

Key Points
Distinct gene expression signatures are associated with genetic and clinical subtypes of hemophagocytic lymphohistiocytosis.
Abstract
We performed gene-expression profiling of PBMCs obtained from patients with familial hemophagocytic lymphohistiocytosis (FHL) to screen for biologic correlates with the genetic and/or clinical forms of this disease. Unsupervised hierarchical clustering of 167 differentially expressed probe sets, representing 143 genes, identified 3 groups of patients corresponding to the genetic forms and clinical presentations of the disease. Two clusters of up- and down-regulated genes separated patients with perforin-deficient FHL from those with unidentified genetic cause(s) of the disease. The clusterscomprised genes involved in defense/immune responses, apoptosis, zinc homeostasis, and systemic inflammation. Unsupervised hierarchical clustering partitioned patients with unknown genetic cause(s) of FHL into 2 well-distinguished subgroups. Patterns of up- and down-regulated genes separated patients with “late-onset” and “relapsing” forms of FHL from patients with an “early onset and rapidly evolving” form of the disease. A cluster was identified in patients with “late onset and relapsing” form of FHL related to B- and T-cell differentiation/survival, T-cell activation, and vesicular transport. The resulting data suggest that unique gene-expression signatures can distinguish between genetic and clinical subtypes of FHL. These differentially expressed genes may represent biomarkers that can be used as predictors of disease progression.
Introduction
Familial hemophagocytic lymphohistiocytosis (FHL) is a collection of autosomal-recessive disorders of the immune system characterized by the uncontrolled activation of T cells and macrophages and by the overproduction of inflammatory cytokines secondary to defects in genes coding for proteins involved in the granule-dependent cytolytic pathway.1 Linkage studies in patients with FHL have identified a candidate region containing a still unknown gene on chromosome 9q21 (FHL1; MIM 603552).2 In a separate category of patients, those with FHL2 (MIM 603553), defects in PRF1 on chromosome 10q21 lead to a significant reduction or complete absence of perforin, resulting in impaired cytolytic activity of T cells and NK cells.3–5 Patients with FHL type 3 (MIM 608898) carry mutations in the UNC13D gene on chromosome 17q25.6 Recent studies have identified further mutations in a gene coding for syntaxin 11 (STX11) that define FHL type 4 (MIM 60352).7 More recently, mutations in the STXBP2 gene encoding a syntaxin-binding protein have been described in patients with FHL5 (MIM63101).8 Mutations in PRF1, UNC13D, STX11, and STXBP2 account for less than 50% of North-American FHL cases.9
The principal underlying defect in FHL is impaired T-cell and natural killer (NK) cell cytotoxicity.1 Characteristic laboratory findings include elevated serum levels of ferritin, triglycerides, transaminases, bilirubin, and lactate dehydrogenase, along with decreased levels of fibrinogen.1 Elevated blood levels of proinflammatory cytokines, including IL-6, IL-8, IL-18, MIP-1α, M-CSF, IFNγ, and TNFα, as well as elevated plasma levels of soluble IL-2 receptor (CD25), sCD95 ligand, and sCD163, have also been reported. Other studies have revealed elevated plasma levels of IL-12 and IL-10.1,10 Hemophagocytosis is an indicator of cytokine-driven macrophages and histiocytes.1,10 All genetic forms of FHL can be rapidly fatal if left untreated.1,9,10 Affected patients die of overwhelming infections or uncontrolled systemic inflammation and multiorgan failure. Although FHL is a monogenic defect of immune regulation, it is not a homogenous disease in the traditional sense and is more akin to a syndrome or a broad heterogeneous disease.1,9 The concept of FHL subclasses is clinically relevant because it could have significant implications for the design of therapeutic strategies. FHL is known to affect children in early childhood.1,9 However, exceptions to this general rule have been observed, because there is an increasing number of reports of familial cases with an age of onset of 18 years and older.11 The common denominator in FHL is the development of an accelerated phase characterized by acute, unremitting systemic inflammation, fever, hepatosplenomegaly, CNS involvement, coagulation abnormalities, and highly elevated serum levels of proinflammatory cytokines. Marked hemophagocytosis is usually found in BM and other tissues. Treatment with a combination of immunosuppressive agents usually leads to control of manifestations of accelerated phases and reinduction of remissions.
Although immune and chemotherapeutic regimens targeting activated macrophages/histiocytes and T cells are effective in achieving remission of symptoms, relapse may occur during continuing therapy or after stopping the initial treatment.9,12,13 Deterioration of liver function and blood counts, along with steady increases in serum levels of ferritin, soluble CD25, and soluble CD163, may be indicators of relapse.14 Relapses occur in patients with severe deficiencies of cytotoxic function and the long-term prognosis for patients is death unless hematopoietic stem cell transplantation (HSCT) is administered. HSCT is currently the only available treatment to cure FHL and thus represents the definitive therapy of choice for many patients. It is not uncommon, however, for patients to develop recurrence of active disease before a suitable donor is identified. Physicians are also faced with the task of monitoring the patient's response to therapy toward either remission of illness or its progression to CNS involvement, multisystem organ failure, and eventually death.10 Clinical management guidelines provide a comprehensive picture of patient status, but fail to provide prognostic information essential to guiding patient stratification and therapeutic intervention.
The aim of our present study was to use genome-wide expression profiling to identify gene-expression signatures that may be useful in distinguishing the clinical subtypes of FHL and in predicting outcome.
Methods
Patients and sample preparation
Patients with active FHL diagnosed according to current diagnostic guidelines of the Histiocyte Society (ie, fever, splenomegaly, cytopenia, hypertriglyceridemia, hemophagocytosis, low or absent NK function, and elevated ferritin and soluble CD25) were included in this study.12,13 Blood samples from 11 patients who had been enrolled in an institutional review board–approved study were collected for microarray analysis between 2003 and 2005 (Gene Expression Omnibus series accession no. GSE26050). An independent cohort of 21 patients applying the same diagnostic guidelines was selected for the validation studies. Blood samples from these patients were collected between 2008 and 2010. Routine laboratory tests, such as WBC count, hemoglobin level, platelet count, erythrocyte sedimentation rate, and C-reactive protein, were available for the majority of patients and were obtained at the time of sampling. Mutational analysis of PRF1, UNC13D, STX11, STXBP2, and RAB27A were performed as described previously.11 For patients in this study with no specific genetic diagnosis, the genetic basis was established based on positive family history with previously affected siblings. The patients' clinical and laboratory characteristics are shown in Tables 1 and 2. Twelve controls were recruited for the validation studies from the outpatient department of Cincinnati Children's Hospital Medical Center (CCHMC) using the following exclusion criteria: recent febrile illness and recent use of anti-inflammatory medications. Clinical, genetic, and laboratory data were reviewed by A.H.F., K.Z., and J.V. to select cases for this study. There were no significant differences in WBC, lymphocyte, or neutrophil counts between patients.
Table 1.
Clinical and laboratory characteristics of the FHL patients subjected to gene-expression profiling
| No. | Patients | Age of onset | Mutation | NK function (LU) | siL2R | Remarks |
|---|---|---|---|---|---|---|
| 1 | P33* | 2 mo | PRF1, c.[50delT];c.[1442A > C] (p.L17fsX50);(p.Q481P) | 0.0 | 28 855 | FHL2; consanguinity; rapidly evolving; early onset; deceased |
| 2 | P59* | 7 y | PRF1, c.[148G > A];c.[148G > A], p.V50M | 9.2 | 69 543 | FHL2; rapidly evolving; late onset |
| 3 | P35* | 3 mo | PRF1, c.[50delT];c.[50delT] (p.L17fsX50) | 0.0 | 43 042 | FHL2; consanguinity; rapidly evolving; early onset; received HSCT |
| 4 | P1002† | 9 y | NM | 57.4 | 57 432 | Unknown genetic cause; late onset; relapse |
| 5 | P101† | 6 y | NM | 15.3 | 23 563 | Unknown genetic cause; consanguinity late onset; relapse |
| 6 | P66† | 15 y | STXBP2, c.[511G > T];c[?], p.[168V > L] | 0.1 | 19 743 | Unknown genetic cause; late onset; relapse; received HSCT |
| 7 | P76† | 5 y | NM | 13.3 | NA | Consanguinity; rapidly evolving form; late onset |
| 8 | P92† | 1 mo | NM | 1.0 | 21 030 | Rapidly evolving form; early onset; received HSCT |
| 9 | P94† | 1 mo | NM | 0.0 | 16 203 | Rapidly evolving form; early onset |
| 10 | P96† | 5 y | NM | 0.0 | 48 543 | Rapidly evolving form; late onset; received HSCT |
| 11 | P98† | 16 mo | NM | 7.4 | 3904 | Rapidly evolving form; early onset; received HSCT |
Table 2.
Characteristics of FHL patients subjected to qRT-PCR analysis
| No. | Patients | Age of onset | Mutation | NK function (LU) | siL2R | Remarks |
|---|---|---|---|---|---|---|
| 1 | LH1 | 15 y | ND | 0.0 | NA | Late onset; relapse |
| 2 | LH2 | 2 mo | PRF1, c.[50delT];c.[1442A > C] p.[L17fsX50[;p.[Q481P] | 0.0 | 25 564 | Early onset; FHL2 |
| 3 | LH3 | 3 mo | PRF1, c.[50delT];c.[1442A > C] p.[L17fsX50];p.[Q481P] | 0.0 | 35 947 | Early onset; FHL2 |
| 4 | LH4 | 2 y | NM | 0.0 | 18 627 | Early onset |
| 5 | LH6 | 12 y | UNC13D, g.[1389(+1)G > A];g.[?] | 0.0 | Late onset; relapse | |
| 6 | LH7 | 15 y | UNC13D, c.[847A > G];c.[847A > G], p.I283V | 0.0 | 43 044 | Late onset; FHL3 |
| 7 | LH8 | 2 y | NM | 4.4 | 39 815 | Early onset |
| 8 | LH9 | 2 y | NM | 0.3 | 51 325 | Early onset |
| 9 | LH11 | 13 y | PRF1, c.[272C > T];c.[?], p.[A91V] | 7.9 | 7704 | Late onset; A91V polymorphism |
| 10 | LH12 | 2 y | NM | 0.0 | 20 240 | Early onset |
| 11 | LH13 | 2y | NM | 0.0 | NA | Early onset |
| 12 | LH15 | 11 y | ND | 0.1 | 15 689 | Consanguinity; late onset; relapse |
| 13 | LH16 | 12 y | ND | 0.0 | 119,585 | Late onset |
| 14 | LH17 | 14 y | NM | 0.0 | 11 030 | Late onset; relapse |
| 15 | LH18 | 15 y | NM | 0.1 | 3016 | Late onset |
| 16 | LH19 | 7 y | NM | NA | 20 870 | Late onset |
| 17 | LH20 | 9 y | NM | 0.0 | NA | Late onset |
| 18 | LH21 | 6 y | NM | 0.0 | 87 667 | Late onset; relapse |
| 19 | LH22 | 10 y | NM | 0.0 | NA | Late onset |
| 20 | LH23 | 5 y | NM | 0.0 | NA | Late onset |
| 21 | LH26 | 19 y | NM | 0.0 | NA | Late onset; relapse; received HSCT |
ND indicates not determined; NM, no mutations in PRF1, UNC13D, STX11, STXBP2, or RAB27A were detected; and LU, lytic units.
The ethics committee at the CCHMC approved the studies. Signed informed consent was obtained from all healthy and diseased subjects who provided blood samples, or from their guardians where relevant, in accordance with the Declaration of Helsinki. Blood samples were collected before the onset of hemophagocytic lymphohistiocytosis (HLH)–specific treatments.12 Peripheral blood was collected and PBMCs were isolated over Ficoll. RNA was immediately stabilized in TRIzol reagent (Invitrogen) and stored at −80°C.
RNA isolation and microarray procedures
Total RNA was extracted from PBMCs using TRIzol reagent according to the manufacturer's instructions and was further purified using an RNeasy Mini Kit (QIAGEN). RNA integrity was assessed by electrophoresis using a BioAnalyzer (Agilent Technologies). Gene-expression data were obtained using the Human Genome U 133 Plus 2.0 GeneChip according to the manufacturer's recommendations (Affymetrix) and as described previously.15 Data quality was assessed using the standard metrics of the CCHMC Affymetrix Gene Expression Analysis Core, including assessment of positive and negative controls on the arrays. The expression data that passed the quality test were analyzed using Significant Analysis of Microarray (SAM) to select RNAs that were significantly changed between patients (P < .05). These RNAs were then subjected to the Tukey Honestly Significant Difference (HSD) test (P < .05). The differentially expressed genes were uploaded to the Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resource, where a functional annotation chart tool was used to generate gene ontology terms.16,17
qRT-PCR
Microarray results were validated by quantitative RT-PCR (qRT-PCR) using SYBR Green–based chemistry. For validation, 21 of the most differentially expressed genes by microarray, 10 up-regulated and 11 down-regulated, were analyzed on a custom array made by SABiosciences (part of QIAGEN) for qRT-PCR analysis. Total RNA (500 ng) was reverse transcribed with an SABiosciences RT2 First Strand Kit and applied to the PCR array plates. These plates were then processed in an Applied Biosystems 7500 Real-Time PCR System (Life Technologies) using automated baseline and threshold cycle detection. Data were analyzed using the Web-based PCR array data analysis tool from SABiosciences. Relative changes in the transcript levels of differentially expressed genes compared with the controls were expressed as ΔΔCt values (ΔΔCt = ΔCtpatient × ΔCtcontrol) using SABiosciences software. The custom array contained 2 housekeeping genes, GAPDH and β-2 microglobulin (B2M), as mandatory controls for each experiment.
Results
In a previous study, we identified differentially expressed genes corresponding to various signaling and metabolic pathways relevant to the pathophysiology of FHL by gene-expression profiling patients with FHL and healthy pediatric controls.15 Data suggested patient heterogeneity at the genomic expression level. In the present study, we have further explored gene-expression analysis of patients with FHL and investigated whether peripheral blood expression patterns can discriminate among genetic or clinical forms of the disease. Following the recommendation of Ogilvie et al,18 we reasoned that healthy children are not necessarily an appropriate control group for identifying differentially expressed genes in various forms of FHL, especially when the numbers of patients are small and the differences observed may simply reflect variations between healthy subjects and patients with FHL. By comparing samples obtained from patients with FHL2 directly against patients with positive family history of FHL and an as-yet-unidentified genetic cause of the disease, we expected of targeting gene-expression pattern differences related to subtypes of the disease.
Heterogeneity of FHL based on clustering analysis of differentially expressed genes
PBMCs were derived from whole blood obtained from 11 patients with FHL. Three patients carried disease-causing mutations in the gene encoding perforin and the remaining 8 patients had a wild-type allele(s) of PRF1, UNC13D, STX11, STXBP2, and RAB27A (Table 1). As described previously, these 8 patients did not have EBV infection, malignancy, or rheumatologic disorders prior to the onset of FHL.15,19 They had positive family history of FHL, suggesting that an as-yet-unidentified gene(s) could account for their disease.
Microarray-generated gene-expression levels were analyzed as described in the Methods. Gene-expression data were subjected to RMA preprocessing and then normalized to the medium of all samples. We identified 167 differentially expressed probe sets (a list of the up-regulated and down-regulated genes with fold change differences and P values is shown in supplemental Table 1; see the Supplemental Materials link at the top of the article) representing 143 unique and predicted genes (false discovery rate, 5%) when patients with FHL2 were compared with patients with an as-yet-unidentified genetic cause(s) of disease. Samples and differentially expressed probe sets were then ordered using hierarchical clustering and 3 groups of patients (ha, hb, and hc) could be distinguished (Figure 1). We next analyzed the clinical features of the patients in the clustering tree to determine whether their genotype or clinical phenotypes were correlated with the gene-expression patterns.
Figure 1.

Clustering of FHL patient samples in accordance with the disease phenotype. Patients with FHL type 2 (ha), patients with unknown genetic cause of FHL and relapse (hb), and patients with rapidly evolving form of FHL (hc) were distinguished by the hierarchical clustering of all samples. The blue and red boxes on the right side of the heat map display down- and up-regulated genes, which are clustered according to disease subtypes.
Patients with inactivating mutations of the perforin gene
Of the patients designated group ha (Figure 1), P35 and P59 harbored the homozygous mutations 148G>A (V50M) and homozygous 50delT (L17fsX50) of PRF1, respectively.15,19 Patient P33 had a compound heterozygous mutation of 50delT (L17fsX50) and 1442A>C (Q481P) in the perforin gene.19 The age of patients at onset of the disease ranged from 2 months to 7 years (average, 2.4 years). Patients P33, P59, and P35 formed at least 2 clusters of differentially expressed genes (Figure 1 gene clusters A-B) that distinguished them from patients with an as-yet-unidentified genetic cause(s) of FHL. Cluster A was composed of down-regulated genes, whereas cluster B highlighted the genes showing increased expression compared with patients with as-yet-unknown genetic cause(s) of this disease. Using the DAVID functional annotation chart tool,16,17 the genes in cluster A were shown to be 2 major functional categories of genes coding for proteins related to immune response proteins such as SEMA4D and DEFB4A and apoptosis-regulatory proteins such as CDKN2C and LRDD. Cluster A was also enriched in genes coding for metal-binding proteins (eg, WBSCR17, CCDC72, PRICKLE2, and ZNF333). Cluster B was found to be enriched in genes coding for proteins involved in the transmembrane receptor protein tyrosine kinase signaling pathway, enzyme-linked receptor protein signaling pathways, cyclic nucleotide phosphodiesterase activities, and genes involved in the regulation of cell cycle and apoptosis.
Patients with as-yet-unidentified genetic cause(s) of disease
Based on the branching patterns of the clustering tree, the 8 patients with as-yet-unidentified genetic cause(s) of FHL formed 2 groups, hb and hc (Figure 1). Patients P76, P92, P94, P96, and P98 of group hc had an early onset of the disease with a median of 2.3 years. Patients in group hc had a rapidly evolving form of FHL, as did the ha patients. These patients developed a rapidly progressive form of FHL that was frequently associated with early CNS involvement. This group of patients responded to treatment with resolution of symptoms and normalization of inflammatory markers. As shown in Figure 1, a cluster of down-regulated genes (cluster C) separates this group of patients from patients in groups ha and hb. Cluster C contains 25 genes (Figure 1 cluster C), which, according to DAVID functional annotation chart tool analysis,16,17 are enriched for genes related to cellular immunity, the myeloid cell lineage, and apoptosis.
A subset of patients (hb) on the clustering tree is visibly separated from group hc patients with a rapidly evolving form of FHL (Figure 1). During the 12-month follow-up period, group hb patients experienced recurrence of one or more episodes of accelerated phases. These patients are distinguished from patients in group ha or hc by 2 gene clusters (D and E) of up- and down-regulated genes (Figure 1). Functional analysis based on the DAVID bioinformatics tool related the up-regulated genes (cluster D) to diverse biologic functions and the down-regulated genes (cluster E) to transcription, immunoregulation, and differentiation/development of B cells, T cells, and dendritic cells.
The 3 patients (P1002, P101, and P66) in group hb who experienced relapse were also classified as having a late-onset form of FHL, with a median age of 10.0 years. Variables other than relapse and age onset were investigated as a possible explanation for the differentially expressed genes. Clustering was found not to be related to the race or sex of the patients.
Validation of gene-expression changes by qRT-PCR using an independent cohort of patients
Confirmatory analysis of the gene-expression array study was performed using an independent method, qRT-PCR, and a new cohort of 21 patients who had not been included in the microarray analysis (Table 2). Six patients (LH1, LH6, LH15, LH17, LH21, and LH26) of the 21 patients had experienced 1 or more episodes of disease recurrence during the study period of 6 months. The remaining 15 patients responded either to HLH therapy, were on “continuation therapy,” or received HSCT (Table 2).
Of the 167 differentially expressed probe sets, 21 were selected for validation experiments (Table 3). The most differentially expressed genes from each gene cluster were selected to compare the 2 methods. Two control housekeeping genes, GAPDH and B2M, were also included for normalization of the target genes. All RNA samples from the patients and controls were converted to cDNA using the same reverse-transcriptase enzyme from SABiosciences following the recommended procedure. Comparisons of average fold changes were calculated using the SABiosciences Web-based software, RT2 Profiler PCR Array Data Analysis Version 3.5.
Table 3.
Genes selected for validation experiments
| Expression | No. | Gene symbol | Protein function |
|---|---|---|---|
| Down-regulated in patients of group ha | 1 | CDKN2C | Cyclin-dependent kinase 4 inhibitor C; member of the INK4 family of cyclin-dependent kinase inhibitors; interacts with CDK4 or CDK6; prevents the activation of the CDK kinases; functions as a cell growth regulator; controls cell-cycle G1 progression |
| 2 | RNF26 | Contains a C3HC5 type of RING finger involved in protein-DNA and protein-protein interactions | |
| 3 | UQCRH | Mitochondrial Hinge protein | |
| Up-regulated in patients of group hb | 4 | LRDD | Contains a leucine-rich repeat and a death domain; interacts with Fas and MAPK activating death domain-containing protein MADD; functions as an adaptor protein in signaling processes |
| 5 | HLA-DRB4 | HLA-DRB4 belongs to the HLA class II beta-chain paralogs | |
| 6 | ADCK2 | Uncharacterized aarF domain-containing protein kinase 2 | |
| 7 | C18orf8 | Uncharacterized protein C18orf8 of 657 aa | |
| 8 | HIAT1 | Putative tetracycline transporter-like protein | |
| Down-regulated in patients of group hb | 9 | GIMAP1 | Belongs to the GTP-binding immuno-associated nucleotide subfamily of nucleotide-binding proteins; is involved in the differentiation of Th cells; is critical for the development of mature B and T lymphocytes |
| 10 | DCAKD | Dephospho-CoA kinase domain-containing protein | |
| 11 | FKBP5 | Member of the immunophilin protein family; plays a role in immunoregulation and basic cellular processes | |
| 12 | GTPBP2 | GTP-binding protein member of superfamily capable of binding GTP or GDP | |
| 13 | FLJ20366 | Syntabulin is a microtubule-associated protein implicated in syntaxin transport | |
| Down-regulated in patients of group hc | 14 | ETV3 | Transcriptional repressor; contributes to growth arrest during terminal macrophage differentiation by repressing target genes |
| 15 | KIF5C | Microtubule-associated force-producing protein; may play a role in organelle transport | |
| 16 | RPS27 | Ribosomal protein | |
| 17 | PDE4B | Involved in many signal transduction pathways; may play an important role in platelet aggregation, hormone secretion, and immune cell activation | |
| 18 | DOCK4 | Membrane-associated cytoplasmic protein; functions as a guanine nucleotide exchange factor; is involved in the regulation of adherens junctions between cells | |
| 19 | RFX7 | Ribosomal protein that is a component of the 40S subunit | |
| 20 | KIAA1128 | Hypothetical protein of 588 aa | |
| Up-regulated in patients of group ha | 21 | MTHFD1L | Involved in the synthesis of tetrahydrofolate in the mitochondrion |
Supplemental Table 2 shows the validated genes, indicating 2∧[×Avg(Delta (Ct)], fold change, and the P value and the number of RNA samples that were included in each comparison. Figures 2 through 4 show the up- and down-regulated genes and fold regulation changes compared with the controls.
Figure 2.

Real-time qRT-PCR validation of microarray data. Relative fold changes in the expression of genes selected from gene clusters A and B (Figure 1). Patients LH1 (1), LH2 (2), LH3 (3), LH4 (4), LH6 (5), LH7 (6), LH8 (7), LH9 (8), LH11 (9), LH12 (10), LH13 (11), LH15 (12), LH16 (13), LH17 (14), LH18 (15), LH19 (16), LH21 (18) LH22 (19), LH23 (20), and LH26 (21) are represented by bars. Red bars indicate patients who relapsed and blue bars those who did not.
Figure 4.
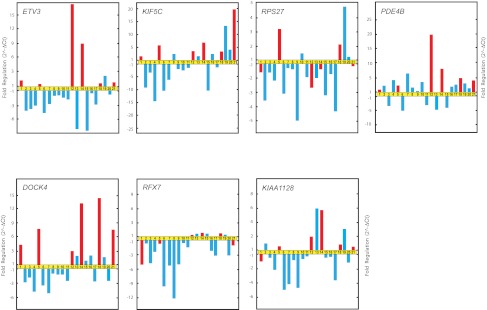
Real-time qRT-PCR validation of microarray data. Relative fold changes in the expression of genes randomly selected from gene cluster C.
qRT-PCR results of RNA samples obtained from patients with FHL2 (LH2 and LH3) confirmed the previous microarray results for the genes CDKN2C, RNF26, UQCRH, and MTHFD1L (Figure 2).
The qRT-PCR data confirmed that the expression of GIMAP1, DCKAD, FKBP5, and FLJ20366 was at lower levels in patients with the “late onset” and “recurrent form” of FHL than in patients without any history of relapse during the defined study period (Figure 3A). However, the qRT-PCR results failed to distinguish any significant correlation between the expression levels of GTPBP2 and the relapsing form of FHL. The qRT-PCR results did reveal that the correlation among the expression levels of GIMAP1, DCKAD, FKBP5, and FLJ20366 and the emergence of relapse was stronger than that among the expression levels of GIMAP1, DCKAD, FKBP5, and FLJ20366 and the age of onset of FHL.
Figure 3.

Real-time qRT-PCR validation of microarray data. (A) Relative fold changes in the expression of genes selected from gene cluster E. (B) Relative fold changes in the expression of genes selected from gene cluster D. The patients and the order of patients are as in Figure 2.
Genes from cluster D (LRDD, HLA-DRB4, ADCK2, C18orf8, and HIAT1) showed variable expression according to real-time qRT-PCR analysis and failed to recapitulate the association with relapses in patients with FHL (Figure 3B).
Additional genes with significant differences in expression between the relapsing and rapidly evolving forms of FHL were ETV3, KIF5C, and DOCK4. These genes were found to be up-regulated in patients with relapse compared with patients without an episode of recurrence of FHL during the study period (Figure 4).
qRT-PCR analysis confirmed the microarray data and validated 7 genes (GIMAP1, DCKAD, FKBP5, FLJ20366, ETV3, KIF5C, and DOCK4) showing significant differences in expression between patients with FHL who experienced recurrence of the acute phase and those who did not have an incidence of relapse within the study period (Figure 5).
Figure 5.
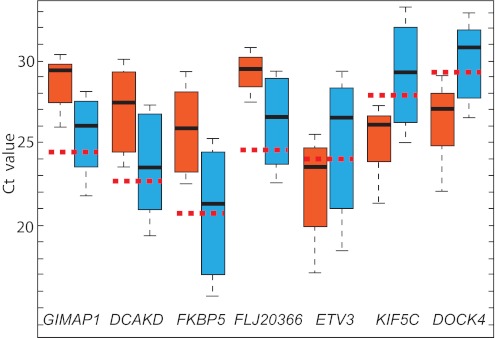
Evaluation of GIMPA1, DCAKD, FKBP5, FLJ20366, ETV3, KIF5C, and DOCK4 gene expression in 6 FHL patients who relapsed (red box) and 15 patients with FHL who did not relapse during the study period (blue box). The vertical lines indicate the minimum and maximum of all of the data; the horizontal boundaries of the boxes the first and the third quartile of data; the thick black lines in the middle of the boxes the median of the data; and the red dotted line the median Ct of controls.
There was one exception to the association of disease form by gene expression. Patient 9 did not experienced relapse within the study period, but showed suppressed GIMAP1, DCKAD, FKBP5, and FLJ20366 expression. This patient also had a pattern of gene expression associated with the rapidly evolving form of the disease.
Discussion
FHL is an aggressive and potentially fatal disease presenting significant challenges for clinicians related to both its diagnosis and treatment. The features of FHL include altered cytolytic functions, systemic release of proinflammatory cytokines, persistent activation of macrophages/histiocytes along with T cells, and multisystem inflammation with characteristic clinical and laboratory findings. The clinical course of FHL is characterized by prolonged fever and hepatosplenomegaly associated with anemia and thrombocytopenia, liver dysfunction, hypofibrinogenemia, hypertriglyceridemia, hypoalbuminemia, and hyponatremia.1,9,20 Neurologic symptoms can dominate the early accelerated phase or may develop later.1,9,20 A significant number of patients die at an early stage during treatment from progressive disease, recurrent infection, or relapse of the accelerated phase. A definitive treatment and a possible cure for FHL are only achieved by HSCT.1,9,20 Clinical outcome for FHL patients depends on how rapidly and accurately the diagnosis is established. Appropriate diagnosis, prognostic information, and prompt treatment with HLH therapy are essential in these patients.12 Relapses are the leading cause of poor outcome in FHL patients before HSCT. Therefore, the early prediction of relapses is beneficial. The focus of the present study was to determine whether there are biologically significant gene-expression signatures that distinguish various genetic or clinical forms of the disease.
Our previous study involving genome-wide expression profiling in children with FHL focused on the differences between patients and healthy controls.15 We demonstrated sustained repression of genes corresponding to innate/adaptive immunity and activation of genes encoding inflammatory cytokines and chemokines. In the present analyses, we identified gene-expression profiles associated with clinical subgroups of FHL. The comparison of PBMC gene-expression profiles obtained from patients with FHL identified 167 differentially expressed probe sets (P = .005). To group patients with similar gene-expression profiles, we used unsupervised hierarchical clustering and demonstrated that the 11 patients with FHL fell into 3 groups displaying different patterns of gene expression (Figure 1).
At least 5 distinct gene clusters contributed to the heterogeneity of patients with FHL. Two clusters of down-regulated (A) and up-regulated (B) genes undoubtedly separated patients with disease-causing PRF1 mutations (group ha) from patients with an as-yet-unknown genetic cause of the disease (groups hb and hc). FHL2 patients more frequently show presentation at an earlier age and have more severe illness and a higher mortality rate compared with FHL patients with other genetic cause(s).21 The 24 down-regulated genes in cluster A included genes coding for proteins involved defense response (DEFB4), negative regulation of cell proliferation and induction of apoptosis (CDKN2C), zinc ion binding (RNF26), proteoglycan metabolic processes (COL11A1 and WBSCR17), mitochondrial electron transport (NDUFB8, UQCRH and NDUFA1), and transcriptional regulation (RHOJ and GCN5L2).
DEFB4 is a member of the gene family coding for β-defensins. The β-defensins are antimicrobial polypeptides that contribute to the immune response against microbial infections.22 In addition to their antimicrobial effects, β-defensins contribute to the adaptive immune response.23 The CKDN2C gene encodes the cyclin-dependent kinase inhibitor p18INK4c (p18), which has been shown to be involved both in early and late B-cell differentiation.
Gene cluster B contains genes that are up-regulated in patients with FHL2 (group ha) compared with patients with an as-yet-unidentified genetic cause of HLH (groups hb and hc). This gene cluster is enriched in genes implicated in the innate immunity response (AXL, BMPR2, SLC30A7) and the regulation of transcription (ARID5B, MNAB, CREM). The receptor-tyrosine-kinase AXL has several cell type–specific roles, including growth induction of endothelial cells, antiapoptotic effects on endothelial cells, the activation of platelets, and the deactivation of APCs.24
The MTHFD1L gene, which is involved in the tetrahydrofolate synthesis pathway catalyzing folate-cofactor interconversion reactions that cause the cells to form and accumulate methotrexate (MTX) polyglutamate, is well recognized as a determinant of MTX cytotoxicity.25,26 MTX is one of the components in contemporary treatment protocols for HLH.9,13 Chemotherapy with the epipodophyllotoxin derivatives etoposide and teniposide, combined with corticosteroids and intrathecal MTX, induce remission in 50% of patients.9 Differences in MTHFD1L expression may point to treatment failures in HLH.
The other gene cluster identified (cluster C), designated the “rapidly evolving form of FHL,” included down-regulated genes encoding transcriptional regulators (ZFYVE9, ETV3, RFXDC2, and JMJD1C), small guanine nucleotide–interacting proteins (ARF1, TRIO, DOCK4, PDE4B, and PDE8), and membrane proteins (DOCK4 and C1orf71[consortin]). ZFYVE9 (SARA) encodes a FYVE domain protein that facilitates signal transduction by promoting the association of SMAD2 and SMAD3 with receptor complexes.27 Phosphorylation of the C-terminal domain of ZFYVE9 (SARA) increases the transcriptional activity of target genes and controls TGF-mediated signaling.27 ZFYVE9 (SARA) also plays an important functional role downstream of Rab5-regulated endosomal trafficking.28 CD55 is markedly down-regulated in patients with the “rapidly evolving form of FHL” compared with the patients in groups ha and hc. The down-regulation of CD55 enhances T-cell proliferation and augments the induced frequency of effector cells.29
ETV3, a member of the ETS family of transcriptional repressors, is expressed during macrophage development.30 SBNO2 and ETV3 function as components of the IL-10–regulated pathway that represses inflammatory gene expression.31 The existence of the IL-10-STAT3-ETV3/SBNO2 pathway is consistent with the hypothesis that IL10 selectively controls inflammatory gene expression through a regulated repression mechanism.32 DOCK4 is a member of the DOCK family of intracellular signaling proteins that are recruited to phosphatidylinositol trisphosphate–rich membranes through their Dock homology region 1 (DHR1) domain and serve as guanine exchange factors for Rho/Rac family GTPases through their DHR2 domain.33 Guanine nucleotide exchange factors have a significant role in many biologic events. They have the ability to restructure the actin cytoskeleton through the activation of specific downstream effectors.33 The deficiency of another member of this family, DOCK8, impairs CD8+ T-cell survival and function in humans and mice.34 The role of DOCK4 in immune cells is unknown. DE4B and PDE8A are the members of cyclic nucleotide phosphodiesterases that control the intracellular levels of cAMP and cGMP. Cyclic nucleotide phosphodiesterases are involved in the differentiation of monocytes, dendritic cells, and macrophages.35,36
Groups ha and hc represent patients with early onset and rapidly accelerated form of FHL with a dissimilar pattern of mRNA expression in PBMCs. In group ha patients, there was an evidence of gross PRF1 protein misfolding, protein degradation, and a severely decreased or absence of lytic function.21 Patients in group hc had a wild-type PRF1 gene with normal level of mRNA expression and impaired lytic function. Based on current knowledge, it seems that there are at least 2 additional FHL-related genes yet to be identified.37 The as-yet-unidentified gene(s) in patients of group hc could induce an early onset, rapidly evolving form of FHL with distinct mRNA patterns.
Cluster D (Figure 1) contains 4 known up-regulated genes coding for proteins with a leucine0rich repeat and a death domain (LRDD), an aarF domain-containing protein kinase (ADCK2), a major histocompatibility complex II DR β 4 (HLA-DRB4), and a member of superfamily of the solute carriers (HIAT1).
Cluster E represents a 12-gene signature associated with recurrence of FHL in patients. This signature is based on genes found to be consistently underexpressed in disease with relapse compared with FHL2 or the rapidly evolving form of FHL of as-yet-unknown genetic cause(s). Our initial analyses revealed that this gene signature predicted recurrence in 3 of the 11 patients analyzed by microarray. The down-regulation of selected genes from the 12-gene signature was also observed by qRT-PCR in 6 patients with a relapse of the accelerated phase of FHL. In addition, patients in subclass hb were older than patients in subclasses ha and hc. Cluster E was also enriched in genes related to B- and T-cell differentiation and survival, T-cell activation, and intracellular vesicular transport, and these patterns of gene regulation were correlated with a distinct and clinically relevant phenotype of FHL.
The GTPase of the immune-associated protein 1 (GIMAP1) gene is a close relative of GIMAP5 and lies adjacent to it within the GIMAP gene cluster.38 GIMAP5 is a key regulator of hematopoietic integrity and lymphocyte homeostasis.39 In GIMAP5–deficient mice, T cells and B cells appear to undergo normal development, but fail to proliferate on Ag-receptor stimulation.39 GIMAP5 deficiency imposes a block in the NK- and NKT-cell differentiation required for the survival of peripheral T cells, NK, and NKT cells.40 GIMAP1, like GIMAP5, encodes a protein that contains a putative transmembrane domain at its carboxyl terminus, which is indicative of the targeting of intracellular membranes. GIMAP1 mRNA expression has been found in all stages of T-cell development and is differentially expressed under conditions of Th1 or Th2 polarization.41 The GIMAP1 protein is required during lymphocyte development in the T- and B-cell lineages, suggesting the involvement of common pathways such as NF-κB.32 Homozygous loss of murine Gimap1 leads to a severe deficiency in mature T and B lymphocytes, indicating the crucial requirement of this gene for mature lymphocyte development and survival.42
Dephospho-CoA kinase domain-containing protein (DCAKD) is a member of the CoA biosynthetic pathway and catalyzes the final step of the biosynthesis of mevalonate, the precursor of cholesterol.43 Very little is known about the regulation of this pathway. Dephospho-CoA kinase forms complexes with various src homology-2 domain proteins in vitro and in vivo.43 Inhibition of the mevalonate biosynthetic pathway down-regulates T-cell proliferation and B-lymphocyte survival.44
The protein encoded by FKBP5 is a member of the family of immunophilins, the FK506-binding proteins.45 The encoded protein is a cis-trans prolyl isomerase that binds to the immunosuppressants FK506, cyclosporine, and rapamycin.46 FKBPs are involved in several biochemical processes, including protein folding, receptor signaling, protein trafficking, transcription, and mediating calcineurin inhibition.46 FKBPs also play important functional roles in T-cell activation when complexed with their ligands.46 This complex of an immunophilin and cyclosporine A inhibits calcineurin, which under normal conditions induces the transcription of IL-2.46 The role of immunophilins in protein transportation and apoptosis through their molecular interactions with receptors or proteins has emerged recently.47
FLJ20366 encodes SYBU, a syntaxin-interacting protein that is expressed in CD56dimCD16+ and at lower levels in CD56brightCD16− NK cells.42 Cytolytic activity is mostly confined to CD56dimCD16+ NK cells, whereas cytokine production is generally assigned to CD56brightCD16+/− NK cells. The expression of SYBU is enhanced after IL-2 treatment.48 The FLJ20366 expression pattern is very similar to that of syntaxin 11 (STX11), which is mutated in FHL4.7 The syntaxins play a role in the intracellular vesicle transport of the phagocytic system.7 Syntaxin-interacting proteins such as STXBP2 are important for controlling intracellular granule/membrane trafficking in polarized epithelial cells, neutrophils, and mast cells.8 STXBP2 belongs to the Sec/Munc family of regulatory proteins involved in the assembly and disassembly of SNARE complexes, as well as control of the specificity and timing of membrane fusion. STXBP2 is also mutated in FHL5.8 The decreased expression of FLJ20366 coding for a syntaxin-interacting protein in patients with recurrent form of FHL may lower the threshold for hemophagocytic lymphohistiocytosis by impairing degranulation and NK-cell cytotoxicity.
Based on our study design, the genes identified in the microarray analysis should be those with the highest differences in expression among the 3 groups of patients. Clustering of the genes that were differently regulated across the 3 groups illustrated at the genomic level that gene-expression signatures can be used to identify FHL patients with different clinical characteristics. One limitation of the present study is the small number of patients: 11 patients analyzed by microarray and 20 patients assessed by qRT-PCR analysis. Caution must be exercised when interpreting the results from a small sample set. However, our results do primarily suggest that gene-expression profiling in PBMCs may be used to predict recurrence of FHL. This approach, if validated in a larger cohort, could have direct implications for future patient management. We speculate that the down-regulation of the genes in cluster E predisposes patients with FHL to relapse and that our findings will serve as a foundation for formulating testable, novel hypotheses regarding the pathophysiology of FHL outcomes.
An alternative interpretation of our present findings is that the down-regulation of the identified genes in FHL patients is simply an epiphenomenon of illness severity rather than being a direct underlying cause of the pathophysiology of the different forms of FHL. Even if this proves to be the case, expression differences could still potentially serve as a gene-expression signature for predicting outcomes in FHL patients. PBMC gene-expression profiling, particularly when combined with the clinical assessment of FHL, may provide a better indication of a patient's risk of relapse or other unwanted outcomes of this disease, and thus is a valuable tool for optimizing the medical management of these patients.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (R21 AI079759 and R21 AI076746) and by the Histiocytosis Association of America.
Footnotes
This article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.S. conceived and designed the experiments, analyzed the data, and wrote the manuscript; S.V.N. performed the research and was responsible for the qRT-PCR experiments; M.G.B. performed the statistical analyses of the expression data and wrote and reviewed the manuscript; J.V. performed the research; K.Z. performed the genetic analyses; A.A.G. contributed vital reagents and wrote and reviewed the manuscript; and A.H.F. was responsible for the selection and clinical evaluation of patients and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Janos Sumegi, Division of Bone Marrow Transplantation and Immunodeficiency, Cincinnati Children's Hospital Medical Center, 3333 Burnett Ave, Cincinnati, OH 45229; e-mail: janos.sumegi@cchmc.org.
References
- 1.Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am. 2008;28(2):293–313. doi: 10.1016/j.iac.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Ohadi M, Lalloz MR, Sham P, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999;64(1):165–171. doi: 10.1086/302187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dufourcq-Lagelouse R, Jabado N, Le Deist, et al. Linkage of familial hemophagocytic lymphohistiocytosis to 10q21-22 and evidence for heterogeneity. Am J Hum Genet. 1999;64(1):172–179. doi: 10.1086/302194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 5.Kogawa K, Lee SM, Villanueva J, et al. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. 2002;99(1):61–66. doi: 10.1182/blood.v99.1.61. [DOI] [PubMed] [Google Scholar]
- 6.Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 7.zur Stadt U, Schmidt S, Kasper B, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHLH) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14(6):827–834. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 8.zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHLH-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–892. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jordan MB, Allen CE, Weitzman S, et al. How we treat hemophagocytic lymphohistocytosis. Blood. 2011;118(15):4041–4052. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henter JI, Andersson B, Elinder G, et al. Elevated circulating levels of interleukin-1 receptor antagonist but not IL-1 agonists in hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 1996;27(1):21–25. doi: 10.1002/(SICI)1096-911X(199607)27:1<21::AID-MPO5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 11.Zhang K, Jordan MB, Marsh RA, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118(22):5794–5798. doi: 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henter JI, Arico M, Egeler M, et al. HLH-94: A treatment protocol for hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 1997;28(5):342–347. doi: 10.1002/(sici)1096-911x(199705)28:5<342::aid-mpo3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 13.Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 14.Lin TF, Ferlic-Stark LL, Allen CE, et al. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154–155. doi: 10.1002/pbc.22774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sumegi J, Barnes MG, Nestheide SV, et al. Gene expression profiling of peripheral blood mononuclear cells from children with active hemophagocytic lymphohistiocytosis. Blood. 2011;117(15):e151–e160. doi: 10.1182/blood-2010-08-300046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dennis G, Jr., Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3. [PubMed] [Google Scholar]
- 17.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Ogilvie EM, Khan A, Hubank M, et al. Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(6):1954–1965. doi: 10.1002/art.22644. [DOI] [PubMed] [Google Scholar]
- 19.Molleran Lee S, Villanueva J, Sumegi J, et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. 2004;41(2):137–144. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166(2):95–109. doi: 10.1007/s00431-006-0258-1. [DOI] [PubMed] [Google Scholar]
- 21.Risma KA, Frayer RW, Filipovich AH, et al. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J Clin Invest. 2006;116(1):182–192. doi: 10.1172/JCI26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehrer RI, Ganz T. Defensins of vertebrate animals. Curr Opin Immunol. 2002;14(1):96–102. doi: 10.1016/s0952-7915(01)00303-x. [DOI] [PubMed] [Google Scholar]
- 23.Funderburg N, Lederman MM, Feng Z, et al. Human -defensin-3 activates professional antigen-presenting cells via Toll-like receptors 1 and 2. Proc Nat Acad Sci U S A. 2007;104(47):18631–18635. doi: 10.1073/PNAS.0702130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki T, Knyazev PG, Clout NJ, et al. Structural basis for Gas6-Axl signaling. EMBO J. 2006;25(1):80–87. doi: 10.1038/sj.emboj.7600912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao R, Goldman ID. Resistance to antifolates. Oncogene. 2003;22(47):7431–7457. doi: 10.1038/sj.onc.1206946. [DOI] [PubMed] [Google Scholar]
- 26.Kager L, Cheok M, Yang W, et al. Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J Clin Invest. 2005;115(1):110–117. doi: 10.1172/JCI22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsukazaki T, Chiang TA, Davison AF, et al. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95(6):779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 28.Hu Y, Chuang JZ, Xu K, et al. SARA, a FYVE domain protein, affects Rab5-mediated endocytosis. J Cell Sci. 2002;115(24):4755–4763. doi: 10.1242/jcs.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heeger PS, Lalli PN, Lin F, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201(10):1523–1530. doi: 10.1084/jem.20041967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawka-Verhelle D, Escoubet-Lozach L, Fong AL, et al. PE-1/METS, an antiproliferative Ets repressor factor, is induced by CREB-1/CREM-1 during macrophage differentiation. J Biol Chem. 2004;279(17):17772–17784. doi: 10.1074/jbc.M311991200. [DOI] [PubMed] [Google Scholar]
- 31.Klappacher GW, Lunyak VV, Sykes DB, et al. An induced Ets repressor complex regulates growth arrest during terminal macrophage differentiation. Cell. 2002;109(2):169–180. doi: 10.1016/s0092-8674(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 32.El Kasmi KC, Smith AM, Williams L, et al. Cutting edge: A transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179(11):7215–7219. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- 33.Côté JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007;17(8):383–393. doi: 10.1016/j.tcb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Randall KL, Chan SS, Ma CS, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. 2011;208(11):2305–2320. doi: 10.1084/jem.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hertz AL, Beavo JA. Cyclic nucleotides and phosphodiesterases in monocytic differentiation. Handb Exp Pharmacol. 2011;204:365–390. doi: 10.1007/978-3-642-17969-3_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hertz AL, Bender AT, Smith KC, et al. Elevated cyclic AMP and PDE4 inhibition induce chemokine expression in human monocyte-derived macrophages. Proc Natl Acad Sci U S A. 2009;106(51):21978–21983. doi: 10.1073/pnas.0911684106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cetica V, Santoro A, Gilmour KC, S, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet. 2010;47(9):595–600. doi: 10.1136/jmg.2009.075341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamm O, Krücken J, Schmitt-Wrede HP, et al. Human ortholog to mouse gene imap38 encoding an ER-localizable G-protein belongs to a gene family clustered on chromosome 7q32-36. Gene. 2002;282(1-2):159–167. doi: 10.1016/s0378-1119(01)00837-x. [DOI] [PubMed] [Google Scholar]
- 39.Barnes MJ, Aksoylar H, Krebs P, et al. Loss of T cell and B cell quiescence precedes the onset of microbial flora-dependent wasting disease and intestinal inflammation in Gimap5-deficient mice. J Immunol. 2010;184(7):3743–3754. doi: 10.4049/jimmunol.0903164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schulteis RD, Chu H, Dai X, et al. Impaired survival of peripheral T cells, disrupted NK/NKT cell development, and liver failure in mice lacking Gimap5. Blood. 2008;112(13):4905–4914. doi: 10.1182/blood-2008-03-146555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saunders A, Webb LM, Janas ML, et al. Putative GTPase GIMAP1 is critical for the development of mature B and T lymphocytes. Blood. 2010;115(16):3249–3257. doi: 10.1182/blood-2009-08-237586. [DOI] [PubMed] [Google Scholar]
- 42.Filén JJ, Filén S, Moulder R, et al. Quantitative proteomics reveals GIMAP family proteins 1 and 4 to be differentially regulated during human T helper cell differentiation. Mol Cell Proteomics. 2009;8(1):32–44. doi: 10.1074/mcp.M800139-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yokooji Y, Tomita H, Atomi H, Imanaka T. Pantoate kinase and phosphopantothenate synthetase, two novel enzymes necessary for CoA biosynthesis in the archaea. J Biol Chem. 2009;284(41):28137–28145. doi: 10.1074/jbc.M109.009696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Musso A, Zocchi MR, Poggi A. Relevance of the mevalonate biosynthetic pathway in the regulation of bone marrow mesenchymal stromal cell-mediated effects on T-cell proliferation and B-cell survival. Haematologica. 2011;96(1):16–23. doi: 10.3324/haematol.2010.031633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16(4):318–25. doi: 10.1159/000123041. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Lou Z, Wang L. The role of FKBP5 in cancer aetiology and chemoresistance. Br J Cancer. 2011;104(1):19–23. doi: 10.1038/sj.bjc.6606014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quintá HR, Galigniana NM, Erlejman AG, et al. Management of cytoskeleton architecture by molecular chaperones and immunophilins. Cell Signal. 2011;23(12):1907–1920. doi: 10.1016/j.cellsig.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanna J, Bechtel P, Zhai Y, et al. Novel insights on human NK cells' immunological modalities revealed by gene expression profiling. J Immunol. 2004;173(11):6547–6563. doi: 10.4049/jimmunol.173.11.6547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.