

Summary
The WAVE regulatory complex (WRC) drives the polymerisation of actin filaments located beneath the plasma membrane to generate lamellipodia that are pivotal to cell architecture and movement. By reconstituting WRC-dependent actin assembly at the membrane, we recently discovered that several classes of Arf family GTPases directly recruit and activate WRC in cell extracts, and that Arf cooperates with Rac1 to trigger actin polymerisation. Here, we demonstrate that the Class 1 Arf1 homologue Arf79F colocalises with the WRC at dynamic lamellipodia. We report that Arf79F is required for lamellipodium formation in Drosophila S2R+ cells, which only express one Arf isoform for each class. Impeding Arf function either by dominant-negative Arf expression or by Arf double-stranded RNA interference (dsRNAi)-mediated knockdown uncovered that Arf-dependent lamellipodium formation was specific to Arf79F, establishing that Class 1 Arfs, but not Class 2 or Class 3 Arfs, are crucial for lamellipodia. Lamellipodium formation in Arf79F-silenced cells was restored by expressing mammalian Arf1, but not by constitutively active Rac1, showing that Arf79F does not act via Rac1. Abolition of lamellipodium formation in Arf79F-silenced cells was not due to Golgi disruption. Blocking Arf79F activation with guanine nucleotide exchange factor inhibitors impaired WRC localisation to the plasma membrane and concomitant generation of lamellipodia. Our data indicate that the Class I Arf GTPase is a central component in WRC-driven lamellipodium formation.
Key words: Abi, Cyfip, Nap1, PIR121, Scar, Sra1
Introduction
Lamellipodia are sheet-like protrusions generated at the leading edge of a cell which play crucial roles in cell morphology and motility (Ridley, 2011; Weijer, 2009). The protrusive force is supplied by polymerisation of actin filaments initiated via the ubiquitous Arp2/3 complex which is activated by nucleation promoting factors (NPFs) (Campellone and Welch, 2010). In mammalian and Drosophila cells, the critical NPF underlying lamellipodium formation is the WRC {WAVE [WASP (Wiskott–Aldrich syndrome protein)-family verprolin-homologous protein] regulatory complex}, which is thought to be activated at the plasma membrane by the Rho GTPase Rac1 (Kunda et al., 2003; Miki et al., 1998). Purified mammalian WRC is inactive, but can be activated in buffer by high concentrations of Rac1 (Ismail et al., 2009). The low affinity of Rac1 for WRC (Chen et al., 2010) suggested that additional unknown factors are required to facilitate WRC-dependent actin polymerisation (Davidson and Insall, 2011). We showed that active Rac1 was indeed insufficient for recruitment and activation of the WRC at the membrane in mammalian cell extract (Koronakis et al., 2011). This led to our discovery that Arf family GTPases directly recruit and activate the WRC, which is enhanced when Arf and Rac1 work together to trigger actin polymerisation. Here, we sought to demonstrate that Arf family GTPases play a role in WRC-dependent generation of lamellipodia.
Results and Discussion
WRC is composed of five subunits, namely SCAR/WAVE, Sra1/PIR121, Nap1, Abi and HSPC300 (Gautreau et al., 2004). In human cells, paralogous genes give rise to SCAR/WAVE1, WAVE2 and WAVE3, Sra1 and PIR121, and Abi1 and Abi2 to generate divergent WRC combinations. The mammalian Arf GTPase family is also divergent and can be divided into three subfamilies: Class 1 (Arf1, Arf3), Class 2 (Arf4, Arf5) and Class 3 (Arf6) (Donaldson and Jackson, 2011), which display significant functional redundancy (Volpicelli-Daley et al., 2005). In Drosophila cells however, each Arf class is represented by a single isoform, namely Arf79F (Class 1), Arf102F (Class 2) and Arf51F (Class 3). Therefore, to bypass this additional layer of complexity, we used Drosophila S2R+ cells where only single WRC subunits (SCAR, Sra1, Kette/Nap1, Abi and HSPC300) are encoded, all of which are required for lamellipodium formation, which is very prominent in this cell type (Kunda et al., 2003; Rogers et al., 2003; Zallen et al., 2002). We first examined whether the Drosophila Arf1 homologue Arf79F localised at lamellipodia of S2R+ cells by immunofluorescence using antibodies against human Arf1 and Drosophila Sra1 (Fig. 1A). While Arf79F was predominantly found at the perinuclear Golgi apparatus, Arf79F was also observed enriched at the actin-rich cell edge where it colocalised with the WRC component Sra1 (inset). Anti-Arf1 specifically recognised Arf79F as detection of Arf at the Golgi and plasma membrane was abolished when Arf79F gene expression was silenced by double-stranded RNA interference (dsRNAi) (supplementary material Fig. S1A; Fig. S2A). Like endogenous Arf79F, recombinant RFP-tagged Arf79F was also detected by immunoblotting with Arf1 antibody (supplementary material Fig. S2B) and was visualised at the cell edge where it colocalised with Sra1 and actin (insets) (Fig. 1B). This was not the case in cells expressing RFP alone which was predominantly cytosolic relative to Arf79F (supplementary material Fig. S2C,D). The dynamics of fluorescent Arf79F was also monitored in real time where it was enriched at the plasma membrane and the Golgi (supplementary material Movie 1) which was further corroborated when Arf79F–RFP cells were stained with anti-Arf1 antibody (supplementary material Fig. S2E).
Fig. 1.
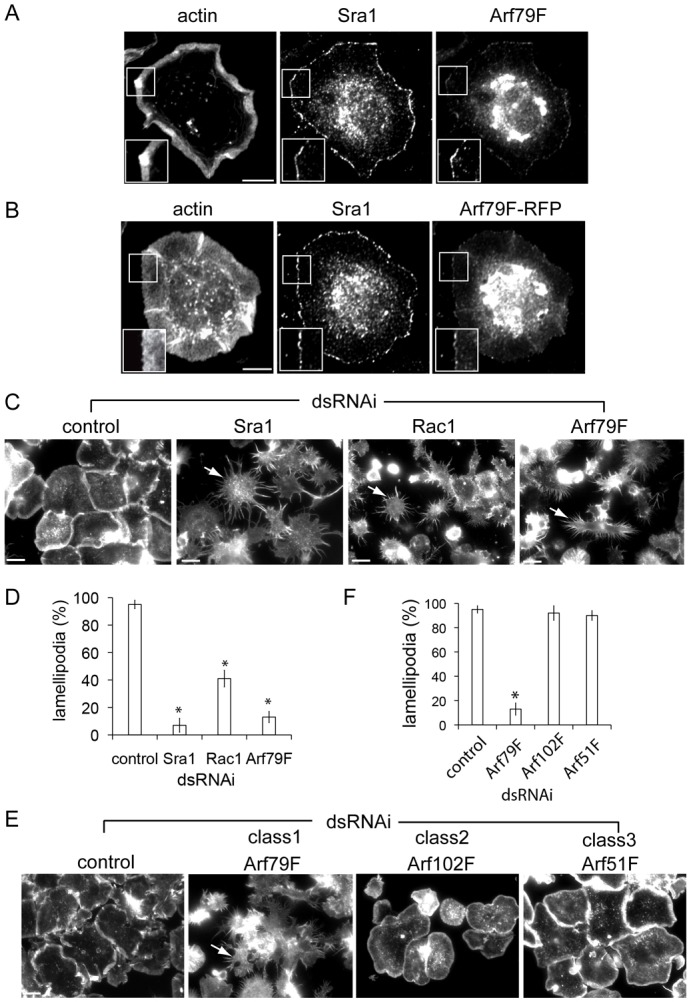
Arf79F is required for lamellipodium formation in S2R+ cells. (A) Arf79F and control Sra1 visualised at lamellipodia (insets) using anti-Sra1 or anti-Arf1 antibodies and fluorescent phalloidin (actin). (B) Cells expressing RFP-tagged Arf79F labelled as in panel A. (C) Lamellipodia in control-, Sra1-, Rac1- and Arf79F-dsRNAi cells visualised by staining actin (arrows indicate starfish phenotype) then quantified microscopically (D). Asterisks indicate a significant difference from the control. Error bars represent ±s.e.m. (E) Lamellipodia in control-, Arf79F-, Arf102F- and Arf51F-dsRNAi cells visualised as in panel B and quantified in (F), as in panel D. Scale bars: 10 μm (A,B), 20 μm (C,E).
We next investigated the requirement for Arf79F in lamellipodium formation. Actin-rich lamellipodia were clearly apparent in ∼95% of control cells (Fig. 1C,D) but Sra1 dsRNAi (supplementary material Fig. S1B) impaired lamellipodia in ∼93% of cells giving rise to a characteristic “starfish” phenotype (Kunda et al., 2003), typified by multiple thin extensions and disruption of cortical actin filaments (Fig. 1C,D). This was also seen in SCAR or Kette dsRNAi cells (data not shown). Rac1-silencing also impaired lamellipodium formation in ∼59% of cells (Fig. 1C,D), a much milder phenotype than in WRC-silenced cells as previously reported (D'Ambrosio and Vale, 2010; Kunda et al., 2003; Rohn et al., 2011). In Arf79F-silenced cells, lamellipodia were abolished in ∼87% of cells showing that Arf79F is critical for lamellipodium formation (Fig. 1C,D).
We next tested the requirement for specific classes of Arf GTPases in lamellipodium formation using dsRNAi (supplementary material Fig. S1C). In contrast to Arf79F dsRNAi, lamellipodia were unaffected by Arf102F and Arf51F dsRNAi (Fig. 1E,F). To confirm a role for Class 1 Arf GTPases in lamellipodium formation, we examined the morphology of cells equivalently expressing a readily available bank of dominant-negative human Arf GTPases (hArfTN) (supplementary material Fig. S3A). These are known to bind and sequester endogenous Arf guanine nucleotide exchange factors (GEF), thereby blocking Arf activation (Dascher and Balch, 1994; Hart et al., 1994). As human and Drosophila Arfs retain high amino acid identity, we speculated that hArfTN would also sequester class specific Drosophila Arf GEFs. While lamellipodia were not compromised in cells expressing hArf3T31N, hArf4T31N, hArf5T31N or hArf6T27N, their formation was impaired in ∼84% of hArf1T31N cells (Fig. 2A,B; supplementary material Fig. S3B) further supporting a specific role for class 1 Arf79F.
Fig. 2.

Rescue of lamellipodium formation in Arf79F-silenced cells by Arf1 and Rac1. (A) Lamellipodia in cells expressing RFP-tagged human dominant-negative Arf GTPases (hArf), hArf1T31N or hArf5 T31N, visualised by staining actin then quantified in (B), as Fig. 1D. Lamellipodia in control- or Arf79F-dsRNAi cells expressing either (C) hArf1–RFP or (E) HA-tagged Rac1 G12V visualised by staining actin and Rac1 with anti-HA antibody (arrows indicate transfected cells). Lamellipodia quantified in non-expressing (−) and either (D) hArf1–RFP or (F) Rac1G12V cells from experiments in panels C and E were quantified as in panel B. Scale bars: 10 μm.
We next sought to restore lamellipodium formation in Arf79F-silenced cells. Human Arf1 shares 95% amino acid identity with Arf79F, but only possesses 77% mRNA identity making it ideal for Arf79F dsRNAi rescue experiments. Like Arf79F (Fig. 1B), RFP-tagged hArf1 was observed at the Golgi and at actin-rich lamellipodia (Fig. 2C, arrows) which were formed equivalently in non-transfected (–; ∼91%) and hArf1-transfected cells (hArf1; ∼93%) (Fig. 2D, control dsRNAi). When hArf1 was expressed in Arf79F-silenced cells, lamellipodium formation was restored from ∼15% to ∼62% (Fig. 2C, arrows; Fig. 2D, Arf79F dsRNAi). Importantly, while ∼93% of control cells expressing constitutively active Rac1G12V formed lamellipodia, Rac1G12V failed to restore lamellipodium formation in Arf79F-silenced cells (Fig. 2E, arrows; Fig. 2F) showing that Arf79F-dependent lamellipodium formation is not mediated via upstream activation of Rac1.
Arf GTPases are predominantly observed at the perinuclear Golgi apparatus (Fig. 1A,B) where they are best characterised as having roles in membrane trafficking and Golgi organisation (Donaldson and Jackson, 2011). We therefore addressed whether it is Arf79F-mediated membrane trafficking from the Golgi that is imperative for lamellipodia by examining the Golgi apparatus in Arf79F-silenced cells (Fig. 3A). Immunofluorescence of the cis-Golgi marker GM130 following Arf79F dsRNAi revealed a clearly defined Golgi network (∼84% of cells) comparable to that seen in control cells (∼93%) with lamellipodia. This was not the case in cells expressing dominant-negative hArf4T31N where the Golgi was impaired in ∼75% of cells (Fig. 3B). Despite this Golgi disruption, cells expressing hArf4T31N still displayed lamellipodia and Sra1 localisation at the cell cortex in ∼90% of cells (Fig. 3C). These results suggest that Arf79F may facilitate lamellipodium formation independently of its role in Golgi organisation and membrane trafficking.
Fig. 3.

Arf79F is required for Sra1 localisation to the plasma membrane. (A) cis-Golgi in control- or Arf79F-dsRNAi cells visualised with anti-GM130 antibody and actin as a control. Cells expressing RFP-tagged dominant-negative hArf44T31N labelled with antibodies to (B) GM130 for the cis-Golgi or (C) Sra1 (magnified in insets) with actin as a control. (D) Arf79F recruitment of WRC components to the membrane. Silica beads coated with phospholipid bilayers alone (−) or with anchored myristoylated Arf79F loaded with GTPγS (+) were isolated from Drosophila cell extract, then the recruited proteins were analysed by SDS-PAGE and immunoblotting with antibodies to Sra1 and Kette. (E) Sra1 in control-, Sra1- or Arf79F-dsRNAi cells were visualised at the cell cortex with anti-Sra1 antibody and actin as a control, then cell cortex Sra1 and lamellipodia were quantified (F) as in Fig. 1D (P<0.001, Student's t-test). Scale bars: 10 μm (A-C); 20 μm (E).
We recently demonstrated that mammalian Arf1 directly binds the structural homologues Sra1 and Nap1 (Koronakis et al., 2011) and therefore wondered whether Arf79F drives lamellipodium formation by recruiting WRC to the plasma membrane. First, purified in vitro myristoylated Arf79F was anchored to silica beads coated in a phospholipid bilayer consisting of equal amounts of phosphatidylcholine and phosphatidylinositol (PC∶PI). Anchored Arf79F was locked in an active state by loading with non-hydrolysable GTPγS before addition of the beads to S2R+ cell extract. The beads were isolated from the extract, washed and the recruited proteins extracted from the membrane before analysis by SDS-PAGE and immunoblotting to detect WRC components Sra1 and Kette (Nap1 homologue) (Fig. 3D). In contrast to control beads with no Arf79F (−), active Arf79F (+) recruited Sra1 and Kette demonstrating active Arf79F acts like mammalian Arf GTPases by recruiting the WRC to the membrane. We next tested whether Arf79F could also recruit the WRC to the cell plasma membrane by examining Sra1 localisation in Arf79F-silenced cells by immunofluorescence. In control cells, Sra1 was observed enriched at the perinuclear region and at the plasma membrane (arrows) in ∼92% of cells (Fig. 3E,F). Sra1 was virtually non-detectable in control Sra1-silenced cells confirming the specificity of the Sra1 antibodies (Fig. 3E). In Arf79F-silenced cells, Sra1 was still found at the perinuclear area but cell cortex Sra1 was abolished in ∼79% of cells.
WRC recruitment from cell extract by GTP bound Arf79F (Fig. 3D) indicated that Arf79F activation would be key to Sra1 plasma membrane localisation and therefore lamellipodium formation. To test this hypothesis, we employed Arf GEF inhibitors SecinH3 and BrefeldinA (BFA) (Fig. 4A,B). Lamellipodia were generated by ∼95% of control cells which was reflected in Sra1 plasma membrane localisation in ∼92% of cells. SecinH3 treatment abrogated lamellipodia and cell cortex Sra1 by ∼35% (insets), while ∼50% of remaining SecinH3-treated cells displayed an abnormal cell edge resulting in disjointed lamellipodia (white arrows), likely representing incomplete pathway inhibition as indicated by Sra1 plasma localisation in these cells (white arrows). BFA eliminated lamellipodia and Sra1 at the cell cortex in ∼84% of cells. Differences in effectiveness between inhibitors is probably due to their mechanism where SecinH3 targets the cytohesin-family of Arf GTPase activators, Steppke in Drosophila (Donaldson and Jackson, 2011; Fuss et al., 2006; Hafner et al, 2006), while BFA forms a ternary complex with Arf1–GDP and Arf GEFs that would sequester class 1 Arf GTPase(s) (Mossessova et al., 2003). To dissect whether the reduction in Sra1 localisation ensues directly from inhibiting Arf or indirectly as a consequence of inhibiting lamellipodia, we uncoupled the two with the actin polymerisation inhibitors latrunculin B (LatB) and cytochalasin D (CytoD) (Fig. 4A,B). While both inhibitors abrogated lamellipodium formation, cell cortex Sra1 still remained in ∼44% of LatB-treated cells and ∼51% of CytoD-treated cells (insets) demonstrating that Sra1 still localised to the cell periphery upon inhibition of lamellipodial actin polymerisation. Consistent with our finding, WRC components have been previously observed at the plasma membrane of mammalian cell treated with LatB (Weiner et al., 2007).
Fig. 4.
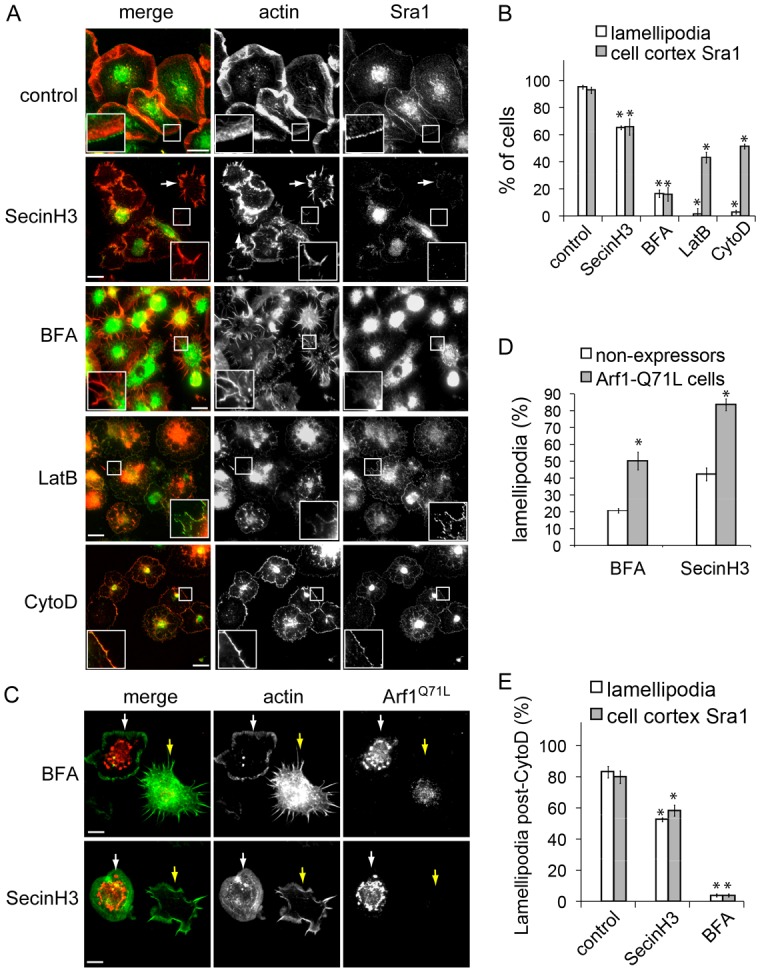
Active Arf79F recruits Sra1 to the cell membrane. (A) Sra1 in control-, SecinH3-, BFA-, LatB- and CytoD-treated cells visualised at the cell cortex (magnified in insets) with anti-Sra1 antibody and actin as a control (arrows indicate disjointed lamellipodia), then cell cortex Sra1 and lamellipodia quantified (B) as in Fig. 1A. (C) Lamellipodia in cells expressing constitutively active Arf1Q71L (white arrows) or adjacent non-expressing cells (yellow arrows) visualised by staining actin then quantified microscopically (D) as in Fig. 1D (P<0.05, Student's t-test). (E) Lamellipodia and cell cortex Sra1 in cells treated with CytoD (30 min) that were subsequently incubated with either growth medium (control), SecinH3 or BFA (1 h) were quantified as in panel B. Scale bars: 20 μm (A); 10 μm (C).
To address whether impaired lamellipodia following BFA- and SecinH3-treatment was due to inhibiting Arf79F, we examined whether expressing constitutively active Arf1Q71L would restore lamellipodia (Fig. 4C,D). Relative to non-expressers (yellow arrows), cells expressing Arf1Q71L generated ∼2.5-fold more lamellipodia in BFA-treated cells (white arrows) and ∼2-fold more in SecinH3-treated cells demonstrating that the Arf GEF inhibitors target Drosophila Arf1 (i.e. Arf79F).
Having established that Arf79F and its activation is required for maintaining lamellipodia, we investigated whether active Arf79F triggers re-generation of lamellipodia following their destruction by CytoD (Fig. 4E). When CytoD-treated cells were washed then subsequently incubated with either growth media (control), SecinH3 or BFA, lamellipodia were produced in ∼83% of control cells but only in ∼52% SecinH3-treated cells and ∼4% of BFA-treated cells demonstrating a role for active Arf79F in establishing new lamellipodia. The impairment in lamellipodium formation caused by Arf GEF inhibition was mirrored in the cell cortex localisation of Sra1 indicating that recruitment of Sra1 to the plasma membrane by Arf79F is key for triggering lamellipodia.
Arf GTPases are best known for their roles in membrane trafficking and organelle organisation (Donaldson and Jackson, 2011) but they have also been implicated in actin-dependent processes at the plasma membrane (Boulay et al., 2008; Cohen et al., 2007; Myers and Casanova, 2008; Stalder et al., 2011). Here, we establish that Arf79F not only localises at the cell edge with the WRC component Sra1 but is also essential for lamellipodium formation. This builds on our recent demonstration that Arf1 directly recruits and activates mammalian WRC in vitro (Koronakis et al., 2011).
High-throughput RNAi screening of Drosophila S2R+ cells has enabled the discovery of cellular factors driving lamellipodia (D'Ambrosio and Vale, 2010; Kiger et al., 2003; Rogers et al., 2003; Rohn et al., 2011); though as yet, Arf79f has not been implicated. This could be due to a 8% false negative rate in RNAi screens (Booker et al., 2011) and the relatively long time required for complete Arf79F dsRNAi (Fig. 1C; supplementary material Fig. S1A). Our discovery that Arf79F is key to lamellipodia establishes a new platform to dissect the Arf–WRC pathway. This role appears to be specific for Arf79F as Class 1 but not Class 2 or 3 Arfs were required for lamellipodia. This was surprising as WRC can be activated in cell extract by divergent Arf GTPases such as Arf5 (Arf102F homologue) and Arl1 (Koronakis et al., 2011). However, Gautier and colleagues showed that Arf79F, but not related Drosophila Arf isoforms, was found bound to immunoprecipitated WRC (Gautier et al., 2011) underlining Arf specificity for the WRC. Since Arf1 alone is insufficient for WRC activation in vitro (Koronakis et al., 2011) and Arf must cooperate with Rac1 for WRC-driven actin assembly (Humphreys et al., 2012; Koronakis et al., 2011), it seems that it is only active Arf79F which is temporally and spatially coincident with Rac1 activation in Drosophila cells.
Recent structural studies of WRC indicate that its plasma membrane localisation is mediated by Sra1–Rac1 interactions that would release the SCAR/WAVE VCA domain for Arp2/3 activation (Chen et al., 2010). However, WRC–Rac1 interactions are of low affinity suggesting recruitment and activation of WRC requires additional factors (Davidson and Insall, 2011). This is supported by a recent report revealing a role for clathrin in WRC recruitment which is independent of its connection to Arf–AP1/AP2 trafficking (Gautier et al., 2011). However, we demonstrated that Rac1 is insufficient for WRC activation (Koronakis et al., 2011) so while clathrin could promote the recruitment of WRC to the plasma membrane, its activation would only be triggered when both active Arf and Rac1 cooperate. We showed the role for Arf79F in lamellipodium formation is independent of Rac1 activation and trafficking. Consistent with this, we confirmed effects on lamellipodia were not due to altered Golgi function implying direct Arf–WRC interactions and not indirect Arf-mediated membrane trafficking triggers WRC-signalling. A direct mechanism involving Arf79F is further supported by its ability to recruit WRC components in vitro which was reflected in cells where active Arf79F was critical for Sra1 localisation and concomitant generation of lamellipodia. The data indicate Arf GTPase is a key component of the WRC signalling pathway in cells.
Materials and Methods
Plasmids and recombinant proteins
The following plasmids were generated by Invitrogen Gateway methodology: pAHW-Rac-V12 (Rac1G12V) and pMT-RFP (Liu et al., 2010) were used to create RFP-tagged Arf79F, human Arf1, Arf1Q71L Arf1T31N, Arf3T31N, Arf4T31N, Arf5T31N and Arf6T27N. Arf79F was cloned into pET20b, expressed and purified as previously for human Arf1 (Koronakis et al., 2011).
Drosophila cell culture, transfection and dsRNAi
S2R+ cells were cultured at 25°C in M3 insect medium (Sigma) with 10% heat-inactivated fetal bovine serum (JRH Biosciences) and 1% penicillin/streptomycin (Sigma–Aldrich) as described (Liu et al., 2010). When appropriate, cells were incubated with 0.5 µM Latrunculin B or cytochalasin D, 5 µM Brefeldin A (Sigma–Aldrich) or 25 µM SecinH3 (Merck) for 1 h. Drosophila cell transfection and RNAi experiments were carried out on concanavalin A-coated coverslips as previously described (Liu et al., 2010). RNAi was performed with double-stranded RNA generated from the following primer pairs: Arf79F (Arf79F_01_f: 5′-TAATACGACTCACTATAGGGTGTGATTGGTTGGGCTTTT-3′, Arf79F_01_r: 5′-TAATACGACTCACTATAGGGACATCAGAAGGAATTTTCACAAA-3′ and Arf79F_02-f: 5′-TAATACGACTCACTATAGGAGGGGTAGGGAGAGGACAAA-3′, Arf79F_02_r: 5′-TAATACGACTCACTATAGGAACTGGTACATCCAGGCGAC-3′); Arf102 (Arf102F_f: 5′-TAATACGACTCACTATAGGATGCTGCTGGAAAAACGACT-3′, Arf102F_r: 5′-TAATACGACTCACTATAGGCCATGTTCTGTAGTTCTCTTTCAGC-3′); Arf51 (Arf51F_f: 5′-TAATACGACTCACTATAGGTGAAACCCCATGAAATCCAG-3′, Arf51F_r: 5′-TAATACGACTCACTATAGGCTTGCGGTTGGTTTTCTTTG-3′); SCAR (Scar_f: 5′-TAATACGACTCACTATAGGTCGATGAGGACGCACTAATTT-3′, Scar_r: 5′-TAATACGACTCACTATAGGCACGTCGCGGGCCAGTTC-3′); Sra1 (Sra1_f: 5′-TAATACGACTCACTATAGGAGTTATGGGCTTTGGCCTGT-3′, Sra1_r: 5′-TAATACGACTCACTATAGGAAATGCACCATTAGATCCGC-3′); Kette (Kette_f: 5′-TAATACGACTCACTATAGGATTGAATCCAGTAGGCGGG-3′, Kette_r: 5′-TAATACGACTCACTATAGGATTCGATGCCACGGATCTTA-3′); Rac1 (Rac1_f: 5′-TAATACGACTCACTATAGGTCGAACACGGTGGGTATGTA-3′, Rac1_r: 5′-TAATACGACTCACTATAGGAGCAACAGTCCTCCGCCG-3′). RNAi was quantified in each case by EXPRESS One-Step SYBR® GreenER qRT-PCR according to manufacturer's instructions (Invitrogen) with RP49 as an internal control.
Immunofluorescence
Immunofluorescence microscopy and images assembled as described (Humphreys et al., 2009). RFP intensity (arbitrary units) within regions-of-interest at the cell edge were compared to equivalent regions in the cytoplasm (4 regions per cell) using Volocity 3D Image Analysis Software (PerkinElmer). Antibodies used were Sra1 and Kette (gifts from Buzz Baum), anti-Arf1 (Abcam-ab58578) and anti-GM130 (Abcam-ab30637). Alexa Fluor-labelled secondary antibodies, Alexa Fluor- and TxRed-labelled phalloidin (Invitrogen).
WRC recruitment to Arf79F anchored phospholipid beads
Drosophila S2R+ cell extract was prepared by passage through a 22-gauge needle in 10 mM HEPES (pH 7.4), 100 mM KCL, 1 mM MgCl and Complete EDTA-free protease inhibitor cocktail (Roche) then ultracentrifugation (180,000 g, 20 min) separated membranes from the high-speed supernatant cytoplasm which was used for experiments. Preparation of phospholipid-coated beads and pulldowns have been described in detail (Koronakis et al., 2011).
Statistics
In all experiments (n≥3), at least 300 cells were analysed for quantification per experiment. Comparisons between two data points were made using Student's t-test, between three or more data points using ANOVA combined with post-hoc Bonferroni's multiple comparison test. Asterisks indicate P<0.01 unless stated otherwise. Error bars represent ±s.e.m.
Supplementary Material
Acknowledgments
We thank Buzz Baum (University College London) for providing the SR+ cells, Sra1 antibodies and the template plasmid pUAS-RacV12.
Footnotes
Funding
This work was funded by The Wellcome Trust [grant number 070266]. Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.108092/-/DC1
References
- Booker M., Samsonova A. A., Kwon Y., Flockhart I., Mohr S. E., Perrimon N. (2011). False negative rates in Drosophila cell-based RNAi screens: a case study. BMC Genomics 12, 50 10.1186/1471-2164-12-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay P. L., Cotton M., Melançon P., Claing A. (2008). ADP-ribosylation factor 1 controls the activation of the phosphatidylinositol 3-kinase pathway to regulate epidermal growth factor-dependent growth and migration of breast cancer cells. J. Biol. Chem. 283, 36425–36434 10.1074/jbc.M803603200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone K. G., Welch M. D. (2010). A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 11, 237–251 10.1038/nrm2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Borek D., Padrick S. B., Gomez T. S., Metlagel Z., Ismail A. M., Umetani J., Billadeau D. D., Otwinowski Z., Rosen M. K. (2010). Structure and control of the actin regulatory WAVE complex. Nature 468, 533–538 10.1038/nature09623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L. A., Honda A., Varnai P., Brown F. D., Balla T., Donaldson J. G. (2007). Active Arf6 recruits ARNO/cytohesin GEFs to the PM by binding their PH domains. Mol. Biol. Cell 18, 2244–2253 10.1091/mbc.E06-11-0998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio M. V., Vale R. D. (2010). A whole genome RNAi screen of Drosophila S2 cell spreading performed using automated computational image analysis. J. Cell Biol. 191, 471–478 10.1083/jcb.201003135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascher C., Balch W. E. (1994). Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J. Biol. Chem. 269, 1437–1448 [PubMed] [Google Scholar]
- Davidson A. J., Insall R. H. (2011). Actin-based motility: WAVE regulatory complex structure reopens old SCARs. Curr. Biol. 21, R66–R68 10.1016/j.cub.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Donaldson J. G., Jackson C. L. (2011). ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 12, 362–375 10.1038/nrm3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss B., Becker T., Zinke I., Hoch M. (2006). The cytohesin Steppke is essential for insulin signalling in Drosophila. Nature 444, 945–948 10.1038/nature05412 [DOI] [PubMed] [Google Scholar]
- Gautier J. J., Lomakina M. E., Bouslama–Oueghlani L., Derivery E., Beilinson H., Faigle W., Loew D., Louvard D., Echard A., Alexandrova A. Y.et al. (2011). Clathrin is required for Scar/Wave-mediated lamellipodium formation. J. Cell Sci. 124, 3414–3427 10.1242/jcs.081083 [DOI] [PubMed] [Google Scholar]
- Gautreau A., Ho H. Y., Li J., Steen H., Gygi S. P., Kirschner M. W. (2004). Purification and architecture of the ubiquitous Wave complex. Proc. Natl. Acad. Sci. USA 101, 4379–4383 10.1073/pnas.0400628101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M., Schmitz A., Grüne I., Srivatsan S. G., Paul B., Kolanus W., Quast T., Kremmer E., Bauer I., Famulok M.2006). Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature 444, 941–944 10.1038/nature05415 [DOI] [PubMed] [Google Scholar]
- Hart M. J., Eva A., Zangrilli D., Aaronson S. A., Evans T., Cerione R. A., Zheng Y. (1994). Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the dbl oncogene product. J. Biol. Chem. 269, 62–65 [PubMed] [Google Scholar]
- Humphreys D., Hume P J., Koronakis V. (2009). The Salmonella effector SptP dephosphorylates host AAA+ ATPase VCP to promote development of its intracellular replicative niche. Cell. Host Microbe. 5, 225–233 10.1016/j.chom.2009.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys D., Davidson A., Hume P. J., Koronakis V. (2012). Salmonella virulence effector SopE and Host GEF ARNO cooperate to recruit and activate WAVE to trigger bacterial invasion. Cell Host Microbe 11, 129–139 10.1016/j.chom.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail A. M., Padrick S. B., Chen B., Umetani J., Rosen M. K. (2009). The WAVE regulatory complex is inhibited. Nat. Struct. Mol. Biol. 16, 561–563 10.1038/nsmb.1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiger A. A., Baum B., Jones S., Jones M. R., Coulson A., Echeverri C., Perrimon N. (2003). A functional genomic analysis of cell morphology using RNA interference. J. Biol. 2, 27 10.1186/1475-4924-2-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V., Hume P. J., Humphreys D., Liu T., Hørning O., Jensen O. N., McGhie E. J. (2011). WAVE regulatory complex activation by cooperating GTPases Arf and Rac1. Proc. Natl. Acad. Sci. USA 108, 14449–14454 10.1073/pnas.1107666108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunda P., Craig G., Dominguez V., Baum B. (2003). Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr. Biol. 13, 1867–1875 10.1016/j.cub.2003.10.005 [DOI] [PubMed] [Google Scholar]
- Liu T., Rohn J. L., Picone R., Kunda P., Baum B. (2010). Tao-1 is a negative regulator of microtubule plus-end growth. J. Cell Sci. 123, 2708–2716 10.1242/jcs.068726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H., Suetsugu S., Takenawa T. (1998). WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 17, 6932–6941 10.1093/emboj/17.23.6932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova E., Corpina R. A., Goldberg J. (2003). Crystal structure of ARF1*Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 12, 1403–1411 10.1016/S1097-2765(03)00475-1 [DOI] [PubMed] [Google Scholar]
- Myers K. R., Casanova J. E. (2008). Regulation of actin cytoskeleton dynamics by Arf-family GTPases. Trends Cell Biol. 18, 184–192 10.1016/j.tcb.2008.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. J. (2011). Life at the leading edge. Cell 145, 1012–1022 10.1016/j.cell.2011.06.010 [DOI] [PubMed] [Google Scholar]
- Rogers S. L., Wiedemann U., Stuurman N., Vale R. D. (2003). Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J. Cell Biol. 162, 1079–1088 10.1083/jcb.200303023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn J. L., Sims D., Liu T., Fedorova M., Schöck F., Dopie J., Vartiainen M. K., Kiger A. A., Perrimon N., Baum B. (2011). Comparative RNAi screening identifies a conserved core metazoan actinome by phenotype. J. Cell Biol. 194, 789–805 10.1083/jcb.201103168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder D., Barelli H., Gautier R., Macia E., Jackson C. L., Antonny B. (2011). Kinetic studies of the Arf activator Arno on model membranes in the presence of Arf effectors suggest control by a positive feedback loop. J. Biol. Chem. 286, 3873–3883 10.1074/jbc.M110.145532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli–Daley L. A., Li Y., Zhang C. J., Kahn R. A. (2005). Isoform-selective effects of the depletion of ADP-ribosylation factors 1-5 on membrane traffic. Mol. Biol. Cell 16, 4495–4508 10.1091/mbc.E04-12-1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijer C. J. (2009). Collective cell migration in development. J. Cell Sci. 122, 3215–3223 10.1242/jcs.036517 [DOI] [PubMed] [Google Scholar]
- Weiner O. D., Marganski W. A., Wu L. F., Altschuler S. J., Kirschner M. W. (2007). An actin-based wave generator organizes cell motility. PLoS Biol. 5, e221 10.1371/journal.pbio.0050221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen J. A., Cohen Y., Hudson A. M., Cooley L., Wieschaus E., Schejter E. D. (2002). SCAR is a primary regulator of Arp2/3-dependent morphological events in Drosophila. J. Cell Biol. 156, 689–701 10.1083/jcb.200109057 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.