

Abstract
Traumatic brain injury (TBI) is a major health and socioeconomic problem throughout the world. It is a complicated pathological process that consists of primary insults and a secondary insult characterized by a set of biochemical cascades. The imbalance between a higher energy demand for repair of cell damage and decreased energy production led by mitochondrial dysfunction aggravates cell damage. At the cellular level, the main cause of the secondary deleterious cascades is cell damage that is centred in the mitochondria. Excitotoxicity, Ca2+ overload, reactive oxygen species (ROS), Bcl-2 family, caspases and apoptosis inducing factor (AIF) are the main participants in mitochondria-centred cell damage following TBI. Some preclinical and clinical results of mitochondria-targeted therapy show promise. Mitochondria- targeted multipotential therapeutic strategies offer new hope for the successful treatment of TBI and other acute brain injuries.
Keywords: traumatic brain injury, mitochondrion, mitochondrial membrane permeabilization, apoptosis, necrosis, energy
Traumatic brain injury (TBI) constitutes a major health and socioeconomic problem throughout the world. According to the report (2002–2006) of Centers for Disease Control and Prevention, at least 1.7 million people sustain a TBI in the United States each year. About 1.365 million of those individuals are treated in an emergency department, 275 000 are hospitalized and 52 000 die. Although many preclinical studies have shown positive results in treating TBI, almost none have proved effective in clinical settings. This might reflect the diverse nature of clinical TBI and/or an incomplete understanding of the mechanisms of secondary neuronal loss.
TBI is a heterogeneous disorder with different forms of presentation. In past decades, research of TBI made great progress in clarifying the pathophysiological mechanisms. These consist of a primary insult resulting from the direct biomechanical forces and a secondary insult that plays an important role in the brain damage and death following TBI. At the cellular level, two initiating events related to energy depletion and Ca2+ homeostasis are of particular importance in the response to primary injury. The first is an ‘ischaemia-like’ pattern that is characterized by direct tissue damage and impaired regulation of cerebral blood flow (CBF) and metabolism. The ATP stores are depleted, and failure of energy-dependent membrane ion pumps occurs. The second is characterized by nerve terminal membrane depolarization along with excessive release of excitatory neurotransmitters (i.e. glutamate, aspartate), activation of NMDA, AMPA and voltage-dependent Ca2+ and Na+ channels. This, in turn, releases additional Ca2+ from intracellular stores, thus producing abnormally high levels of free intracellular and mitochondrial Ca2+. The consecutive Ca2+ overload leads to self-digesting (catabolic) intracellular processes that involve overproduction of free radicals, activation of cell death signalling pathways and up-regulation of inflammatory mediators (Figure 1). Together, these events lead to membrane degradation of vascular and cellular structures and ultimately cell death (Werner and Engelhard, 2007).
Figure 1.
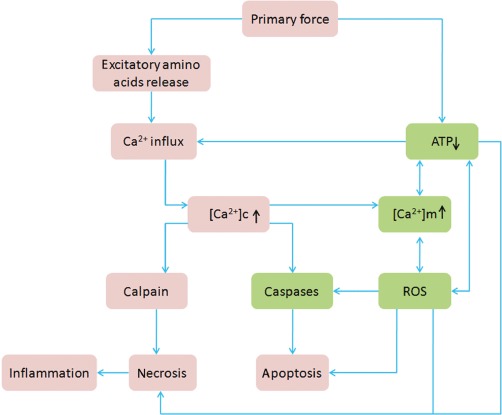
Pathological changes of TBI. The primary force (trauma) will cause direct tissue damage, impaired regulation of CBF and metabolism and excessive release of excitatory neurotransmitters. This results in an abnormally high level of free intracellular and mitochondrial Ca2+. The consequent Ca2+ overload leads to self-digesting (catabolic) intracellular processes that involve overproduction of free radicals, activation of cell death signalling pathways and up-regulation of inflammatory mediators. Together, these events lead ultimately to cell death. Note: the green boxes show the main pathological changes in mitochondria. [Ca2+]c indicates intracellular Ca2+ concentration. [Ca2+]m indicates mitochondrial Ca2+ concentration.
As the ‘power plant of the cell’, ATP production via oxidative phosphorylation is the primary function of mitochondria, and Ca2+ is the characteristic stimulatory signal for activation of numerous mitochondrial enzymes (Graier et al., 2007). Several studies in recent years have indicated that mitochondria play a pivotal role in neuronal cell survival. Mitochondrial dysfunction is considered to be an early event in the CNS injury that can cause neuronal cell death.
Structure and function of mitochondria
The widely accepted model of mitochondria is the ‘crista junction’ model that was first introduced by Daems and Wisse in 1966 (Figure 2) (Daems and Wisse, 1966). In this model, there are three membrane systems – an outer membrane (OM) and inner membrane (IM) that is further divided into an inner boundary membrane (IBM) and a cristae membrane (criM) – and three distinct compartments. The latter are the intermembrane space (IMS), the matrix and the additional intracristal space (intracriS).
Figure 2.
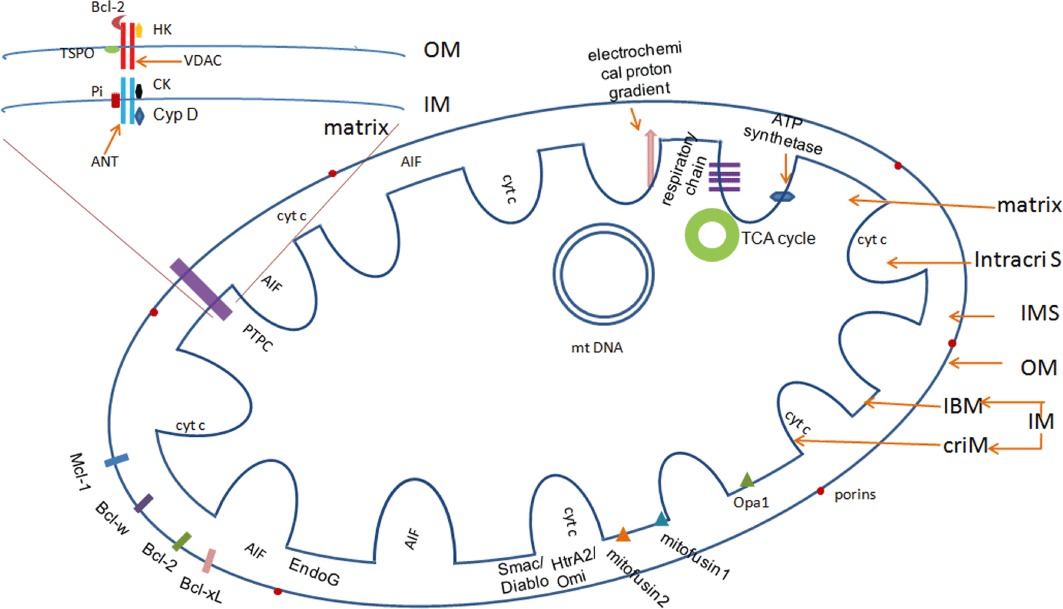
The structure of a mitochondrion. According to the ‘crista junction’ model, each mitochondrion includes three membrane systems and three compartments. The outer membrane (OM) contains large numbers of porins that allow free diffusion of small molecules. In physiological conditions, there are many anti-apoptotic Bcl-2 family members attached to the OM. The inner membrane (IM) contains compartmentalized proteins with different functions, such as those that perform the redox reactions of oxidative phosphorylation, ATP synthase, mitochondria fusion and fission protein. There is an electrochemical proton gradient across the IM. The permeability transition pore complex (PTPC) is a supramolecular channel that is assembled at the junctions between the IM and the OM, which is proposed to be composed of the adenine nucleotide translocase (ANT), cyclophilin D (Cyp D), voltage-dependent anion channels (VDAC), creatine kinase (CK), hexokinase (HK), the translocator protein TSPO and the mitochondrial phosphate carrier Pi. The IMS contains many proteins that play important roles in cell death, such as cyt c and AIF. Enzymes that carry out the citric acid cycle and mitochondrial DNA (mtDNA) are located in the matrix.
The OM contains large numbers of integral proteins that are called porins. Molecules of less than 5 kDa can diffuse freely through the channels that are formed by these porins, but larger proteins must be actively translocated by a translocase of the OM (Herrmann and Neupert, 2000). The voltage-dependent anion channel (VDAC) is the most common pathway. Under physiological conditions, this channel is closed and there is free exchange of metabolites of molecular mass of up to 5 kDa in size and cations, like Ca2+, K+ and Na+, through the OM (Colombini, 2004). Three isoforms (VDAC1, VDAC2 and VDAC3) have been identified in multicellular organisms (Lemasters and Holmuhamedov, 2006).
The IM contains compartmentalized proteins with five types of functions: those that perform the redox reactions of oxidative phosphorylation, ATP synthase, specific transport proteins that regulate metabolite passage into and out of the matrix, protein import machinery, mitochondria fusion and fission protein (Walter, 1994). The IBM is enriched in proteins that are involved in mitochondrial fusion and in mitochondrial protein import, whereas the protein complexes of the respiratory chain and the proteins that are involved in the iron/sulfur cluster biogenesis accumulate in the criM. Moreover, the proteins redistribute dynamically in response to changes in the physiological state of mitochondria (Vogel et al., 2006). The IM is highly impermeable to all molecules. Almost all ions and molecules require special membrane transporters to enter or exit the matrix. Cardiolipin (CL), the only membrane component that is synthesized by the IM and it acts as a platform for multiple apoptotic signals that converge to coordinate apoptotic cell death (Cristea and Degli Esposti, 2004; Mari et al., 2008; Schlame, 2008; Schug and Gottlieb, 2009; Zaltsman et al., 2010). There is an electrochemical proton gradient across the IM that is ctically involved in the import of mitochondrial proteins and regulation of metabolite transport across the mitochondrial membrane (Magder, 2006).
The permeability transition pore complex (PTPC) is a supramolecular channel that is assembled at the junction of the IM and the OM. Although the exact stoichiometry and molecular architecture of the PTPC remains elusive, it is proposed that it is composed of ADP/ATP carrier adenine nucleotide translocase (ANT: IM channel), cyclophilin D (Cyp D: matrix) and VDAC (OM channel) (Sullivan et al., 2005; Garrido et al., 2006; Tsujimoto and Shimizu, 2007). Creatine kinase (CK, periplasmic space), VDAC-associated hexokinase (HK, cytoplasm) and the 18 kDa translocator protein (TSPO) (also named as the peripheral benzodiazepine receptor), as well as Bcl-2 family proteins (Colombini, 2004; Rostovtseva et al., 2004), may also have roles (Castedo et al., 2002; Belizario et al., 2007). The OM-located protein TSPO could modulate PTPC both directly and indirectly (Castedo et al., 2002; Soustiel et al., 2008). More recently, it was proposed that the mitochondrial phosphate carrier (Pi) is a PTPC constituent (Leung et al., 2008) that undergoes a Ca2+-induced conformational change to induce pore formation (Figure 2) (Varanyuwatana and Halestrap, 2011).
The IMS contains many proteins that play important roles in cell death, such as cytochrome (cyt) c and the apoptosis inducing factor (AIF). The matrix contains about two-thirds of the total protein in a mitochondrion, including enzymes that carry out the oxidation of pyruvate and fatty acids, and the citric acid cycle. A published human mitochondrial DNA sequence revealed 16 569 base pairs that encode a total of 37 genes: 22 tRNA, 2 rRNA and 13 peptide genes (Anderson et al., 1981).
In order to provide enough ATP for the proper functioning of the cells in response to local changes, mitochondria are in constant fusion and fission (Rube and van der Bliek, 2004; Okamoto and Shaw, 2005; Chan, 2006; Cheung et al., 2007; Parone et al., 2008; Santel and Frank, 2008), both of which entail the participation of different proteins – Fission1 (Fis1), dynamin-related protein 1 and Endophilin B1 are three mammalian orthologues that are known to be required for mitochondrial fission in mammals (Karbowski et al., 2004). Mitochondrial fusion requires both outer and inner mitochondrial components, such as mitofusin 1 and 2 and optical atrophy protein 1 (Opa1).
Close cooperation between mitochondria and other intracellular organelles is also important in maintaining normal function of the cells (Mattson and Kroemer, 2003; Green and Kroemer, 2004; Wasilewski and Scorrano, 2009). The mitochondria-associated endoplasmic reticulum(ER) membrane (MAM) is the structure that associates the mitochondria with the ER membrane. The MAM can integrate signal transduction with metabolic pathways to regulate the communication and functional interactions between the ER and mitochondrion (Hayashi et al., 2009), especially in lipid transport and Ca2+ exchanges (Pizzo and Pozzan, 2007). In pathological conditions, Ca2+ exchanges between mitochondria and ER mediated by MAM is an important apoptotic control point. The mitochondrial–lysosomal axis is also involved in the cell death cascade in TBI pathology. Lysosomal membrane permeabilization (LMP) results in the release of cathepsin proteases into the cytosol, which facilitates the release of AIF and EndoG from the IMS and results in caspase-independent DNA degradation. Such cathepsins can also trigger mitochondrial outer membrane permeabilization (MOMP), thereby stimulating the mitochondrial pathway of apoptosis (Turk et al., 2001; Boya et al., 2003; Luo et al., 2010).
As mitochondria play a central role in the fate of the cells, the damage to mitochondria in pathological conditions, such as TBI and other acute brain injury, will have serious consequences for brain tissue. More and more evidence shows that mitochondria are among the key players in neuronal damage in the pathological processes of TBI.
Mitochondria provide a common pathway of cell death
Neuronal and glial cell death and traumatic axonal injury contribute to the overall pathology of TBI in both humans and animals. Although it is difficult to develop a generally acceptable taxonomy of cell death, apoptosis, necrosis and autophagy are three major forms of cell death that are widely recognised (Kroemer et al., 2009). Today, necrosis is believed to be a programmed event, termed necroptosis, which is characterized by necrotic cell death morphology and the activation of autophagy (Li et al., 2008). All three forms of cell death participate in cell damage after TBI.
The major link that connects different death programs seems to be the mitochondrion and, in particular, its membranes (Jacotot et al., 1999; Vander Heiden and Thompson, 1999; Scorrano and Korsmeyer, 2003; Breckenridge and Xue, 2004; Lucken-Ardjomande and Martinou, 2005; Zoratti et al., 2005; Chipuk et al., 2006; Kroemer et al., 2007; Galluzzi et al., 2010). The mitochondrial membrane permeabilization (MMP) and subsequent release of death effectors will lead to cell death (Ferri and Kroemer, 2001; Li et al., 2001; Lemasters, 2005; Zoratti et al., 2005; Garrido et al., 2006; Galluzzi et al., 2008). Essentially, two models of the formation of pores in the mitochondrial membranes have been proposed: (1) direct MOMP through pre-existing pores and ex novo pore formation, and (2) mitochondrial membrane permeability transition (MPT) (a process of a sudden increase in the permeability of the IM to solutes with a molecular mass of less than 1500 Da.) that occurs following the opening of the PTPC in the IM (Figure 3) (Galluzzi and Kroemer, 2008).
Figure 3.
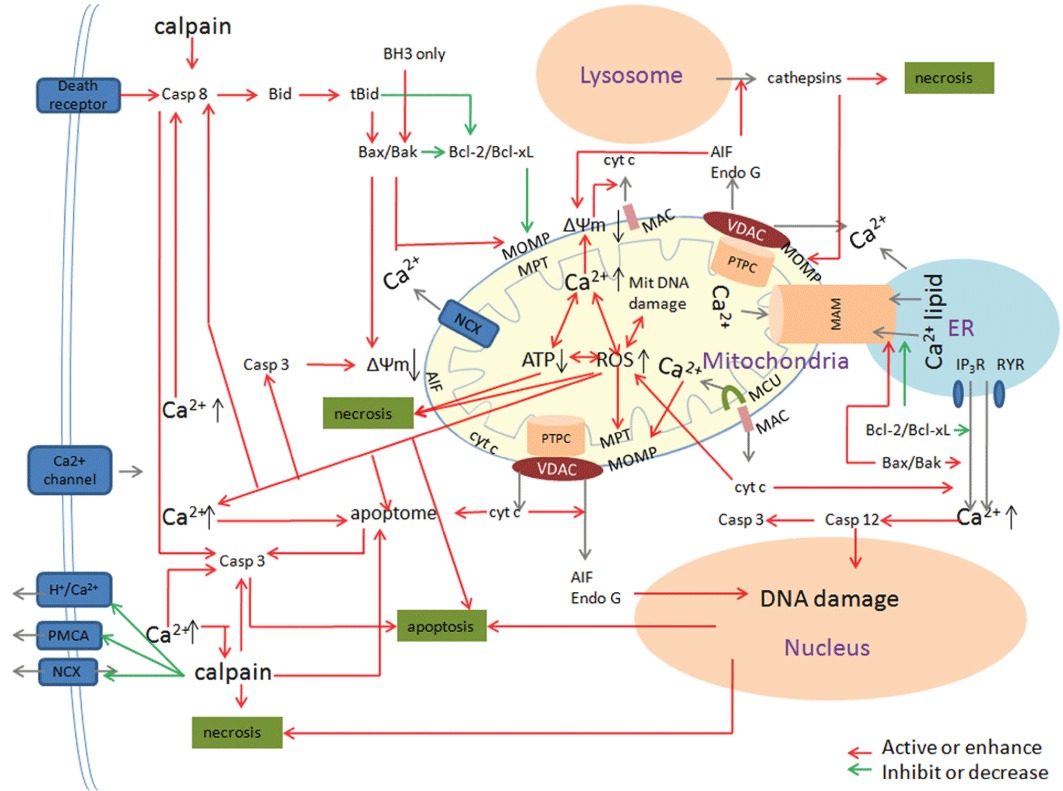
The role of mitochondria in cell death in TBI pathology. In TBI pathology, mitochondria provide the main platform of many intertwined factors that direct the cell to live or die. Three major pathways – the extrinsic pathway, the mitochondrial pathway and the caspase-12-mediated ER apoptotic pathway participate in cell death. In the extrinsic apoptotic pathway, mitochondria amplify the apoptotic signal or are essential for execution of the apoptotic program. The BH3-only protein Bid is one of the major links between extrinsic and mitochondrial apoptosis. The mitochondrial apoptotic pathway is classified as caspase-dependent or caspase-independent. In a caspase-dependent pathway, cyt c is the essential component whereas AIF is critically involved in the caspase-independent pathway. There are synergistic effects between mitochondrial membrane, Ca2+ and ROS in mediating cell damage after TBI. MOMP and MPT are two models of the formation of pores in the mitochondrial membranes. The Bcl-2 family is a critical regulator of mitochondria and cell fate. There are many amplifying loops among different pro-apoptotic effectors. In addition, there is coordination among mitochondria, ER and lysosome. In pathological conditions, Ca2+ exchanges between mitochondria and ER mediated by MAM is an important apoptotic control point. The release of cathepsin proteases from lysosome has a positive effect on mitochondria-mediated cell death.
Structural changes in VDACs are the main reason for MOMP. Ex novo formatted pores include ceramide-formed, lipid channel, mitochondrial apoptosis-induced channel (MAC) (Siskind, 2005; Dejean et al., 2006; Kinnally and Antonsson, 2007) and ruptured OM (Sesso et al., 2004). Mitochondrial dynamics also contributes to MOMP (Cassidy-Stone et al., 2008). The incomplete MOMP (iMOMP) found during apoptosis may relate to the process of mitochondrial fission. Inhibition of mitochondrial fission reduced the incidence of iMOMP, whereas the promotion of fission had the opposite effect. iMOMP may provide a critical cohort of healthy mitochondria that permits cellular recovery from MOMP, which may depend on Bax or Bak activation (Tait et al., 2010).
PTPC is a highly tunable mechanism that integrates a multiplicity of signals to maintain the viability of a cell or to commit it to death (Rasola and Bernardi, 2011). In physiological conditions, the PTPC has a ‘flickering’ status that is characterized by reduced conductance. In the presence of Ca2+ overload or oxidative stress, cyclophilin D (Cyp D) can induce a conformational change in the adenine nucleotide translocator (ANT) that results in MPT (Tsujimoto and Shimizu, 2007). It is generally believed that Cyp D, a mitochondrial member of the cyclophilin family, is an indispensable component of PTPC (Galat and Metcalfe, 1995); and that the modulation of the interaction of Cyp D with ANT is a suitable target in modulating mitochondrial cell death (Temkin et al., 2006). The putative role of Cyp D in regulating the MPT is derived from the observation that cyclosporin A (CsA), a specific inhibitor of the cyclophilin family, blocks the MPT (Broekemeier et al., 1989). However, other evidence showed that not all apoptosis can be inhibited by CsA (Nakagawa et al., 2005). Basso et al. (2005) proposed that there might be a CsA-insensitive MPT. Both the CsA-sensitive MPT and CsA-insensitive MPT may share a common mechanism, because both forms of MPT are inhibited by ubiquinone 0.
MOMP is always viewed as the hallmark of apoptosis, whereas MPT will lead to necrosis (Baines et al., 2005; Nakagawa et al., 2005). Apoptosis can be initiated by three major pathways – the extrinsic pathway, the mitochondrial pathway and the caspase-12-mediated ER apoptotic pathway. In the extrinsic apoptotic pathway, mitochondria amplify the apoptotic signal or are essential for execution of the apoptotic program (Barnhart et al., 2004). The mitochondrial apoptotic pathway is classified as caspase-dependent or caspase-independent. In a caspase-dependent pathway, cyt c is the necessary participant, whereas AIF is involved in the caspase-independent pathway. During apoptosis, death signals are relayed via BH3-only proteins to Bax and Bak, which will lead to MOMP (Chipuk et al., 2006). The resulting release of cyt c into the cytosol triggers apoptosome assembly and the subsequent caspase activation and apoptosis. There is some evidence that cristae remodelling is necessary for the complete release of cyt c during cell death because about 85% of cyt c is in the intracriS and only 15% is in the IMS (Bernardi and Azzone, 1981). Cristae morphology and the diameter of cristae junctions are regulated, at least in part, by the GTPase activity of Opa1 and the protease presenilin-associated rhomboid-like protein (PARL) (Gottlieb, 2006). In addition to Opa1 and PARL, the Bcl-2 family members have been shown to affect cristae remodelling. However, another study found that cristae remodelling is not required for the efficient release of cyt c, as swelling occurs only late in apoptosis after the release of cyt c and loss of the mitochondrial membrane potential (ΔΨm) (Sun et al., 2007). Arnoult et al. (2003) proposed that the release of different mitochondrial apoptogenic factors is a sequential process. First, Bax/Bak-mediated MOMP leads to the release of a significant part of the cyt c, Smac/Diablo and HtrA2/Omi proteins. Then ΔΨm loss occurs, which may be required for the release of the last pool of cyt c, Smac/Diablo and HtrA2/Omi. In a third step, cyt c, Smac/Diablo and HtrA2/Omi, which were released into the cytosol, trigger caspase activation. This is necessary to alter the physical association of AIF and EndoG with the IM to enable their relocation to the cytosol. Thus, EndoG and AIF seem to define a ‘caspase-dependent’, mitochondria-initiated apoptotic DNA degradation pathway. Severe mitochondrial cyt c release is also a predictor of necrotic cell death (Lewen et al., 2001). ER-stress leading to caspase-12 activation is the third route of apoptosis that is involved in TBI pathology. It can act either as an executioner caspase or activate caspase-3 directly or indirectly (Morishima et al., 2002).
MPT will lead to profound cellular consequences in necrosis. There is a loss of Δψm that is needed to drive production of ATP from ADP. The result is rapid mitochondrial dysfunction and excessive production of ROS that ultimately lead to necroptosis. Moreover, MPT results in marked mitochondrial swelling and potentially the outright rupture of the OM and severe release of death effectors (Kitsis and Molkentin, 2010).
In TBI pathology, mitochondria serve as signalling platforms for numerous biomolecules that are produced inside and outside the mitochondria. The coordinative role of different regulators and effectors in mitochondria determine cell death or life (Figure 3).
Excitotoxicity – one of the initiating factors in secondary insult
During TBI, building up of excitatory neurotransmitters due to synaptic release and impaired/reversed uptake mechanisms will lead to excitotoxicity. Studies have shown that excitotoxicity is location-dependent. Glutamate is one of the main excitatory neurotransmitters in CNS. AMPA receptors, NMDA receptors, voltage-dependent Ca2+ channels (VDCC) and metabotropic glutamate (mGlu) receptors are the main routes that mediate excitotoxicity by different mechanisms (Palmer et al., 1994; Wu and Saggau, 1997; Ertel et al., 2000). For example, Na+- and Cl--dependent influx is related to immediate cell swelling (Hossmann, 1994; Siesjo et al., 1995; Seiler et al., 2008; Brennan et al., 2009; Forder and Tymianski, 2009). Extrasynaptic NMDA receptor activation can activate a CREB (cAMP response element binding) protein shut-off that, in turn, causes loss of ΔΨm and apoptosis (Hardingham et al., 2002). Stress-activated protein kinases (SAPKs) are another class of signalling molecules that are implicated in NMDA receptor-dependent cell death (Legos et al., 2002; Borsello et al., 2003; Cao et al., 2004; Soriano et al., 2008).
Compared with other mechanisms, Ca2+-dependent influx is the main factor that is responsible for cell death induced by excitoxicity. In physiological conditions, Ca2+ is an important messenger in maintaining cellular bioactivtity. In neurons, the departure of Ca2+ from the cell is achieved through the plasma membrane Ca2+ ATPase pump (PMCA), Na+/ Ca2+ exchangers (NCXs) and H+/ Ca2+ uniporter (Cross et al., 2010). The cellular homeostasis of Ca2+ is maintained mainly by mitochondria via a calcium uniporter (MCU) on the IM, uncoupling proteins 2 and 3, Letm1 mitochondrial Ca2+/H+ antiporter (Pan et al., 2011), NCXs or via ‘calcium-induced-calcium-release’ pathways (Rossier, 2006), and the interplay between the mitochondria and ER (Pizzo and Pozzan, 2007). During brain injury, intracellular Ca2+ influx is mediated by many deeply interconnected signalling pathways. These include the MAPK/ERK kinase (MEK)–ERK–p38 MAPK signalling axis and protein phosphatase 2A activation (Kikuchi et al., 2000; Mori et al., 2002; Xu et al., 2006). mGluRs activation coupled to the GTP-binding protein Gq11 stimulates the release of inositol triphosphate (IP3), which activates Ca2+ channels in the ER (Mattson, 2007). Mitochondria serve as very efficient Ca2+ buffers, taking up substantial amounts of cytosolic Ca2+ at the expense of ΔΨm. As a consequence of Ca2+ uptake, mitochondria can suffer Ca2+ overload. According to the ‘two-hit’ hypothesis, a concurrent pathological stimulus can turn Ca2+ from a physiological to a pathological effector (Brookes et al., 2004).
Ca2+ overload can cause damage to the cell by several different, cross-amplifying, cascades. First, Ca2+ activates (either directly or indirectly) cysteine proteases called calpains and caspases that degrade a variety of substrates, including cytoskeletal proteins, membrane receptors and metabolic enzymes (Nixon, 2003; Bano et al., 2005). Second, Ca2+ induces oxidative stress through different mechanisms (Mattson and Sherman, 2003). Third, Ca2+ triggers apoptosis by induction/activation of various pro-apoptotic proteins (Ankarcrona et al., 1995; Dargusch et al., 2001; Culmsee and Mattson, 2005). Furthermore, excessive Ca2+ influx relates to a decreased ATP level and activated caspases and calpains, which contribute to impairment and inactivation of both PMCA and NCXs, thus exacerbating intracellular Ca2+ overload (Schwab et al., 2002; Bano et al., 2005; Pottorf et al., 2006; Bruce, 2010).
Mitochondrial dysfunction that is caused by excessive Ca2+ uptake by the mitochondria through the potential-driven uniporter is a key event in severe excitotoxicity, which leads to depolarization of ΔΨm (Vergun et al., 2003), ROS generation, depletion of ATP and the permeabilization of mitochondrial membrane that leads to apoptosis or necrosis due to mitochondrial damage. Recent studies suggest a link between mitochondrial dynamics and excitotoxic injury. NMDA-induced toxicity results in a fragmented mitochondrial phenotype and an impairment of mitochondrial fusion. Opa1 is a key regulator of mitochondrial integrity. Calpain activation by Ca2+ overload may impair Opa1 and result in mitochondrial morphology defects. The protective effect of inhibition of calpains can preserve mitochondrial morphology and protect neurons against excitotoxic cell death (Jahani-Asl et al., 2011).
ROS-mitochondrial dysfunction-related initiating factors in secondary insult
ROS are natural byproducts of the normal metabolism of oxygen. Categorically, ROS include free radicals, such as superoxide, hydroxyl radical and singlet oxygen, as well as non-radical species, such as hydrogen peroxide. Under physiological conditions, numerous endogenous antioxidants prevent oxidative damage, including superoxide dismutase, glutathione peroxidase, catalase, low-molecular-weight antioxidants and vitagenes (Calabrese et al., 2008). In the setting of TBI, each of those neuroprotective systems in the brain becomes overwhelmed and results in oxidative cell damage.
The post-traumatic sequelae establish conditions of increased metabolic demands on the reduced normal mitochondrial population resulting in abundant ROS production. The excessive production of ROS is due in part to excitotoxicity, free iron and interactions among ROS. Glutamate-mediated excitotoxicity leads to an increase in intracellular Ca2+ and the subsequent induction of enzymes, such as nitric oxide synthase and xanthine oxidase, that produce free radicals. Mitochondrial Ca2+ uptake can stimulate the net production of ROS through activation of MPT, release of cyt c, respiratory inhibition, release of pyridine nucleotides and loss of intramitochondrial glutathione necessary for detoxification of peroxides (Starkov et al., 2004).
Although there are non-mitochondrial sources of ROS (Stout et al., 1998; Vesce et al., 2004; Nicholls et al., 2007), the mitochondrion is a major intracellular source of ROS (Starkov, 2008; Murphy, 2009). To date, approximately 10 potential mitochondrial ROS-generating systems have been identified (Andreyev et al., 2005; Choi et al., 2009). In the mitochondria, superoxide can be produced by respiratory complexes and individual enzymes on the OM, on both sides of the IM and in the matrix. The respiratory chain complexes I and III are the primary mitochondrial sources of univalent reduction of O2 into superoxide (Mustafa et al., 2010). Superoxide is very unstable and quickly reacts with nearby molecules. It naturally dismutes into hydrogen peroxide (H2O2) and O2. In contrast to superoxide, H2O2 is membrane-permeable. H2O2 can, in turn, react with metal ions to form the highly reactive hydroxyl radical via the Fenton reaction (Feissner et al., 2009). Each ROS has an unpaired electron in its outer electron shell and thus is highly reactive and unstable. Essentially, ROS cause cell injury by compromising the integrity of the cell membrane by lipid peroxidation, protein and DNA oxidation and inhibition of the mitochondrial electron-transport chain (Kowaltowski et al., 2001; Skulachev, 2006).
Besides being the major source of ROS production, mitochondria are also targets of oxidative stress (Tavazzi et al., 2005). Overproduction of ROS in the mitochondria is one of the very early events that precede the collapse of ΔΨm, release of pro-apoptotic factors and activation of caspases. Most importantly, oxidative structural changes that are induced by ROS can impair mitochondrial energy metabolism. ROS also acts as a key contributor to necrotic cell death and also a promoter of apoptosis. Mitochondrial DNA (mtDNA) is prone to oxidative damage because it lacks introns and is close to an ROS source. mtDNA damage-induced decreased respiratory function enhances ROS generation, thus eliciting a vicious cycle of ROS-mtDNA damage that ultimately triggers apoptosis (Van Houten et al., 2006). Evidence suggests possible direct roles for ROS in mediating death receptor activation and apoptotic induction through ROS-induced receptor clustering and the formation of lipid raft-derived signalling platforms. ROS–NO interaction in controlling FLICE inhibitory protein (FLIP) down-regulation was considered to be a key regulatory mechanism of Fas-induced apoptosis (Wang et al., 2008). ROS are known triggers of the mitochondrial apoptotic cascade via interactions with proteins of the PTPC (Tsujimoto and Shimizu, 2007). Peroxynitrites can directly activate MPT (Vieira et al., 2001; Bernardi et al., 2006). Peroxidation of CL may initiate cyt c release by loosening cyt c from the IM (Kirkland et al., 2002). Furthermore, oxidized CL is distributed to the outer leaflet of the mitochondrial membrane and functions as a docking platform for truncated Bid (tBid), enabling MPT and cyt c movement across the OM into the cytosol (Gonzalvez et al., 2005). A significant mitochondrial loss of cyt c will lead to further ROS increase due a disrupted electron transport chain (Circu and Aw, 2010).
There are synergistic effects between mitochondrial membrane, Ca2+ and ROS in mediating cell damage after TBI. Ca2+ overload enhances ROS generation (Festjens et al., 2006; Gunter and Sheu, 2009). Oxidative stress can aggravate Ca2+ overload and sensitize the bioactivity of Ca2+ (Crompton and Costi, 1990; Connern and Halestrap, 1996; Halestrap et al., 1997; Kim et al., 2006; Juhaszova et al., 2008). Ca2+-induced injury cascade can be amplified by the interaction of oxidative stress and Ca2+ (Camello-Almaraz et al., 2006). Both Ca2+ and ROS can directly activate caspase or facilitate the release of caspases by increasing MPT during apoptosis (Circu and Aw, 2010). Ca2+ is the most important cellular permissive factor for MPT (Jambrina et al., 2003). Its effect is greatly enhanced by oxidative stress (Kowaltowski et al., 2001; Bernardi et al., 2006). ROS or increased cytosolic Ca2+ concentration ([Ca2+]c) can oxidize CL and results in the detachment of cyt c (Kagan et al., 2005). In this respect, strategies, such as Ca2+ channel blockade, reduction of oxidative stress and inhibition of the mitochondrial pore opening, appear to be promising tools for the management of acute brain-injured patients.
As the platform of death or life during the second insult in TBI pathology, Ca2+ and ROS are two main players, but not the only determiners of the fate of the mitochondria and the cell. The Bcl-2 family is a critical regulator in the death or life game.
Bcl-2 family- critical regulator of mitochondria and cell fate
The Bcl-2 family consists of proteins that contain Bcl-2 homology (BH) domains. The Bcl-2 family is divided into anti-apoptotic Bcl-2-like proteins according to structure and function that carry the BH1-4 domains (e.g. Bcl-2 and Bcl-xL) and pro-apoptotic Bcl-2-like proteins that contain the BH1-3 domains (e.g. Bax and Bak) or just a single BH3 domain (e.g. the so-called BH3-only proteins).
In TBI pathology, the performance of the anti-apoptotic and pro-apoptotic Bcl-2 family on the OM determines the fate of neurons (Kroemer and Reed, 2000; Green and Kroemer, 2004). The mitochondrial apoptosis-induced channel (MAC) is a pathway for cyt c release, the proposed components of which include the oligomeric Bax/Bax, Bax/Bak and/or Bak/Bak (Guihard et al., 2004; Dejean et al., 2006). In normal healthy cells, the anti-apoptotic proteins, such as Bcl-2, Bcl-xL, Bcl-w and Mcl-1, are found on the OM where they inhibit both MOMP (by sequestering Bax and Bak) and the MPT (through their interaction with the PTPC) (Scorrano and Korsmeyer, 2003; Scorrano et al., 2003; Rostovtseva et al., 2004; Rong and Distelhorst, 2008). Under pro-apoptotic conditions, Bax and Bak undergo conformational modifications and enter the OM fully, thereby creating MOMP (Kroemer et al., 2007). Apoptotic signals also up-regulate the expression of BH3-only proteins (Shimizu et al., 1999; Crompton et al., 2002). Once they are at the mitochondria, the BH3-only proteins either directly activate the pro-apoptotic proteins Bax or Bak or inhibit the anti-apoptotic members, such as Bcl-2 or Bcl-xL. However, the exact mechanism is still elusive (Green and Kroemer, 2004; Kroemer et al., 2007; Galluzzi et al., 2008; Lindsay et al., 2011). It is tempting to speculate that BH3-only proteins, such as Bmf, might promote apoptosis by relieving the Bcl-2/Bcl-xL-mediated inhibition of MOMP, and cause necrosis by interfering with the Bcl-2/Bcl-xL-mediated blockage of the MPT.
The BH3-only protein Bid is one of the major links between extrinsic and mitochondrial apoptosis that can be cleaved as tBid by caspase-8 and promote MOMP (Li et al., 1998; Luo et al., 1998). Bcl-2 and Bcl-xL are also cleaved by caspases that enable the new fragments to promote apoptosis (Scorrano et al., 2003; Lucken-Ardjomande and Martinou, 2005). A self-amplifying feed forward loop is involved in caspases, Bcl-2 and mitochondria, which may help to establish an irreversible commitment to apoptosis (Shelton et al., 2009). The CL is an important platform for multiple apoptotic signals, including the Bcl-2 family. For example, the selective degradation of mitochondrial CL impairs the pro-apoptotic action of tBid plus Bax (Lucken-Ardjomande et al., 2008). Furthermore, the binding of a pro-apoptotic Bcl-2 family (such as tBid, Bax and Bak) to mitochondria also plays critical roles in other apoptotic associated processes, such as cristae remodelling (Frezza et al., 2006), mitochondrial fission and fragmentation interacting with proteins such as mitofusin 2 and Endophilin B1 (Youle and Karbowski, 2005; Suen et al., 2008; Grohm et al., 2010). A number of studies have demonstrated that members of the Bcl-2 family reside in the ER where they have opposing actions in regulating the transfer of ER Ca2+ to mitochondria and thus direct the cell to live or die (Breckenridge et al., 2003; Forte and Bernardi, 2006).
The BH4 domain is specific for the anti-apoptotic proteins of the Bcl-2 family and, hence, is a suitable candidate in designing agents that have anti-apoptotic effect (Shimizu et al., 2000). HIV-TAT BH4 is a cell-permeable MOMP inhibitory recombinant fusion protein that is composed of the HIV-TAT plasma membrane translocation domain and the anti-apoptotic Bcl-xL-derived BH4 domain, that inhibits neuronal apoptosis in various models. The anti-apoptotic mechanisms of HIV-TAT BH4 peptide include efficient inhibition of caspase-3 activation, prevention of AIF translocation and suppression of AIF and cyt c translocation (Asoh et al., 2002; Dietz et al., 2002; Kilic et al., 2002; Yin et al., 2006). It is proposed that such BH4-like peptides can inhibit both caspase-dependent and caspase-independent apoptosis. Recently, studies found that HIV protease inhibitors can also simultaneously block caspase-dependent (activation of caspase-9, -3) and caspase-independent cell death pathways (AIF translocation) by blocking MOMP, presumably by inhibiting the ANT (Phenix et al., 2001; Wan and DePetrillo, 2002; Matarrese et al., 2003; Weaver et al., 2005; Hisatomi et al., 2008).
The fate of mitochondria is determined by the struggle between life supporters and death effectors. Once the death effectors become dominant, mitochondria will invariably direct the cell to apoptosis or necrosis mainly by caspases or AIF.
Caspases – main executors of apoptosis
Cellular caspases belong to a highly conserved family of cysteine proteases that cleave aspartate residues of caspase substrates and are the main players in the execution phase of apoptosis (Fischer et al., 2003; Shi, 2004). Under physiological conditions, the activation and activity of caspases can be inhibited at different stages. First, members of the inhibitor of the apoptosis protein (IAP) family constitute an endogenous barrier against improper or excessive caspase activation (Verhagen et al., 2001). Second, members like XIAP, c-IAP1, c-IAP2 and survivin bind and suppress enzyme catalytic activity (Kugler et al., 2000; Perrelet et al., 2000). Finally, post-translational modification of catalytic site cysteine residues is a potentially important redox mechanism in the control of caspase activity (Tzeng et al., 1997; Rossig et al., 1999; Kim et al., 2000; 2002; Chung et al., 2001; Zech et al., 2003; Huang et al., 2008). ROS, NO and glutathione are involved in redox regulation of caspase activity (Mannick et al., 1999; 2001; Kim et al., 2004; Mitchell et al., 2007; Pan and Berk, 2007; Sykes et al., 2007).
During apoptotic signalling, disturbance of the inhibitory mechanisms will lead to the activation of caspases and their cleavage activity. IAPs are antagonized by mitochondria-derived Smac/Diablo and Omi/HtrA2 proteins, allowing caspase-mediated execution (Du et al., 2000; Yang et al., 2003; Suzuki et al., 2004b). Calpains may also play an important role in the triggering of apoptotic cascades by virtue of their ability to activate caspases (Stefanis, 2005). Additionally, activated caspase-3 promotes caspases-2 and -6 activation in an amplification loop that enhances caspase-9 processing (Nicholson, 1999; Slee et al., 1999; Degterev et al., 2003).
Depending on their roles, the caspases are divided into initiator and executioner caspases. Under the pro-apoptotic stimuli, initiator caspases are recruited at the death receptor by the death effector domain (caspases-8 and -10) or in the cytosol by the recruiting domain (caspases-2 and -9). Then executioner caspases, such as caspases-3, and -7, are cleaved by the activated initiator caspases to execute apoptosis by the cleavage of protein substrates (Knoblach et al., 2002). More than 300 proteins have been characterized as caspase substrates (Marzo et al., 1998). In a large-scale analysis using CaSPredictor software, Garay-Malpartida et al. (2005) identified 1600 predicted caspase substrates with a score >0.57, with 60% sensitivity and 97% specificity. For example, brain injury is accompanied by cell cycle progression of neurons that, it has been suggested, leads to apoptosis in a caspase-dependent manner (Di Giovanni et al., 2005).
In TBI pathology, mitochondria are key targets of caspase cleavage and locations for the regulation of caspases. The cleavage of plasma membrane PMCA and NCX results in the increase of intracellular Ca2+ pools that may precede the opening of mitochondrial death decision pores (Schwab et al., 2002; Bano et al., 2005). Furthermore, activated caspases are localized in, or translocate to, mitochondria to trigger the extrinsic or mitochondrial pathways of apoptosis (Lemasters, 2005; Chipuk et al., 2006). For example, pro-caspase-8 was shown to be predominantly localized within the cytosol but has also been shown to be loosely associated with the OM. The translocation of caspase-8 to the mitochondria was necessary for efficient caspase-8 processing and activation (Stegh et al., 2000; 2002; Henshall et al., 2001). In vitro studies have shown an important role for caspase-2 in regulation of cyt c release (Lassus et al., 2002; Robertson et al., 2004). The inhibition of caspase-2 by the pan-caspase inhibitor boc-aspartyl (OMe)-fluoromethylketone reduces acute cell death after TBI (Clark et al., 2007). Caspases-3 and -7 are two highly related effector caspases. Active caspase-3 is required for the sustained loss of the ΔΨm (Waterhouse et al., 2006). Mice that lack both enzymes have exhibited a resistance to drugs that induce the mitochondrial and extrinsic pathways to apoptosis (Lakhani et al., 2006). The close cooperation between mitochondria and caspases makes mitochondria the central player in both the extrinsic and mitochondrial apoptosis pathways in acute brain injury.
AIF – main participant of necrosis
In contrast to capases, AIF, a ubiquitously expressed flavoprotein, plays a critical role in caspase-independent apoptosis and necrosis in areas with less energy supply. Under normal physiological conditions, the NADH oxidase activity of AIF is required for the functioning of the respiratory chain. AIF deficiency results in reduced expression of complexes I and II and inefficient oxidative phosphorylation (Vahsen et al., 2004). Under pro-apoptotic stimulation, AIF and EndoG are released from IMS and translocate to the nucleus to promote caspase-independent DNA degradation (Yamashita et al., 2004; Slemmer et al., 2008).
Moubarak et al. (2007) have reported that the sequential activation of PARP-1, calpain and Bax is essential in AIF-mediated programmed necrosis. PARP-1 is a nuclear enzyme that acts as a DNA damage sensor. If it is overactivated, it mediates cell death (Yu et al., 2002; Cande et al., 2004). There are also active PARPs within mitochondria (mtPARP) that induce intramitochondrial poly (ADP) ribosylation (Du et al., 2003). Various poly (ADP)-ribosylated mitochondrial proteins have been identified. mtPARP over-activation may influence cellular energy stores both by depleting NAD+ and decreasing ATP production, which can lead to cell dysfunction and death (Lai et al., 2008). Recently, a novel AIF release and cell death by superoxide production and prolonged ERK1/2 phosphorylation were found (Kondo et al., 2010). The cell death was not associated with Bax, cyt c, caspase-3 or PARP-1. The significance of PARP-1-independent AIF release in post-traumatic neuronal damage needs to be established.
Yu et al. found that about 30% of AIF loosely associates with the OM on the cytosolic side. This outer mitochondrial pool of AIF is sufficient to cause cell death (Yu et al., 2006b; Wang et al., 2009). Additionally, cytosolic AIF acts on the mitochondria to collapse the ΔΨm and initiates the release of cyt c, thus initiating caspase-dependent apoptosis (Susin et al., 1999). AIF, itself, does not have any DNase activity and it induces DNA degradation only when it binds and translocates to the nucleus together with CsA (Zhu et al., 2007). Therefore, pharmacological agents that prevent the interaction of AIF and CsA might act as neuroprotectors. The DNase activity of EndoG can be suppressed by the heat-shock protein (HSP) 70 in an ATP-dependent manner (Kalinowska et al., 2005).
It is clear that mitochondria are the main platform and one of the main participants in determining the death or life of cells in TBI pathology. Thus, mitochondria-centred therapy is of vital importance in reducing tissue damage following TBI. Although mitochondria displayed bioenergetic deficits after one hour following injury, the damage was not exacerbated after 3 h, with peak mitochondrial dysfunction occurring at approximately 12–24 h, which provides an opportunity for effective intervention of the mitochondrial dysfunction in a clinical setting (Gilmer et al., 2009). As ATP depletion led by mitochondrial dysfunction is the fundamental cause of cell damage, a combination of compounds that target different metabolic pathways of mitochondrial could diminish cellular disturbance and thereby improve or stabilize clinical features. Second, considering the complexity of the process and central role of mitochondria in the process of secondary brain damage, multipotential therapeutic strategies are necessary in mitochondria-targeted protection (Royo et al., 2003; Pitkänen et al., 2005; Schouten, 2007; Beauchamp et al., 2008).
Energy supply – the first level in mitochondrial-targeted therapy
The brain is highly dependent on a continuous supply of oxygen and glucose to maintain cellular integrity. In TBI pathology, energy depletion is the result of mitochondria impairment and an important cause of mitochondria damage. Inadequate O2 supply to the traumatized brain results in the conversion of aerobic metabolism to anaerobic metabolism that results in acidosis and depletion of cellular energy. Excitotoxicity, Ca2+ overload and ROS overproduction that follow the primary insult and energy failure will bring severe damage to mitochondria. The impairment of mitochondrial respiratory chain-linked oxidative phosphorylation will lead to further functional failure of aerobic metabolism. On the other hand, the repair of damaged tissue needs more energy than its normal physiological condition. This results in what has been termed a ‘flow/metabolism mismatch’, which is the most important unfavourable factor in TBI (Rockswold et al., 2007). Studies investigating the metabolic fate of glucose after TBI have shown that, after the initial hours following injury, glucose utilization changes from energy production to cellular repair mechanisms by the pentose phosphate pathway. This results in a higher temperature in the lesion core. In an energy-deprived environment, before an irreversible event,such as MOMP occurs, cells respond by maintaining a minimal survival level while awaiting a rescue in the event of a growth factor or nutrient re-addition (Chipuk et al., 2006). Therefore, impaired energy metabolism is a potential target of TBI therapy.
It is generally agreed that with a reduction in temperature, the injured brain decreases its consumption of energy-producing substrates. Clinically induced hypothermia is the only therapy that demonstrates improved neurological outcomes following cardiac arrest (Bernard et al., 2002). Hypothermia has been widely accepted as the gold standard method by which the body can protect the brain (Choi et al., 2012). During the pathological process following TBI, induced hypothermia can protect mitochondria in at least three ways: (1) reducing the energy demand of mitochondria by reducing the cerebral metabolic rate of oxygen (CMRO2); (2) direct protection of mitochondria via maintenance of ΔΨm and decreasing production of ROS (Shao et al., 2010); (3) indirect protective effect such as the reduction of excitotoxic neurotransmitters, facilitation of anti-inflammatory responses and anti-apoptotic pathways, reduction of intracranial pressure (ICP) and stabilization of the blood–brain barrier (BBB).
Increasing the metabolic support is another important aspect in mitochondrial protection. Supporting the aerobic processes of the threatened cells could possibly preserve viable, but non-functioning, tissue. It has been proved that hyperoxia can increase the partial pressure of oxygen (pO2) within the blood and the subsequent, improved mitochondrial metabolism/tissue oxygenation. In a rat model of fluid percussion injury (FPI), 1 h 1.5 ATA with 3 h 100% normobaric oxygen treatments significantly improved the recovery of synaptosomal mitochondrial metabolic activity (Azbill et al., 1997). Palzur et al. (2008) hypothesized that hyperoxia-induced increases in Bcl-2 expression, and that the resultant increase in intracellular oxygen bioavailability may contribute both to preserve mitochondrial integrity and to reduce the activation of the mitochondrial mediated apoptotic pathway following TBI. Work by several investigators suggests that hyperbaric oxygen allows the injured brain to use baseline amounts of O2 more efficiently following treatment and has a persistent effect on the injured brain tissue (Contreras et al., 1988; Rockswold et al., 2001). The main controversy about hyperoxia therapy concerns the increased production of ROS. In fact, brief periods of normobaric hyperoxia do not produce oxidative stress and/or change antioxidant reserves in CSF (Puccio et al., 2009).
Mitochondria dysfunction in TBI makes it impossible to make use of glucose effectively. However, lactate can be used directly through the tricarboxylic acid cycle. Over the past two decades, mounting clinical and experimental evidence suggests that lactate may appropriately fuel aerobic brain energy metabolism (Schurr et al., 1999; Ide et al., 2000; Smith et al., 2003; Gallagher et al., 2009) and provide definite therapeutic effects (Izumi et al., 1994; Maran et al., 1994). Recently, Arun et al. (2010) found that glyceryl triacetate (GTA), an FDA-approved food and drug additive, is more effective than lactate in treating TBI. In several TBI models, lactate per se is not enough to improve the altered cerebral energy state because of the disturbance of other co-substrates, such as NAD+ (Prieto et al., 2011). However, this does not exclude the definite benefit provided by lactate.
Besides oxygen and glucose, damage to mitochondria could occur at any number of sites in the molecular machinery that might be responsible for a decline in respiration and the production of ATP. In the brain, the acetate can be converted to acetyl CoA, which can be utilized for energy production, lipid synthesis and other metabolic processes. In addition, acetate metabolism bypasses both glucose and N-acetylaspartate metabolism, which helps to compensate for the substantial reductions in N-acetylaspartate and ATP levels observed after TBI (Arun et al., 2010).
In fact, the preservation of energy supply and mitochondria protection are two closely connected processes. Enough energy supply is the prerequisite for mitochondria to cope with various vicious agents and to maintain the integrity and normal function. For example, creatine not only can maintain ATP levels, but can ameliorate neuronal cell death and reduces mitochondrial ROS production following TBI (Sullivan et al., 2000). Exogenous ketone bodies can improve the mitochondrial function and increase cortical levels of ATP after injury (Davis et al., 2008a). Fasting and a ketogenic diet can also decrease ROS production by uncoupling protein (UCP)-mediated uncoupling (Sullivan et al., 2004a; Davis et al., 2008b).
Mitochondrial – targeted multipotential therapeutic strategies
The mitochondrial membrane is the last barrier against cell death. From this point on, mitochondria-centred, especially mitochondrial membrane-targeted combined therapy will hold ‘the last line’ and help to prevent subsequent waves of secondary cascade events. A secondary insult after TBI is a complicated pathological process that involves various stimuli, and thus one is naive to expect that patients can benefit from a single treatment that is active on a specific mechanism and administered at a single stage of the unfolding process (Saatman et al., 2008). The preservation of energy supply alone is not enough for mitochondria protection. In recent years, based on new knowledge about mitochondria, some innovative strategies have shown good prospects in mitochondrial-targeted multipotential therapy (Figure 4).
Figure 4.
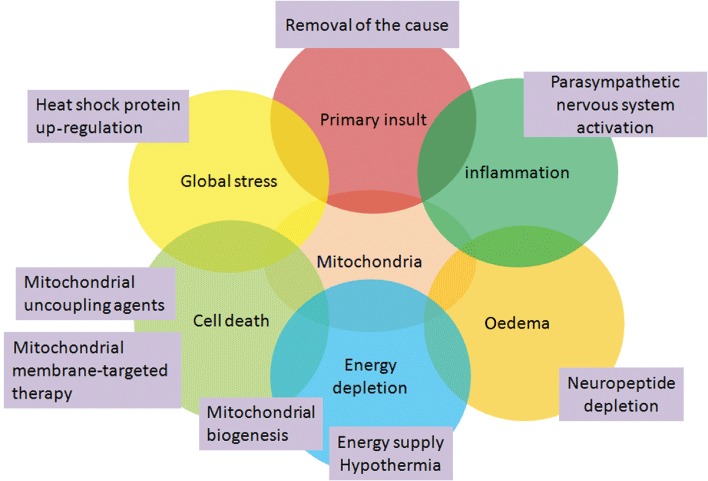
Mitochondrial-targeted multipotential therapeutic strategies. TBI is a pathological process with many intertwined participants that are involved in mitochondria. Combined strategies that target different pathological process will obtain the best outcome in mitochondrial-targeted multipotential therapy.
A 2009 workshop that was sponsored by the National Institute of Neurological Disorders and Stroke, recommended a combined approach for the treatment of TBI, focusing on therapeutics with complementary targets and effects (Margulies and Hicks, 2009). First, the early removal of the cause of injury is known to limit the area of neuronal damage by reducing the duration of the insult. In our previous study, we found that gross-total haematoma removal was an effective way to decrease intracranial haemorrhage-induced injury to brain tissue. This effect was related to decreased perihaematomal oedema formation and secondary injury by an inflammatory cascade activated by coagulation end products (Zuo et al., 2009).
The development of cerebral oedema with brain swelling is the most significant predictor of outcome. Mitochondrial dysfunction, triggered by various death stimuli, is the main cause of propagation of cytotoxic brain oedema (Marmarou et al., 2000; Unterberg et al., 2004) and is a leading event in the cascade of cell death in TBI (Fiskum et al., 2000; Sullivan et al., 2005; Wang et al., 2010). Although vasogenic oedema is transient in comparison to cytotoxic oedema, vasogenic oedema is permissive for cytotoxic oedema formation (Beaumont et al., 2000). Neurogenic inflammation, mediated mainly by calcitonin gene-related peptide and substance P, is the main cause of vasogenic oedema (Geppetti et al., 1995; Donkin et al., 2009). Neuropeptide depletion or post-traumatic administration of the neurokinin-1 receptor antagonist N-acetyl-L-tryptophan inhibits BBB permeability and any subsequent oedema formation. Early inhibition of neurogenic inflammation may present a novel approach to the treatment of post-traumatic oedema formation (Donkin and Vink, 2010).
A systemic inflammatory response syndrome is a persistent pathological process that worsens patient outcome (Kemp et al., 2008). Activation of the sympathetic nervous system is critical in CNS-induced immunodepression and organ inflammation. Phagocytic catecholamine production also contributes to a release of the pro-inflammatory cytokines (Flierl et al., 2009). Therefore, therapeutic strategies that are aimed at activating the parasympathetic nervous system could counterbalance the effects of the activated sympathetic system (Catania et al., 2009). The release of chemokines by the liver is a significant aspect of the acute-phase response that is associated with CNS injury (Campbell et al., 2003), and the suppression of Kupffer cell activity prevents secondary damage after acute brain injury (Campbell et al., 2008).
Following acute injury, neurons and glia activate a global stress response that involves the transcriptional and translational upregulation of several several heat shock proteins (HSPs) (Wagstaff et al., 1996), which contribute generally to survival by virtue of their chaperone function (Yenari et al., 2005). Several HSPs (e.g.HSP27, HSP70 and HSP90) and derived molecular chaperones act as specific anti-apoptotic factors through multiple mechanisms, including the following: inhibiting both caspase-dependent and caspase-independent pathways (Saleh et al., 2000; Gurbuxani et al., 2003; Kalinowska et al., 2005; Ruchalski et al., 2006), apoptosome inhibition by direct apoptotic peptidase activating factor 1 binding (Saleh et al., 2000), blockage of AIF mitochondrial release (Ruchalski et al., 2006), cytosolic AIF sequestration and inhibition of EndoG DNase activity (Ravagnan et al., 2001; Kalinowska et al., 2005) and possible antioxidant property (Choi et al., 2005).
At the cellular level, five principles of metabolic manipulation are proposed that target oxidative damage, lipid peroxidation, altered ΔΨm, Ca2+ imbalance and transcription regulation (Koene and Smeitink, 2011). In this respect, pharmacological agents that offer ‘mitochondrial uncoupling’ ability provide new hope because they can increase proton leakage, thereby reducing oxygen stress by lowering ΔΨm (Cannon and Nedergaard, 2004; Jarmuszkiewicz et al., 2004; Sullivan et al., 2004a, b,Sluse et al., 2006; Pandya et al., 2007; 2009). Furthermore, because ΔΨm is the driving force for cytoplasmic Ca2+ entry to mitochondria, a mild ‘mitochondrial uncoupling’ can reduce ΔΨm and attenuate mitochondrial Ca2+ cycling (Gunter et al., 1994; Mattiasson et al., 2003). Stable nitroxide radicals have the ability to combine radical scavenging action with recycling capacities, which provide a new strategy to reduce ROS overproduction (Borisenko et al., 2004; Mustafa et al., 2010). Some stable nitroxide radical agents have shown positive results in preclinical studies (Kwon et al., 2003; Macias et al., 2007). ROS is produced only briefly, but the chain reaction of ROS-related lipid peroxidation (LP) persists for several days. Indirectly-acting antioxidant mechanisms that stop the ‘chain reaction’ propagation of LP once it has begun are a suitable choice. The combination of different antioxidant mechanistic strategies may improve the extent of neuroprotective efficacy, lessen the variability of the effect and possibly provide a longer therapeutic window of opportunity (Hall et al., 2010).
As an important mediator of neuronal apoptosis and necrosis, PTPC represents a crucial target for neuroprotection (Nieminen et al., 1996). An abundance of experiments have proved the efficiency of CsA in ameliorating mitochondrial functions in TBI. In an impact acceleration TBI model (Friberg et al., 1998; Buki et al., 1999; Crompton, 1999; Scheff and Sullivan, 1999; Sullivan et al., 1999; Albensi et al., 2000; Alessandri et al., 2002; Friberg and Wieloch, 2002; Hansson et al., 2003; Signoretti et al., 2004; Mazzeo et al., 2008), Okonkwo and Povlishock (1999) found that CsA protects both mitochondria and the related axonal shaft. phase II clinical trials found that CsA can improve cerebral perfusion pressure and cerebral metabolism. Considering the proven safe and CSF pharmacokinetics of CsA, a phase III clinical trial is underway (Empey et al., 2006; Mazzeo et al., 2006; Merenda and Bullock, 2006). Because immunosupression is one of the possible side effects of CsA, other CsA derivatives, such as NIM-811 and 2-aminoethoxydiphenylborate, have been designed and show promising results (Waldmeier et al., 2002; Chinopoulos et al., 2003; Merenda and Bullock, 2006; Mbye et al., 2008; Readnower et al., 2011). The progesterone metabolite allopregnanolone is also a suitable choice to inhibit MPT and cyt c release (Sayeed et al., 2009). The slow-onset kinetics of necroptosis suggests that this pathway might provide a new target for neuroprotecitve intervention with an extended therapeutic window. Thus, the combination of anti-apoptotic and anti-necroptotic treatment might be suitable. Necrostatin-1 can inhibit all published examples of necrotic cell death induced by activation of death domain receptors in the presence of caspase inhibitors (Degterev et al., 2005). Under stress, mitochondrial biogenesis becomes an essential endogenous neuroprotective response that creates new functional mitochondria. Therefore, the stimulation or enhancement of mitochondrial biogenesis is a novel neuroprotective strategy.
TBI is a multi-factorial pathological change that affects the whole body. Thus, any potential benefits gained by targeting a single molecule or pathway may be masked by the plethora of simultaneously activated cascades. Therapies that serve to modulate multiple pathophysiological pathways and target multiple cell types may prove to be more effective. In preclinical studies, improved levels of neuroprotection have been obtained using therapeutic agents with multifunctional activities (Faden and Stoica, 2007; Vink and Nimmo, 2009), such as small cyclized dipeptides (Stoica et al., 2009), progesterone (De Nicola et al., 2009), PPAR activators (Semple and Noble-Haeusslein, 2011) and erythropoietin, among others (Stoica and Faden, 2010).
At the present, there are still many unanswered questions about the structure and function of the mitochondrion that need to be resolved. For example, the lipid composition and structural organization of the OM are among the central players in the regulation of Bcl-2 family member activity. However, the precise 3D structure that many Bcl-2 proteins adopt in this membrane is still unknown. Without this essential information, it is impossible to understand precisely how MOMP occurs (Lindsay et al., 2011). Yet it has been made clear that, by providing a sufficient energy supply as soon as possible, a mitochondrial-targeted multipotential therapeutic strategy will provide the best protection for TBI patients.
Conclusion
Mitochondria play a pivotal role in the secondary insult in TBI pathology. The dysfunction of mitochondria is the main cause of energy failure of damaged tissue and the platform of death. The coordinative role of different regulators and effectors in mitochondria, including excitotoxicity, ROS, caspases, the Bcl-2 family and AIF, determine cell death or life. Some preclinical and clinical results of mitochondria-targeted therapy show promise. With the clarification of the function and structure of mitochondria, mitochondrial-targeted multipotential therapeutic strategies will provide new hope for the treatment of TBI and other acute brain injury states.
Glossary
- AIF
apoptosis inducing factor
- ANT
ADP/ATP carrier adenine nucleotide translocase
- BH
Bcl-2 homology
- CBF
cerebral blood flow
- CK
creatine kinase
- CL
cardiolipin
- CsA
cyclosporin A
- Cyp
D, cyclophilin D
- cyt
cytochrome
- ER
endoplasmic reticulum
- ETC
electron transport chain
- Fis1
fission1
- HSPs
heat-shock proteins
- IAP
inhibitor of the apoptosis protein
- IBM
inner boundary membrane
- IM
inner membrane
- IMS
intermembrane space
- iNOS
inducible NOS
- HK
hexokinase
- LP
lipid peroxidation
- MAC
mitochondrial apoptosis-induced channel
- MAM
mitochondria-associated endoplasmic reticulum-membrane
- MEK
MAPK/ERK kinase
- mGlu
receptor, metabotropic glutamate receptor
- MMP
mitochondrial membrane permeabilization
- MOMP
mitochondrial outer membrane permeabilization
- MPT
membrane permeability transition
- mtDNA
mitochondrial DNA
- mtPARP
PARPs within mitochondria
- NCXs
Na+/Ca2+ exchangers
- OM
outer membrane
- Opa
optical atrophy protein
- PARL
presenilin associated rhomboid-like protein
- Pi
mitochondrial phosphate carrier
- PMCA
plasma membrane Ca2+ ATPase pump
- PTPC
permeability transition pore complex
- ROS
reactive oxygen species
- SAPKs
stress-activated protein kinases
- TBI
traumatic brain injury
- tBid
truncated Bid
- TSPO
translocator protein
- UCP
uncoupling protein
- VDAC
voltage-dependent anion channel
- VDCC
voltage-dependent Ca2+ channels
- ΔΨm
membrane potential
Statement of conflict of interest
No conflicts of interest exist.
References
- Albensi BC, Sullivan PG, Thompson MB, Scheff SW, Mattson MP. Cyclosporin ameliorates traumatic brain-injury-induced alterations of hippocampal synaptic plasticity. Exp Neurol. 2000;162:385–389. doi: 10.1006/exnr.1999.7338. [DOI] [PubMed] [Google Scholar]
- Alessandri B, Rice AC, Levasseur J, DeFord M, Hamm RJ, Bullock MR. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma. 2002;19:829–841. doi: 10.1089/08977150260190429. [DOI] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003;22:4385–4399. doi: 10.1093/emboj/cdg423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Ariyannur PS, Moffett JR, Xing G, Hamilton K, Grunberg NE, et al. Metabolic acetate therapy for the treatment of traumatic brain injury. J Neurotrauma. 2010;27:293–298. doi: 10.1089/neu.2009.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asoh S, Ohsawa I, Mori T, Katsura K, Hiraide T, Katayama Y, et al. Protection against ischemic brain injury by protein therapeutics. Proc Natl Acad Sci U S A. 2002;99:17107–17112. doi: 10.1073/pnas.262460299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997;765:283–290. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004;23:3175–3185. doi: 10.1038/sj.emboj.7600325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF. Pharmacology of traumatic brain injury: where is the ‘golden bullet’? Mol Med. 2008;14:731–740. doi: 10.2119/2008-00050.Beauchamp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont A, Marmarou A, Hayasaki K, Barzo P, Fatouros P, Corwin F, et al. The permissive nature of blood brain barrier (BBB) opening in edema formation following traumatic brain injury. Acta Neurochir Suppl. 2000;76:125–129. doi: 10.1007/978-3-7091-6346-7_26. [DOI] [PubMed] [Google Scholar]
- Belizario JE, Alves J, Occhiucci JM, Garay-Malpartida M, Sesso A. A mechanistic view of mitochondrial death decision pores. Braz J Med Biol Res. 2007;40:1011–1024. doi: 10.1590/s0100-879x2006005000109. [DOI] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Azzone GF. Cytochrome c as an electron shuttle between the outer and inner mitochondrial membranes. J Biol Chem. 1981;256:7187–7192. [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, et al. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- Borisenko GG, Martin I, Zhao Q, Amoscato AA, Kagan VE. Nitroxides scavenge myeloperoxidase-catalyzed thiyl radicals in model systems and in cells. J Am Chem Soc. 2004;126:9221–9232. doi: 10.1021/ja0495157. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T, et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003;22:3927–3936. doi: 10.1038/sj.onc.1206622. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Xue D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol. 2004;16:647–652. doi: 10.1016/j.ceb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–7830. [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Bruce J. Plasma membrane calcium pump regulation by metabolic stress. World J Biol Chem. 2010;1:221–228. doi: 10.4331/wjbc.v1.i7.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Povlishock JT. Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma. 1999;16:511–521. doi: 10.1089/neu.1999.16.511. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Mancuso C, Pennisi G, Calafato S, Bellia F, et al. Cellular stress response: a novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem Res. 2008;33:2444–2471. doi: 10.1007/s11064-008-9775-9. [DOI] [PubMed] [Google Scholar]
- Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol. 2006;291:C1082–C1088. doi: 10.1152/ajpcell.00217.2006. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Hughes PM, Iredale JP, Wilcockson DC, Waters S, Docagne F, et al. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J. 2003;17:1168–1170. doi: 10.1096/fj.02-0757fje. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M, et al. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology. 2008;55:780–787. doi: 10.1016/j.neuropharm.2008.06.074. [DOI] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Cao J, Semenova MM, Solovyan VT, Han J, Coffey ET, Courtney MJ. Distinct requirements for p38alpha and c-Jun N-terminal kinase stress-activated protein kinases in different forms of apoptotic neuronal death. J Biol Chem. 2004;279:35903–35913. doi: 10.1074/jbc.M402353200. [DOI] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Kroemer G. Mitochondrial apoptosis and the peripheral benzodiazepine receptor: a novel target for viral and pharmacological manipulation. J Exp Med. 2002;196:1121–1125. doi: 10.1084/jem.20021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania A, Lonati C, Sordi A, Gatti S. Detrimental consequences of brain injury on peripheral cells. Brain Behav Immun. 2009;23:877–884. doi: 10.1016/j.bbi.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Cheung EC, McBride HM, Slack RS. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis. 2007;12:979–992. doi: 10.1007/s10495-007-0745-5. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Starkov AA, Fiskum G. Cyclosporin A-insensitive permeability transition in brain mitochondria: inhibition by 2-aminoethoxydiphenyl borate. J Biol Chem. 2003;278:27382–27389. doi: 10.1074/jbc.M303808200. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- Choi S, Park KA, Lee HJ, Park MS, Lee JH, Park KC, et al. Expression of Cu/Zn SOD protein is suppressed in hsp 70.1 knockout mice. J Biochem Mol Biol. 2005;38:111–114. doi: 10.5483/bmbrep.2005.38.1.111. [DOI] [PubMed] [Google Scholar]
- Choi K, Kim J, Kim GW, Choi C. Oxidative stress-induced necrotic cell death via mitochondira-dependent burst of reactive oxygen species. Curr Neurovasc Res. 2009;6:213–222. doi: 10.2174/156720209789630375. [DOI] [PubMed] [Google Scholar]
- Choi HA, Badjatia N, Mayer SA. Hypothermia for acute brain injury-mechanisms and practical aspects. Nat Rev Neurol. 2012;8:241–222. doi: 10.1038/nrneurol.2012.21. [DOI] [PubMed] [Google Scholar]
- Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun. 2001;282:1075–1079. doi: 10.1006/bbrc.2001.4670. [DOI] [PubMed] [Google Scholar]
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Nathaniel PD, Zhang X, Dixon CE, Alber SM, Watkins SC, et al. boc-Aspartyl(OMe)-fluoromethylketone attenuates mitochondrial release of cytochrome c and delays brain tissue loss after traumatic brain injury in rats. J Cereb Blood Flow Metab. 2007;27:316–326. doi: 10.1038/sj.jcbfm.9600338. [DOI] [PubMed] [Google Scholar]
- Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem. 2004;256-257:107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- Connern CP, Halestrap AP. Chaotropic agents and increased matrix volume enhance binding of mitochondrial cyclophilin to the inner mitochondrial membrane and sensitize the mitochondrial permeability transition to [Ca2+] Biochemistry. 1996;35:8172–8180. doi: 10.1021/bi9525177. [DOI] [PubMed] [Google Scholar]
- Contreras FL, Kadekaro M, Eisenberg HM. The effect of hyperbaric oxygen on glucose utilization in a freeze-traumatized rat brain. J Neurosurg. 1988;68:137–141. doi: 10.3171/jns.1988.68.1.0137. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Degli Esposti M. Membrane lipids and cell death: an overview. Chem Phys Lipids. 2004;129:133–160. doi: 10.1016/j.chemphyslip.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341((Pt 2)):233–249. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Costi A. A heart mitochondrial Ca2(+)-dependent pore of possible relevance to re-perfusion-induced injury. Evidence that ADP facilitates pore interconversion between the closed and open states. Biochem J. 1990;266:33–39. doi: 10.1042/bj2660033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84:143–152. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- Cross JL, Meloni BP, Bakker AJ, Lee S, Knuckey NW. Modes of neuronal calcium entry and homeostasis following cerebral ischemia. Stroke Res Treat. 2010;2010:316862. doi: 10.4061/2010/316862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochem Biophys Res Commun. 2005;331:761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- Daems WT, Wisse E. Shape and attachment of the cristae mitochondriales in mouse hepatic cell mitochondria. J Ultrastruct Res. 1966;16:123–140. doi: 10.1016/s0022-5320(66)80027-8. [DOI] [PubMed] [Google Scholar]
- Dargusch R, Piasecki D, Tan S, Liu Y, Schubert D. The role of Bax in glutamate-induced nerve cell death. J Neurochem. 2001;76:295–301. doi: 10.1046/j.1471-4159.2001.00035.x. [DOI] [PubMed] [Google Scholar]
- Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008a;86:1812–1822. doi: 10.1002/jnr.21628. [DOI] [PubMed] [Google Scholar]
- Davis LM, Rho JM, Sullivan PG. UCP-mediated free fatty acid uncoupling of isolated cortical mitochondria from fasted animals: correlations to dietary modulations. Epilepsia. 2008b;49(Suppl 8):117–119. doi: 10.1111/j.1528-1167.2008.01854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nicola AF, Labombarda F, Deniselle MC, Gonzalez SL, Garay L, Meyer M, et al. Progesterone neuroprotection in traumatic CNS injury and motoneuron degeneration. Front Neuroendocrinol. 2009;30:173–187. doi: 10.1016/j.yfrne.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Dejean LM, Martinez-Caballero S, Kinnally KW. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ. 2006;13:1387–1395. doi: 10.1038/sj.cdd.4401949. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, et al. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A. 2005;102:8333–8338. doi: 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz GP, Kilic E, Bahr M. Inhibition of neuronal apoptosis in vitro and in vivo using TAT-mediated protein transduction. Mol Cell Neurosci. 2002;21:29–37. doi: 10.1006/mcne.2002.1165. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010;23:293–299. doi: 10.1097/WCO.0b013e328337f451. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Nimmo AJ, Cernak I, Blumbergs PC, Vink R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1388–1398. doi: 10.1038/jcbfm.2009.63. [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Empey PE, McNamara PJ, Young B, Rosbolt MB, Hatton J. Cyclosporin A disposition following acute traumatic brain injury. J Neurotrauma. 2006;23:109–116. doi: 10.1089/neu.2006.23.109. [DOI] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, et al. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- Faden AI, Stoica B. Neuroprotection: challenges and opportunities. Arch Neurol. 2007;64:794–800. doi: 10.1001/archneur.64.6.794. [DOI] [PubMed] [Google Scholar]
- Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri KF, Kroemer G. Mitochondria – the suicide organelles. Bioessays. 2001;23:111–115. doi: 10.1002/1521-1878(200102)23:2<111::AID-BIES1016>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta. 2006;1757:1371–1387. doi: 10.1016/j.bbabio.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Kowaltowksi AJ, Andreyev AY, Kushnareva YE, Starkov AA. Apoptosis-related activities measured with isolated mitochondria and digitonin-permeabilized cells. Methods Enzymol. 2000;322:222–234. doi: 10.1016/s0076-6879(00)22023-5. [DOI] [PubMed] [Google Scholar]
- Flierl MA, Rittirsch D, Nadeau BA, Sarma JV, Day DE, Lentsch AB, et al. Upregulation of phagocyte-derived catecholamines augments the acute inflammatory response. PLoS ONE. 2009;4:e4414. doi: 10.1371/journal.pone.0004414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Forte M, Bernardi P. The permeability transition and BCL-2 family proteins in apoptosis: co-conspirators or independent agents? Cell Death Differ. 2006;13:1287–1290. doi: 10.1038/sj.cdd.4401957. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–250. doi: 10.1016/s0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci. 1998;18:5151–5159. doi: 10.1523/JNEUROSCI.18-14-05151.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galat A, Metcalfe SM. Peptidylproline cis/trans isomerases. Prog Biophys Mol Biol. 1995;63:67–118. doi: 10.1016/0079-6107(94)00009-x. [DOI] [PubMed] [Google Scholar]
- Gallagher CN, Carpenter KL, Grice P, Howe DJ, Mason A, Timofeev I, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain. 2009;132((Pt 10)):2839–2849. doi: 10.1093/brain/awp202. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Kepp O, Seror C, Hangen E, Perfettini JL, et al. Methods to dissect mitochondrial membrane permeabilization in the course of apoptosis. Methods Enzymol. 2008;442:355–374. doi: 10.1016/S0076-6879(08)01418-3. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E, et al. Mitochondrial gateways to cancer. Mol Aspects Med. 2010;31:1–20. doi: 10.1016/j.mam.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Garay-Malpartida HM, Occhiucci JM, Alves J, Belizario JE. CaSPredictor: a new computer-based tool for caspase substrate prediction. Bioinformatics. 2005;21(Suppl 1):i169–i176. doi: 10.1093/bioinformatics/bti1034. [DOI] [PubMed] [Google Scholar]
- Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423–1433. doi: 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- Geppetti P, Bertrand C, Ricciardolo FL, Nadel JA. New aspects on the role of kinins in neurogenic inflammation. Can J Physiol Pharmacol. 1995;73:843–847. doi: 10.1139/y95-115. [DOI] [PubMed] [Google Scholar]
- Gilmer LK, Roberts KN, Joy K, Sullivan PG, Scheff SW. Early mitochondrial dysfunction after cortical contusion injury. J Neurotrauma. 2009;26:1271–1280. doi: 10.1089/neu.2008.0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalvez F, Pariselli F, Dupaigne P, Budihardjo I, Lutter M, Antonsson B, et al. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ. 2005;12:614–626. doi: 10.1038/sj.cdd.4401571. [DOI] [PubMed] [Google Scholar]
- Gottlieb E. OPA1 and PARL keep a lid on apoptosis. Cell. 2006;126:27–29. doi: 10.1016/j.cell.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Graier WF, Frieden M, Malli R. Mitochondria and Ca(2+) signaling: old guests, new functions. Pflugers Arch. 2007;455:375–396. doi: 10.1007/s00424-007-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Grohm J, Plesnila N, Culmsee C. Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain Behav Immun. 2010;24:831–838. doi: 10.1016/j.bbi.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Guihard G, Bellot G, Moreau C, Pradal G, Ferry N, Thomy R, et al. The mitochondrial apoptosis-induced channel (MAC) corresponds to a late apoptotic event. J Biol Chem. 2004;279:46542–46550. doi: 10.1074/jbc.M405153200. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267((2 Pt 1)):C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E, et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene. 2003;22:6669–6678. doi: 10.1038/sj.onc.1206794. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurother. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson MJ, Persson T, Friberg H, Keep MF, Rees A, Wieloch T, et al. Powerful cyclosporin inhibition of calcium-induced permeability transition in brain mitochondria. Brain Res. 2003;960:99–111. doi: 10.1016/s0006-8993(02)03798-8. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall DC, Bonislawski DP, Skradski SL, Lan JQ, Meller R, Simon RP. Cleavage of bid may amplify caspase-8-induced neuronal death following focally evoked limbic seizures. Neurobiol Dis. 2001;8:568–580. doi: 10.1006/nbdi.2001.0415. [DOI] [PubMed] [Google Scholar]
- Herrmann JM, Neupert W. Protein transport into mitochondria. Curr Opin Microbiol. 2000;3:210–214. doi: 10.1016/s1369-5274(00)00077-1. [DOI] [PubMed] [Google Scholar]
- Hisatomi T, Nakazawa T, Noda K, Almulki L, Miyahara S, Nakao S, et al. HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest. 2008;118:2025–2038. doi: 10.1172/JCI34267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann KA. Glutamate-mediated injury in focal cerebral ischemia: the excitotoxin hypothesis revised. Brain Pathol. 1994;4:23–36. doi: 10.1111/j.1750-3639.1994.tb00808.x. [DOI] [PubMed] [Google Scholar]
- Huang Z, Pinto JT, Deng H, Richie JP., Jr Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem Pharmacol. 2008;75:2234–2244. doi: 10.1016/j.bcp.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide K, Schmalbruch IK, Quistorff B, Horn A, Secher NH. Lactate, glucose and O2 uptake in human brain during recovery from maximal exercise. J Physiol. 2000;522((Pt 1)):159–164. doi: 10.1111/j.1469-7793.2000.t01-2-00159.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Benz AM, Zorumski CF, Olney JW. Effects of lactate and pyruvate on glucose deprivation in rat hippocampal slices. Neuroreport. 1994;5:617–620. doi: 10.1097/00001756-199401000-00021. [DOI] [PubMed] [Google Scholar]
- Jacotot E, Costantini P, Laboureau E, Zamzami N, Susin SA, Kroemer G. Mitochondrial membrane permeabilization during the apoptotic process. Ann N Y Acad Sci. 1999;887:18–30. doi: 10.1111/j.1749-6632.1999.tb07919.x. [DOI] [PubMed] [Google Scholar]
- Jahani-Asl A, Pilon-Larose K, Xu W, MacLaurin JG, Park DS, McBride HM, et al. The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J Biol Chem. 2011;286:4772–4782. doi: 10.1074/jbc.M110.167155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambrina E, Alonso R, Alcalde M, del Carmen Rodriguez M, Serrano A, Martinez AC, et al. Calcium influx through receptor-operated channel induces mitochondria-triggered paraptotic cell death. J Biol Chem. 2003;278:14134–14145. doi: 10.1074/jbc.M211388200. [DOI] [PubMed] [Google Scholar]
- Jarmuszkiewicz W, Navet R, Alberici LC, Douette P, Sluse-Goffart CM, Sluse FE, et al. Redox state of endogenous coenzyme q modulates the inhibition of linoleic acid-induced uncoupling by guanosine triphosphate in isolated skeletal muscle mitochondria. J Bioenerg Biomembr. 2004;36:493–502. doi: 10.1023/B:JOBB.0000047331.25248.7a. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Wang S, Zorov DB, Nuss HB, Gleichmann M, Mattson MP, et al. The identity and regulation of the mitochondrial permeability transition pore: where the known meets the unknown. Ann N Y Acad Sci. 2008;1123:197–212. doi: 10.1196/annals.1420.023. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Kalinowska M, Garncarz W, Pietrowska M, Garrard WT, Widlak P. Regulation of the human apoptotic DNase/RNase endonuclease G: involvement of Hsp70 and ATP. Apoptosis. 2005;10:821–830. doi: 10.1007/s10495-005-0410-9. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–1039. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CD, Johnson JC, Riordan WP, Cotton BA. How we die: the impact of nonneurologic organ dysfunction after severe traumatic brain injury. Am Surg. 2008;74:866–872. [PubMed] [Google Scholar]
- Kikuchi M, Tenneti L, Lipton SA. Role of p38 mitogen-activated protein kinase in axotomy-induced apoptosis of rat retinal ganglion cells. J Neurosci. 2000;20:5037–5044. doi: 10.1523/JNEUROSCI.20-13-05037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic E, Dietz GP, Hermann DM, Bahr M. Intravenous TAT-Bcl-Xl is protective after middle cerebral artery occlusion in mice. Ann Neurol. 2002;52:617–622. doi: 10.1002/ana.10356. [DOI] [PubMed] [Google Scholar]
- Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- Kim KM, Kim PK, Kwon YG, Bai SK, Nam WD, Kim YM. Regulation of apoptosis by nitrosative stress. J Biochem Mol Biol. 2002;35:127–133. doi: 10.5483/bmbrep.2002.35.1.127. [DOI] [PubMed] [Google Scholar]
- Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, et al. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Kim TH, Chung HT, Talanian RV, Yin XM, Billiar TR. Nitric oxide prevents tumor necrosis factor alpha-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. 2000;32((4 Pt 1)):770–778. doi: 10.1053/jhep.2000.18291. [DOI] [PubMed] [Google Scholar]
- Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007;12:857–868. doi: 10.1007/s10495-007-0722-z. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002;115:587–602. doi: 10.1016/s0306-4522(02)00512-2. [DOI] [PubMed] [Google Scholar]
- Kitsis RN, Molkentin JD. Apoptotic cell death ‘Nixed’ by an ER-mitochondrial necrotic pathway. Proc Natl Acad Sci U S A. 2010;107:9031–9032. doi: 10.1073/pnas.1003827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblach SM, Nikolaeva M, Huang X, Fan L, Krajewski S, Reed JC, et al. Multiple caspases are activated after traumatic brain injury: evidence for involvement in functional outcome. J Neurotrauma. 2002;19:1155–1170. doi: 10.1089/08977150260337967. [DOI] [PubMed] [Google Scholar]
- Koene S, Smeitink J. Metabolic manipulators: a well founded strategy to combat mitochondrial dysfunction. J Inherit Metab Dis. 2011;34:315–325. doi: 10.1007/s10545-010-9162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Obitsu S, Ohta S, Matsunami K, Otsuka H, Teshima R. Poly(ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by alpha-tocopherol and MEK inhibition. J Biol Chem. 2010;285:13079–13091. doi: 10.1074/jbc.M109.044206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler S, Straten G, Kreppel F, Isenmann S, Liston P, Bahr M. The X-linked inhibitor of apoptosis (XIAP) prevents cell death in axotomized CNS neurons in vivo. Cell Death Differ. 2000;7:815–824. doi: 10.1038/sj.cdd.4400712. [DOI] [PubMed] [Google Scholar]
- Kwon TH, Chao DL, Malloy K, Sun D, Alessandri B, Bullock MR. Tempol, a novel stable nitroxide, reduces brain damage and free radical production, after acute subdural hematoma in the rat. J Neurotrauma. 2003;20:337–345. doi: 10.1089/089771503765172291. [DOI] [PubMed] [Google Scholar]
- Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, et al. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 2008;104:1700–1711. doi: 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- Legos JJ, McLaughlin B, Skaper SD, Strijbos PJ, Parsons AA, Aizenman E, et al. The selective p38 inhibitor SB-239063 protects primary neurons from mild to moderate excitotoxic injury. Eur J Pharmacol. 2002;447:37–42. doi: 10.1016/s0014-2999(02)01890-3. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ. Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology. 2005;129:351–360. doi: 10.1053/j.gastro.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator – thinking outside the box. Biochim Biophys Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewen A, Fujimura M, Sugawara T, Matz P, Copin JC, Chan PH. Oxidative stress-dependent release of mitochondrial cytochrome c after traumatic brain injury. J Cereb Blood Flow Metab. 2001;21:914–920. doi: 10.1097/00004647-200108000-00003. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang X, Ma C, Qiao J, Zhang C. Necroptosis contributes to the NMDA-induced excitotoxicity in rat's cultured cortical neurons. Neurosci Lett. 2008;447:120–123. doi: 10.1016/j.neulet.2008.08.037. [DOI] [PubMed] [Google Scholar]
- Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria–specificity in membrane targeting for death. Biochim Biophys Acta. 2011;1813:532–539. doi: 10.1016/j.bbamcr.2010.10.017. [DOI] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Martinou JC. Newcomers in the process of mitochondrial permeabilization. J Cell Sci. 2005;118:473–483. doi: 10.1242/jcs.01654. [DOI] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Montessuit S, Martinou JC. Contributions to Bax insertion and oligomerization of lipids of the mitochondrial outer membrane. Cell Death Differ. 2008;15:929–937. doi: 10.1038/cdd.2008.9. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, et al. Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J Neurosci Res. 2010;88:2847–2858. doi: 10.1002/jnr.22453. [DOI] [PubMed] [Google Scholar]
- Macias CA, Chiao JW, Xiao J, Arora DS, Tyurina YY, Delude RL, et al. Treatment with a novel hemigramicidin-TEMPO conjugate prolongs survival in a rat model of lethal hemorrhagic shock. Ann Surg. 2007;245:305–314. doi: 10.1097/01.sla.0000236626.57752.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magder S. Reactive oxygen species: toxic molecules or spark of life? Crit Care. 2006;10:208. doi: 10.1186/cc3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, et al. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, et al. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maran A, Cranston I, Lomas J, Macdonald I, Amiel SA. Protection by lactate of cerebral function during hypoglycaemia. Lancet. 1994;343:16–20. doi: 10.1016/s0140-6736(94)90876-1. [DOI] [PubMed] [Google Scholar]
- Margulies S, Hicks R. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26:925–939. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Colell A, Morales A, Caballero F, Moles A, Fernandez A, et al. Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kappaB activation. Gastroenterology. 2008;134:1507–1520. doi: 10.1053/j.gastro.2008.01.073. [DOI] [PubMed] [Google Scholar]
- Marmarou A, Fatouros PP, Barzo P, Portella G, Yoshihara M, Tsuji O, et al. Contribution of edema and cerebral blood volume to traumatic brain swelling in head-injured patients. J Neurosurg. 2000;93:183–193. doi: 10.3171/jns.2000.93.2.0183. [DOI] [PubMed] [Google Scholar]
- Marzo I, Susin SA, Petit PX, Ravagnan L, Brenner C, Larochette N, et al. Caspases disrupt mitochondrial membrane barrier function. FEBS Lett. 1998;427:198–202. doi: 10.1016/s0014-5793(98)00424-4. [DOI] [PubMed] [Google Scholar]
- Matarrese P, Gambardella L, Cassone A, Vella S, Cauda R, Malorni W. Mitochondrial membrane hyperpolarization hijacks activated T lymphocytes toward the apoptotic-prone phenotype: homeostatic mechanisms of HIV protease inhibitors. J Immunol. 2003;170:6006–6015. doi: 10.4049/jimmunol.170.12.6006. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med. 2003;9:196–205. doi: 10.1016/s1471-4914(03)00046-7. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Sherman M. Perturbed signal transduction in neurodegenerative disorders involving aberrant protein aggregation. Neuromolecular Med. 2003;4:109–132. doi: 10.1385/NMM:4:1-2:109. [DOI] [PubMed] [Google Scholar]
- Mazzeo AT, Kunene NK, Gilman CB, Hamm RJ, Hafez N, Bullock MR. Severe human traumatic brain injury, but not cyclosporin a treatment, depresses activated T lymphocytes early after injury. J Neurotrauma. 2006;23:962–975. doi: 10.1089/neu.2006.23.962. [DOI] [PubMed] [Google Scholar]
- Mazzeo AT, Alves OL, Gilman CB, Hayes RL, Tolias C, Niki Kunene K, et al. Brain metabolic and hemodynamic effects of cyclosporin A after human severe traumatic brain injury: a microdialysis study. Acta Neurochir (Wien) 2008;150:1019–1031. doi: 10.1007/s00701-008-0021-7. discussion 1031. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp Neurol. 2008;209:243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Merenda A, Bullock R. Clinical treatments for mitochondrial dysfunctions after brain injury. Curr Opin Crit Care. 2006;12:90–96. doi: 10.1097/01.ccx.0000216573.26686.27. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Morton SU, Fernhoff NB, Marletta MA. Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc Natl Acad Sci U S A. 2007;104:11609–11614. doi: 10.1073/pnas.0704898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Wang X, Jung JC, Sumii T, Singhal AB, Fini ME, et al. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Metab. 2002;22:444–452. doi: 10.1097/00004647-200204000-00008. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, et al. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 2010;114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin d-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Johnson-Cadwell L, Vesce S, Jekabsons M, Yadava N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res. 2007;85:3206–3212. doi: 10.1002/jnr.21290. [DOI] [PubMed] [Google Scholar]
- Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-D-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The calpains in aging and aging-related diseases. Ageing Res Rev. 2003;2:407–418. doi: 10.1016/s1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Marion DW, Botscheller ML, Bowen DM, DeKosky ST. Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport. 1994;6:153–156. doi: 10.1097/00001756-199412300-00039. [DOI] [PubMed] [Google Scholar]
- Palzur E, Zaaroor M, Vlodavsky E, Milman F, Soustiel JF. Neuroprotective effect of hyperbaric oxygen therapy in brain injury is mediated by preservation of mitochondrial membrane properties. Brain Res. 2008;1221:126–133. doi: 10.1016/j.brainres.2008.04.078. [DOI] [PubMed] [Google Scholar]
- Pan S, Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- Pan S, Ryu SY, Sheu SS. Distinctive characteristics and functions of multiple mitochondrial Ca2+ influx mechanisms. Sci China Life Sci. 2011;54:763–769. doi: 10.1007/s11427-011-4203-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, et al. Post-Injury Administration of Mitochondrial Uncouplers Increases Tissue Sparing and Improves Behavioral Outcome following Traumatic Brain Injury in Rodents. J Neurotrauma. 2007;24:798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Sullivan PG. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Exp Neurol. 2009;218:381–389. doi: 10.1016/j.expneurol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE. 2008;3:e3257. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrelet D, Ferri A, MacKenzie AE, Smith GM, Korneluk RG, Liston P, et al. IAP family proteins delay motoneuron cell death in vivo. Eur J Neurosci. 2000;12:2059–2067. doi: 10.1046/j.1460-9568.2000.00098.x. [DOI] [PubMed] [Google Scholar]
- Phenix BN, Lum JJ, Nie Z, Sanchez-Dardon J, Badley AD. Antiapoptotic mechanism of HIV protease inhibitors: preventing mitochondrial transmembrane potential loss. Blood. 2001;98:1078–1085. doi: 10.1182/blood.v98.4.1078. [DOI] [PubMed] [Google Scholar]
- Pitkänen ALL, Marklund N, Morales DM, McIntosh TK. Neurodegeneration and neuroprotective strategies after traumatic brain injury. Drug Discov Today Dis Mech. 2005;2:409–418. [Google Scholar]
- Pizzo P, Pozzan T. Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol. 2007;17:511–517. doi: 10.1016/j.tcb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Pottorf WJ, 2nd, Johanns TM, Derrington SM, Strehler EE, Enyedi A, Thayer SA. Glutamate-induced protease-mediated loss of plasma membrane Ca2+ pump activity in rat hippocampal neurons. J Neurochem. 2006;98:1646–1656. doi: 10.1111/j.1471-4159.2006.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto R, Tavazzi B, Taya K, Barrios L, Amorini AM, Di Pietro V, et al. Brain energy depletion in a rodent model of diffuse traumatic brain injury is not prevented with administration of sodium lactate. Brain Res. 2011;1404:39–49. doi: 10.1016/j.brainres.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puccio AM, Hoffman LA, Bayir H, Zullo TG, Fischer M, Darby J, et al. Effect of short periods of normobaric hyperoxia on local brain tissue oxygenation and cerebrospinal fluid oxidative stress markers in severe traumatic brain injury. J Neurotrauma. 2009;26:1241–1249. doi: 10.1089/neu.2008.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–843. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- Readnower RD, Pandya JD, McEwen ML, Pauly JR, Springer JE, Sullivan PG. Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J Neurotrauma. 2011;28:1845–1853. doi: 10.1089/neu.2011.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JD, Gogvadze V, Kropotov A, Vakifahmetoglu H, Zhivotovsky B, Orrenius S. Processed caspase-2 can induce mitochondria-mediated apoptosis independently of its enzymatic activity. EMBO Rep. 2004;5:643–648. doi: 10.1038/sj.embor.7400153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockswold SB, Rockswold GL, Vargo JM, Erickson CA, Sutton RL, Bergman TA, et al. Effects of hyperbaric oxygenation therapy on cerebral metabolism and intracranial pressure in severely brain injured patients. J Neurosurg. 2001;94:403–411. doi: 10.3171/jns.2001.94.3.0403. [DOI] [PubMed] [Google Scholar]
- Rockswold SB, Rockswold GL, Defillo A. Hyperbaric oxygen in traumatic brain injury. Neurol Res. 2007;29:162–172. doi: 10.1179/016164107X181798. [DOI] [PubMed] [Google Scholar]
- Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91. doi: 10.1146/annurev.physiol.70.021507.105852. [DOI] [PubMed] [Google Scholar]
- Rossier MF. T channels and steroid biosynthesis: in search of a link with mitochondria. Cell Calcium. 2006;40:155–164. doi: 10.1016/j.ceca.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A, et al. Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J Biol Chem. 1999;274:6823–6826. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM. Bid, but not Bax, regulates VDAC channels. J Biol Chem. 2004;279:13575–13583. doi: 10.1074/jbc.M310593200. [DOI] [PubMed] [Google Scholar]
- Royo NC, Shimizu S, Schouten JW, Stover JF, McIntosh TK. Pharmacology of traumatic brain injury. Curr Opin Pharmacol. 2003;3:27–32. doi: 10.1016/s1471-4892(02)00006-1. [DOI] [PubMed] [Google Scholar]
- Rube DA, van der Bliek AM. Mitochondrial morphology is dynamic and varied. Mol Cell Biochem. 2004;256-257:331–339. doi: 10.1023/b:mcbi.0000009879.01256.f6. [DOI] [PubMed] [Google Scholar]
- Ruchalski K, Mao H, Li Z, Wang Z, Gillers S, Wang Y, et al. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem. 2006;281:7873–7880. doi: 10.1074/jbc.M513728200. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- Santel A, Frank S. Shaping mitochondria: the complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life. 2008;60:448–455. doi: 10.1002/iub.71. [DOI] [PubMed] [Google Scholar]
- Sayeed I, Parvez S, Wali B, Siemen D, Stein DG. Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism for better neuroprotective effects of allopregnanolone over progesterone. Brain Res. 2009;1263:165–173. doi: 10.1016/j.brainres.2009.01.045. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res. 2008;49:1607–1620. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr Opin Crit Care. 2007;13:134–142. doi: 10.1097/MCC.0b013e3280895d5c. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788:2022–2031. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Schurr A, Miller JJ, Payne RS, Rigor BM. An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J Neurosci. 1999;19:34–39. doi: 10.1523/JNEUROSCI.19-01-00034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab BL, Guerini D, Didszun C, Bano D, Ferrando-May E, Fava E, et al. Cleavage of plasma membrane calcium pumps by caspases: a link between apoptosis and necrosis. Cell Death Differ. 2002;9:818–831. doi: 10.1038/sj.cdd.4401042. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304:437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Semple BD, Noble-Haeusslein LJ. Broad-spectrum neuroprotection against traumatic brain injury by agonism of peroxisome proliferator-activated receptors. Exp Neurol. 2011;229:195–197. doi: 10.1016/j.expneurol.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesso A, Marques MM, Monteiro MM, Schumacher RI, Colquhoun A, Belizario J, et al. Morphology of mitochondrial permeability transition: morphometric volumetry in apoptotic cells. Anat Rec A Discov Mol Cell Evol Biol. 2004;281:1337–1351. doi: 10.1002/ar.a.20134. [DOI] [PubMed] [Google Scholar]
- Shao ZH, Sharp WW, Wojcik KR, Li CQ, Han M, Chang WT, et al. Therapeutic hypothermia cardioprotection via Akt- and nitric oxide-mediated attenuation of mitochondrial oxidants. Am J Physiol Heart Circ Physiol. 2010;298:H2164–H2173. doi: 10.1152/ajpheart.00994.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton SN, Shawgo ME, Robertson JD. Cleavage of Bid by executioner caspases mediates feed forward amplification of mitochondrial outer membrane permeabilization during genotoxic stress-induced apoptosis in Jurkat cells. J Biol Chem. 2009;284:11247–11255. doi: 10.1074/jbc.M809392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Caspase activation, inhibition, and reactivation: a mechanistic view. Protein Sci. 2004;13:1979–1987. doi: 10.1110/ps.04789804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo BK, Zhao Q, Pahlmark K, Siesjo P, Katsura K, Folbergrova J. Glutamate, calcium, and free radicals as mediators of ischemic brain damage. Ann Thorac Surg. 1995;59:1316–1320. doi: 10.1016/0003-4975(95)00077-x. [DOI] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Tavazzi B, Dunbar J, Amorini AM, Lazzarino G, et al. The protective effect of cyclosporin A upon N-acetylaspartate and mitochondrial dysfunction following experimental diffuse traumatic brain injury. J Neurotrauma. 2004;21:1154–1167. doi: 10.1089/neu.2004.21.1154. [DOI] [PubMed] [Google Scholar]
- Siskind LJ. Mitochondrial ceramide and the induction of apoptosis. J Bioenerg Biomembr. 2005;37:143–153. doi: 10.1007/s10863-005-6567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006;11:473–485. doi: 10.1007/s10495-006-5881-9. [DOI] [PubMed] [Google Scholar]
- Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slemmer JE, Shacka JJ, Sweeney MI, Weber JT. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr Med Chem. 2008;15:404–414. doi: 10.2174/092986708783497337. [DOI] [PubMed] [Google Scholar]
- Sluse FE, Jarmuszkiewicz W, Navet R, Douette P, Mathy G, Sluse-Goffart CM. Mitochondrial UCPs: new insights into regulation and impact. Biochim Biophys Acta. 2006;1757:480–485. doi: 10.1016/j.bbabio.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Smith D, Pernet A, Hallett WA, Bingham E, Marsden PK, Amiel SA. Lactate: a preferred fuel for human brain metabolism in vivo. J Cereb Blood Flow Metab. 2003;23:658–664. doi: 10.1097/01.WCB.0000063991.19746.11. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Martel MA, Papadia S, Vaslin A, Baxter P, Rickman C, et al. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci. 2008;28:10696–10710. doi: 10.1523/JNEUROSCI.1207-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soustiel JF, Zaaroor M, Vlodavsky E, Veenman L, Weizman A, Gavish M. Neuroprotective effect of Ro5-4864 following brain injury. Exp Neurol. 2008;214:201–208. doi: 10.1016/j.expneurol.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–264. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Stefanis L. Caspase-dependent and -independent neuronal death: two distinct pathways to neuronal injury. Neuroscientist. 2005;11:50–62. doi: 10.1177/1073858404271087. [DOI] [PubMed] [Google Scholar]
- Stegh AH, Herrmann H, Lampel S, Weisenberger D, Andra K, Seper M, et al. Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol Cell Biol. 2000;20:5665–5679. doi: 10.1128/mcb.20.15.5665-5679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegh AH, Barnhart BC, Volkland J, Algeciras-Schimnich A, Ke N, Reed JC, et al. Inactivation of caspase-8 on mitochondria of Bcl-xL-expressing MCF7-Fas cells: role for the bifunctional apoptosis regulator protein. J Biol Chem. 2002;277:4351–4360. doi: 10.1074/jbc.M108947200. [DOI] [PubMed] [Google Scholar]
- Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurother. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica B, Byrnes K, Faden AI. Multifunctional drug treatment in neurotrauma. Neurother. 2009;6:14–27. doi: 10.1016/j.nurt.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Geiger JD, Mattson MP, Scheff SW. Dietary supplement creatine protects against traumatic brain injury. Ann Neurol. 2000;48:723–729. [PubMed] [Google Scholar]
- Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. 2004a;55:576–580. doi: 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Springer JE, Hall ED, Scheff SW. Mitochondrial uncoupling as a therapeutic target following neuronal injury. J Bioenerg Biomembr. 2004b;36:353–356. doi: 10.1023/B:JOBB.0000041767.30992.19. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, et al. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–1065. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Takahashi-Niki K, Akagi T, Hashikawa T, Takahashi R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004b;11:208–216. doi: 10.1038/sj.cdd.4401343. [DOI] [PubMed] [Google Scholar]
- Sykes MC, Mowbray AL, Jo H. Reversible glutathiolation of caspase-3 by glutaredoxin as a novel redox signaling mechanism in tumor necrosis factor-alpha-induced cell death. Circ Res. 2007;100:152–154. doi: 10.1161/01.RES.0000258171.08020.72. [DOI] [PubMed] [Google Scholar]
- Tait SW, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Munoz-Pinedo C, et al. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 2010;18:802–813. doi: 10.1016/j.devcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazzi B, Signoretti S, Lazzarino G, Amorini AM, Delfini R, Cimatti M, et al. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery. 2005;56:582–589. doi: 10.1227/01.neu.0000156715.04900.e6. discussion 582–589. [DOI] [PubMed] [Google Scholar]
- Temkin V, Huang Q, Liu H, Osada H, Pope RM. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol. 2006;26:2215–2225. doi: 10.1128/MCB.26.6.2215-2225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 2001;20:4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng E, Kim YM, Pitt BR, Lizonova A, Kovesdi I, Billiar TR. Adenoviral transfer of the inducible nitric oxide synthase gene blocks endothelial cell apoptosis. Surgery. 1997;122:255–263. doi: 10.1016/s0039-6060(97)90016-7. [DOI] [PubMed] [Google Scholar]
- Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Thompson CB. Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- Varanyuwatana P, Halestrap AP. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion. 2011;12:120–125. doi: 10.1016/j.mito.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Votyakova TV, Reynolds IJ. Spontaneous changes in mitochondrial membrane potential in single isolated brain mitochondria. Biophys J. 2003;85:3358–3366. doi: 10.1016/S0006-3495(03)74755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen AM, Coulson EJ, Vaux DL. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol. 2001;2:REVIEWS3009. doi: 10.1186/gb-2001-2-7-reviews3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesce S, Kirk L, Nicholls DG. Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J Neurochem. 2004;90:683–693. doi: 10.1111/j.1471-4159.2004.02516.x. [DOI] [PubMed] [Google Scholar]
- Vieira HL, Belzacq AS, Haouzi D, Bernassola F, Cohen I, Jacotot E, et al. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene. 2001;20:4305–4316. doi: 10.1038/sj.onc.1204575. [DOI] [PubMed] [Google Scholar]
- Vink R, Nimmo AJ. Multifunctional drugs for head injury. Neurother. 2009;6:28–42. doi: 10.1016/j.nurt.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel F, Bornhovd C, Neupert W, Reichert AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol. 2006;175:237–247. doi: 10.1083/jcb.200605138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff MJ, Collaco-Moraes Y, Aspey BS, Coffin RS, Harrison MJ, Latchman DS, et al. Focal cerebral ischaemia increases the levels of several classes of heat shock proteins and their corresponding mRNAs. Brain Res Mol Brain Res. 1996;42:236–244. doi: 10.1016/s0169-328x(96)00127-1. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- Walter ABAJJLMRKRP. 1994. Molecular Biology of the Cell.
- Wan W, DePetrillo PB. Ritonavir protects hippocampal neurons against oxidative stress-induced apoptosis. Neurotoxicology. 2002;23:301–306. doi: 10.1016/s0161-813x(02)00057-8. [DOI] [PubMed] [Google Scholar]
- Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH, et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol. 2008;180:3072–3080. doi: 10.4049/jimmunol.180.5.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Li X, Greer PA, Koehler RC, Dawson VL, et al. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos) J Neurochem. 2009;110:687–696. doi: 10.1111/j.1471-4159.2009.06167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Zhang S, Du TY, Wang B, Sun XQ. Hyperbaric oxygen preconditioning reduces ischemia-reperfusion injury by stimulating autophagy in neurocyte. Brain Res. 2010;1323:149–151. doi: 10.1016/j.brainres.2010.01.074. [DOI] [PubMed] [Google Scholar]
- Wasilewski M, Scorrano L. The changing shape of mitochondrial apoptosis. Trends Endocrinol Metab. 2009;20:287–294. doi: 10.1016/j.tem.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Waterhouse NJ, Sedelies KA, Sutton VR, Pinkoski MJ, Thia KY, Johnstone R, et al. Functional dissociation of DeltaPsim and cytochrome c release defines the contribution of mitochondria upstream of caspase activation during granzyme B-induced apoptosis. Cell Death Differ. 2006;13:607–618. doi: 10.1038/sj.cdd.4401772. [DOI] [PubMed] [Google Scholar]
- Weaver JG, Tarze A, Moffat TC, Lebras M, Deniaud A, Brenner C, et al. Inhibition of adenine nucleotide translocator pore function and protection against apoptosis in vivo by an HIV protease inhibitor. J Clin Invest. 2005;115:1828–1838. doi: 10.1172/JCI22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wang BR, Wang X, Kuang F, Duan XL, Jiao XY, et al. ERK1/2 and p38 mitogen-activated protein kinase mediate iNOS-induced spinal neuron degeneration after acute traumatic spinal cord injury. Life Sci. 2006;79:1895–1905. doi: 10.1016/j.lfs.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Yamashita D, Miller JM, Jiang HY, Minami SB, Schacht J. AIF and EndoG in noise-induced hearing loss. Neuroreport. 2004;15:2719–2722. [PubMed] [Google Scholar]
- Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17:1487–1496. doi: 10.1101/gad.1097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenari MA, Liu J, Zheng Z, Vexler ZS, Lee JE, Giffard RG. Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann N Y Acad Sci. 2005;1053:74–83. doi: 10.1196/annals.1344.007. [DOI] [PubMed] [Google Scholar]
- Yin W, Cao G, Johnnides MJ, Signore AP, Luo Y, Hickey RW, et al. TAT-mediated delivery of Bcl-xL protein is neuroprotective against neonatal hypoxic-ischemic brain injury via inhibition of caspases and AIF. Neurobiol Dis. 2006;21:358–371. doi: 10.1016/j.nbd.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006b;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaltsman Y, Shachnai L, Yivgi-Ohana N, Schwarz M, Maryanovich M, Houtkooper RH, et al. MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat Cell Biol. 2010;12:553–562. doi: 10.1038/ncb2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech B, Kohl R, von Knethen A, Brune B. Nitric oxide donors inhibit formation of the Apaf-1/caspase-9 apoptosome and activation of caspases. Biochem J. 2003;371:1055–1064. doi: 10.1042/BJ20021720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Wang X, Deinum J, Huang Z, Gao J, Modjtahedi N, et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med. 2007;204:1741–1748. doi: 10.1084/jem.20070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim Biophys Acta. 2005;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Cheng G, Gao DK, Zhang X, Zhen HN, Zhang W, et al. Gross-total hematoma removal of hypertensive basal ganglia hemorrhages: a long-term follow-up. J Neurol Sci. 2009;287:100–104. doi: 10.1016/j.jns.2009.08.046. [DOI] [PubMed] [Google Scholar]