

Summary
Stimulation of epidermal growth factor receptor (EGFR) initiates RAS signaling simultaneously with EGFR internalization. Endocytosed EGFR is then either recycled or degraded. EGFR fate is determined in part by the RAS effector RIN1, a guanine nucleotide exchange factor (GEF) for RAB5 GTPases. EGFR degradation was slowed by RIN1 silencing, enhanced by RIN1 overexpression and accelerated by RIN1 localization to the plasma membrane. RIN1 also directly activates ABL tyrosine kinases, which regulate actin remodeling, a function not previously connected to endocytosis. We report that RIN1-RAB5 signaling favors EGFR downregulation over EGFR recycling, whereas RIN1-ABL signaling stabilizes EGFR and inhibits macropinocytosis. RIN1QM, a mutant that blocks ABL activation, caused EGF-stimulated membrane ruffling, actin remodeling, dextran uptake and EGFR degradation. An ABL kinase inhibitor phenocopied these effects in cells overexpressing RIN1. EGFR activation also promotes RIN1 interaction with BIN1, a membrane bending protein. These findings suggest that RIN1 orchestrates RAB5 activation, ABL kinase activation and BIN1 recruitment to determine EGFR fate.
Key words: Endocytosis, EGFR, RIN1, RAB5, ABL, BIN1, 14-3-3
Introduction
Epidermal growth factor receptor (EGFR) is expressed on multiple cell types and EGFR signaling contributes to cell survival, proliferation and migration. EGF binding to EGFR induces receptor dimerization that promotes intrinsic tyrosine kinase activity (Yarden and Schlessinger, 1987). Phospho-tyrosines in the EGFR cytoplasmic region provide docking sites for adaptor proteins that engage RAS activators. This, in turn, triggers signal transduction through RAS effectors, including RAF protein kinases, PI3K lipid kinases and RIN proteins (reviewed by Colicelli, 2004).
EGFR is internalized primarily via clathrin-mediated endocytosis (CME) (Bertelsen et al., 2011), but may also enter through clathrin-independent endocytosis (CIE) including macropinocytosis (Sigismund et al., 2008). RAB5 GTPases are involved in early trafficking steps during both CME and CIE (Gorvel et al., 1991; Schnatwinkel et al., 2004). Other common early endocytic components include membrane-bending BAR domain proteins (Kishimoto et al., 2011). Less is known about the proteins that direct receptors through CME versus CIE pathways.
Endosome-localized EGFR may continue to activate downstream proteins (Sorkin and Goh, 2009), but in a manner that is distinct from plasma membrane localized EGFR signaling (Cheng et al., 2011; Hyatt and Ceresa, 2008). Within the endocytic pathway, the engagement of alternate adaptor and effector proteins dictates EGFR signal output (Casar et al., 2009). Attenuation of endosomal EGFR signaling occurs through dephosphorylation (Monteleone et al., 2006) and ligand dissociation (Maeda et al., 2002). Ultimately, internalized EGFR follows one of two possible paths. Recycling endosomes return EGFR to the plasma membrane for reuse (Medts et al., 2010), a pathway that allows continued EGF detection. The alternative path, favored at high EGF concentrations (Sigismund et al., 2008), engages late endosomes (Woodman and Futter, 2008) and leads to EGFR degradation in the lysosome (Roepstorff et al., 2009).
Activated EGFR recruits CBL (Okutani et al., 1994), an E3 ubiquitin ligase. CBL-mediated ubiquitylation is not required for internalization of EGFR (Huang et al., 2007; Huang et al., 2006), but is a major factor in determining EGFR stability. CBL overexpression increases the EGF-induced degradation of EGFR (Levkowitz et al., 1998), while a dominant negative CBL mutant can reduce EGF-induced degradation of EGFR (Longva et al., 2002) and mutation of ubiquitin attachment sites in EGFR disrupts the normal degradation pathway (Huang et al., 2006). Sustained poly-ubiquitylation by CBL is required for EGFR tethering to HGS and STAM (Bache et al., 2003; Hirano et al., 2006) and trafficking to the lysosome (Longva et al., 2002; Umebayashi et al., 2008). The death march of EGFR to lysosomes is negatively regulated by USP8-mediated de-ubiquitylation (Berlin et al., 2010).
A key step in regulating EGFR endocytosis is the activation of RAB5 GTPases by guanine nucleotide exchange factors (GEFs) of the VPS9 family, including the RAS effector RIN1 (Chen et al., 2001). The RAB5 GEF activity of RIN1 promotes early endosome fusion (Galvis et al., 2009a), an early event in transit to the lysosome. In addition, the SH2 domain of RIN1 binds to tyrosine phosphorylated EGFR and other receptor tyrosine kinases (RTKs) (Barbieri et al., 2003; Hu et al., 2008), providing a mechanism for prolonged RIN1 engagement through endocytic pathways. During the final stages of endocytosis, an association between RIN1 and STAM has been implicated in promoting RTK degradation through a late endosome to lysosome shunt (Kong et al., 2007).
RIN1 also binds and activates ABL proteins (ABL1, a.k.a c-Abl, and ABL2, a.k.a. Arg). These closely related non-receptor tyrosine kinases regulate actin remodeling through phosphorylation of actin-associated proteins and through direct F-actin binding (Colicelli, 2010). Although previous studies have implicated ABL proteins in promoting EGFR phosphorylation (Bauer et al., 2009; Plattner et al., 1999; Srinivasan et al., 2008) and actin remodeling has been demonstrated in specialized cases of mammalian cell endocytosis (Boulant et al., 2011), there is no clear mechanism connecting EGF stimulation of EGFR to the activation of ABL and actin remodeling.
We examined whether EGFR endocytosis requires RIN1 signaling through both its RAB5 GTPase and ABL tyrosine kinase pathways. Our results suggest that RIN1 coordinates the activation of RAB5 and ABL to determine the mode of receptor endocytosis, the rate of receptor ubiquitylation and degradation, and the amount of receptor recycling. In particular, an excess of RAB5 over ABL signaling triggers membrane ruffling and macropinocytosis with increased EGFR downregulation. Our data also reveal an EGF-inducible association of RIN1 with BIN1, a membrane bending protein that promotes endocytosis.
Results
Ligand concentration and RIN1 function determine EGFR ubiquitylation and degradation rate
HeLa cells express physiological levels of EGFR and are a well-characterized model for the study of EGFR trafficking (Ley and Ellem, 1992; Rush et al., 2012). Stimulation of HeLa cells using a range of EGF concentrations caused EGFR activation, as assessed by an increase in phosphorylated ERK1 and ERK2, and high concentration EGF elicited a marked downregulation of EGFR (supplementary material Fig. S1A). Subsequent experiments used EGF at 100 ng/ml (favors degradation) or 1.5 ng/ml (favors recycling), except where indicated. Intermediate EGF concentrations were used when needed to slow EGFR degradation kinetics.
RIN1 overexpression increased the EGF-induced degradation rate (supplementary material Fig. S1B,C), while RIN1 silencing decreased the degradation rate (Fig. 1A; supplementary material Fig. S1C). These results are consistent with a role for RIN1 in determining EGFR stability. EGFR stabilization in RIN1-silenced HeLa cells was less dramatic at a higher EGF concentration (100 ng/ml), leading us to consider the contribution of redundant factors. The closest RIN1 paralogs, RIN2 and RIN3, were undetectable in HeLa cells (supplementary material Fig. S1D). We did, however, detect RABGEF1 (Rabex5). This protein is the next-closest RAB5-directed GEF family member, as well as a RAS-directed E3 ubiquitin ligase. RABGEF1 silencing enhanced the stability of endogenous EGFR following high concentration EGF treatment (Fig. 1B) and the combined silencing of RIN1 and RABGEF1 caused an even greater stabilization of EGFR (supplementary material Fig. S1E,F). This is consistent with RIN1 and RABGEF1 both contributing to EGFR downregulation in response to EGF stimulation. RABGEF1 silenced cells also had an increase in EGF-induced ERK1/2 phosphorylation (Fig. 1B), which is in line with the established role of RABGEF1 in RAS→RAF→MEK→ERK pathway repression through RAS ubiquitylation (Xu et al., 2010; Yan et al., 2010). The human RAB5 GEF domain, also called VPS9 domain, family has ten members including RIN1-3 and RABGEF1 (http://www.ensembl.org). Although we did not examine the six remaining members, our analysis indicates that RIN1 plays a major role in determining EGFR fate in HeLa cells. We next examined whether the increased rate of EGFR degradation in RIN1 overexpressing cells correlated with the rate of receptor ubiquitylation following EGF treatment. Indeed, there was a marked increase in ubiquitylation at five minutes post-stimulation with 100 ng/ml EGF (Fig. 1C), suggesting that RIN1 induces EGFR degradation at least in part by promoting receptor ubiquitylation. The lysosome inhibitor bafilomycin A stabilized EGFR levels in RIN1 overexpressing cells (supplementary material Fig. S1G), consistent with a lysosome-mediated mechanism for RIN1-induced EGFR degradation.
Fig. 1.
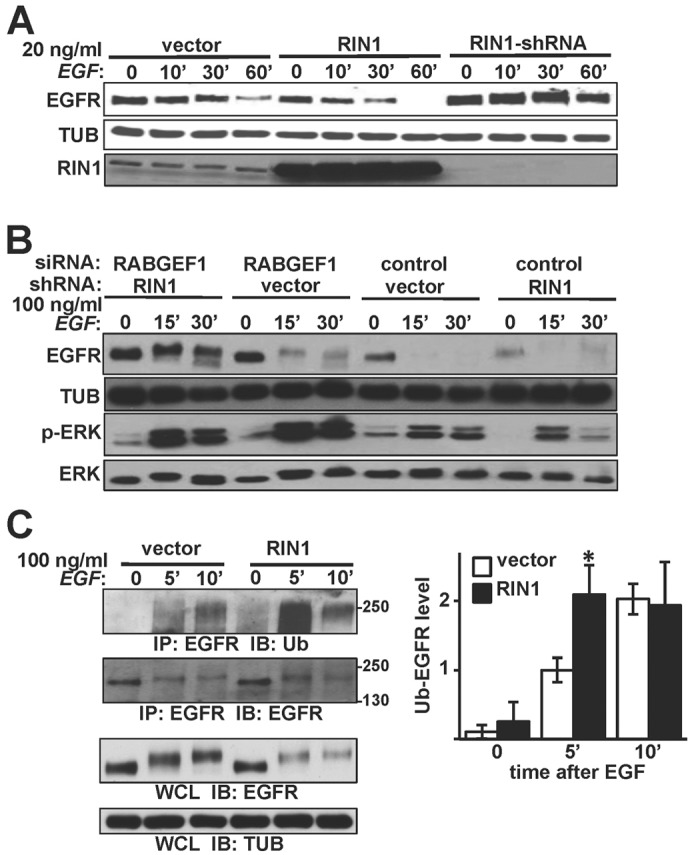
RIN1 promotes EGFR degradation after EGF stimulation. (A) HeLa cells stably transduced with vector, RIN1 or RIN1-shRNA were treated with 20 ng/ml EGF for the indicated time (minutes) and lysates immunoblotted for EGFR, RIN1 or α-tubulin (TUB). (B) Vector and RIN1-shRNA cells transfected with control or RABGEF1 siRNA were treated with 100 ng/ml EGF for the indicated time and lysates immunoblotted as indicated. (C) Left: Vector- and RIN1-transduced HeLa cells were stimulated with 100 ng/ml EGF. EGFR immunoprecipitates were blotted for EGFR or ubiquitin. Whole cell lysates (WCL) were immunoblotted for EGFR and tubulin. Right: Ubiquitin-EGFR levels normalized to total immunoprecipitated EGFR using NIH ImageJ. Mean ± s.d. of three experiments; *P<0.05. EGFR activation was confirmed by p-ERK immunoblot in A and C (data not shown).
Activated RAS can stimulate RIN1's GEF function towards RAB5 in cells overexpressing these components (Tall et al., 2001). We tested whether an EGFR→RAS→RIN1→RAB5 pathway was operational in control HeLa cells, and found that EGFR stimulation increased endogenous RAB5(GTP) relative to total RAB5 (Fig. 2A). RIN1 overexpression increased both resting and EGF-stimulated RAB5(GTP) level but a RIN1 mutant with diminished GEF activity, RIN1E574A (supplementary material Table S1) (Galvis et al., 2009b; Hu et al., 2008; Xu et al., 2010), reduced baseline and EGF-induced RAB5(GTP) to levels below detection (Fig. 2A). This dominant negative effect suggested that RIN1E574A competes with endogenous RIN1 for efficient activation of RAB5. This analysis does not distinguish among RAB5 paralogs, although RIN1 has been shown to preferentially activate RAB5A (Chen et al., 2009). Because RIN1E574A binds poorly to RAB5 (Galvis et al., 2009b), however, the limiting factor may not be RAB5 itself. These results also reinforce the model that positions RIN1 upstream of RABGEF1 in the activation of RAB5 (Xu et al., 2010).
Fig. 2.
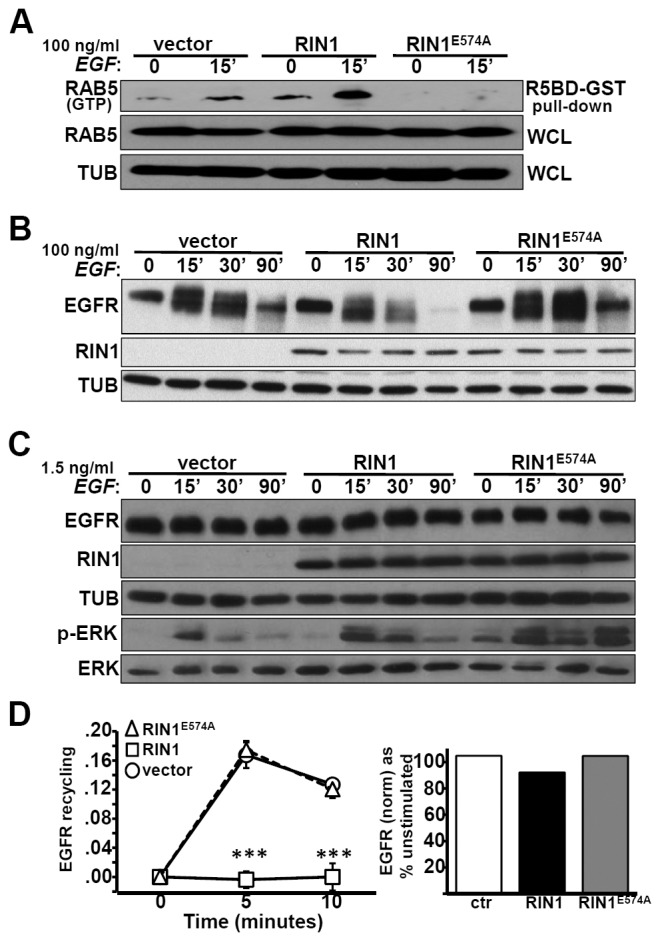
The RIN1→RAB5 signal pathway favors EGFR downregulation. (A) HeLa cells stably transduced with vector, RIN1 or RIN1E574A were treated with 100 ng/ml EGF for 0 or 15 minutes. Active RAB5 was isolated using a RAB5 binding domain fused to GST (R5BD-GST). Pull-down material and whole cell lysate (WCL) were immunoblotted with anti-RAB5 to quantify RAB5(GTP) and total RAB5, respectively. (B) Transduced HeLa cells (as in A) were stimulated with 100 ng/ml EGF for the time indicated and lysates immunoblotted for EGFR and RIN1. (C) Transduced HeLa cells (as in A) were stimulated with 1.5 ng/ml EGF for the time indicated and lysates immunoblotted for EGFR, RIN1, ERK1/2 and p-ERK1/2. (D) Left: Transduced HeLa cells were pulsed with 10 ng/ml EGF-Alexa Fluor 647 for 5 minutes. Cells were acid-stripped and chased in serum-free medium at 37°C for the time indicated. The data represent fluorescence of acid-stripped recycled EGF-AF647 normalized to bound EGF at time 0 (data from two independent experiments, each performed in duplicate); ***P<0.0001. Right: Total EGFR in transduced HeLa cells stimulated with 10 ng/ml EGF for 10 minutes. Final EGFR levels (normalized to tubulin) are shown as a percentage of starting EGFR (unstimulated). EGFR activation was confirmed by p-ERK immunoblot in all experiments (data not shown).
RIN1E574A slowed the rate of EGFR degradation following high concentration EGF treatment (Fig. 2B), consistent with a required role for active RAB5 in receptor downregulation. Cells expressing RIN1E574A had smaller endosomes than control or RIN1 cells (supplementary material Fig. S2), reflecting the contribution of RIN1-RAB5 signaling in early endosome fusion (Galvis et al., 2009b). In addition, the RIN1E574A mutant moderately prolonged downstream signaling, as judged by ERK phosphorylation (Fig. 2C). We reasoned that reduced RAB5 activity might favor receptor recycling. Indeed, while EGFR recycling was blocked in cells overexpressing RIN1, recycling was observed at control levels in cells expressing the RIN1E574A mutant (Fig. 2D). These results strongly support the conclusion that RIN1→RAB5 signaling promotes EGFR degradation over recycling.
RIN1-mediated ABL activation stabilizes EGFR
The RAS effector functions of RIN1 include the activation of ABL tyrosine kinases (Cao et al., 2008; Hu et al., 2005; Ziegler et al., 2011), which regulate actin remodeling (reviewed by Colicelli, 2010). Initial weak binding leads to RIN1 phosphorylation by ABL. RINI phosphorylated at Tyr36 (pY36-RIN1) then binds the ABL SH2 domain, creating a stable interaction that derepresses ABL autoinhibition and increases catalytic efficiency (Cao et al., 2008; Hu et al., 2005). EGF treatment caused a marked increase in the phosphorylation of RIN1 as well as CRKL, an established ABL substrate (Fig. 3A). This is consistent with signal transduction from EGFR through RAS to RIN1, which then directly activates ABL tyrosine kinases concurrent with RAB5 stimulation. Overexpression of wild-type RIN1 greatly boosted the pY36-RIN1 signal at all time points, as expected, with a more modest increase in the pY-CRKL signal (Fig. 3A). We next employed a RIN1 mutant that carries Y→F mutations at codon 36 as well as three secondary phosphorylation sites, greatly diminishing the capacity of RIN1 to bind and activate ABL kinases (Hu et al., 2008) without altering its RAB5 GEF domain (supplementary material Table S1). Expression of this mutant, RIN1QM (supplementary material Table S1), eliminated the tyrosine phosphorylation signal from endogenous RIN1 and reduced the phosphorylation of CRKL (Fig. 3A). This repression of ABL-mediated phosphorylation was most likely attributable to dominant interference with endogenous RIN1. We observed a modest decrease in ABL1, but not ABL2, in RIN1QM expressing cells (data not shown), and this may have contributed to the diminished pY signal.
Fig. 3.
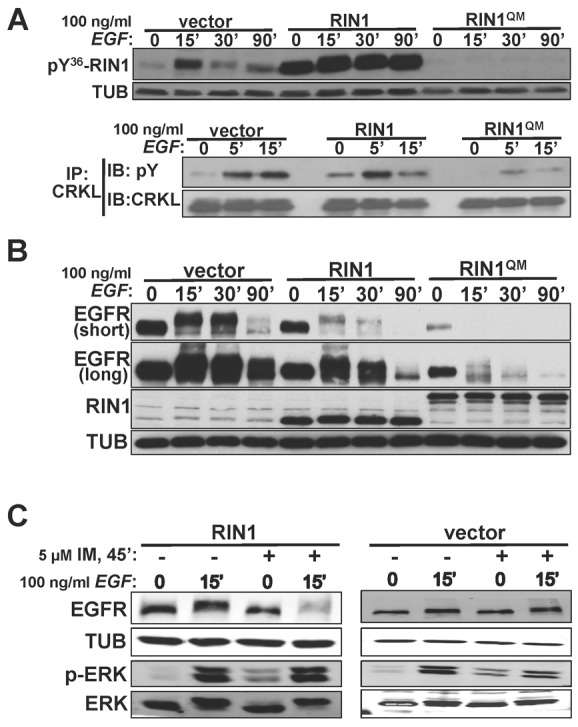
EGF activates RIN1→ABL signaling to regulate EGFR downregulation. (A) Top: HeLa cells transduced with vector, RIN1 or RIN1QM were stimulated with 100 ng/ml EGF for the indicated times, lysated and immunoblotted with anti-pY36-RIN1. Bottom: Cells were treated as shown and lysates subjected to anti-CRKL immunoprecipitation. After immunoblotting this material with pY36, the blot was stripped and reprobed with anti-CRKL. (B) Transduced HeLa cells were EGF-stimulated (as in A) and lysates immunoblotted. Two exposures of the EGFR blot are shown. (C) HeLa cells stably transduced with vector or RIN1 were pre-treated (or not) with 5 µM imatinib and stimulated (or not) with EGF. Cell lysates were immunoblotted for EGFR, ERK1/2, p-ERK1/2 and tubulin (TUB). EGFR activation was confirmed by p-ERK immunoblot in A, B and C (data not shown).
Notably, stable RIN1QM cells had a reduced steady state level of EGFR protein (Fig. 3B), which correlated with reduced EGFR mRNA (supplementary material Fig. S3B). This apparent feedback repression of EGFR transcription may be due to persistent EGFR downregulation (see below). For this reason we focused our analysis primarily on normalized, EGF-induced changes in receptor levels.
Expression of RIN1QM accelerated the rate of ligand-induced EGFR degradation beyond that resulting from wild-type RIN1 overexpression (Fig. 3B; supplementary material Fig. S3A). This finding suggested that RIN1 functions downstream of EGFR to promote ABL pathways antagonistic to receptor degradation. We therefore tested the effect of imatinib, an ABL kinase inhibitor, on EGF-induced EGFR downregulation. Imatinib did not enhance EGFR degradation in control cells, but did have a pronounced effect in cells overexpressing wild-type RIN1 (Fig. 3C). Hence the more rapid EGFR degradation was likely due to an imbalance between elevated RAB5 activation (by RIN1QM or RIN1) without a commensurate increase in ABL activation (blocked by QM mutation or imatinib).
EGF induced multiple membrane ruffles in RIN1QM expressing cells. These structures had an underlying F-actin network and were enriched for the RIN1QM protein (Fig. 4A). Membrane ruffling was not observed in cells overexpressing wild-type RIN1 unless they were pre-treated with the ABL kinase inhibitor imatinib prior to EGF treatment (Fig. 4B). These structures are reminiscent of those seen in macropinocytosis, a clathrin- and dynamin-independent endocytosis mechanism favoring receptor degradation and characterized by F-actin supported membrane ruffles at cholesterol-rich domains (Donaldson et al., 2009; Lee and Knecht, 2002). To test if the membrane perturbations we observed were indicative of macropinocytosis, we treated RIN1QM expressing cells with filipin. Over the course of an hour this cholesterol-stripping drug partially rescued the basal EGFR downregulation observed in RIN1QM expressing cells (Fig. 5A). These results suggested that activating RAB5 while blocking ABL kinases (by the QM mutation or by imatinib), promotes macropinocytosis of EGFR.
Fig. 4.
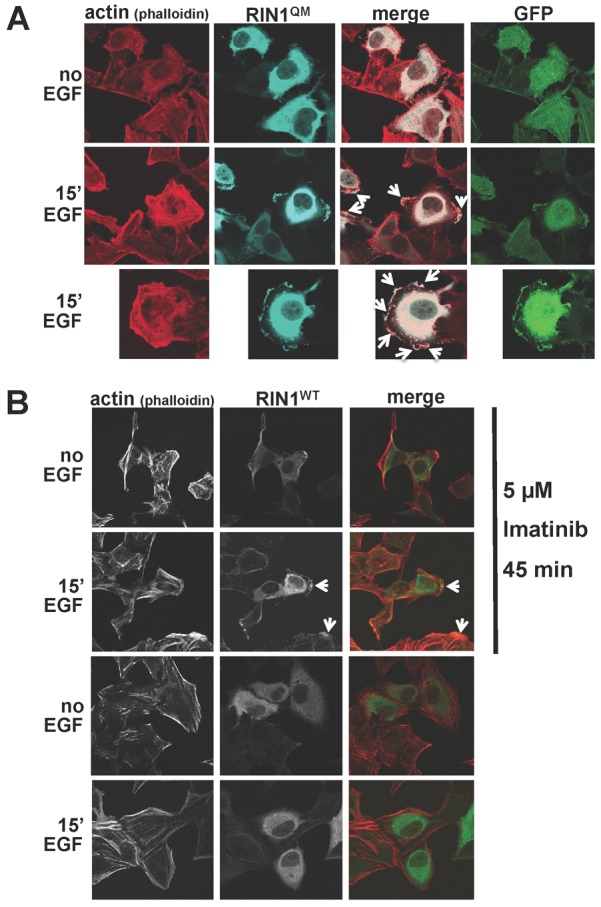
Strong RAB5 activation without ABL activation causes EGF-induced membrane ruffles. (A) HeLa cells transduced with RIN1QM were stimulated or not with 100 ng/ml EGF for 15 minutes. Fixed and permeabilized cells were stained with phalloidin (red) and anti-RIN1 (light blue). Arrows point to membrane ruffles. Untransfected cells in the same field (no anti-RIN1 or GFP signal) serve as internal controls. (B) RIN1-transduced HeLa cells were pre-treated, or not, with 5 µM imatinib for 45 minutes. and stimulated, or not, with 100 ng/ml EGF for 15 minutes. Fixed and permeabilized cells were stained with phalloidin (red) or anti-RIN1 (green). Arrows point to membrane ruffles.
Fig. 5.
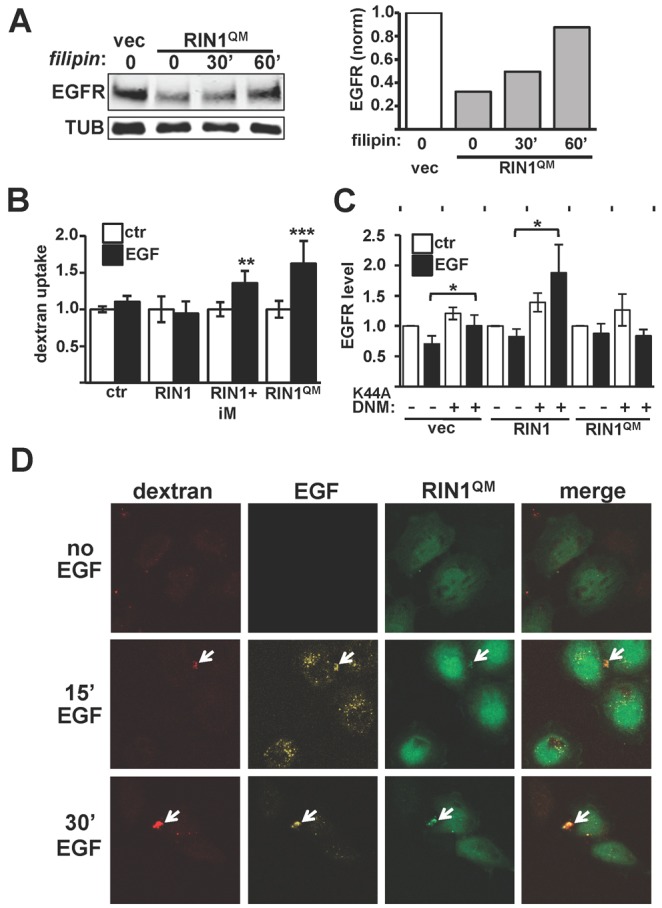
Enhanced RAB5 signaling without ABL signaling induces macropinocytosis. (A) Left: HeLa cell transduced with vector or RIN1QM were treated, or not, with 5 µg/ml filipin for the indicated time and lysates probed with anti-EGFR. Right: EGFR levels normalized to TUB. (B) Fluorescent dextran uptake in transduced HeLa cells; iM indicates pre-treated with 5 µM imatinib for 45 minutes. Other samples were mock-treated. Values are means ± s.d. of 5–7 data points. (C) HeLa cells transduced with vector, RIN1 or RIN1QM were transfected with control vector (−) or dominant-negative dynamin (K44A-DNM). EGFR levels, normalized to TUB, were determined for control and EGF-treated (20 ng/ml, 60 minutes) cells. Values are means ± s.d. of three experiments. (D) RIN1QM-transduced HeLa cells were pulsed with 20 ng/ml EGF and 1 mg/ml dextran for 5 minutes and chased in serum-free medium for the indicated time. RIN1QM-transduced cells are GFP-positive (untransduced cells serve as internal controls). Arrows indicate co-localized dextran and EGF. Red, dextran; yellow, EGF; green, GFP. *P<0.05, **P<0.005, ***P<0.0005.
F-actin based membrane ruffles and cholesterol dependence are, together, strongly indicative of macropinocytosis. We next assayed for macropinocytosis-associated large solute uptake. HeLa cells overexpressing wild-type RIN1 showed no EGF-induced dextran incorporation. In contrast, RIN1QM cells and RIN1 overexpression cells treated with an ABL inhibitor, both of which showed EGF-induced membrane ruffling, had significant dextran uptake (Fig. 5B). In addition, EGFR stability was increased by dominant negative dynamin (DNMK44A) in cells transduced with vector or RIN1 but not RIN1QM (Fig. 5C; supplementary material Fig. S3D). This dyamin-independence is also consistent with macropinocytosis in the EGF-stimulated RIN1QM cells. Co-localization of fluorescent dextran and EGF on RIN1QM cells, but not RIN1 cells, indicated that a significant portion of EGFR was entering cells via macropinosomes (Fig. 5D; supplementary material Fig. S4A). Together, these results demonstrate that strong activation of RAB5 by RIN1, without the concomitant activation of ABL tyrosine kinases, leads to EGF-induced macropinocytosis.
Because the receptor-targeted E3 ligase CBL plays a critical role in directing activated EGFR through a degradation pathway, we considered whether changes in CBL levels might contribute to the effect of RIN1QM on EGFR stability. RIN1QM cells showed elevated levels of CBL compared to control HeLa cells, while wild type RIN1 overexpressing cells had an intermediate increase in CBL (supplementary material Fig. S4B). There was also an increase in EGFR-associated CBL in cells overexpressing RIN1 or RIN1QM (supplementary material Fig. S4C). EGF stimulation had little effect on total CBL protein levels. These findings suggest that RIN1-mediated ABL signaling might normally reduce EGFR degradation in part by de-stabilizing CBL.
Plasma membrane localization of RIN1 facilitates EGFR downregulation
Post receptor internalization, signaling events continue to take place at endosome membranes where RAS proteins engage effectors, such as RIN1, and where RAB5 proteins are localized by C-terminal prenylation. Translocation of RIN1 between cytoplasmic and membrane compartments is controlled in part by Ser351 phosphorylation-dependent binding to 14-3-3 proteins. The RIN1S351A mutant (supplementary material Table S1) shows reduced 14-3-3 binding and enhanced membrane residence (Wang et al., 2002) as well as increased association with EGFR (Hu et al., 2008). We reasoned that greater membrane association should enhance RIN1-mediated effects on EGFR downregulation. Indeed, expression of RIN1S351A increased ligand-induced EGFR downregulation, when compared with cells that overexpress wild-type RIN1 (Fig. 6A). Even using a low EGF concentration that had little effect on endogenous EGFR levels in control HeLa cells, RIN1S351A expressing cells showed marked receptor downregulation (supplementary material Fig. S5A). As with RIN1QM cells, RIN1S351A cells had lower basal EGFR protein levels, but they showed no change in the amount of transferrin receptor (supplementary material Fig. S5B), indicating that RIN1-induced downregulation of basal receptor levels is receptor type specific.
Fig. 6.
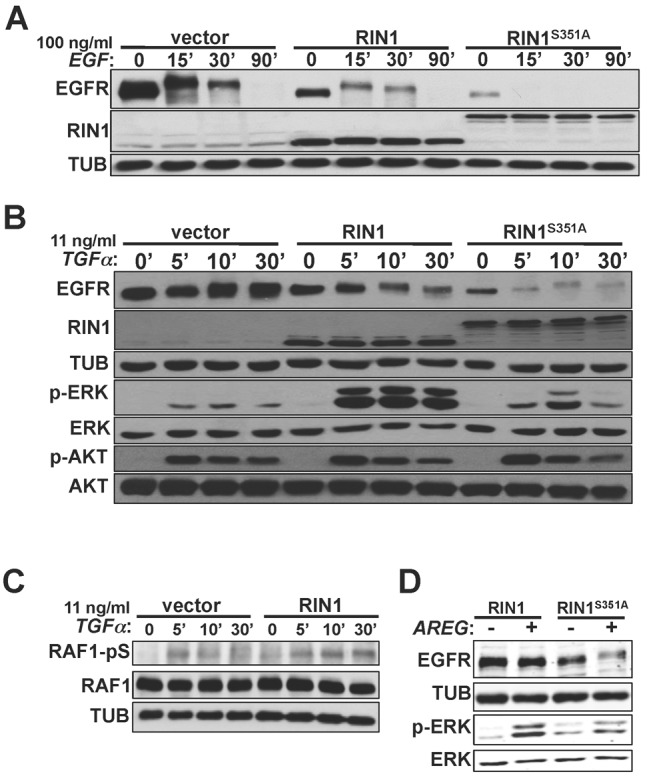
Plasma membrane localization of RIN1 promotes EGFR degradation in a ligand-independent manner. (A) HeLa cells transduced with vector, RIN1 or RIN1S351A were stimulated with 100 ng/ml EGF for the indicated times, and cell lysates immunoblotted for EGFR and RIN1. EGFR activation was confirmed by p-ERK immunoblot (data not shown). (B) HeLa cells transduced with vector, RIN1 or RIN1S351A were stimulated with 11 ng/ml TGFα for the indicated times, and lysates immunoblotted for EGFR, RIN1, tubulin, total ERK1/2, p-ERK1/2, total AKT and p-AKT. (C) Transduced HeLa cells were stimulated with 11 ng/ml TGFα for the indicated times, and lysates immunoblotted for total RAF1 and RAF1 phosphorylated at Ser338 (RAF1-pS). (D) HeLa cells transduced with RIN1 or RIN1S351A were transfected with a vector (−) or amphiregulin (AREG) construct (+). Cell lysates prepared 48 hours later were immunoblotted for EGFR, tubulin, p-ERK1/2 and total ERK1/2.
Surprisingly, we observed relatively normal induction of phosphorylated ERK (p-ERK) levels following low concentration EGF treatment of cells expressing RIN1S351A (supplementary material Fig. S5A) or RIN1QM (supplementary material Fig. S3C), despite lower levels of EGFR in these cells. These results suggest that the functions disrupted in these RIN1 mutants are not required for a normal ERK signaling response to EGF, and may even have helped to compensate for lower receptor levels.
RIN1 contributes to EGFR fate following activation by ligands other than EGF
EGFR can be activated by several non-EGF ligands with distinct effects on downstream signaling and receptor degradation (reviewed by Harris et al., 2003). Transforming growth factor alpha (TGFα) binds EGFR and triggers many of the same downstream signaling pathways as EGF, including those mediated by phosphorylation of ERK and AKT (Martínez-Arca et al., 2005). Once in the early endosome, however, TGFα dissociates from EGFR more readily than EGF, causing most TGFα-activated EGFR to be recycled rather than degraded (Roepstorff et al., 2009). TGFα levels that gave no discernible EGFR degradation in control cells led to pronounced EGFR downregulation in RIN1 overexpressing cells and RIN1S351A expressing cells (Fig. 6B), consistent with a RIN1 contribution that is receptor activation dependent but ligand independent.
A surprisingly robust and prolonged p-ERK signal was observed following TGFα treatment of RIN1 overexpressing cells, compared to control cells (Fig. 6B). This enhanced and sustained burst of p-ERK was muted in RIN1S351A expressing cells, suggesting that RIN1 translocation from the membrane may be required for normal downstream signaling from TGFα-stimulated EGFR. The increased levels of p-ERK in RIN1 overexpressing cells correlated with prolonged levels of RAF1-S338 phosphorylation (Fig. 6C). This modification is associated with enhanced kinase activity for MEK, which in turn phosphorylates ERK. Reduced dephosphorylation might also contribute to higher p-ERK levels. This enhanced signaling effect of TGFα seemed to be pathway specific, however, as only normal levels of p-AKT were detected in the RIN1 overexpressing cells.
Amphiregulin (AREG) is another physiological ligand with low affinity for EGFR. Compared to EGF, AREG produces less EGFR phosphorylation (Willmarth et al., 2009). AREG treatment activates RAS signaling pathways but is not normally accompanied by EGFR degradation, leading instead to the accumulation of cell surface EGFR (Willmarth et al., 2009). AREG treatment produced little apparent change in EGFR levels in RIN1 overexpressing cells, but did cause a drop in EGFR levels in RIN1S351A cells (Fig. 6D). Our results suggest that RIN1 signaling, enhanced by plasma membrane localization in the RIN1S351A mutant, promotes degradation of EGFR following activation by a broad range of stimuli.
Additional RIN1 partners contribute to EGFR signaling
RIN1 encodes a proline-rich (PR) sequence with consensus motifs for binding SH3 and/or WW domains (Li, 2005). Following treatment with high EGF, cells expressing a RIN1 mutant missing the largest proline-rich motif (RIN1ΔPR) showed a pattern of EGFR degradation more similar to control cells than to cells overexpressing wild-type RIN1 (Fig. 7A). This result suggested that the RIN1ΔPR mutant is compromised for interactions that promote EGFR degradation.
Fig. 7.
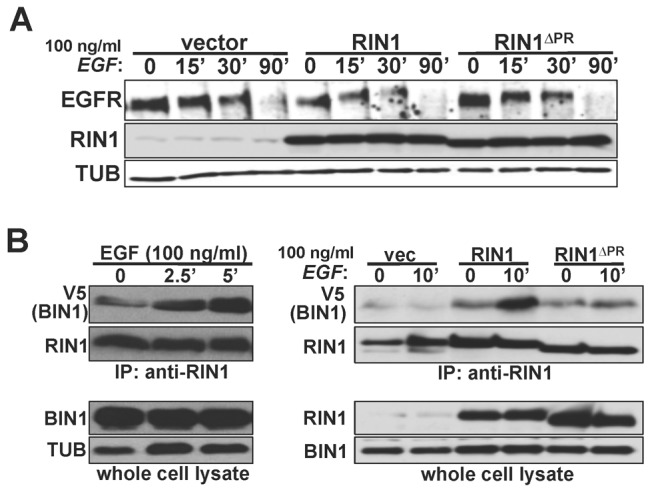
EGF triggers BIN1 recruitment to RIN1. (A) HeLa cells transduced with vector, RIN1 or RIN1ΔPR were stimulated with 100 ng/ml EGF for the time indicated. Cell lysates were immunoblotted for EGFR, RIN1 and tubulin (TUB). (B) Left: RIN1-transduced cells were transfected with BIN1-V5 and stimulated with 100 ng/ml EGF for the time indicated. Anti-RIN1 immunoprecipitates and whole cell lysates were blotted for V5 (BIN1) and RIN1. Right: HeLa cells transduced with vector, RIN1 or RIN1ΔPR were stimulated, or not, with EGF (100 ng/ml, 10 minutes). Anti-RIN1 immunoprecipitates and whole cell lysates were blotted for V5 (BIN1) and RIN1. RIN1ΔPR brought down 60% less BIN1 from unstimulated cells than did wild-type RIN1 (normalized to RIN1 in immunoprecipitate; n = 3; P<0.05). EGFR activation was confirmed by p-ERK immunoblot for A and B (data not shown).
The PR domain of RIN1 has been implicated in binding to the SH3 domain of STAM2 (Kong et al., 2007). STAM proteins are components of ESCRT0 complexes that facilitate receptor transit from endosomes to multi-vesicular bodies and lysosomes. In agreement with an earlier study using a similar but not identical deletion of the RIN1 proline-rich motif, RIN1ΔPR was compromised for association with STAM2 (supplementary material Fig. S7). This may account for the normal EGFR downregulation rate, even when RIN1ΔPR was expressed at a level similar to that at which wild type RIN1 enhanced EGFR degradation.
A similar PR sequence in RIN3, a RIN1 paralog, has been reported to bind BIN1 (a.k.a. amphiphysin 2) in two-hybrid assays (Kajiho et al., 2003). Because BIN1 is a membrane bending protein implicated in trafficking, we tested whether RIN1 also interacts with BIN1. Indeed, the association of RIN1 with BIN1 was readily apparent and was markedly enhanced within five minutes of EGF treatment (Fig. 7B). The RIN1::BIN1 association was reduced by 2.5-fold for the RIN1ΔPR mutant (Fig. 7B). This is consistent with an interaction between the RIN1 PR domain and the BIN1 SH3 domain, analogous to the proposed interaction of RIN3 with BIN1 (Kajiho et al., 2003). These findings suggest a model in which RIN1 recruits BIN1 to activated receptors at an early stage of endosome formation when the membrane bending properties of BIN1 might aid the formation or resolution of early endosome structures.
Because the RIN1 SH2 domain mediates binding to activated EGFR (Barbieri et al., 2003) and other receptor tyrosine kinases (Hu et al., 2008), we next determined the effect of mutating a residue required for phosphotyrosine binding. Although RIN1R94N has a reduced affinity for activated EGFR (Hu et al., 2008), expression of this mutant had little effect on EGFR degradation or p-ERK levels after high concentration EGF treatment (supplementary material Fig. S6A,B). This suggests that RIN1R94N (unlike RIN1E574A and RIN1QM) does not interfere in a dominant manner with endogenous RIN1, at least in this system.
Discussion
RIN1 is a RAS effector protein that regulates endocytosis and signaling of receptor tyrosine kinases such as EGFR. RIN1 encodes a GEF domain that activates RAB5 GTPases governing early endosome traffic (Barbieri et al., 2004; Barbieri et al., 2003). A separate RIN1 domain activates ABL tyrosine kinases and actin remodeling, but this had not been implicated in endocytosis. Here we demonstrate that EGF binding to EGFR triggers ABL activation, and we dissect out the unique contributions of RAB5 and ABL during EGFR endocytosis. We propose that a balance of RIN1→RAB5 and RIN1→ABL signaling controls the internalization route and eventual fate of activated receptors (Fig. 8). These studies also suggest that efficient receptor trafficking requires the coordination of RIN1 subcellular localization and BIN1 binding.
Fig. 8.
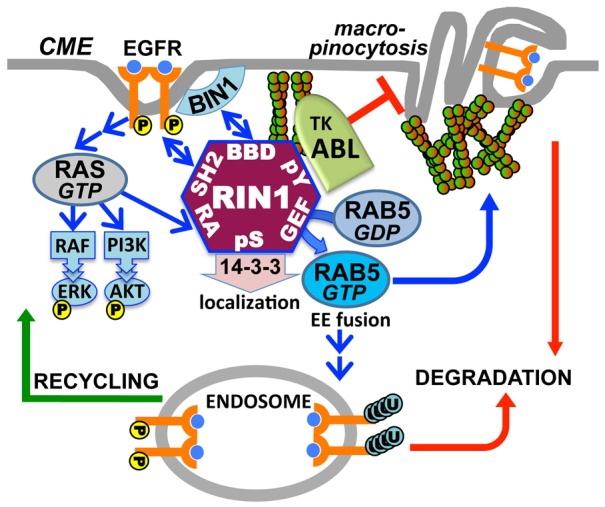
Model of coordinated RIN1 signal pathways in EGFR endocytosis. Upon EGF stimulation, the RAS effector RIN1 activates RAB5 to drive early endosome fusion and promote EGFR degradation. Concomitant ABL activation regulates actin remodeling to inhibit macropinocytosis, which otherwise results from unchecked RAB5 signaling. Strong ABL activation in the absence of RAB5 activation favors EGFR stability and recycling. Recruitment of BIN1 to the BIN1 binding domain (BBD) facilitates membrane bending and endocytosis, whereas 14-3-3 binding inhibits plasma membrane access and endocytosis.
RIN1-ABL signaling regulates the route of EGFR internalization
ABL proteins have been implicated in receptor tyrosine kinase signaling and endocytosis (Tanos and Pendergast, 2006). ABL1 directly phosphorylates several receptor tyrosine kinases as well as CAV1 (caveolin) and the ubiquitin ligase CBL (Andoniou et al., 1996). The combined effects of ABL activation normally promote EGFR stability (reduced degradation). This may result in part from blocking CBL recruitment and disrupting an EGF-mediated CBL-Abi1 interaction (Tanos and Pendergast, 2006; Tanos and Pendergast, 2007). Consistent with a role for ABL in protecting RTKs from downregulation, RIN1QM (mutant that cannot activate ABL) blocked EGF-induced ABL tyrosine kinase activity and accelerated the EGF-induced receptor degradation rate. We also noted lower EGFR mRNA levels in RIN1QM cells. Regulation of EGFR gene transcription has been previously noted (Clark et al., 1985), and in this circumstance persistent EGFR downregulation may lead to EGFR gene repression in order to avoid futile cycles of receptor synthesis.
RIN1QM induced membrane ruffling and macropinocytosis, suggesting that elevating RAB5(GTP) levels without a commensurate increase in ABL activity can alter the route of internalization. This interpretation is supported by the fact that treatment of wild-type RIN1 overexpressing cells with the ABL inhibitor imatinib phenocopied what was observed in RIN1QM cells. Dextran uptake, EGFR and dextran co-localization, cholesterol dependence and dynamin indpendence all support a macropinocytosis mechanism, which is known to favor receptor degradation over recycling (Sigismund et al., 2008). Consistent with our findings, macropinocytosis is promoted by RAB5 (Gorvel et al., 1991; Lanzetti et al., 2004; Schnatwinkel et al., 2004) and CBL (Chakraborty et al., 2011) but inhibited by the ABL substrate Abi1 (Dubielecka et al., 2010). Conversely, blocking RIN1's GEF function (RIN1E574A) restored the EGFR recycling that was greatly diminished in RIN1 overexpression cells. Hence, the coordination of RIN1→ABL and RIN1→RAB5 pathways appears necessary to regulate receptor internalization in a way that balances degradation and recycling.
RIN1 localization and additional partners regulate early stages of receptor endocytosis
Upon RTK stimulation, RIN1 translocates from the cytoplasm to plasma membrane through a mechanism that involves 14-3-3 binding (Wang et al., 2002) and is similar to the localization control of other RAS effectors such as RAF. The 14-3-3 binding mutant RIN1S351A dramatically enhanced EGFR degradation. This may result from the proximity of RIN1 to RAS, EGFR and the endocytosis machinery, highlighting the importance of properly timed RIN1 localization in endocytosis.
This study also implicates the membrane bending protein BIN1 in early endocytosis by RIN1, because the RIN1::BIN1 interaction was markedly increased by EGF stimulation. The RIN1ΔPR mutant showed reduced binding to BIN1, suggesting an association through proline-rich (RIN1) to SH3 (BIN1) domains. Recruitment of BIN1 could facilitate membrane deformations associated with early endosome fusion, a function that RIN1 is known to regulate. A report that BIN1 interacts with the RIN family member RIN3 (Kajiho et al., 2003) implies a conserved connection between RIN proteins and membrane bending. Notably, a proline-rich segment of RIN1 also interacts with the STAM protein complex that mediates late stage EGFR trafficking and degradation (Kong et al., 2007). The extent to which BIN1 directly participates in RIN1-mediated receptor trafficking remains uncertain, however, and will require further study.
RIN1 overexpression caused higher CBL levels and more EGFR-associated CBL compared to control cells. The RIN1QM mutant showed even higher levels of total CBL and EGFR-associated CBL, suggesting that ABL activation is not required for – and may actually antagonize – these effects. Aside from its role as a major E3-ubiquitin ligase for receptor tyrosine kinases, CBL functions as an adaptor protein that regulates actin cytoskeleton dynamics (reviewed by Schmidt and Dikic, 2005). Our results are consistent with CBL participation at multiple levels in RTK endocytosis.
RIN1 plays multiple roles in EGFR trafficking
RTK activation triggers downstream signal transduction through RAS proteins and their effectors including RIN1. Our data demonstrate that RIN1 regulates RTK endocytosis not only through RAB5 activation and RTK binding, but also through its ABL tyrosine kinase activating function. This highlights the need to further define the role played by ABL tyrosine kinases and cytoskeleton remodeling in endocytosis. Importantly, it is the balance of RAB5 and ABL activation that determines receptor fate (Fig. 8). Additional interactions with BIN1 and STAM suggest that RIN1 orchestrates multiple effectors and binding partners to shepherd activated RTKs through successive stages of endocytosis and determine the degree of receptor downregulation versus recycling. More broadly, our results illustrate the capacity of multi-domain proteins to coordinate seemingly disparate pathways to regulate alternate outcomes.
Materials and Methods
Expression constructs
All RIN1 expression constructs were made in lentivirus vectors. RIN1 wild type, RIN1E574A in pM4-blastR vector (Milstein et al., 2007); RIN1QM and RIN1S351A were in the M4-IRES-GFP vector (Hu et al., 2008). Virus production and transduction were performed as previously described (Wang et al., 2002). To create RIN1ΔPR, 149 base pairs surrounding the proline-rich region of RIN1 were removed by restriction digest with KpnI and HindIII and replaced with 89 bp of overlapping oligonucleotides for a precise deletion of 20 codons. The final construct was cloned into pM4-blastR. For stable RIN1 knockdown, RIN1 shRNA containing lentivirus vector pLKO.1-puroR was used as described previously (Thai et al., 2011). The RIN1 mutants and their properties are summarized in supplementary material Table S1.
The RAB5-GTP pulldown construct was created using the Zn2+ finger domain of rabenosyn (ZFYVE20), which functions as a RAB5 binding domain (R5BD) (Merithew et al., 2003). The sequence encoding aa 1–40 of human ZFYVE20 was amplified using forward primer 5′-ATGCGCTAGCAGATCTACTAGTATGGCTTCTCTGGACGACCC and reverse primer 5′-ATGCGGATCCTTATCTAGATTCCCCTGAGTGTTCTTCCT. Ligation compatible restriction sites in each primer (XbaI/SpeI and BamHI/BglII) were used for stepwise head-to-tail additions in pKS bluescript. The 4x concatemer was then inserted into the BamHI-EcoRI sites of pGEX-2T to create pGEX-4xZFYVE (R5BD-GST).
HA-STAM2 (Kong et al., 2007) (a gift from Dr Philip D. Stahl, Department of Cell Biology and Physiology, Washington University School of Medicine) was used to amplify the STAM2 gene and this was introduced into the pKS vector along with an N-terminal V5 tag, added using adaptor oligos. The final construct V5-STAM2 was introduced into the M4-neoR lentivirus vector. The BIN1 cDNA was amplified out of pcDNA3-BIN1 (a gift from Dr Corrine Leprince) and introduced into a M4-neoR-V5 lentivirus vector to obtain M4-neoR-BIN1-V5 construct. pcDNA3-RIN2-HTM and M4-blastR-RIN3-Flag tagged constructs were used for transient transfection of RIN2 and RIN3, respectively, in HeLa cells.
Cell culture and reagents
HeLa cells were cultured in DMEM (Media Tech) with 10% Fetal Bovine Serum (Hyclone) and 1% penicillin streptomycin (Invitrogen). HeLa cells stably expressing M4-blastR constructs were established by lentivirus infection followed by selection with 4 µg/ml blasticidin (Invitrogen). All transfection experiments were performed using Polyfect reagent (Qiagen).
EGF (Invitrogen) was used at 100 ng/ml, 20 ng/ml or 1.5 ng/ml as indicated. TGFα (Invitrogen) was used at 11 ng/ml. pcDNA3-amphiregulin-neomycin (a gift from Dr Anson, Stanford University) was transfected into HeLa cells and the cells were selected with 400 µg/ml G418 (Gibco). Filipin (Sigma Aldrich) pretreatment of cells was carried out using 10 µg/ml for 4 or 12 hours at 37°C. Bafilomycin (Sigma Aldrich) treatment was done on cells at 0.25 µM for 1 hour at 37°C. Imatinib treatment was performed at 5 µM for 45 minutes at 37°C.
Growth factor stimulation was performed by serum starving cells overnight followed by addition of serum-free medium with the indicated concentration of the growth factor. The cells were then incubated at 37°C for the indicated time, washed with cold PBS and lysed.
Gene silencing
HeLa cells with stable silencing of RIN1 were established by infection with pLKO.1-shRIN1-puroR and selection with 2 µg/ml puromycin (Invivogen) (Thai et al., 2011). Expression levels were evaluated by western blotting. RABGEF1 silencing was achieved by transfection of the targeted or scramble vectors- pcDNA3.Super-Rabex5 shRNA/pCDNA3.Super-scramble shRNA (a gift from Dr Dafna Bar-Sagi, NYU) (Xu et al., 2010) followed by selection with 4 µg/ml blasticidin in naïve HeLa cells or with 4 µg/ml blasticidin and 2 µg/ml puromycin in RIN1-silenced HeLa cells.
RAB5 pull-down assay
RAB5-GTP binding domain coated glutathione-Sepharose (GE Healthcare) beads were prepared by expressing 4xZFYVE-GST (R5BD-GST) in BL21 bacterial cells which were induced with 1 mM IPTG (Fisher) at 30°C for 3 hours. The cells were then pelleted, washed in ice cold PBS and resuspended in lysis buffer (250 mM NaCl, 20 mM Tris pH 8.0, 10% glycerol, 0.01% Triton-X-100, 1 mM PMSF (Sigma Aldrich), 10 µg/ml leupeptin (Sigma Aldrich), 1 µM pepstatin (Sigma Aldrich) and EDTA-free Protease Inhibitor Cocktail tablet (Roche). The lysate was incubated with glutathione Sepharose beads (GE Health) for one hour at 4°C. The beads coated with 4X-ZFYVE-GST were then washed three times with the lysis buffer and resuspended in the lysis buffer.
HeLa cells, unstimulated or stimulated, were lysed in NP-40 lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40) containing 1 mM PMSF, 10 µg/ml leupeptin, 1 µM pepstatin, 1 mM sodium orthovanadate (Sigma Aldrich) and 10 mM MgCl2 to stop the exchange reaction. 1000 µg of total lysate protein was used for pull-down of activated RAB5 and added to 4X-ZFYVE bound glutathione-Sepharose beads described above. After overnight incubation at 4°C, the beads were washed with NP-40 lysis buffer (without MgCl2) and boiled in 1× Laemmli loading buffer, followed by gel electrophoresis and immunoblotting for RAB5.
EGFR recycling assay
HeLa cells were seeded in duplicate on 6-well plates. The cells were serum starved overnight. Cells were stimulated with EGF conjugated to Alexa Fluor 647 (Invitrogen) at 10 ng/ml, a concentration favoring EGFR recycling over degradation, to allow internalization for 5 minutes at 37°C, then washed with cold medium and stripped twice with 0.2 M sodium acetate pH 4.5 buffer. The cells were rinsed in cold serum-free medium and excess unlabeled EGF and chased at 37°C for the indicated amounts of time. Following the chase, the cells were stripped twice in the sodium acetate buffer. Signal from stripped fluorescent EGF (EGF-AF647) from the acid washes and the medium was read on a Wallac 1420 mutilabel counter (PerkinElmer) at 635/665 nm absorption/emission channels.
Dextran uptake assays
HeLa cells were plated at 2×104 cells per well in a 96-well plate and serum starved overnight at 37°C. Cells were then pre-treated with control or imatinib containing medium and placed on ice for 30 minutes. This was followed by treatment with serum-free DMEM containing 1 mg/ml 70,000 MW Dextran conjugated to Texas Red (Invitrogen, no. D1830), in the presence or absence of 100 ng/ml EGF and 5 µM imatinib. The cells were incubated at 37°C for 30 minutes after which they were placed on ice and quickly rinsed several times in ice-cold PBS. The cells were kept in 100 µl ice cold PBS and fluorescence emission from Dextran uptaken was read using a Wallac 1420 multilabel counter (PerkinElmer) at 560/615 Absorption/Emission channels.
Immunoprecipitation, immunoblotting and immunofluorescence
Antibodies used for immunoblotting and their sources were- EGFR 1∶2000 (Santa Cruz, SC-03), tubulin 1∶5000 (Sigma Aldrich, T6074-200 ul), RIN1 1∶1000 (Mouse mAb, clone C9E11, Colicelli lab, AbPro), RIN1 1∶1000 (Rabbit pAb anti-99, Colicelli lab, 21st century Biochemicals), p-ERK1/2 pY204 1∶1500 (Epitomics, 2219-1), ERK1/2 1∶5000 (BD, 610123), p-AKT pS473 1∶250 (Cell Signaling, 9271-S), AKT 1∶1000 (Cell Signaling, 9272), p-Raf1 pS338 1∶200 (Millipore, 05-538), c-Raf 1∶200 (BD Transduction Laboratories, R19120), V5 1∶2000 (Invitrogen, R960-25), Transferrin Receptor 1∶2000 (Invitrogen, 13-6800), ubiquitin 1∶1000 (Cell Signaling, 3936), CRKL 1∶1000 (Santa Cruz, SC-319), 4G10 1∶1000 (Millipore,. 05-321), pY36-RIN1 (Hu et al., 2005) 1∶1000 (Rabbit pAb, Colicelli Lab, Biosource International), Pan-RAB5 1∶1000 (Abcam, ab18211), CBL 1∶1000 (BD, 610441), c-ABL 1∶1000 (Santa Cruz, SC-131), sheep-anti-mouse-HRP 1∶3000 (Amersham Biosciences, NA931), goat-anti-rabbit-HRP 1∶3000 (Kirkegaard and Perry, 4741506), Protein-A-HRP 1∶1000 (Invitrogen, 10-1023), goat-anti-rabbit-IRDye 800 1∶5000 (Li-Cor Biosciences, 926-32211) and goat-anti-mouse-IRDye 680 1∶5000 (Li-Cor Biosciences, 926-32220).
For immunoblotting, proteins were transferred to Nitrocellulose membranes overnight. The membranes were blocked with 5% milk in TBST (0.1% Tween-20) followed by incubation with primary and secondary antibodies at room temperature. The membranes were washed with TBST between the incubations and developed using the ECL plus western blotting reagent (VWR) or scanned using a Li-Cor Odyssey scanner.
For immunoprecipitation experiments, the antibody sources were- EGFR 1∶1000 (Santa Cruz, SC-03), RIN1 1∶500 (Rabbit pAb anti-99, Colicelli lab, 21st Century Biochemicals) or CRKL 1∶1000 (Santa Cruz, SC-319). Cells were lysed in NP-40 lysis buffer containing protease and phosphatase inhibitors. Lysates were incubated with the antibody and protein A-agarose (Fisher) beads overnight at 4°C. Following incubation, beads were washed in lysis buffer, boiled in SDS sample loading buffer and run on 8% SDS PAGE, followed by immunoblotting.
The antibody sources for immunofluorescence experiments were- RIN1 1∶200 (Mouse mAb, clone C9E11, Colicelli lab, AbPro), Phalloidin-rhodamine 0.2 uM (Gift of Dr Margot Quinlan, UCLA), EGF-Alexa Fluor 647 (Invitrogen, E35351), Dextran-Texas Red 70000 MW (Invitrogen), goat-anti-rabbit Alexa-Fluor 568 1∶200 (Invitrogen, A11036) and goat-anti-mouse Alexa Fluor 647 1∶200 (Invitrogen, A21245). For imaging experiments, 4×105 cells were seeded on 35 mm MatTek glass bottom tissue culture plates (MatTek corporation). The cells were treated with growth factor as indicated and were fixed in 4% paraformaldehyde, permeabilized in 0.1% TritonX-100 detergent, quenched in 50 mM NH4Cl and blocked in 10% goat serum (Gibco). For EGF internalization experiments, 100 ng/ml EGF-Alexa Fluor 647 was added to cells and bound on ice for 2 hours. The cells were then transferred to 37°C and incubated for 15 minutes, followed by fixation and staining. Cells were then incubated with primary and secondary antibodies at room temperature. Phalloidin was added along with the secondary antibody staining. For EGF-Dextran colocalization studies, cells were pulsed with 20 ng/ml EGF and 1 mg/ml Dextran-Texas Red for five minutes at 37°C. The cells were then chased for the indicated amounts of time at 37°C in serum-free medium, followed by fixation and staining. Confocal microscopy (Zeiss Pascal) was used to image the cells.
RNA isolation and qRT-PCR
RNA was isolated from vector, RIN1 and RIN1QM HeLa cells using the RNeasy mini kit (Qiagen). 1 µg of RNA was converted to cDNA using the SuperScriptIII First-Strand Synthesis SuperMix kit (Invitrogen). qPCR was performed on 2 µl of the diluted cDNA (1∶5) template using LightCycler 480 SYBR Green I Master Mix (Roche Applied Science) and Stratagene MX3000 real-time PCR cycler. The data was analyzed using MXPro software (Agilent Technologies).
The PCR primers used were based on a previous study (Kung et al., 2011) and are as follows: EGFR forward 5′-CTGCGTCTCTTGCCGGAATG-3′, EGFR reverse 5′-TTGGCTCACCCTCCAGAAGG-3′; and GAPDH forward 5′-TGCACCACCAACTGCTTAGC-3′, GAPDH reverse 5′-GAGGGGCCATCCACAGTCTT-3′.
Microscopy
Fixed and stained cells were examined on a laser scanning confocal microscope (Axiovert 200M, Carl Zeiss LSM 5 Pascal). Cells were imaged with a plan/neofluar 100× oil lens, NA 1.3 (Carl Zeiss) and 8-bit digital images were captured using a cooled charge-coupled device (transmitted light channel for lsm5 camera, Zeiss). LSM 5 Pascal software (release version 4.2 sp1) was used to process the images.
Supplementary Material
Acknowledgments
The authors would like to thank the following colleagues for generously sharing key reagents: Dr Dafna Bar-Sagi (RABGEF1 siRNA), Dr Anson (amphiregulin), Dr Philip D. Stahl (HA-STAM), Dr Corrine Leprince (pCDNA3-BIN1), Dr Elizabeth Neufeld (anti-transferrin receptor) and Dr Margot Quinlan (phalloidin-rhodamine). We are also grateful to Nadia Sellami for technical advice and to the following for helpful discussion and comments: Greg Payne, Gerry Weinmaster, Esteban Dell'Angelica and Alex van der Bliek. The authors declare they have no competing financial activities or conflicts of interests.
Footnotes
Funding
This work was supported by the National Institutes of Health [grant number CA136699 to J.C.]; the UCLA Jonsson Comprehensive Cancer Center (to J.C.); the Ruth L. Kirschstein National Research Service Award [grant number GM007185 to C.J. and J.M.B.]; and the Whitcome Training Fellowship (to K.B.). Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.113688/-/DC1
References
- Andoniou C. E., Thien C. B., Langdon W. Y. (1996). The two major sites of cbl tyrosine phosphorylation in abl-transformed cells select the crkL SH2 domain. Oncogene 12, 1981–1989 [PubMed] [Google Scholar]
- Bache K. G., Raiborg C., Mehlum A., Stenmark H. (2003). STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J. Biol. Chem. 278, 12513–12521 10.1074/jbc.M210843200 [DOI] [PubMed] [Google Scholar]
- Barbieri M. A., Kong C., Chen P. I., Horazdovsky B. F., Stahl P. D. (2003). The SRC homology 2 domain of Rin1 mediates its binding to the epidermal growth factor receptor and regulates receptor endocytosis. J. Biol. Chem. 278, 32027–32036 10.1074/jbc.M304324200 [DOI] [PubMed] [Google Scholar]
- Barbieri M. A., Fernandez–Pol S., Hunker C., Horazdovsky B. H., Stahl P. D. (2004). Role of Rab5 in EGF receptor-mediated signal transduction. Eur. J. Cell Biol. 83, 305–314 10.1078/0171-9335-00381 [DOI] [PubMed] [Google Scholar]
- Bauer B., Bartfeld S., Meyer T. F. (2009). H. pylori selectively blocks EGFR endocytosis via the non-receptor kinase c-Abl and CagA. Cell. Microbiol. 11, 156–169 10.1111/j.1462-5822.2008.01246.x [DOI] [PubMed] [Google Scholar]
- Berlin I., Schwartz H., Nash P. D. (2010). Regulation of epidermal growth factor receptor ubiquitination and trafficking by the USP8·STAM complex. J. Biol. Chem. 285, 34909–34921 10.1074/jbc.M109.016287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen V., Sak M. M., Breen K., Rødland M. S., Johannessen L. E., Traub L. M., Stang E., Madshus I. H. (2011). A chimeric pre-ubiquitinated EGF receptor is constitutively endocytosed in a clathrin-dependent, but kinase-independent manner. Traffic 12, 507–520 10.1111/j.1600-0854.2011.01162.x [DOI] [PubMed] [Google Scholar]
- Boulant S., Kural C., Zeeh J. C., Ubelmann F., Kirchhausen T. (2011). Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol. 13, 1124–1131 10.1038/ncb2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X., Tanis K. Q., Koleske A. J., Colicelli J. (2008). Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J. Biol. Chem. 283, 31401–31407 10.1074/jbc.M804002200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casar B., Arozarena I., Sanz–Moreno V., Pinto A., Agudo–Ibáñez L., Marais R., Lewis R. E., Berciano M. T., Crespo P. (2009). Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol. Cell. Biol. 29, 1338–1353 10.1128/MCB.01359-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., ValiyaVeettil M., Sadagopan S., Paudel N., Chandran B. (2011). c-Cbl-mediated selective virus-receptor translocations into lipid rafts regulate productive Kaposi's sarcoma-associated herpesvirus infection in endothelial cells. J. Virol. 85, 12410–12430 10.1128/JVI.05953-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. R., Kang Y., Massagué J. (2001). Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor β growth arrest program. Proc. Natl. Acad. Sci. USA 98, 992–999 10.1073/pnas.98.3.992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. I., Kong C., Su X., Stahl P. D. (2009). Rab5 isoforms differentially regulate the trafficking and degradation of epidermal growth factor receptors. J. Biol. Chem. 284, 30328–30338 10.1074/jbc.M109.034546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C. M., Li H., Gasman S., Huang J., Schiff R., Chang E. C. (2011). Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol. Cell. Biol. 31, 983–997 10.1128/MCB.00137-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. J., Ishii S., Richert N., Merlino G. T., Pastan I. (1985). Epidermal growth factor regulates the expression of its own receptor. Proc. Natl. Acad. Sci. USA 82, 8374–8378 10.1073/pnas.82.24.8374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli J. (2004). Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, re13 10.1126/stke.2502004re13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli J. (2010). ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci. Signal. 3, re6 10.1126/scisignal.3139re6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson J. G., Porat–Shliom N., Cohen L. A. (2009). Clathrin-independent endocytosis: a unique platform for cell signaling and PM remodeling. Cell. Signal. 21, 1–6 10.1016/j.cellsig.2008.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döppler H., Storz P., Li J., Comb M. J., Toker A. (2005). A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J. Biol. Chem. 280, 15013–15019 10.1074/jbc.C400575200 [DOI] [PubMed] [Google Scholar]
- Dubielecka P. M., Machida K., Xiong X., Hossain S., Ogiue–Ikeda M., Carrera A. C., Mayer B. J., Kotula L. (2010). Abi1/Hssh3bp1 pY213 links Abl kinase signaling to p85 regulatory subunit of PI-3 kinase in regulation of macropinocytosis in LNCaP cells. FEBS Lett. 584, 3279–3286 10.1016/j.febslet.2010.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvis A., Balmaceda V., Giambini H., Conde A., Villasana Z., Fornes M. W., Barbieri M. A. (2009a). Inhibition of early endosome fusion by Rab5-binding defective Ras interference 1 mutants. Arch. Biochem. Biophys. 482, 83–95 10.1016/j.abb.2008.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvis A., Giambini H., Villasana Z., Barbieri M. A. (2009b). Functional determinants of ras interference 1 mutants required for their inhbitory activity on endocytosis. Exp. Cell Res. 315, 820–835 10.1016/j.yexcr.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel J. P., Chavrier P., Zerial M., Gruenberg J. (1991). rab5 controls early endosome fusion in vitro. Cell 64, 915–925 10.1016/0092-8674(91)90316-Q [DOI] [PubMed] [Google Scholar]
- Harris R. C., Chung E., Coffey R. J. (2003). EGF receptor ligands. Exp. Cell Res. 284, 2–13 10.1016/S0014-4827(02)00105-2 [DOI] [PubMed] [Google Scholar]
- Hirano S., Kawasaki M., Ura H., Kato R., Raiborg C., Stenmark H., Wakatsuki S. (2006). Double-sided ubiquitin binding of Hrs-UIM in endosomal protein sorting. Nat. Struct. Mol. Biol. 13, 272–277 10.1038/nsmb1051 [DOI] [PubMed] [Google Scholar]
- Hu H., Bliss J. M., Wang Y., Colicelli J. (2005). RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr. Biol. 15, 815–823 10.1016/j.cub.2005.03.049 [DOI] [PubMed] [Google Scholar]
- Hu H., Milstein M., Bliss J. M., Thai M., Malhotra G., Huynh L. C., Colicelli J. (2008). Integration of transforming growth factor β and RAS signaling silences a RAB5 guanine nucleotide exchange factor and enhances growth factor-directed cell migration. Mol. Cell. Biol. 28, 1573–1583 10.1128/MCB.01087-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F., Kirkpatrick D., Jiang X., Gygi S., Sorkin A. (2006). Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 21, 737–748 10.1016/j.molcel.2006.02.018 [DOI] [PubMed] [Google Scholar]
- Huang F., Goh L. K., Sorkin A. (2007). EGF receptor ubiquitination is not necessary for its internalization. Proc. Natl. Acad. Sci. USA 104, 16904–16909 10.1073/pnas.0707416104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. C., Ceresa B. P. (2008). Cellular localization of the activated EGFR determines its effect on cell growth in MDA-MB-468 cells. Exp. Cell Res. 314, 3415–3425 10.1016/j.yexcr.2008.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiho H., Saito K., Tsujita K., Kontani K., Araki Y., Kurosu H., Katada T. (2003). RIN3: a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 116, 4159–4168 10.1242/jcs.00718 [DOI] [PubMed] [Google Scholar]
- Kishimoto T., Sun Y., Buser C., Liu J., Michelot A., Drubin D. G. (2011). Determinants of endocytic membrane geometry, stability, and scission. Proc. Natl. Acad. Sci. USA 108, E979–E988 10.1073/pnas.1113413108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong C., Su X., Chen P. I., Stahl P. D. (2007). Rin1 interacts with signal-transducing adaptor molecule (STAM) and mediates epidermal growth factor receptor trafficking and degradation. J. Biol. Chem. 282, 15294–15301 10.1074/jbc.M611538200 [DOI] [PubMed] [Google Scholar]
- Kung C. P., Meckes D. G., Jr and Raab–Traub N. (2011). Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCδ. J. Virol. 85, 4399–4408 10.1128/JVI.01703-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetti L., Palamidessi A., Areces L., Scita G., Di Fiore P. P. (2004). Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature 429, 309–314 10.1038/nature02542 [DOI] [PubMed] [Google Scholar]
- Lee E., Knecht D. A. (2002). Visualization of actin dynamics during macropinocytosis and exocytosis. Traffic 3, 186–192 10.1034/j.1600-0854.2002.030304.x [DOI] [PubMed] [Google Scholar]
- Levkowitz G., Waterman H., Zamir E., Kam Z., Oved S., Langdon W. Y., Beguinot L., Geiger B., Yarden Y. (1998). c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 12, 3663–3674 10.1101/gad.12.23.3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K. D., Ellem K. A. (1992). UVC modulation of epidermal growth factor receptor number in HeLa S3 cells. Carcinogenesis 13, 183–187 10.1093/carcin/13.2.183 [DOI] [PubMed] [Google Scholar]
- Li S. S. (2005). Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem. J. 390, 641–653 10.1042/BJ20050411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longva K. E., Blystad F. D., Stang E., Larsen A. M., Johannessen L. E., Madshus I. H. (2002). Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 156, 843–854 10.1083/jcb.200106056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K., Kato Y., Sugiyama Y. (2002). pH-dependent receptor/ligand dissociation as a determining factor for intracellular sorting of ligands for epidermal growth factor receptors in rat hepatocytes. J. Control. Release 82, 71–82 10.1016/S0168-3659(02)00126-8 [DOI] [PubMed] [Google Scholar]
- Martínez–Arca S., Bech–Serra J. J., Hurtado–Küttner M., Borroto A., Arribas J. (2005). Recycling of cell surface pro-transforming growth factor-α regulates epidermal growth factor receptor activation. J. Biol. Chem. 280, 36970–36977 10.1074/jbc.M504425200 [DOI] [PubMed] [Google Scholar]
- Medts T., de Diesbach P., Cominelli A., N'Kuli F., Tyteca D., Courtoy P. J. (2010). Acute ligand-independent Src activation mimics low EGF-induced EGFR surface signalling and redistribution into recycling endosomes. Exp. Cell Res. 316, 3239–3253 10.1016/j.yexcr.2010.09.001 [DOI] [PubMed] [Google Scholar]
- Merithew E., Stone C., Eathiraj S., Lambright D. G. (2003). Determinants of Rab5 interaction with the N terminus of early endosome antigen 1. J. Biol. Chem. 278, 8494–8500 10.1074/jbc.M211514200 [DOI] [PubMed] [Google Scholar]
- Milstein M., Mooser C. K., Hu H., Fejzo M., Slamon D., Goodglick L., Dry S., Colicelli J. (2007). RIN1 is a breast tumor suppressor gene. Cancer Res. 67, 11510–11516 10.1158/0008-5472.CAN-07-1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone G., Franchi L., Fina D., Caruso R., Vavassori P., Monteleone I., Calabrese E., Naccari G. C., Bellinvia S., Testi R.et al. (2006). Silencing of SH-PTP2 defines a crucial role in the inactivation of epidermal growth factor receptor by 5-aminosalicylic acid in colon cancer cells. Cell Death Differ. 13, 202–211 10.1038/sj.cdd.4401733 [DOI] [PubMed] [Google Scholar]
- Okutani T., Okabayashi Y., Kido Y., Sugimoto Y., Sakaguchi K., Matuoka K., Takenawa T., Kasuga M. (1994). Grb2/Ash binds directly to tyrosines 1068 and 1086 and indirectly to tyrosine 1148 of activated human epidermal growth factor receptors in intact cells. J. Biol. Chem. 269, 31310–31314 [PubMed] [Google Scholar]
- Plattner R., Kadlec L., DeMali K. A., Kazlauskas A., Pendergast A. M. (1999). c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 13, 2400–2411 10.1101/gad.13.18.2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepstorff K., Grandal M. V., Henriksen L., Knudsen S. L., Lerdrup M., Grøvdal L., Willumsen B. M., van Deurs B. (2009). Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10, 1115–1127 10.1111/j.1600-0854.2009.00943.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J. S., Quinalty L. M., Engelman L., Sherry D. M., Ceresa B. P. (2012). Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J. Biol. Chem. 287, 712–722 10.1074/jbc.M111.294470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. H., Dikic I. (2005). The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 6, 907–918 10.1038/nrm1762 [DOI] [PubMed] [Google Scholar]
- Schnatwinkel C., Christoforidis S., Lindsay M. R., Uttenweiler–Joseph S., Wilm M., Parton R. G., Zerial M. (2004). The Rab5 effector Rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol. 2, e261 10.1371/journal.pbio.0020261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund S., Argenzio E., Tosoni D., Cavallaro E., Polo S., Di Fiore P. P. (2008). Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 15, 209–219 10.1016/j.devcel.2008.06.012 [DOI] [PubMed] [Google Scholar]
- Sorkin A., Goh L. K. (2009). Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 315, 683–696 10.1016/j.yexcr.2008.07.029 [DOI] [PubMed] [Google Scholar]
- Srinivasan D., Sims J. T., Plattner R. (2008). Aggressive breast cancer cells are dependent on activated Abl kinases for proliferation, anchorage-independent growth and survival. Oncogene 27, 1095–1105 10.1038/sj.onc.1210714 [DOI] [PubMed] [Google Scholar]
- Tall G. G., Barbieri M. A., Stahl P. D., Horazdovsky B. F. (2001). Ras-activated endocytosis is mediated by the Rab5 guanine nucleotide exchange activity of RIN1. Dev. Cell 1, 73–82 10.1016/S1534-5807(01)00008-9 [DOI] [PubMed] [Google Scholar]
- Tanos B., Pendergast A. M. (2006). Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 281, 32714–32723 10.1074/jbc.M603126200 [DOI] [PubMed] [Google Scholar]
- Tanos B. E., Pendergast A. M. (2007). Abi-1 forms an epidermal growth factor-inducible complex with Cbl: role in receptor endocytosis. Cell. Signal. 19, 1602–1609 10.1016/j.cellsig.2007.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai M., Ting P. Y., McLaughlin J., Cheng D., Müschen M., Witte O. N., Colicelli J. (2011). ABL fusion oncogene transformation and inhibitor sensitivity are mediated by the cellular regulator RIN1. Leukemia 25, 290–300 10.1038/leu.2010.268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umebayashi K., Stenmark H., Yoshimori T. (2008). Ubc4/5 and c-Cbl continue to ubiquitinate EGF receptor after internalization to facilitate polyubiquitination and degradation. Mol. Biol. Cell 19, 3454–3462 10.1091/mbc.E07-10-0988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Waldron R. T., Dhaka A., Patel A., Riley M. M., Rozengurt E., Colicelli J. (2002). The RAS effector RIN1 directly competes with RAF and is regulated by 14-3-3 proteins. Mol. Cell. Biol. 22, 916–926 10.1128/MCB.22.3.916-926.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmarth N. E., Baillo A., Dziubinski M. L., Wilson K., Riese D. J., 2nd, Ethier S. P. (2009). Altered EGFR localization and degradation in human breast cancer cells with an amphiregulin/EGFR autocrine loop. Cell. Signal. 21, 212–219 10.1016/j.cellsig.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman P. G., Futter C. E. (2008). Multivesicular bodies: co-ordinated progression to maturity. Curr. Opin. Cell Biol. 20, 408–414 10.1016/j.ceb.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Lubkov V., Taylor L. J., Bar–Sagi D. (2010). Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr. Biol. 20, 1372–1377 10.1016/j.cub.2010.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Jahanshahi M., Horvath E. A., Liu H. Y., Pfleger C. M. (2010). Rabex-5 ubiquitin ligase activity restricts Ras signaling to establish pathway homeostasis in Drosophila. Curr. Biol. 20, 1378–1382 10.1016/j.cub.2010.06.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y., Schlessinger J. (1987). Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry 26, 1434–1442 10.1021/bi00379a034 [DOI] [PubMed] [Google Scholar]
- Ziegler S., Eiseler T., Scholz R. P., Beck A., Link G., Hausser A. (2011). A novel protein kinase D phosphorylation site in the tumor suppressor Rab interactor 1 is critical for coordination of cell migration. Mol. Biol. Cell 22, 570–580 10.1091/mbc.E10-05-0427 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.