

Summary
Efficient wound repair is essential for the maintenance of the integrity of the skin. The repair process is controlled by a variety of growth factors and cytokines, and their abnormal expression or activity can cause healing disorders. Here, we show that wound repair is severely delayed in mice lacking fibroblast growth factor receptors (FGFR) 1 and 2 in keratinocytes. As the underlying mechanism, we identified impaired wound contraction and a delay in re-epithelialization that resulted from impaired keratinocyte migration at the wound edge. Scratch wounding and transwell assays demonstrated that FGFR1/2-deficient keratinocytes had a reduced migration velocity and impaired directional persistence owing to inefficient formation and turnover of focal adhesions. Underlying this defect, we identified a significant reduction in the expression of major focal adhesion components in the absence of FGFR signaling, resulting in a general migratory deficiency. These results identify FGFs as key regulators of keratinocyte migration in wounded skin.
Key words: FGF, Focal adhesion, Migration, Re-epithelialization, Wound
Introduction
The skin comprises the largest organ in humans and covers the entire body surface. Owing to its essential role in the formation of a barrier against the environment, every defect in the skin needs to be rapidly and efficiently repaired. Upon injury, a complex healing process is initiated, which involves blood clotting, inflammation, re-epithelialization, granulation tissue formation and, finally, tissue remodelling. Under normal conditions, this leads to complete repair of the injured body site, although a scar remains that lacks all epidermal appendages and exhibits reduced tensile strength and elasticity (Gurtner et al., 2008; Martin, 1997; Menke et al., 2007; Sen et al., 2009). Unfortunately, the healing process is frequently impaired, resulting in the formation of chronic, nonhealing ulcers. This is a particularly common problem in the elderly population, in diabetic patients, and in patients treated with anti-inflammatory steroids or chemotherapy (Menke et al., 2007). Because impaired healing causes significant morbidity and generates an enormous financial burden for the health care system, it is of major importance to identify the factors that control the normal wound healing process and the mechanisms underlying impaired healing.
The wound repair process is orchestrated by a large number of growth factors and cytokines (Werner and Grose, 2003). Of particular importance are fibroblast growth factors (FGFs), which comprise a family of 22 polypeptides. Most of them exert their functions through activation of four receptor tyrosine kinases, designated FGFR1–4 (Beenken and Mohammadi, 2009; Ornitz and Itoh, 2001). Further complexity is achieved by alternative splicing of FGFR transcripts. Most importantly, alternative splicing in the third immunoglobulin-like domain of FGFR1–3 generates two alternative FGFR variants, designated IIIb and IIIc. Epithelial cells, including keratinocytes, express predominantly or even exclusively the IIIb variants, whereas stromal cells produce mainly the IIIc variants. Significantly, the IIIb and IIIc variants of each receptor are characterized by different ligand binding specificities (Beenken and Mohammadi, 2009; Ornitz and Itoh, 2001).
We and others previously demonstrated that different members of the FGF family contribute to the wound repair process. Whereas FGF2 is particularly important for wound angiogenesis and granulation tissue formation (Broadley et al., 1989; Ortega et al., 1998), ligands activating FGF receptors on keratinocytes control re-epithelialization. This was reflected by the severe delay in this process in mice expressing a dominant-negative FGFR2-IIIb mutant in keratinocytes (Werner et al., 1994). FGFR2-IIIb expressed by keratinocytes is activated by FGF1, FGF7, FGF10 and FGF22, which are expressed in normal and particularly in wounded skin (Steiling and Werner, 2003). In addition, FGF1, FGF10 and FGF22 activate FGFR1-IIIb, another receptor expressed on keratinocytes (Beer et al., 2000; Zhang et al., 2006). Through its ability to form heterodimers with FGFR2-IIIb in response to common ligands, FGFR1-IIIb might also be inhibited by a dominant-negative FGFR2-IIIb mutant. Therefore, the contributions of specific FGF receptor(s) and their ligands to wound re-epithelialization remain to be determined, and the underlying mechanisms have not been characterized.
To unravel the function of FGFR1-IIIb, FGFR2-IIIb and their ligands in the skin, we recently generated and characterized mice lacking one or both receptors in keratinocytes. The double mutant mice showed a complete loss of epidermal appendages and they developed mild, but progressive cutaneous inflammation. This was caused by a defect in the epidermal barrier as a consequence of reduced expression of different claudins and of occludin and concomitant formation of abnormal tight junctions. The chronic inflammation caused keratinocyte hyperproliferation through induction of a double paracrine loop that involved production of pro-inflammatory cytokines and chemokines by epidermal cells and secretion of keratinocyte mitogens by cells of the underlying dermis (Yang et al., 2010). These findings revealed important roles of FGFR1 and FGFR2 in appendage regeneration, epidermal barrier function and cutaneous homeostasis. In this study we determined the consequences of the loss of these receptors in keratinocytes for cutaneous wound repair and for keratinocyte migration in vitro.
Results
To determine the role of FGFR1 and FGFR2 in keratinocytes for the wound healing process, we generated full-thickness excisional wounds on the back of mice lacking FGFR1 (K5-R1 mice), FGFR2 (K5-R2 mice) or both receptors (K5-R1/R2 mice) in keratinocytes. Given that the double mutant mice develop mild but progressive inflammation (Yang et al., 2010), we performed the experiments with mice at the age of 1.5, 3 or 5 months to determine whether the inflammation affected the healing process.
K5-R1 mice showed normal wound healing at all ages, confirming previous results with mice lacking FGFR1-IIIb in all cells (Zhang et al., 2004). K5-R2 mice showed a slight, but nonsignificant delay in healing (supplementary material Fig. S1A). However, wound healing was severely impaired in K5-R1/R2 mice (Fig. 1A–C). The phenotype was already observed in young mice (6 weeks of age) and did not increase in severity upon aging (data not shown), indicating that the mild, but progressive inflammation seen in these mice (Yang et al., 2010) is not responsible for the healing defect.
Fig. 1.

Delayed wound closure in K5-R1/R2 mice. (A) Paraffin sections from the middle of 3-day-old (3dW), 5-day-old (5dW) and 14-day-old (14dW) wounds were stained with H/E. Cl, clot; D, dermis; E, epidermis; Es, eschar; G, granulation tissue; HE, hyperproliferative wound epidermis; HF, hair follicle. Scale bars: 500 µm. Arrows point to the tip of the migrating tongues of the wound epidermis. (B,C) H/E-stained wound sections were analyzed morphometrically, and wound closure (B) and wound diameter (distance between the borders to the noninjured dermis) (C) were determined. An average of two wounds, each from at least six mice per genotype, were analyzed per time point. Bars represent mean ± s.e.m.
Histological analysis with subsequent histomorphometry confirmed the impaired healing in K5-R1/R2 mice (Fig. 1B,C). The different components of the wound are schematically shown in supplementary material Fig. S1B. At day 3 after injury, the appearance of the wounds was similar in control and K5-R1/R2 mice. In mice of both genotypes, wounds were filled with a clot, and the granulation tissue comprised only a small area at the wound edge (Fig. 1A). However, a delay in wound closure was detected in K5-R1/R2 mice as determined by morphometric measurement of the distance between the epithelial tongues at both wound edges in relation to the wound diameter (distance between the borders of the noninjured dermis) (Fig. 1B). This was obviously because of impaired re-epithelialization, given that the wound diameter, which reflects wound contraction, was similar in mice of both genotypes (Fig. 1C).
At day 5 after wounding, a dense granulation tissue had formed in control mice, whereas wounds in K5-R1/R2 mice were still predominantly filled with a clot (Fig. 1A). Most importantly, wound closure was strongly delayed at this time point (Fig. 1B, middle panel). This was at least in part a result of impaired wound contraction. Thus, reduction of the wound diameter occurred between day 3 and day 5 in control mice, demonstrating the onset of contraction. However, this was not the case in K5-R1/R2 mice where the wound diameter even increased further owing to the movements of the mice and obvious lack of contraction. Given the important role of myofibroblasts in wound contraction (Hinz, 2007), we stained sections of 5-day wounds with an antibody against α-smooth muscle actin (α-SMA). However, we could not detect obvious differences in the area of granulation tissue populated by myofibroblasts and in myofibroblast location in the wounds of K5-R1/R2 mice (supplementary material Fig. S1C), indicating that the abnormal contraction is not the consequence of myofibroblast abnormalities.
We also did not observe an excessive inflammatory response at any stage of healing as revealed by immunostaining with an antibody against the neutrophil marker Ly6G (supplementary material Fig. S1D). The number and distribution of these inflammatory cells was not obviously altered in the FGFR mutant mice. Most importantly, there was no granulomatous reaction that could impair contraction.
At day 14 after wounding, mice of all genotypes were fully healed (Fig. 1A,B, right panel), and the density of the late granulation tissue/early scar tissue was comparable between mice of both genotypes as revealed by Masson Goldner staining, where a similar intensity of collagen staining was observed (supplementary material Fig. S1E, green). However, the wounds were still much larger in the double mutant animals, and they had an extended area of granulation tissue (Fig. 1A,C).
Taken together, wound healing was significantly delayed in K5-R1/R2 mice. During the early phase of repair the delay was predominantly caused by impaired re-epithelialization, whereas reduction of wound contraction was responsible for the delay at later stages.
We next focused on the impaired re-epithelialization, because this defect was likely to be a direct consequence of the loss of FGFR1 and FGFR2 in keratinocytes. Interestingly, the area of the hyperproliferative wound epithelium was similar in control and K5-R1/R2 mice at day 3 after wounding and showed only a mild, but nonsignificant reduction at day 5 after injury (Fig. 2A). The rate of keratinocyte proliferation was even increased within the hyperproliferative wound epithelium of 5-day wounds as determined by 5-bromo-2′-deoxyuridine (BrdU) incorporation studies (Fig. 2B,C). This increase was already observed in nonwounded skin and resulted from the enhanced expression of keratinocyte mitogens in the dermis of K5-R1/R2 mice (Yang et al., 2010). In spite of the similar area of the wound epithelium in K5-R1/R2 mice, its length was significantly reduced in double-knockout mice at day 3 after injury (Fig. 2D, left panel). At this time point, the length of the wound epidermis reflects the ability of the keratinocytes to migrate into the wound tissue, because only few keratinocytes in the wound epidermis proliferate at this early stage of repair. At day 5 after wounding, the length of the wound epidermis was only slightly reduced in K5-R1/R2 mice compared to control animals, probably reflecting compensation by enhanced proliferation (Fig. 2D, right panel). These results suggest that keratinocyte migration is impaired in the healing wounds of K5-R1/R2 mice. This hypothesis is further supported by the morphological appearance of the keratinocytes at the tip of the migrating epidermal tongue: Whereas keratinocytes from control mice formed a flat epithelial tongue with elongated keratinocytes at the front, the epithelial tongue in K5-R1/R2 mice was generally thicker, and the cells at the tip did not have the flattened appearance (Fig. 2E, indicated by arrows).
Fig. 2.

Keratinocyte migration is impaired in wounded skin of K5-R1/R2 mice. (A) The area of the hyperproliferative wound epidermis was determined morphometrically using H/E-stained sections from 3-day-old and 5-day-old wounds. An average of two wounds from at least six mice were analyzed per time point and genotype. (B) Cell proliferation was analyzed by BrdU incorporation at day 5 after wounding. The wound is on the right-hand side of the sections. Representative sections from control and K5-R1/R2 mice are shown for 5-day-old wounds. (C) The number of BrdU-positive cells per area of wound epidermis was determined at both wound edges. An average of two wounds from at least three mice was analyzed per time point and genotype. (D) Sections from 3-day-old and 5-day-old wounds of control and K5-R1/R2 mice were analyzed morphometrically. The length of the wound epidermis (distance from the wound edge to the tip of the migrating tongue) was determined at both wound edges. An average of two wounds from at least seven mice was analyzed per time point and genotype. (E) Representative H/E-stained sections from 3-day-old wounds showing the tip of the wound epidermis. Note the flattened wound tongue in control mice, but the broad, nonflattened tongue in K5-R1/R2 mice (indicated by arrows). Bars in A–D represent mean ± s.e.m. Scale bars: 200 µm (B); 50 µm (E). G, granulation tissue; HE, hyperproliferative wound epidermis.
FGFR1 and FGFR2 are required for efficient keratinocyte migration
To further study the migratory capacity of keratinocytes lacking FGFR1 and FGFR2, we performed migration experiments with primary and immortalized keratinocytes from control and K5-R1/R2 mice (Fig. 3A–C; supplementary material Figs S2, S4). In a modified transwell assay, primary (Fig. 3A) and immortalized keratinocytes (supplementary material Fig. S2A) from K5-R1/R2 mice showed a strongly reduced migration rate in defined keratinocyte serum-free medium. Addition of FGF7 and FGF10 further stimulated migration of keratinocytes from wild-type, but not K5-R1/R2 mice. Surprisingly, cells from K5-R1/R2 mice even showed a reduced migratory capacity in the presence of high concentrations of epidermal growth factor (EGF), demonstrating that EGF can only partially substitute for FGFs (Fig. 3A). This was not because of a reduced responsiveness of the FGFR1/2-deficient cells to EGF (Yang et al., 2010), but rather reflects the intrinsic migratory deficiency of the cells that was also seen in the absence of an exogenous growth factor.
Fig. 3.
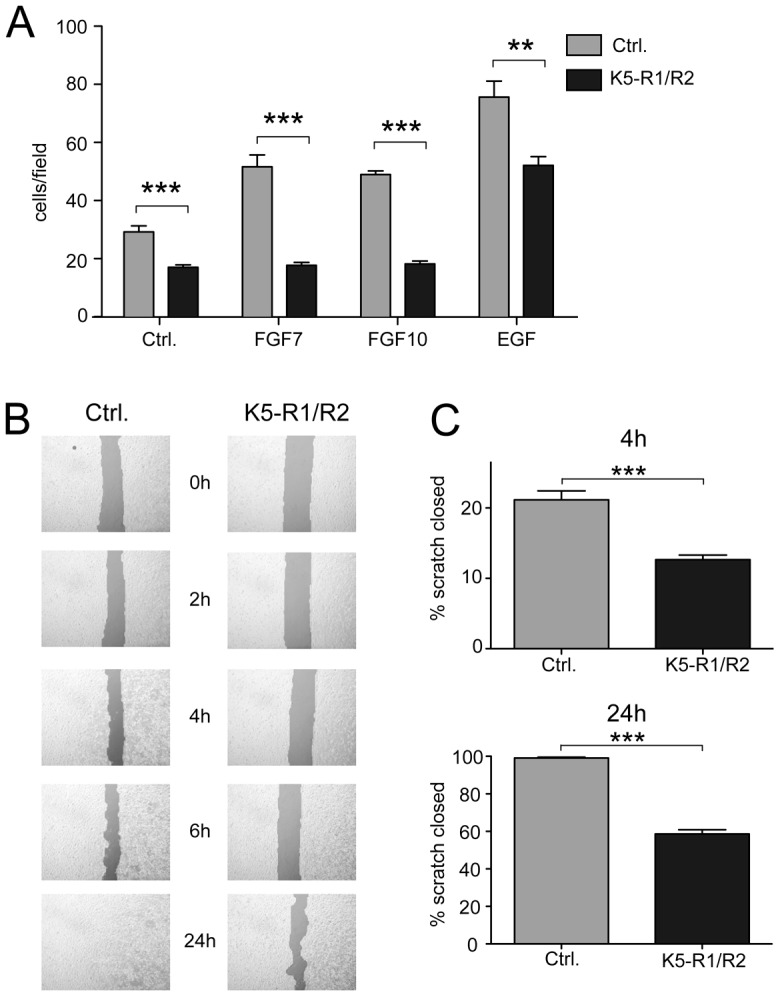
Loss of FGFR1 and FGFR2 affects migration of cultured keratinocytes. (A) Primary keratinocytes from control and K5-R1/R2 mice were analyzed for their migratory capacity using a modified transwell assay. FGF7, FGF10, EGF or vehicle (Ctrl.) was added to the culture medium. Cells at the bottom side of the membrane were counted 24 hours after seeding of cells onto the upper side. Bars represent mean ± s.e.m. (B). Immortalized keratinocytes from control and K5-R1/R2 mice were grown to confluency. A scratch wound was inserted into the monolayer, and the cells were then incubated in the presence of mitomycin C. Pictures were taken at different time points after scratch wounding. (C) The area devoid of cells was determined 4 hours and 24 hours after wounding. Bars represent mean ± s.e.m. N≥12.
The severe migratory defect of primary and immortalized K5-R1/R2 keratinocytes was confirmed in scratch assays (Fig. 3B,C; supplementary material Figs S2B and S3). The difference between FGFR-deficient and control keratinocytes was still observed in the presence of mitomycin C, which inhibits cell proliferation (shown for immortalized keratinocytes in Fig. 3B,C) as well as on collagen I-coated dishes (supplementary material Fig. S2B). Given that the scratch closure depends on the migratory speed (velocity), the direction of migration, and the directional persistence (Hartwig et al., 2007), we analyzed these parameters in our in vitro system (Fig. 4A–E). Live-cell recordings of migrating, immortalized keratinocytes were taken after scratching the cell layer using two independent lines of immortalized keratinocytes from control and K5-R1/R2 keratinocytes (supplementary material Movies 1, 2). K5-R1/R2 cells consistently migrated with a reduced velocity compared to control cells (Fig. 4B). Furthermore, their directional persistence was severely impaired, and they did not migrate into the scratch as efficiently as control cells (Fig. 4C–E). These differences were observed for cells at the migrating front as well as for cells in the fifth row behind the front, with the exception of velocity, which was only significantly different for cells in the front row (supplementary material Fig. S3). The defect in directional migration was further reflected by the impaired Golgi polarization upon scratch wounding as determined by immunostaining with an antibody against the Golgi marker protein giantin. This protein was concentrated in front of the nucleus of most migrating cells from wild-type mice (Fig. 4F, left panel, indicated by arrows), but it was evenly distributed around the nucleus of most cells from K5-R1/R2 mice (Fig. 4F, right panel and Fig. 4G, indicated by asterisks).
Fig. 4.

Loss of FGFR1 and FGFR2 reduces velocity and impairs directional migration of cultured keratinocytes. Immortalized keratinocytes from control and K5-R1/R2 mice were subjected to scratch wounding and analyzed by live cell imaging for 13 hours. Eight cells from a minimum of five movies were analyzed for each genotype, including four cells from the front row and four from the fifth row behind the front. All results were reproduced with an independent cell line from control and K5-R1/R2 mice. (A) Parameters analyzed for the quantification of migration. The migrating cells were analyzed for (B) velocity, (C) displacement (linear distance between starting and end point; ‘b’ in A), (D) persistence coefficient (‘b’/‘a’ in A) (Hartwig et al., 2007) and (E) perpendicular movement (‘c’ in A). Bars represent mean ± s.e.m. (F,G) Migrating keratinocytes were stained with an antibody against giantin to label the Golgi apparatus (green) together with rhodamine-conjugated phalloidin (red) and counterstained with Hoechst (blue). Note the polarization of the Golgi apparatus in cells from wild-type mice, but the equal distribution of giantin around the nucleus in cells from K5-R1/R2 mice. Examples of polarized cells are indicated with an arrow, examples of nonpolarized cells are indicated with an asterisk. The scratch is at the top of the pictures. Scale bar: 50 µm. The percentage of cells with polarized Golgi among all cells is shown in G. Bars represent mean ± s.e.m. A minimum of six different pictures from two different cell lines per genotype was analyzed. (H) A representative picture of a migrating cell with a prominent ruffle is shown on the right-hand side. Ruffles are indicated by a white dot. Pictures of a scratch assay experiment were taken to count cells with ruffles. Four time points of the live imaging experiment were chosen. The percentage of cells with ruffles among all cells was determined at the indicated time points. Bars represent mean ± s.e.m.; n≥5.
It has previously been shown for cultured human keratinocytes that enhanced formation of membrane ruffles reflects inefficient migration (Borm et al., 2005). Consistent with this observation, there were significantly more cells from K5-R1/R2 mice with ruffles compared to cells from wild-type mice, and this was particularly obvious at later time points after scratching of the monolayer (Fig. 4H).
Impaired attachment and focal adhesion formation of K5-R1/R2 keratinocytes
Upon passaging of primary and immortalized keratinocytes from K5-R1/R2 mice we noticed that their adhesion to the substrate and subsequent spreading was delayed compared to control cells. To quantify this finding, we performed adhesion assays with immortalized keratinocytes. Consistent with our preliminary data, adhesion of FGFR1/R2-deficient keratinocytes on PBS-coated dishes or on dishes coated with different concentrations of fibronectin was significantly reduced compared to cells from control mice (Fig. 5A). Only a minor reduction was observed on collagen I-coated dishes.
Fig. 5.

Impaired adhesion and FA formation of K5-R1/R2 keratinocytes. (A) Immortalized keratinocytes from control and K5-R1/R2 mice were analyzed for adhesion on BSA, collagen I, or fibronectin 45 minutes after seeding as described in Materials and Methods. Three different concentrations of fibronectin were used (1, 2.5 and 5 µg/ml, as indicated). Bars represent mean ± s.e.m.; n = 4 wells per genotype. (B) Immortalized keratinocytes from control and K5-R1/R2 mice were allowed to attach for 20 or 90 minutes on collagen I- and/or fibronectin-coated dishes. Cells were stained with antibodies against total or phosphorylated paxillin (green). The actin cytoskeleton was visualized using rhodamine-conjugated phalloidin (red). Cells were analyzed by confocal microscopy. Scale bars: 50 µm. Cells representative of the different genotypes are shown. FAs in control cells are indicated with arrows. (C) The percentage of cells with large FAs (between one and ten per cell; indicated by asterisks on cells from K5-R1/R2 mice) among all cells was determined; n = 7 photomicrographs per cell line, including at least 680 cells. The result was reproduced with independent cell lines. Bars represent mean ± s.e.m. Scale bars: 100 µm.
We next analyzed the actin cytoskeleton of cells from control and mutant mice at different time points after seeding into collagen I/fibronectin coated plates by staining with rhodamine-coupled phalloidin (Fig. 5B, red). Concomitantly, formation of focal adhesions (FA) was analyzed by immunofluorescence staining with antibodies against total or phosphorylated paxillin (Fig. 5B, green). Twenty minutes after seeding, control cells reproducibly showed efficient attachment and spreading, and FAs were uniformly distributed at the cell periphery. By contrast, most cells from K5-R1/R2 mice were still rounded and exhibited only a few FAs. Representative examples are shown in Fig. 5B. Ninety minutes after seeding, cells of both genotypes had attached, but there were fewer FAs in K5-R1/R2 cells compared with controls. The FAs that had formed were unevenly distributed, and ∼25% of the cells had between one and ten very large FAs (Fig. 5B,C). By contrast, less than 10% of control cells formed such large FAs. This finding suggests that FA turnover is also impaired. Given that spreading and migration require the rapid formation and turnover of FAs, these findings provide a probable explanation for the migratory deficiency.
Reduced expression of focal adhesion kinase and paxillin in K5-R1/R2 keratinocytes
We next determined the mechanisms underlying the impaired adhesion and FA formation of keratinocytes from K5-R1/R2 mice. For this purpose we first determined if loss of FGFR1 and FGFR2 on keratinocytes affects the expression of major keratinocyte integrins. Flow cytometry analysis of immortalized keratinocytes revealed no significant difference in surface expression levels of the integrin subunits αv, α6 and β1 in control versus K5-R1/R2 keratinocytes, and there was also no difference in integrin β1 activation as assessed by binding of the activation-specific monoclonal antibody 9EG7 (Fig. 6A). Integrin subunits α1, α2, α5, and β3 were undetectable or barely detectable, indicating low expression levels (data not shown). The similar expression of the α6 subunit in cells from control and K5-R1/R2 mice was confirmed for immortalized and primary keratinocytes using western blotting (supplementary material Fig. S4C).
Fig. 6.

FGFs control expression of FAK and paxillin, but not of integrins in keratinocytes. (A) Immortalized keratinocytes from control and K5-R1/R2 keratinocytes were stained with antibodies against the integrin subunits αv, α6, β1, or active integrin β1 (β1*) or unspecific isotype controls and analyzed by flow cytometry. Numbers indicate the ratio of the mean fluorescence intensity (±relative s.e.m.) between cells from K5-R1/R2 mice and those from control mice. Results are representative of triplicate measurements repeated at least three times. (B) Protein lysates were prepared from immortalized keratinocytes from K5-R1/R2 and control mice grown in dk-SFM (see Materials and Methods). 30 µg of the lysates was analyzed by western blotting for the levels of total and phosphorylated paxillin and FAK. β-Actin was used as a loading control. Band intensities from triplicate experiments (using samples from three independent wells) were scanned. Bars represent mean ± s.e.m. (C,D) Immortalized keratinocytes from wild-type mice were grown to confluency, starved for 20 hours in serum-free medium and subsequently treated for 12 hours or 24 hours with 10 ng/ml FGF7 or vehicle. (C) RNA from the treated cells was analyzed by qRT-PCR for the expression of paxillin and Fak. Expression of the housekeeping gene rps29 was used for normalization. Expression levels in vehicle-treated cells were arbitrarily set to 1. (D) Protein lysates were analyzed for expression of FAK and paxillin by western blotting. Probing of the membrane with an antibody against GAPDH was used as a loading control. Quantification of the band intensities is shown in the bar graph. (E) Human HaCaT keratinocytes were grown to confluency, starved for 24 hours in serum-free medium and subsequently treated for 26 hours with 10 ng/ml FGF7 or vehicle. Protein lysates were analyzed by western blotting for expression of FAK and paxillin. GAPDH was used as loading control. Quantification of the band intensities is shown in the bar graph. Expression levels of vehicle-treated cells were arbitrarily set to 1. Results from representative experiments are shown (C–E). The results were reproduced in three independent experiments.
Laminin-332 is deposited by keratinocytes and mediates efficient keratinocyte adhesion and migration (Margadant et al., 2009; Nguyen et al., 2000) (Hartwig et al., 2007). Therefore, we next determined if the lack of FGFR1 and FGFR2 in keratinocytes affects expression of this extracellular matrix protein. Total lysates from freshly trypsinized primary or immortalized keratinocytes were used for western blot analysis using an antibody against the γ2-chain of laminin-332. There was some variability in the expression of this extracellular matrix protein, but the expression levels did not correlate with the genotype of the cells (supplementary material Fig. S4A).
Furthermore, immunofluorescence analysis of attached and permeabilized cells revealed only a weak staining for γ2 in immortalized keratinocytes from mice of both genotypes at the time point that we used for the adhesion assay (45 minutes) (supplementary material Fig. S4B), and upon detachment of the cells we could not detect any deposited laminin 332 (γ2-chain) (data not shown). Therefore, other mechanisms are likely to be predominantly responsible for the impaired adhesion and migration of FGFR1/R2-deficient keratinocytes. In particular, the impaired formation and possibly turnover of FAs could result from reduced expression and/or phosphorylation of major FA components. We focused on paxillin and FA kinase (FAK), because these proteins were shown to be required for hepatocyte growth factor induced cell spreading and migration (Ishibe et al., 2004). Western blot analysis of protein lysates from immortalized keratinocytes revealed a strongly reduced expression of these FA components in the absence of FGFR1 and FGFR2 (Fig. 6B). This result was confirmed with three independent cell lines per genotype. The reduction in the levels of FAK and paxillin proteins in the absence of FGFR1 and FGFR2 was confirmed for primary keratinocytes (supplementary material Fig. S4D). As a consequence of the reduced expression of these proteins, levels of phosphorylated paxillin (Y-118) and phosphorylated FAK (Y-397) were reduced to a similar extent. When immortalized keratinocytes from control mice or human HaCaT keratinocytes were treated with recombinant FGF7 and analyzed for the levels of total FAK and paxillin, we reproducibly found an increase in the expression of these genes at the RNA and protein level within 12–24 hours after addition of FGF7 (Fig. 6C–E). These findings demonstrate that FGF7 controls their expression in keratinocytes.
Discussion
Chronic, nonhealing wounds are a major health problem in an aging society (Sen et al., 2009). Although rarely life threatening, they severely affect the quality of life of the affected individuals and cause enormous costs to the health care system. Therefore, there is a strong need to develop novel and efficient strategies for the treatment of these disorders, and this requires a thorough understanding of the underlying cellular and molecular mechanisms.
Here we show that loss of FGFR1 and FGFR2 in keratinocytes results in a wound-healing defect, which resembles the abnormalities seen in chronic human skin ulcers. This phenotype is characterized by severely impaired re-epithelialization combined with reduced wound contraction (Usui et al., 2008). The latter effect is obviously indirect in the K5-R1/R2 mice, because the loss of FGFR1 and FGFR2 occurred specifically in keratinocytes and not in cells of the dermis/granulation tissue. It seems probable that the loss of FGFR1 and FGFR2 in keratinocytes results in the production of factors that affect the underlying dermis/granulation tissue. Indeed, we previously showed a strong upregulation of S100A8 and S100A9 as well as of interleukin-1 family member 8 in the nonwounded epidermis of K5-R1/R2 mice, which strongly affected immune cells and fibroblasts in the dermis (Yang et al., 2010). Therefore, these and possibly additional factors that are abnormally expressed in the wound epidermis of K5-R1/R2 mice and control animals are likely to affect wound contraction in a paracrine manner. However, these factors did not obviously affect differentiation of myofibroblasts, a cell type with a particularly important function in wound contraction (Hinz, 2007). Furthermore, we also did not observe a granulomatous reaction that could impair contraction. Rather, it seems probable that the alterations in the connective tissue that develop in these mice (Meyer et al., 2011) are responsible for the impaired wound contraction.
Whereas the impaired contraction is obviously a noncell-autonomous phenotype, the delayed re-epithelialization seen in K5-R1/R2 mice is a direct consequence of the loss of FGFR1 and FGFR2 in keratinocytes. Impaired re-epithelialization was also observed in transgenic mice expressing a dominant-negative FGFR mutant in keratinocytes (Werner et al., 1994), although this approach did not allow the identification of the responsible FGF receptor(s). The results of our new study revealed that loss of a single FGFR does not significantly affect the wound healing process, whereas the combined loss of FGFR1 and FGFR2 is deleterious. We previously showed that keratinocytes express mainly or exclusively the IIIb splice variants of these receptors (Yang et al., 2010), which are activated by FGF1, FGF7, FGF10 and FGF22 (Zhang et al., 2006). All of these growth factors are expressed in healing skin wounds, and at least FGF7 and FGF22 are strongly upregulated at the wound site (Werner et al., 1992; Beer et al., 1997; Beyer et al., 2003). This expression pattern, together with the results presented in this study, strongly suggest that activation of FGFR1-IIIb and in particular of the more abundant FGFR2-IIIb by the above-mentioned FGFs is crucial for efficient wound re-epithelialization.
Re-epithelialization is achieved by a combination of keratinocyte proliferation and migration. Interestingly, the rate of keratinocyte proliferation was not impaired in wounded skin of K5-R1/R2 mice, and even a slight hyperproliferation was observed. The latter was already seen in nonwounded skin and shown to result from the progressive inflammation that develops in these animals (Yang et al., 2010). Consistent with this finding, mice expressing a dominant-negative FGFR2-IIIb mutant in keratinocytes, which did not show cutaneous inflammation, exhibited reduced keratinocyte proliferation at the wound site (Werner et al., 1994). Furthermore, cultured keratinocytes from K5-R1/R2 mice did not show hyperproliferation (Yang et al., 2010), demonstrating that this phenotype is not cell autonomous. In spite of the normal or even enhanced keratinocyte proliferation in K5-R1/R2 mice, wound re-epithelialization was delayed as a result of impaired keratinocyte migration. At later stages of the repair process, however, the strong proliferation of keratinocytes obviously compensated for the impaired migration.
The data presented in this manuscript reflect the importance of FGF receptor signaling for keratinocyte migration and are consistent with the potent effect of exogenous FGF7 on migration of human keratinocytes (Ceccarelli et al., 2007). Surprisingly, keratinocytes from K5-R1/R2 mice did not only show impaired migration in response to FGFs but also to other mitogens, including EGF, suggesting nonredundant functions of FGFs in keratinocyte migration. The reduced pro-migratory effect of EGF was not the result of a generally reduced responsiveness to this mitogen, because EGF strongly activated the Erk1/2 and PI3K signaling pathways in keratinocytes from control and K5-R1/R2 mice (Yang et al., 2010). Finally, migration of cells from K5-R1/R2 mice in a transwell assay was still reduced in the absence of exogenous growth factors. This finding could be explained by ligand-independent activation of FGFR2, as previously shown for mechanically wounded keratinocytes (Li et al., 2009) and/or by the difference in the expression levels of paxillin and FAK.
Our functional studies revealed that the migratory defect of keratinocytes from K5-R1/R2 mice did not result from impaired expression or alterations in the activation of keratinocyte integrins. Furthermore, expression and activation of major keratinocyte matrix metalloproteinases was not obviously affected by the loss of FGFR1 and FGFR2 (supplementary material Fig. S4E). Rather, we identified a defect in cell attachment and formation of FAs as a consequence of reduced levels of total and phosphorylated FAK and paxillin.
Interestingly, treatment with FGF7 increased the total mRNA and protein levels of FAK and paxillin (Fig. 6), demonstrating that FGFs control the expression of these major FA components in keratinocytes. It remains to be determined if the reduction in FAK and paxillin is also responsible for the impaired keratinocyte migration in wounded skin. This seems probable, because preliminary experiments revealed reduced expression of FAK and paxillin in the epidermis of K5-R1/R2 mice at day 12 after birth when phenotypic abnormalities are not yet present in the skin of these animals (Yang et al., 2010) (supplementary material Fig. S4F). In future studies, it will be important to analyze the expression of these FA components in the wound epidermis and to perform wound-healing studies with mice lacking different FA components in keratinocytes. It has previously been reported that mice lacking FAK in keratinocytes do not have an obvious defect in wound closure (McLean et al., 2004; Essayem et al., 2006), although re-epithelialization had not been specifically analyzed in these studies, and minor differences might have been missed. Furthermore, only one FA component was affected in the FAK-knockout mice, whereas a reduction of at least two FA components was observed in our FGFR1/R2-deficient keratinocytes. Our preliminary data also revealed a reduction in the levels of vinculin (data not shown). Therefore, it will be important to analyze the consequences of a combined knockdown of several FA components for wound healing in vitro and in vivo.
The reduced levels of major FA proteins provide a probable explanation for the abnormalities in FA formation and turnover that we observed in the absence of FGFR1 and FGFR2. Thus, FA formation was delayed upon seeding of the cells, and the FAs that formed were strongly enlarged. Large FAs were shown to inhibit cell migration through formation of less dynamic anchoring structures and limited generation of propulsive forces (Beningo et al., 2001), a finding that is consistent with the migratory defect of K5-R1/R2 keratinocytes. Such enlarged adhesions were shown to result from reduced tyrosine phosphorylation of paxillin (Zaidel-Bar et al., 2007), and we indeed observed reduced paxillin-Y118 levels in FGFR1/R2-deficient keratinocytes.
It has previously been shown that the presence of activated FAK at FAs is required for FA disassembly at the base of extending lamellipodia, a process required for efficient directed cell migration (Webb et al., 2004). This process requires concomitant activation of Rac (Ishibe et al., 2004), and our preliminary data suggest that this is also impaired in K5-R1/R2 keratinocytes (data not shown).
A defect in keratinocyte migration was also observed in mice lacking other growth factors or their receptors, and a particularly severe phenotype was seen in the absence of the hepatocyte growth factor receptor c-Met. In mice lacking c-Met in keratinocytes, only keratinocytes that had escaped recombination were able to contribute to wound re-epithelialization, demonstrating that c-Met signaling is essential for this process (Chmielowiec et al., 2007). Such a severe phenotype was not observed in K5-R1/R2 mice, indicating that FGFR1/FGFR2-deficient keratinocytes are still able to re-epithelialize the wound. Indeed, migrating cells from K5-R1/R2 mice were tested for recombination in cell culture and shown to be true knockouts for the receptors as determined by qRT-PCR (data not shown). Therefore, FGFR1 and FGFR2 are obviously not essential for keratinocyte migration, but their loss strongly impairs this process.
Impaired keratinocyte migration combined with normal or even enhanced proliferation of these cells is a hallmark of chronic human skin ulcers (Harsha et al., 2008; Usui et al., 2008). Therefore, it will be interesting to determine if reduced expression of FGF receptors or their ligands occurs in these ulcers or if FGFR signaling is impaired in the keratinocytes of these patients, in particular at the wound edge. This would be a prerequisite for the development of novel and more efficient strategies for the improvement of these severe wound-healing disorders by activating FGFR signaling pathways.
Materials and Methods
FGFR-knockout mice
Mice lacking FGFR1 and FGFR2 in keratinocytes were generated by mating of mice with floxed alleles of these receptors (Trokovic et al., 2003; Yu et al., 2003) with transgenic mice expressing Cre recombinase under control of the keratin 5 promoter (Ramirez et al., 2004) and genotyped as previously described (Yang et al., 2010). All mice were in a C57BL/6 genetic background. They were housed and fed according to Federal guidelines.
Wounding and preparation of wound tissues
Mice at the age of 6 weeks, 3 or 5 months were anaesthetized with a single intraperitoneal injection of ketamine/xylazine. Two full-thickness excisional wounds, 5 mm in diameter, were made on either side of the dorsal midline by excising skin and panniculus carnosus as described previously (Thorey et al., 2001). Mice were sacrificed at different time points after injury. For histological analysis, the complete wounds were excised and either fixed overnight in 95% ethanol/1% acetic acid or in 4% paraformaldehyde (PFA)/phosphate buffered saline (PBS) followed by paraffin embedding, or frozen in tissue freezing medium (Leica Microsystems, Heerbrugg, Switzerland). Sections (7 µm) from the middle of the wound were stained with hematoxylin/eosin (HE) or by the Masson Goldner procedure or used for immunofluorescence analysis. Morphometric analysis of different parameters of the wound healing process was previously described (Thorey et al., 2001; Kümin et al., 2007).
All procedures with mice were approved by the local veterinary authorities of Zurich, Switzerland.
Cell culture
Keratinocytes were isolated from single mice with different genotype as described previously (Braun et al., 2002) with the difference that cells were seeded at a density of 5×104 cells per cm2 on dishes treated with coating medium: 25 ml DMEM, 2.5 ml BSA Fraction V (1 mg/ml; sterile filtered) (both from Sigma), 500 µl Hepes pH 7.3 (1 M), 250 µl Vitrogen 100 collagen (Cohesion, Palo Alto, CA), 290 µl CaCl2 (100 mM sterile filtered, Sigma), 250 µl fibronectin (1 mg/ml, Invitrogen, Carlsbad, CA).
The freshly isolated cells were incubated for 14 hours at 37°C. Thereafter the medium was replaced and cells were grown in defined keratinocyte serum-free medium (dK-SFM) (Invitrogen) supplemented with 10 ng/ml EGF and 10−10 M cholera toxin and penicillin/streptomycin (Sigma). Spontaneously immortalized keratinocytes from control and K5-R1/R2 mice were generated (Braun et al., 2002), and three independent lines per genotype were used for some experiments and cultured in the same medium on uncoated dishes.
The immortalized human HaCaT keratinocyte cell line was cultured in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% FCS and penicillin/streptomycin.
Isolation of RNA and qRT-PCR
Separation of mouse dermis and epidermis, RNA isolation and qRT-PCR were performed as previously described (Yang et al., 2010). All PCR reactions were performed in duplicates.
Primers used for qRT-PCR: Rps29 forward: 5′-GGTCACCAGCAGCTCTACTG-3′; Rps29 reverse: 5′-GTCCAACTTAATGAAGCCTATGTCC-3′; Fak forward: 5′-TCAAAGCTGGCAGGGAGGTGAACA-3′; Fak reverse: 3′-CCATTGCACCAGGGGACCGTT-3′; Paxillin forward: 5′-GGCCCTCAATGGCACGGT-CC-3′; and Paxillin reverse: 3′-AACAAAGGGAGCCCACGCCG-3′.
Preparation of keratinocyte lysates and western blot analysis
Keratinocytes were lysed in 70 µl (per 3 cm dish) of cell lysis buffer (T-Per; Pierce, Rockford, IL), including 10 µg/µl aprotinin, 50 µg/µl leupeptin, 100 µg/µl pepstatin, 0.525 mM AEBSF, 1 mM EDTA, 1 mM Na4VO3, 20 µM PAO, 50 mM NaF, and 10 mM Na4P2O7. Cells were scraped off the dish and sonicated. After centrifugation, the protein concentration was determined using the bicinchoninic acid (BCA) kit (Pierce). Proteins were then separated by SDS–PAGE and transferred to a nitrocellulose membrane. Primary antibody incubations were performed in 5% BSA in TBS-T (10 mM Tris/HCl pH 8.0, 150 mM NaCl, 0.05% Tween 20), secondary antibodies were used in 5% nonfat dry milk in TBS-T. Membranes were probed with antibodies listed below. Equal loading and transfer efficiency were controlled by Ponceau S staining of the membrane before antibody treatment.
Immunofluorescence and immunohistochemistry
After deparaffinization, skin sections were blocked with PBS containing 3% BSA and 0.025% NP-40 for 1 hour at room temperature, and then incubated overnight at 4°C with the primary antibodies (see below) diluted in the same buffer. After three washes with PBST (1×PBS/0.1% Tween 20), slides were incubated at room temperature for 1 hour with secondary antibodies and DAPI (1 µg/ml) as counter-stain, washed with PBST again and mounted with Mowiol (Hoechst, Frankfurt, Germany). Sections were photographed using a Zeiss Imager.A1 microscope equipped with an Axiocam MRm camera and EC Plan-Neofluar objectives (10×/0.3, 20×/0.5). For data acquisition we used the Axiovision 4.6 software (all from Carl Zeiss, Inc., Oberkochen, Germany).
For immunofluorescence analysis of cultured cells, cells were washed in PBS, fixed for 30 minutes with 4% PFA at room temperature, washed with PBS and incubated for 30 minutes with 0.1% Triton X-100 in PBS. After another washing step in PBS the staining was performed as described above for immunohistochemistry. For actin staining, sections were incubated with rhodamine-coupled phalloidin (Invitrogen). For analysis of laminin-332 in the extracellular matrix, cells were seeded onto uncoated dishes and allowed to adhere. Forty-five minutes after seeding, cells were detached with EDTA and the extracellular matrix was analyzed by immunofluorescence (Margadant et al., 2009).
Stained cells were photographed using a Leica SP1-2 confocal microscope equipped with a 63×0.6–1.32 NA (Iris) PL Apo Oil objective. For data acquisition we used the Leica Confocal Software (Leica, Wetzlar, Germany).
Flow cytometry
Immortalized keratinocytes were trypsinized and stained with primary antibodies for 10 minutes on ice, washed, and stained with secondary antibodies or fluorophore-coupled streptavidin (for detection of biotinylated antibodies) for 10 minutes on ice. After washing cells were measured on a Calibur flow cytometer (BD). Active integrin β1 was detected with rat monoclonal antibody clone 9EG7.
BrdU incorporation assay
Mice were injected intraperitoneally (i.p.) with BrdU (250 mg/kg in 0.9% NaCl; Sigma) at different time points after injury and sacrificed 2 hours after injection. Skin samples were fixed in 95% ethanol/1% acetic acid. Sections were incubated with a peroxidase-conjugated monoclonal antibody directed against BrdU (Roche) and stained with diaminobenzidine. Stained sections were photographed with a Zeiss Axioskop 2 microscope equipped with an Axiocam HRc camera and Plan-Neofluor (10×/0.3, 20×/0.5) objectives. For data acquisition we used the Axiovision 4.2 software (all from Carl Zeiss Inc.).
In vitro wound healing assay
Primary or immortalized murine keratinocytes were grown to confluency in defined keratinocyte serum-free medium (dK-SFM) (Invitrogen) supplemented with 10 ng/ml EGF and 10−10 M cholera toxin. Primary keratinocyte growth medium (KGM), which included 8% Chelex-treated fetal calf serum (Chrostek et al., 2006), was added 4 hours before the experiment in a ratio of 1:3. Cells were kept in this medium throughout the migration experiment. A scratch was made within the cell layer with a sterile pipette tip. The same area was photographed under phase contrast directly after scratching and at different time points thereafter. For live cell imaging we used a Zeiss 200 M microscope with a 10×0.3NA Plan NeoFluar Objective with an incubator box. Areas, which were not covered by cells, were measured using the TScratch software (Gebäck et al., 2009). Tracks of cells in the front row and in the fifth row behind the front were recorded, and their velocity and direction were determined from photomicrographs taken at 15-minute intervals during live cell imaging using ImageJ and the manual tracking plugin-in (Iwadate and Yumura, 2009). Ruffles were counted in images taken with the same system using a 20×NeoFluar Objective at different time points.
Transwell migration assay
Transwell migration assays were performed with matrix-coated transwell plates (8 µm pore size, Corning Life Sciences, Lowell, MA). Filters were coated with BSA (100 µg/ml), vitrogen and fibronectin (10 µg/ml) in PBS for 1 hour at 37°C. Murine keratinocytes were trypsinized, suspended in dK-SFM and seeded onto the filters at a density of 4×104 cells per well. Cells that had adhered on the upper part of the membrane were counted to verify equal cell numbers. For primary keratinocytes, a larger number of cells from K5-R1/R2 mice than from control mice were seeded to compensate for the impaired adhesion. Different cytokines were added to the lower compartment prior to seeding of the cells. After 48 hours incubation, cells in the upper chamber were removed, and cells on the lower surface were fixed with 4% PFA and stained with H/E. Multiple fields were counted at high power magnification. Data represent results from at least three individual experiments.
Cell adhesion assay
Adhesion assays were performed as previously described (Czuchra et al., 2005). In brief, keratinocytes were seeded into 96-well plates that were either noncoated (PBS) or coated with BSA, fibronectin (Invitrogen), or collagen I (Serva, Heidelberg, Germany). Coating was performed using 50 µg/ml per well of the different substrates. All wells were blocked with 1% BSA in PBS pH 7.4, containing 1 mM Ca2+ and 1 mM Mg2+ for 2 h at room temperature. Cells were allowed to attach to the wells for 45 minutes at 37°C. Nonadherent cells were removed in a washing step. Remaining cells were washed with PBS and then incubated with 50 µl substrate buffer (7.5 mM NPAG, 0.1 M sodium citrate, 0.5% Triton X-100 in H2O) overnight at 37°C. 75 µl stop buffer (0.375% glycine, 5 mM EDTA in H2O) was added, and the plate was analyzed for absorption at 405 nm in a plate reader.
For analysis of FA formation, dishes were coated with the above-mentioned coating medium. Cells were seeded into the coated dishes after trypsinization and allowed to adhere for 20 or 90 minutes.
Statistical analysis
Statistical analysis was performed using the PRISM software (Graph Pad Software Inc., La Jolla, CA). Mann–Whitney U test was used for experiments examining differences between groups. *P≤0.05, **P≤0.005, ***P≤0.001.
Supplementary Material
Acknowledgments
We thank Christiane Born-Berclaz and Andreia Fernandes, ETH Zurich, Switzerland for excellent technical assistance, Dr José L. Jorcano, CIEMAT, Madrid, Spain, for the K5-Cre transgenic mice, Dr Juha Partanen, University of Helsinki, Finland, for the floxed fgfr1 mice, Dr Reinhard Fässler, Max Planck Institute of Biochemistry, Martinsried, Germany, and Dr Monique Aumailley, University of Cologne, Germany, for helpful suggestions, Dr Petra Boukamp, German Cancer Research Center Heidelberg, for kindly providing HaCaT keratinocytes.
Footnotes
Funding
This work was supported by ETH Zurich [grant number TH-08 06-3]; the Swiss National Science Foundation [grant number 310030_132884/1 to. S.W.]; and the National Institutes of Health [grant number HL105732-01 to D.M.O.]. M.M. and A-K.M. were/are members of the Zurich graduate program in Molecular Life Sciences. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.108167/-/DC1
References
- Beenken A., Mohammadi M. (2009). The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8, 235–253 10.1038/nrd2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer H. D., Florence C., Dammeier J., McGuire L., Werner S., Duan D. R. (1997). Mouse fibroblast growth factor 10: cDNA cloning, protein characterization, and regulation of mRNA expression. Oncogene 15, 2211–2218 10.1038/sj.onc.1201383 [DOI] [PubMed] [Google Scholar]
- Beer H. D., Vindevoghel L., Gait M. J., Revest J. M., Duan D. R., Mason I., Dickson C., Werner S. (2000). Fibroblast growth factor (FGF) receptor 1-IIIb is a naturally occurring functional receptor for FGFs that is preferentially expressed in the skin and the brain. J. Biol. Chem. 275, 16091–16097 10.1074/jbc.275.21.16091 [DOI] [PubMed] [Google Scholar]
- Beningo K. A., Dembo M., Kaverina I., Small J. V., Wang Y. L. (2001). Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153, 881–888 10.1083/jcb.153.4.881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer T. A., Werner S., Dickson C., Grose R. (2003). Fibroblast growth factor 22 and its potential role during skin development and repair. Exp. Cell Res. 287, 228–236 10.1016/S0014-4827(03)00139-3 [DOI] [PubMed] [Google Scholar]
- Borm B., Requardt R. P., Herzog V., Kirfel G. (2005). Membrane ruffles in cell migration: indicators of inefficient lamellipodia adhesion and compartments of actin filament reorganization. Exp. Cell Res. 302, 83–95 10.1016/j.yexcr.2004.08.034 [DOI] [PubMed] [Google Scholar]
- Braun S., Hanselmann C., Gassmann M. G., auf dem Keller U., Born–Berclaz C., Chan K., Kan Y. W., Werner S. (2002). Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol. 22, 5492–5505 10.1128/MCB.22.15.5492-5505.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadley K. N., Aquino A. M., Woodward S. C., Buckley–Sturrock A., Sato Y., Rifkin D. B., Davidson J. M. (1989). Monospecific antibodies implicate basic fibroblast growth factor in normal wound repair. Lab. Invest. 61, 571–575 [PubMed] [Google Scholar]
- Ceccarelli S., Cardinali G., Aspite N., Picardo M., Marchese C., Torrisi M. R., Mancini P. (2007). Cortactin involvement in the keratinocyte growth factor and fibroblast growth factor 10 promotion of migration and cortical actin assembly in human keratinocytes. Exp. Cell Res. 313, 1758–1777 10.1016/j.yexcr.2007.03.013 [DOI] [PubMed] [Google Scholar]
- Chmielowiec J., Borowiak M., Morkel M., Stradal T., Munz B., Werner S., Wehland J., Birchmeier C., Birchmeier W. (2007). c-Met is essential for wound healing in the skin. J. Cell Biol. 177, 151–162 10.1083/jcb.200701086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrostek A., Wu X., Quondamatteo F., Hu R., Sanecka A., Niemann C., Langbein L., Haase I., Brakebusch C. (2006). Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol. Cell. Biol. 26, 6957–6970 10.1128/MCB.00075-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuchra A., Wu X., Meyer H., van Hengel J., Schroeder T., Geffers R., Rottner K., Brakebusch C. (2005). Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol. Biol. Cell 16, 4473–4484 10.1091/mbc.E05-01-0061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essayem S., Kovacic–Milivojevic B., Baumbusch C., McDonagh S., Dolganov G., Howerton K., Larocque N., Mauro T., Ramirez A., Ramos D. M.et al. (2006). Hair cycle and wound healing in mice with a keratinocyte-restricted deletion of FAK. Oncogene 25, 1081–1089 10.1038/sj.onc.1209130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebäck T., Schulz M. M., Koumoutsakos P., Detmar M. (2009). TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 46, 265–274 [DOI] [PubMed] [Google Scholar]
- Gurtner G. C., Werner S., Barrandon Y., Longaker M. T. (2008). Wound repair and regeneration. Nature 453, 314–321 10.1038/nature07039 [DOI] [PubMed] [Google Scholar]
- Harsha A., Stojadinovic O., Brem H., Sehara–Fujisawa A., Wewer U., Loomis C. A., Blobel C. P., Tomic–Canic M. (2008). ADAM12: a potential target for the treatment of chronic wounds. J. Mol. Med. 86, 961–969 10.1007/s00109-008-0353-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig B., Borm B., Schneider H., Arin M. J., Kirfel G., Herzog V. (2007). Laminin-5-deficient human keratinocytes: defective adhesion results in a saltatory and inefficient mode of migration. Exp. Cell Res. 313, 1575–1587 10.1016/j.yexcr.2007.02.003 [DOI] [PubMed] [Google Scholar]
- Hinz B. (2007). Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 10.1038/sj.jid.5700613 [DOI] [PubMed] [Google Scholar]
- Ishibe S., Joly D., Liu Z. X., Cantley L. G. (2004). Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 16, 257–267 10.1016/j.molcel.2004.10.006 [DOI] [PubMed] [Google Scholar]
- Iwadate Y., Yumura S. (2009). Cyclic stretch of the substratum using a shape-memory alloy induces directional migration in Dictyostelium cells. Biotechniques 47, 757–767 10.2144/000113217 [DOI] [PubMed] [Google Scholar]
- Kümin A., Schäfer M., Epp N., Bugnon P., Born–Berclaz C., Oxenius A., Klippel A., Bloch W., Werner S. (2007). Peroxiredoxin 6 is required for blood vessel integrity in wounded skin. J. Cell Biol. 179, 747–760 10.1083/jcb.200706090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Firth J. D., Putnins E. E. (2009). An in vitro analysis of mechanical wounding-induced ligand-independent KGFR activation. J. Dermatol. Sci. 53, 182–191 10.1016/j.jdermsci.2008.10.008 [DOI] [PubMed] [Google Scholar]
- Margadant C., Raymond K., Kreft M., Sachs N., Janssen H., Sonnenberg A. (2009). Integrin alpha3beta1 inhibits directional migration and wound re-epithelialization in the skin. J. Cell Sci. 122, 278–288 10.1242/jcs.029108 [DOI] [PubMed] [Google Scholar]
- Martin P. (1997). Wound healing–aiming for perfect skin regeneration. Science 276, 75–81 10.1126/science.276.5309.75 [DOI] [PubMed] [Google Scholar]
- McLean G. W., Komiyama N. H., Serrels B., Asano H., Reynolds L., Conti F., Hodivala–Dilke K., Metzger D., Chambon P., Grant S. G.et al. (2004). Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 18, 2998–3003 10.1101/gad.316304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke N. B., Ward K. R., Witten T. M., Bonchev D. G., Diegelmann R. F. (2007). Impaired wound healing. Clin. Dermatol. 25, 19–25 10.1016/j.clindermatol.2006.12.005 [DOI] [PubMed] [Google Scholar]
- Meyer M., Müller A. K., Yang J., Ŝulcová J., Werner S. (2011). The role of chronic inflammation in cutaneous fibrosis: fibroblast growth factor receptor deficiency in keratinocytes as an example. J. Investig. Dermatol. Symp. Proc. 15, 48–52 10.1038/jidsymp.2011.1 [DOI] [PubMed] [Google Scholar]
- Nguyen B. P., Ryan M. C., Gil S. G., Carter W. G. (2000). Deposition of laminin 5 in epidermal wounds regulates integrin signaling and adhesion. Curr. Opin. Cell Biol. 12, 554–562 10.1016/S0955-0674(00)00131-9 [DOI] [PubMed] [Google Scholar]
- Ornitz D. M., Itoh N. (2001). Fibroblast growth factors. Genome Biol. 2, REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S., Ittmann M., Tsang S. H., Ehrlich M., Basilico C. (1998). Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc. Natl. Acad. Sci. USA 95, 5672–5677 10.1073/pnas.95.10.5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A., Page A., Gandarillas A., Zanet J., Pibre S., Vidal M., Tusell L., Genesca A., Whitaker D. A., Melton D. W.et al. (2004). A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis 39, 52–57 10.1002/gene.20025 [DOI] [PubMed] [Google Scholar]
- Sen C. K., Gordillo G. M., Roy S., Kirsner R., Lambert L., Hunt T. K., Gottrup F., Gurtner G. C., Longaker M. T. (2009). Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 17, 763–771 10.1111/j.1524-475X.2009.00543.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiling H., Werner S. (2003). Fibroblast growth factors: key players in epithelial morphogenesis, repair and cytoprotection. Curr. Opin. Biotechnol. 14, 533–537 10.1016/j.copbio.2003.08.003 [DOI] [PubMed] [Google Scholar]
- Thorey I. S., Roth J., Regenbogen J., Halle J. P., Bittner M., Vogl T., Kaesler S., Bugnon P., Reitmaier B., Durka S.et al. (2001). The Ca2+-binding proteins S100A8 and S100A9 are encoded by novel injury-regulated genes. J. Biol. Chem. 276, 35818–35825 10.1074/jbc.M104871200 [DOI] [PubMed] [Google Scholar]
- Trokovic R., Trokovic N., Hernesniemi S., Pirvola U., Vogt Weisenhorn D. M., Rossant J., McMahon A. P., Wurst W., Partanen J. (2003). FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J. 22, 1811–1823 10.1093/emboj/cdg169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui M. L., Mansbridge J. N., Carter W. G., Fujita M., Olerud J. E. (2008). Keratinocyte migration, proliferation, and differentiation in chronic ulcers from patients with diabetes and normal wounds. J. Histochem. Cytochem. 56, 687–696 10.1369/jhc.2008.951194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T., Horwitz A. F. (2004). FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154–161 10.1038/ncb1094 [DOI] [PubMed] [Google Scholar]
- Werner S., Grose R. (2003). Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 83, 835–870 [DOI] [PubMed] [Google Scholar]
- Werner S., Duan D. S., de Vries C., Peters K. G., Johnson D. E., Williams L. T. (1992). Differential splicing in the extracellular region of fibroblast growth factor receptor 1 generates receptor variants with different ligand-binding specificities. Mol. Cell. Biol. 12, 82–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S., Smola H., Liao X., Longaker M. T., Krieg T., Hofschneider P. H., Williams L. T. (1994). The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science 266, 819–822 10.1126/science.7973639 [DOI] [PubMed] [Google Scholar]
- Yang J., Meyer M., Müller A. K., Böhm F., Grose R., Dauwalder T., Verrey F., Kopf M., Partanen J., Bloch W.et al. (2010). Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J. Cell Biol. 188, 935–952 10.1083/jcb.200910126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K., Xu J., Liu Z., Sosic D., Shao J., Olson E. N., Towler D. A., Ornitz D. M. (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074 10.1242/dev.00491 [DOI] [PubMed] [Google Scholar]
- Zaidel–Bar R., Milo R., Kam Z., Geiger B. (2007). A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 120, 137–148 10.1242/jcs.03314 [DOI] [PubMed] [Google Scholar]
- Zhang H., Dessimoz J., Beyer T. A., Krampert M., Williams L. T., Werner S., Grose R. (2004). Fibroblast growth factor receptor 1-IIIb is dispensable for skin morphogenesis and wound healing. Eur. J. Cell Biol. 83, 3–11 10.1078/0171-9335-00355 [DOI] [PubMed] [Google Scholar]
- Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M., Ornitz D. M. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281, 15694–15700 10.1074/jbc.M601252200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.