- Li D, Dammer EB, Sewer MB. Resveratrol stimulates cortisol biosynthesis by activating SIRT-dependent deacetylation of P450scc. Endocrinology. 2012;153:3258–3268. doi: 10.1210/en.2011-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, Sherr DH, Tilly JL. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat. Genet. 2001;28:355–360. doi: 10.1038/ng575. [DOI] [PubMed] [Google Scholar]
- Matocha MF, Waterman MR. Synthesis and processing of mitochondrial steroid hydroxylases. In vivo maturation of the precursor forms of cytochrome P-450scc, cytochrome P-450(11)beta, and adrenodoxin. J. Biol. Chem. 1985;260:12259–12265. [PubMed] [Google Scholar]
- Matsuda F, Inoue N, Manabe N, Ohkura S. Follicular growth and atresia in mammalian ovaries: regulation by survival and death of granulosa cells. J. Reprod. Dev. 2012;58:44–50. doi: 10.1262/jrd.2011-012. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Burns KH, Viveiros MM, Eppig JJ. Intercellular communication in the mammalian ovary: oocytes carry the conversation. Science. 2002;296:2178–2180. doi: 10.1126/science.1071965. [DOI] [PubMed] [Google Scholar]
- Miller KP, Gupta RK, Flaws JA. Methoxychlor metabolites may cause ovarian toxicity through estrogen-regulated pathways. Toxicol. Sci. 2006;93:180–188. doi: 10.1093/toxsci/kfl034. [DOI] [PubMed] [Google Scholar]
- Miniero R, De Felip E, Ferri F, di Domenico A. An overview of TCDD half-life in mammals and its correlation to body weight. Chemosphere. 2001;43:839–844. doi: 10.1016/s0045-6535(00)00442-2. [DOI] [PubMed] [Google Scholar]
- Mizuyachi K, Son DS, Rozman KK, Terranova PF. Alteration in ovarian gene expression in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin: reduction of cyclooxygenase-2 in the blockage of ovulation. Reprod. Toxicol. 2002;16:299–307. doi: 10.1016/s0890-6238(02)00024-2. [DOI] [PubMed] [Google Scholar]
- Moore RW, Jefcoate CR, Peterson RE. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits steroidogenesis in the rat testis by inhibiting the mobilization of cholesterol to cytochrome P450scc. Toxicol. Appl. Pharmacol. 1991;109:85–97. doi: 10.1016/0041-008x(91)90193-i. [DOI] [PubMed] [Google Scholar]
- Morán FM, Conley AJ, Corbin CJ, Enan E, VandeVoort C, Overstreet JW, Lasley BL. 2,3,7,8-tetrachlorodibenzo-p-dioxin decreases estradiol production without altering the enzyme activity of cytochrome P450 aromatase of human luteinized granulosa cells in vitro. Biol. Reprod. 2000;62:1102–1108. doi: 10.1095/biolreprod62.4.1102. [DOI] [PubMed] [Google Scholar]
- Morán FM, Lohstroh P, Vandevoort CA, Chen J, Overstreet JW, Conley AJ, Lasley BL. Exogenous steroid substrate modifies the effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on estradiol production of human luteinized granulosa cells in vitro. Biol. Reprod. 2003a;68:244–251. doi: 10.1095/biolreprod.102.007161. [DOI] [PubMed] [Google Scholar]
- Morán FM, Vandevoort CA, Overstreet JW, Lasley BL, Conley AJ. Molecular target of endocrine disruption in human luteinizing granulosa cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin: inhibition of estradiol secretion due to decreased 17 alpha-hydroxylase/17,20-lyase cytochrome P450 expression. Endocrinology. 2003b;144:467–473. doi: 10.1210/en.2002-220813. [DOI] [PubMed] [Google Scholar]
- Myllymäki SA, Haavisto TE, Brokken LJ, Viluksela M, Toppari J, Paranko J. In utero and lactational exposure to TCDD; steroidogenic outcomes differ in male and female rat pups. Toxicol. Sci. 2005;88:534–544. doi: 10.1093/toxsci/kfi308. [DOI] [PubMed] [Google Scholar]
- Nokelainen P, Puranen T, Peltoketo H, Orava M, Vihko P, Vihko R. Molecular cloning of mouse 17 beta-hydroxysteroid dehydrogenase type 1 and characterization of enzyme activity. Eur. J. Biochem. 1996;236:482–490. doi: 10.1111/j.1432-1033.1996.00482.x. [DOI] [PubMed] [Google Scholar]
- Paulose T, Hannon PR, Peretz J, Craig ZR, Flaws JA. Estrogen receptor alpha overexpressing mouse antral follicles are sensitive to atresia induced by methoxychlor and its metabolites. Reprod. Toxicol. 2012;33:353–360. doi: 10.1016/j.reprotox.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesatori AC, Consonni D, Tironi A, Zocchetti C, Fini A, Bertazzi PA. Cancer in a young population in a dioxin-contaminated area. Int. J. Epidemiol. 1993;22:1010–1013. doi: 10.1093/ije/22.6.1010. [DOI] [PubMed] [Google Scholar]
- Pesonen SA, Haavisto TE, Viluksela M, Toppari J, Paranko J. Effects of in utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on rat follicular steroidogenesis. Reprod. Toxicol. 2006;22:521–528. doi: 10.1016/j.reprotox.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Petroff BK, Gao X, Rozman KK, Terranova PF. Interaction of estradiol and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in an ovulation model: evidence for systemic potentiation and local ovarian effects. Reprod. Toxicol. 2000;14:247–255. doi: 10.1016/s0890-6238(00)00075-7. [DOI] [PubMed] [Google Scholar]
- Petroff BK, Roby KF, Gao X, Son DS, Williamsn S, Johnson D, Rozman KK, Terranova PF. A review of mechanisms controlling ovulation with implications for the anovulatory effects of polychlorinated dibenzo-p-dioxins in rodents. Toxicology. 2001;158:91–107. doi: 10.1016/s0300-483x(00)00367-x. [DOI] [PubMed] [Google Scholar]
- Petroff BK, Croutch CR, Hunter DM, Wierman ME, Gao X. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) stimulates gonadotropin secretion in the immature female Sprague–Dawley rat through a pentobarbital- and estradiol-sensitive mechanism but does not alter gonadotropin-releasing hormone (GnRH) secretion by immortalized GnRH neurons in vitro. Biol. Reprod. 2003;68:2100–2106. doi: 10.1095/biolreprod.102.010439. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitot HC, Goldsworthy T, Campbell HA, Poland A. Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethylnitrosamine. Cancer Res. 1980;40:3616–3620. [PubMed] [Google Scholar]
- Pocar P, Fischer B, Klonisch T, Hombach-Klonisch S. Molecular interactions of the aryl hydrocarbon receptor and its biological and toxicological relevance for reproduction. Reproduction. 2005;129:379–389. doi: 10.1530/rep.1.00294. [DOI] [PubMed] [Google Scholar]
- Pollenz RS. The mechanism of AH receptor protein down-regulation (degradation) and its impact on AH receptor-mediated gene regulation. Chem. Biol. Interact. 2002;141:41–61. doi: 10.1016/s0009-2797(02)00065-0. [DOI] [PubMed] [Google Scholar]
- Pollenz RS, Popat J, Dougherty EJ. Role of the carboxy-terminal transactivation domain and active transcription in the ligand-induced and ligand-independent degradation of the mouse Ahb-1 receptor. Biochem. Pharmacol. 2005;70:1623–1633. doi: 10.1016/j.bcp.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Prokipcak RD, Okey AB. Downregulation of the Ah receptor in mouse hepatoma cells treated in culture with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Can. J. Physiol. Pharmacol. 1991;69:1204–1210. doi: 10.1139/y91-176. [DOI] [PubMed] [Google Scholar]
- Pru JK, Kaneko-Tarui T, Jurisicova A, Kashiwagi A, Selesniemi K, Tilly JL. Induction of proapoptotic gene expression and recruitment of p53 herald ovarian follicle loss caused by polycyclic aromatic hydrocarbons. Reprod. Sci. 2009;16:347–356. doi: 10.1177/1933719108327596. [DOI] [PubMed] [Google Scholar]
- Puga A, Ma C, Marlowe JL. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BJ, Whitelaw ML. Degradation of the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway. J. Biol. Chem. 1999;274:36351–36356. doi: 10.1074/jbc.274.51.36351. [DOI] [PubMed] [Google Scholar]
- Robles R, Morita Y, Mann KK, Perez GI, Yang S, Matikainen T, Sherr DH, Tilly JL. The aryl hydrocarbon receptor, a basic helix-loop-helix transcription factor of the PAS gene family, is required for normal ovarian germ cell dynamics in the mouse. Endocrinology. 2000;141:450–453. doi: 10.1210/endo.141.1.7374. [DOI] [PubMed] [Google Scholar]
- Sakurada Y, Sawai M, Inoue K, Shirota M, Shirota K. Comparison of aryl hydrocarbon receptor gene expression in laser dissected granulosa cell layers of immature rat ovaries. J. Vet. Med. Sci. 2011;73:923–926. doi: 10.1292/jvms.10-0558. [DOI] [PubMed] [Google Scholar]
- Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: an overview. Environ. Res. 2006;101:419–428. doi: 10.1016/j.envres.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sorg O, Zennegg M, Schmid P, Fedosyuk R, Valikhnovskyi R, Gaide O, Kniazevych V, Saurat JH. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: identification and measurement of TCDD metabolites. Lancet. 2009;374:1179–1185. doi: 10.1016/S0140-6736(09)60912-0. [DOI] [PubMed] [Google Scholar]
- Springer LN, Tilly JL, Sipes IG, Hoyer PB. Enhanced expression of bax in small preantral follicles during 4-vinylcyclohexene diepoxide-induced ovotoxicity in the rat. Toxicol. Appl. Pharmacol. 2012;139:402–410. doi: 10.1006/taap.1996.0181. [DOI] [PubMed] [Google Scholar]
- Stocco C. Aromatase expression in the ovary: hormonal and molecular regulation. Steroids. 2008;73:473–487. doi: 10.1016/j.steroids.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Nomura E, Sakuragi N, Fujimoto S. The effect of the arylhydrocarbon receptor on the human steroidogenic acute regulatory gene promoter activity. J. Steroid Biochem. Mol. Biol. 2001;78:253–260. doi: 10.1016/s0960-0760(01)00100-5. [DOI] [PubMed] [Google Scholar]
- Takai Y, Canning J, Perez GI, Pru JK, Schlezinger JJ, Sherr DH, Kolesnick RN, Yuan J, Flavell RA, Korsmeyer SJ, Tilly JL. Bax, caspase-2, and caspase-3 are required for ovarian follicle loss caused by 4-vinylcyclohexene diepoxide exposure of female mice in vivo. Endocrinology. 2003;144:69–74. doi: 10.1210/en.2002-220814. [DOI] [PubMed] [Google Scholar]
- Thompson KE, Bourguet SM, Christian PJ, Benedict JC, Sipes IG, Flaws JA, Hoyer PB. Differences between rats and mice in the involvement of the aryl hydrocarbon receptor in 4-vinylcyclohexene diepoxide-induced ovarian follicle loss. Toxicol. Appl. Pharmacol. 2005;203:114–123. doi: 10.1016/j.taap.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Tsutsumi O, Uechi H, Sone H, Yonemoto J, Takai Y, Momoeda M, Tohyama C, Hashimoto S, Morita M, Taketani Y. Presence of dioxins in human follicular fluid: their possible stage-specific action on the development of preimplantation mouse embryos. Biochem. Biophys. Res. Commun. 2011;250:498–501. doi: 10.1006/bbrc.1998.9340. [DOI] [PubMed] [Google Scholar]
- Tuppurainen K, Asikainen A, Ruokojärvi P, Ruuskanen J. Perspectives on the formation of polychlorinated dibenzo-p-dioxins and dibenzofurans during municipal solid waste (MSW) incineration and other combustion processes. Acc. Chem. Res. 2003;36:652–658. doi: 10.1021/ar020104+. [DOI] [PubMed] [Google Scholar]
- Ulaszewska MM, Zuccato E, Davoli E. PCDD/Fs and dioxin-like PCBs in human milk and estimation of infants’ daily intake: a review. Chemosphere. 2011;83:774–782. doi: 10.1016/j.chemosphere.2011.02.066. [DOI] [PubMed] [Google Scholar]
- Whitlock JP. Induction of cytochrome P4501A1. Annu. Rev. Pharmacol. Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- Wojtowicz A, Tomanek M, Augustowska K, Gregoraszczuk EL. Aromatic hydrocarbon receptor (AhR) in the porcine theca and granulosa cells: effect of TCDD, PCB 126 and PCB 153 on the expression of AhR. Endocr. Regul. 2005;39:109–118. [PubMed] [Google Scholar]
- Worner W, Schrenk D. Influence of liver tumor promoters on apoptosis in rat hepatocytes induced by 2-acetylaminofluorene, ultraviolet light, or transforming growth factor beta 1. Cancer Res. 1996;56:1272–1278. [PubMed] [Google Scholar]
- Wörner W, Schrenk D. 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses apoptosis and leads to hyperphosphorylation of p53 in rat hepatocytes. Environ. Toxicol. Pharmacol. 1998;6:239–247. doi: 10.1016/s1382-6689(98)00040-4. [DOI] [PubMed] [Google Scholar]
- Yanagibashi K, Ohno Y, Nakamichi N, Matsui T, Hayashida K, Takamura M, Yamada K, Tou S, Kawamura M. Peripheral-type benzodiazepine receptors are involved in the regulation of cholesterol side chain cleavage in adrenocortical mitochondria. J. Biochem. 1989;106:1026–1029. doi: 10.1093/oxfordjournals.jbchem.a122958. [DOI] [PubMed] [Google Scholar]
- Zeytun A, McKallip RJ, Fisher M, Camacho I, Nagarkatti M, Nagarkatti PS. Analysis of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced gene expression profile in vivo using pathway-specific cDNA arrays. Toxicology. 2002;178:241–260. doi: 10.1016/s0300-483x(02)00230-5. [DOI] [PubMed] [Google Scholar]