

Abstract
Since its discovery over 20 years ago endothelin-1 (ET-1) has been implicated in a number of physiological and pathophysiological processes. Its role in the development and progression of chronic kidney disease (CKD) is well established and is an area of ongoing intense research. There are now available a number of ET receptor antagonists many of which have been used in trials with CKD patients and shown to reduce BP and proteinuria. However, ET-1 has a number of BP-independent effects. Importantly, and in relation to the kidney, ET-1 has clear roles to play in cell proliferation, podocyte dysfunction, inflammation and fibrosis, and arguably, these actions of ET-1 may be more significant in the progression of CKD than its prohypertensive actions. This review will focus on the potential role of ET-1 in renal disease with an emphasis on its BP-independent actions.
Keywords: endothelin-1, chronic kidney disease, BP-independent, inflammation
Introduction
Since its discovery in 1988 (Yanagisawa et al., 1988b), endothelin (ET) has been widely implicated in the pathophysiology of renal disease. ETs are a family of three 21 amino acid peptides, each with distinct genes and tissue distributions, with powerful vasoconstrictor and pressor properties (Yanagisawa et al., 1988b; Inoue et al., 1989; Arinami et al., 1991). Of the three peptides, ET-1 is the major endothelial isoform and, in the human kidney, the only one so far shown to be expressed at the protein level (Karet and Davenport, 1996). Its main site of vascular production is the endothelial cell but it is also produced by other cell types including vascular smooth muscle cells and epicardial cells (Eid et al., 1994). Within the renal system, it is produced by glomerular epithelial and mesangial cells, renal tubular and medullary collecting duct (CD) cells (Kohan, 1997) and, potentially, resident and infiltrating macrophages (Ehrenreich et al., 1990). Importantly, ET-1 has clear roles to play in cell proliferation, podocyte dysfunction, inflammation and fibrosis (Dhaun et al., 2006), and arguably, these effects of ET-1 may be more significant in the progression of chronic kidney disease (CKD) than its prohypertensive actions. This review will focus on the potential role of ET-1 in renal disease with an emphasis on those actions independent of BP. As these data are largely derived from animal models, the focus will be on preclinical studies.
Biology of the ET system in the kidney
Regulation of ET synthesis occurs at the level of gene transcription, with the gene product being the 212 amino acid preproET-1. ET-1 has limited intracellular stores so is mostly transcriptionally regulated. Enhanced gene transcription occurs with a wide range of stimuli (Wesson et al., 1998; Attina et al., 2005). Those pertinent to CKD include other vasocontrictors (Ang II, vasopressin), pro-inflammatory cytokines (TNF-α, IL-1b), hypoxia, reactive oxygen species, profibrotic cytokines (TGF-β, platelet-derived growth factor), hyperglycaemia, acidosis and thrombin. Thus, an up-regulation in ET-1 synthesis may be viewed as a common renal stress response. By contrast, prostacyclin, NO and the natriuretic peptides all inhibit gene transcription. PreproET-1 is cleaved to big ET-1 (38 amino acids), which is largely biologically inactive (Haynes and Webb, 1994). Endothelin converting enzyme then splits big ET-1 to the biologically active ET-1 and C-terminal fragment. Once synthesized, the secretion of mature ET-1 from endothelial cells is largely abluminal (Yoshimoto et al., 1991), towards the adjacent vascular smooth muscle, suggesting an autocrine or paracrine mechanism of action.
The effects of ET-1 are mediated via two G-protein coupled receptors, the ETA and ETB receptor (Arai et al., 1990; Sakurai et al., 1990). Within blood vessels, both receptors are found on smooth muscle cells and their activation results in vasoconstriction. ETB receptors are, however, predominantly found on the vascular endothelium where their activation results in vasodilatation via prostacyclin and NO (DeNucci et al., 1988). Because most ET-1 is released abluminally, plasma concentrations of ET-1 do not accurately reflect ET-1 production. However, some is released into the circulation and the ETB receptor also acts as a clearance receptor for this circulating ET-1. The half-life of ET-1 in the healthy circulation is ∼1 min (Gasic et al., 1992) with removal through receptor and non-receptor mediated mechanisms. ET-1 binds to ETB receptors, with subsequent ligand-receptor complex internalization and intracellular degradation accounting for the majority of clearance, particularly in the pulmonary circulation (Dupuis et al., 1996), although the splanchnic and renal circulations also contribute (Attina et al., 2005). Therefore, reductions in ETB numbers, or ETB receptor blockade, may reduce ET-1 clearance, increasing plasma concentrations without altering production. ET receptors are widely distributed within the human kidney, with the ETA subtype localized to vascular smooth muscle, notably in the glomeruli, vasa recta and arcuate arteries, whereas ETB receptors are more numerous (ETB to ETA ratio 2:1) and more widespread, with a high concentration in the collecting system (Karet and Davenport, 1996; Kuc and Davenport, 2004).
Effects of ET-1 on renal haemodynamics
ET-1 is a potent vasoconstrictor in vitro and pressor in whole animals (Yanagisawa et al., 1988a). With respect to the kidney, exogenous ET-1 causes renal vasoconstriction and an overall reduction in RBF (Chou and Porush, 1995), effects mediated via the ETA receptor (Evans et al., 2001; Abassi et al., 2002). Indeed, the renal vasculature is more sensitive to the vasoconstricting effects of ET-1 than other vascular beds (Pernow et al., 1989). Although exogenous ET-1 reduces total RBF, a regional difference has been observed, with cortical vasoconstriction that is ETA receptor mediated (Rubinstein et al., 1995; Gurbanov et al., 1996; Denton et al., 2004), and medullary vasodilatation which is ETB and NO dependent (Rubinstein et al., 1995). Furthermore, in vitro studies have shown that combined ETA/B receptor antagonism is required to abolish the vasoconstricting effects of exogenous ET-1 on the afferent arteriole suggesting that both ETA and ETB receptors are involved (Inscho et al., 2005). At the efferent arteriole, the actions of ET-1 is blocked by ETA receptor antagonism alone, and enhanced by ETB receptor blockade, suggesting that ET-1 can modulate efferent arteriolar tone via the ETA receptor and that the balance of ETB receptor effects here is to produce vasodilation (Inscho et al., 2005). By this action on efferent and afferent arterioles, ET has the ability to regulate glomerular capillary pressure and GFR.
In healthy man, a similar peripheral vasoconstrictor (Haynes and Webb, 1994) and systemic pressor response has been demonstrated (Sorensen et al., 1994), as well as renal vasoconstriction, a fall in total RBF (with a consequent reduction in GFR) and increase in filtration fraction (Rabelink et al., 1994). As yet, there are no studies of the effects of ET-1 on intra-renal distribution of blood flow in man. Also, there are few studies using ET receptor antagonists. One has demonstrated an increase in RBF after combined ETA/B receptor blockade (Freed et al., 1999). Most, however, do not demonstrate an effect of selective ETA receptor blockade (Schmetterer et al., 1998; Montanari et al., 2000; 2002; Goddard et al., 2004a, b), or combined ETA/B receptor blockade (Goddard et al., 2004b), on basal renal haemodynamics, suggesting that ET-1 acting via the ETA receptor does not contribute to the maintenance of renal vascular tone in health. Selective and unopposed ETB receptor antagonism can, however, produce profound renal vasoconstriction, suggesting that ET-1 mediated tonic renal vasodilatation via the ETB receptor is important (Goddard et al., 2004b).
Studies in patients with CKD are limited. In the renal circulation, in contrast to healthy controls, acute selective ETA but not combined ETA/B receptor antagonism produces a sustained increase in RBF and a decrease in renal vascular resistance suggesting that ET-1 is important in maintaining renal vascular tone (which is about four times higher at baseline than in healthy controls) via the ETA receptor. Because GFR does not significantly alter, this is accompanied by a reduction in filtration fraction that, in the absence of changes in filtration coefficient, may indicate efferent arteriolar dilatation, consistent with animal data suggesting a role for ET-1 in maintaining efferent arteriolar tone via the ETA receptor (Goddard et al., 2004b). Interestingly, chronic ETA receptor antagonism is associated with no significant changes in RBF but a fall in GFR (Dhaun et al., 2011), effects similar to those seen with chronic angiotensin converting enzyme (ACE) inhibition.
The attenuation of the acute renal vasodilatory effect of ETA receptor antagonism by concomitant ETB receptor antagonism suggests that the ETB receptor is important in maintaining renal vasodilatation in CKD and that the renal vasoconstriction seen after selective ETB receptor blockade alone is not simply due to reduced clearance and displacement of ET-1 onto the unblocked ETA receptor but due to a specific role for ET-1 mediated tonic renal vasodilatation via the ETB receptor (Goddard et al., 2004b). Studies in healthy subjects have suggested that this observation in CKD patients may be accounted for at least in part by concomitant administration of ACE inhibitors as these are synergistic with ETA receptor antagonism via an NO mediated, ETB receptor-dependent mechanism (Schmetterer et al., 1998; Montanari et al., 2000; 2002; Goddard et al., 2004a, b).
Effects of ET-1 on the glomerulus (see Figure 1)
Figure 1.
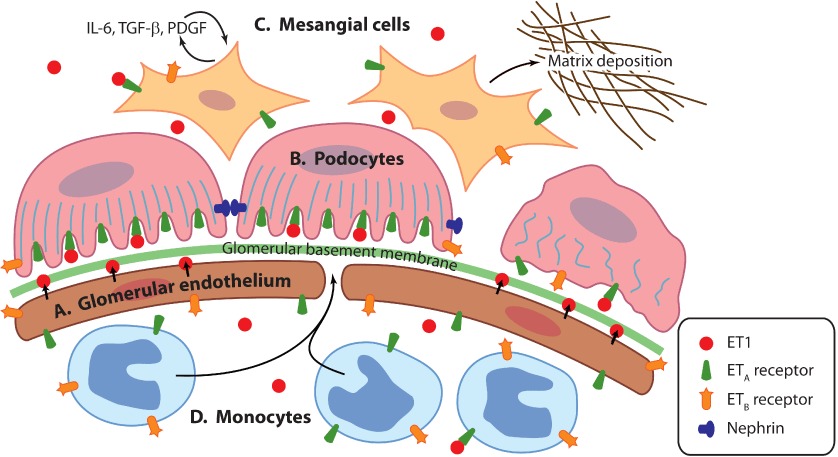
ET-1 effects on the glomerulus. (A) ET-1 is secreted on abluminal surface of glomerular endothelial cells. (B) ET-1 causes contraction of podocyte actin cytoskeleton and loss of slit diaphragm proteins such as nephrin. (C) Mesangial cells are activated to produce pro-inflammatory cytokines and matrix proteins. (D) ET-1 acts as a chemoattractant to monocytes.
The glomerulus is the functional filtration unit of the kidney. The glomerular filtration barrier – composed of two cells types: fenestrated endothelial cells (making up the glomerular capillaries) and podocytes – allow the filtration of water and small molecules whilst excluding large proteins. This barrier is size and charge selective. Sandwiched between the endothelial cells and podocytes is the glomerular basement membrane to which both cell types contribute. It is likely that all components of the glomerular filtration barrier have a role in enabling ultrafiltration and there is considerable crosstalk between the podocyte and endothelium (Mathieson, 2012). Supporting the glomerular capillaries are specialized pericytes known as mesangial cells. These produce extracellular matrix and act as ‘support’ structures within the glomerulus. They also have a contractile function important in changing the filtration coefficient (Guo and Cantley, 2010).
Glomerular injury is frequently associated with progressive CKD. It ranges from the indolent progressive injury of diabetes and hypertension to acute severe inflammation found in systemic vasculitis or lupus nephritis. CKD typically involves glomerular sclerosis and interstitial fibrosis and these may occur regardless of the nature of the initial renal insult. The mechanisms responsible for this continued renal deterioration are not fully understood, but likely involve a number of common pathways and may be distinct from those responsible for the original injury. Glomerular hypertension, glomerular cell hypertrophy and extracellular matrix accumulation are all involved. Significant proteinuria, a marker of CKD, has emerged as a powerful predictor of renal disease progression (Cameron et al., 1978; Mallick et al., 1987) regardless of underlying diagnosis, and proteinuria reduction is an important strategy to retard or prevent loss of renal function (Ruggenenti et al., 2003). Furthermore, reduction of proteinuria confers cardiovascular protection (Ibsen et al., 2005). Once filtered through the glomerulus excess protein is tubulo-toxic and excess protein reabsorption in the tubules can lead to an activation of tubular-dependent pathways of interstitial inflammation and fibrosis, with progressive renal scarring (Remuzzi and Bertani, 1998).
The ET system has been implicated in these processes (Kohan, 1997). In the remnant kidney model, renal ET-1 gene expression and urinary ET-1 excretion correlate with the degree of proteinuria and extent of renal damage (Orisio et al., 1993). Also, transgenic animals in which renal ET pathways have been up-regulated, display glomerulosclerosis and renal tubulointerstitial lesions independent of changes in BP that are usually characteristic of such models (Hocher et al., 1997). These BP-independent effects of ET-1 are supported by antagonist studies where ET receptor antagonists lead to a slowing of progressive renal damage, even in the absence of BP modification (Benigni and Remuzzi, 1999). Furthermore, infusion of ET-1 into rats over 2 weeks increases the permeability of isolated glomeruli to albumin and this effect was blocked by ETA receptor antagonism (Saleh et al., 2010).
Podocytes
Podocytes are complex epithelial cells consisting of a cell body from which major ‘primary’ processes arise. These in turn give rise to smaller ‘secondary’ foot processes. Secondary processes from adjacent podocytes interdigitate and form a ‘zipper-like’ connection known as the slit diaphragm. This slit diaphragm is maintained by multiple protein-protein interactions, which connect and signal with the podocyte actin cytoskeleton (Greka and Mundel, 2012). Podocyte injury is seen in many human glomerular diseases including diabetic nephropathy (DN), minimal change disease, focal segmental glomerulosclerosis and membranous nephropathy, and leads to proteinuria. The earliest pathological sign of podocyte injury is foot process effacement as connections between podocytes are lost. This may be followed by cell detachment and loss with eventual glomerulosclerosis. The presence of podocytes in the urine may be a useful indicator of disease severity (Greka and Mundel, 2012) and there is increasing interest in therapy that specifically targets and protects the podocyte (Mathieson, 2012).
Podocytes have been shown to both express ET-1 and to have ET receptors. Separate data support the presence of ETA (Morigi et al., 2005) and ETB (Yamamoto et al., 2002) receptors on rodent podocytes, although it is likely that they possess both (Davenport et al., 1989). For human podocytes, there are data from agonist-antagonist studies suggesting that ETA receptors are present (Spath et al., 1995; Collino et al., 2008). However, it is likely that human podocytes also possess both ETA and ETB receptors (Rebibou et al., 1992).
Treatment of mouse podocytes with albumin or IgG leads to disruption of the actin cytoskeleton, activation of focal adhesion kinase, which activates transcription of preproET-1 mRNA leading to ET-1 secretion (Morigi et al., 2005). Similarly, treatment of podocytes with shiga toxin leads to ET-1 secretion. Shiga toxin is the offending agent of post-diarrhoeal haemolytic-uraemic syndrome, which is characterized by glomerular ischaemic changes preceding microvascular thrombosis. In turn, ET-1, acting in an autocrine manner via the ETA receptor, causes disruption of the podocyte actin cytoskeleton (Morigi et al., 2006).
One of the key podocyte proteins at the slit diaphragm is nephrin and mutations of the nephrin gene lead to congenital nephrotic syndrome (Kestila et al., 1998). In a number of human glomerular diseases, nephrin is either down-regulated or its cellular localization altered (Welsh and Saleem, 2010). Nephrin is often used in experimental models as a marker of podocyte injury. There are limited in vitro data suggesting that ET-1 may lead to podocyte loss of nephrin. Treatment of podocytes with exogenous ET-1 reduced cell surface nephrin expression. This podocyte shedding of nephrin was blocked by an ETA receptor antagonist (Collino et al., 2008). Similarly, infusion of ET-1 into rats caused nephrin excretion into the urine, again inhibited by ETA receptor antagonism (Saleh et al., 2010).
The role of ET-1 in podocyte injury in CKD has been recently reviewed (Fligny et al., 2011) so will only be briefly discussed here. In a number of experimental models of renal injury, there is evidence that the beneficial effects of ET antagonists may be, in part, due to protection of podocyte structure and function. Aged Wistar rats spontaneously develop glomerulosclerosis and proteinuria associated with electron micrograph evidence of podocyte injury. These changes can be reduced by treatment with an ETA receptor antagonist in the absence of a fall in BP (Ortmann et al., 2004). More recently, in separate studies in streptozocin-induced DN, treatment with an ETA receptor antagonist reduced BP and proteinuria. These effects were associated with either preservation of podocyte number (Gagliardini et al., 2009) or maintenance of glomerular nephrin expression and a reduction of nephrin loss in the urine (Saleh et al., 2011). Future studies are likely to focus on changes in podocyte number and phenotype, as more effective antibodies are available for immunohistochemistry. This will include markers such as nephrin, synaptopodin and podocin, all of which contribute to the machinery of the slit diaphragm (Mathieson, 2012).
Taken together, the current data suggest that podocytes release, bind and respond to ET-1. ET-1 can induce cytoskeletal remodelling in podocytes but how such changes would affect GFR and RBF remains speculative. Furthermore, whilst ET-1 may increase podocyte protein permeability, whether water and small solute filtration will be increased remains speculative. Finally, although putative effects of ET-1 and ET receptor antagonism have been demonstrated in vitro, no direct effects of ET-1 on podocytes have been demonstrated in vivo.
Glomerular endothelium
The glomerular endothelium is a specialized fenestrated capillary bed. Its role in the development of renal disease and proteinuria has been less well studied than that of the podocyte due to difficulty in culturing these cells in vitro and the lack of glomerular endothelial-specific gene targeting approaches. Glomerular endothelial cells (GEnC) are probably the principal source of ET-1 within the glomerulus (Herman et al., 1998), and glomerular ET-1 staining is increased in the presence of proteinuric renal disease including IgA nephropathy, lupus nephritis and membranous nephropathy (Lehrke et al., 2001). It is probable that ET-1 secreted abluminally from GEnC modulates podocyte and mesangial cell structure and function. However, study in this area is limited. Recent data have shown that conditioned media from GEnC stimulated with serum from patients with pre-eclampsia cause podocyte nephrin shedding and changes in the actin cytoskeleton in vitro (Collino et al., 2008). An ETA receptor antagonist was able to inhibit these changes, although the authors did not show evidence for the direct production of ET-1 by GEnC.
Mesangial cells
Mesangial cells are glomerular pericytes and they act to support the other glomerular structures. They synthesize mesangial matrix, which is the ‘filler’ between adjacent mesangial cells. Mesangial cells are able to produce a significant amount of ET-1 (Sakamoto et al., 1990; Herman et al., 1998), which is increased by vasoactive substances such as Ang II and vasopressin (Ikeda et al., 1995), TGF-β (Zoja et al., 2011) and TNF-α (Kohan, 1997). These cells express both ETA and ETB receptors and are ET-1 responsive (Takeda et al., 1994; Herman et al., 1998; Orth et al., 2000). ET-1 can itself stimulate mesangial cell ET-1 production (Iwasaki et al., 1995), and in rats, this has been shown to be mediated via the ETB receptor (Iwasaki et al., 1995). ET-1 stimulates contraction of mesangial cells in vitro (Simonson et al., 1989) as well as in isolated glomeruli (Saleh et al., 2010). These effects of ET-1 would result in changes in glomerular filtration area and intraglomerular haemodynamics.
Many glomerular diseases are associated with increased mesangial matrix and fibrosis (glomerulosclerosis), with DN being the most common. Chronic ET-1 overexpression leads to glomerulosclerosis without systemic hypertension (Hocher et al., 1997). Stimulation of mesangial cells with ET-1 increases expression of collagen types I and IV, fibronectin and versican all of which contribute to extracellular matrix. ET-1 also increases mesangial cell production of tissue inhibitor of matrix metalloproteinase 3 and plasminogen activator inhibitor 2, which inhibit mesangial matrix degradation (Gomez-Garre et al., 1996; Mishra et al., 2003). Furthermore, ET-1 stimulates mesangial cell production of the cytokines TGF-β, IL-6, osteopontin and MCP-1, which results in an autocrine signalling loop increasing collagen synthesis. These effects of ET-1 on mesangial cells appear to be mediated via the ETA receptor (Simonson and Ismail-Beigi, 2011).
Mesangial cell proliferation is common in many glomerulonephritides, such as IgA nephropathy and lupus nephritis. ET-1 stimulates mesangial cell mitogenesis (Simonson et al., 1989; Gomez-Garre et al., 1996). This response is largely mediated via the ETA receptor. However, there are data from cultured human mesangial cells suggesting that the ETB receptor may also be involved (Orth et al., 2000). ET-1 also stimulates mesangial cell production of platelet-derived growth factor, leading to an autocrine loop of cell proliferation and further secretion (Jaffer et al., 1990; Ikeda et al., 1995). Thus, ET-1 can alter the structure and function of glomerular mesangial cells, independent of changes in BP. These effects of the ET system are likely to contribute to renal disease progression. However, it must be noted that any conclusions about the role of ET-1 in mesangial cell biology are based entirely on in vitro data. There remains no conclusive demonstration of a physiological role for ET-1 mediated mesangial cell contraction and so it is likely that any functional or pathological importance of mesangial cell-derived ET-1 will remain speculative.
Effects of ET-1 on the tubule
With respect to renal tubular functions, there is now a substantial body of evidence supporting a role for ET-1 in the regulation of salt and water homeostasis. This has been reviewed extensively recently (Kohan et al., 2011). In vitro, sarafotoxin 6c, a selective ETB receptor agonist, inhibited chloride transport in cells from the thick ascending loop of Henle. This was in an equipotent manner to ET-1 and was inhibited by an ETB receptor antagonist, but not an ETA antagonist (Plato et al., 2000). Animal data also support a role for the ETB receptor in natriuresis and diuresis. A rat model deficient in renal ETB receptors displays a salt-sensitive hypertension, with restoration of normal BP by amiloride, suggesting that the ETB receptor regulates sodium excretion at the epithelial sodium channel in CD cells (Gariepy et al., 2000). Antagonist studies have also proved helpful with ETB antagonist-treated rats developing a sodium-dependent hypertension (Webb et al., 1998; Pollock, 2001).
However, recent in vivo studies have explored the possible roles of the ETA receptor in the effects of ET-1 in the renal medulla. CD ET-1 knockout (KO) (Ahn et al., 2004) and CD ETB KO (Ge et al., 2006) mice show higher BP and salt sensitivity compared with wild type mice. However, the magnitude of these effects in CD ET-1 KO mice is almost double that in CD ETB KO mice, indicating that CD-derived ET-1 may have multiple targets, most likely either ETB receptors on other cell types or ETA receptors. Interestingly, CD ETA KO mice are not hypertensive, even under a high-salt diet (Ge et al., 2005). Surprisingly, the BP of mice with both ETA and ETB receptors knocked out in CD (CD ETA/B KO mice) is almost identical to that of CD ET-1 KO mice (Ge et al., 2008). Although the specific mechanisms for this phenomenon are still under investigation, the authors of that study speculated that ETA receptors in the CD help maintain salt balance if the ETB receptor is dysfunctional. The natriuretic ability of ETB receptors may compensate for (or overwhelm) the lack of ETA activity in CD ETA KO mice. In support of this hypothesis, ET-1 infused into the renal medulla of female ETB receptor-deficient rats caused ETA receptor-dependent natriuresis (Nakano and Pollock, 2009). However, dissecting the different actions of the intrarenal ET system has proved difficult, in part due to an inability to discriminate between effects of ET-1 in vivo on the nephron and on the vasculature.
Effects of ET-1 on renal inflammation
Inflammation contributes to the development and progression of CKD as well as the incident cardiovascular disease with which CKD is associated. In some conditions there is a transient and acute inflammatory response that leads to impaired renal function, for example, anti-neutrophil cytoplasmic antibody-associated vasculitis, lupus nephritis and interstitial nephritis. However, even in more smouldering renal diseases, such as membranous, IgA or DN, there is a correlation between the presence of low level inflammatory infiltrates and progression to end-stage renal disease (Bohle et al., 1992; Radford et al., 1997; Kelly and Dominguez, 2010).
ET-1 likely contributes to the development of inflammation. Systemic overexpression of ET-1 leads to renal, cardiac and pulmonary inflammation and fibrosis (Hocher et al., 1997; 2000). Furthermore, endothelial-restricted ET-1 overexpressing mice develop evidence of vascular inflammation in the absence of hypertension (Amiri et al., 2008). ET-1 also mediates renal inflammation induced by Ang II. In mice, chronic infusion of Ang II causes hypertension and a T-cell-rich renal infiltrate. Treatment with an ETA receptor antagonist reduced BP and attenuated the numbers of T cells in the renal cortex. Interestingly, alternative methods of reducing BP, which achieved similar BP control, did not significantly affect T-cell infiltration (Boesen et al., 2011). Similarly, aldosterone-treated rats developed hypertension and renal macrophage (Mϕ) infiltration that was reduced by ETA receptor antagonism (Tostes et al., 2002). The mechanism for these changes is not clear but should be the focus for future research.
There is evidence that leucocytes are able to synthesize and regulate ET-1 production. Polymorphonuclear neutrophils produce proteolytic enzymes that convert big ET-1 to the active peptide and are subsequently able to break this down (Patrignani et al., 1991; Kaw et al., 1992). Mast cells increase in number in many renal inflammatory kidney diseases and may have a role in limiting renal injury and restoring homeostasis (Blank et al., 2007). Regulating the effects of ET-1 may form part of this mechanism. In support of this hypothesis, mast cells have been shown to limit ET-1 action in vivo both by receptor-mediated uptake and through the action of proteolytic enzymes (Maurer et al., 2004).
There is convincing evidence that Mϕs are able to secrete ET-1. Original studies showed that human monocyte-derived Mϕs produce ET-1 following LPS stimulation (Ehrenreich et al., 1990), although other studies have not confirmed this (Spirig et al., 2009). Human alveolar Mϕs produce ET-1 on stimulation with thrombin or LPS (Kobayashi et al., 1997), and increased ET-1 secretion was found in alveolar Mϕ isolated from patients with scleroderma and pulmonary fibrosis (Odoux et al., 1997). Dendritic cells (DCs) are the major type of antigen presenting cell and can develop from Mϕ. Human DCs produce ET-1 and synthesis is increased by stimulation with LPS and lipotechoic acid [exogenous toll-like receptor (TLR) 4 and TLR2 ligands], and hyaluronic acid and heparan sulphate (endogenous TLR ligands) (Guruli et al., 2004; Spirig et al., 2009). Overall, Mϕs are able to produce ET-1 when activated but there is no convincing evidence that they provide a significant source of ET-1 in renal injury. This is based on the observation that in renal biopsy studies Mϕ or other leucocytes do not significantly express ET-1 or its receptors (Lehrke et al., 2001).
ET-1 exerts a number of pro-inflammatory effects. It acts as a chemo-attractant for PMN (Wright et al., 1994; Cui et al., 2001) and Mϕ (Achmad and Rao, 1992). When assessed, chemokinesis appears to be inhibited by blockade of the ETA receptor. ET-1 can also activate the endothelium to increase leucocyte adhesion and transmigration both in vitro (Zouki et al., 1999) and in vivo (Callera et al., 2004). ET-1 also stimulates secretion of MCP-1 from mesangial cells, which is chemotactic to Mϕ (Ishizawa et al., 2004). Finally, ET-1 may also promote DC differentiation (Guruli et al., 2004). Although both ETA and ETB receptors have been identified on Mϕ (Bacon et al., 1996; Mencarelli et al., 2009), their respective roles remain unclear. There is some evidence that ET-1 can elicit classical pro-inflammatory Mϕ activation with activation of the NF-κB signalling pathway and release of TNF-α (Wilson et al., 2001; Juergens et al., 2008), although this result has not been consistently observed (Speciale et al., 1998; Spirig et al., 2009). ET-1 does stimulate Mϕ production of the chemokine macrophage inflammatory protein-1β. These changes may be part of an ET-1-induced alteration in Mϕ phenotype. However, data to this end are lacking but would be of great interest given the potential role for Mϕ in renal repair (Kluth, 2007). Overall, the main effect of ET-1 on leucocytes is to promote a chemotactic response that serves to increase the leucocyte infiltrate within the kidney.
ET antagonism in models of renal disease
A number of ET receptor antagonists have been used in animal models of renal disease. These show a range of potentially beneficial effects many of which are unlikely to be a result of the BP lowering effects of these drugs. An important point when considering these data is to remember that ET antagonists may selectively block the ETA receptor or act to provide mixed ETA/B receptor antagonism. Benefits of one approach over the other have been previously discussed (Dhaun et al., 2007; Schneider et al., 2007).
Hypertensive nephropathy
Hypertensive nephropathy (or nephrosclerosis) is characterized histologically by vascular, glomerular and tubulointerstitial involvement. The vascular disease consists of intimal thickening and luminal narrowing of the large and small renal arteries and the glomerular arterioles. The glomeruli may show both focal global (involving the entire glomerulus) and focal segmental sclerosis. The vascular and glomerular diseases may be associated with an often severe interstitial nephritis.
The Benigni group was amongst the first to report the benefits of ET receptor blockade in hypertensive nephropathy. They reported reductions in proteinuria and glomerulosclerosis after selective ETA receptor blockade in a renal mass reduction rat model of hypertensive nephropathy (Benigni et al., 1993). Following a different approach, Hocher et al., (1997) showed that systemic overexpression of the preproET gene in mice led to glomerulosclerosis and interstitial fibrosis in the absence of a change in BP (Hocher et al., 1997). Since these early studies, many other preclinical data have emerged confirming the renoprotective effects of both ETA and ETA/B receptor antagonism in several forms of hypertension – Ang II-dependent, renin-dependent, salt-loaded renin-dependent, aldosterone-induced, genetically salt-sensitive, deoxycorticosterone acetate-salt induced – and these are reviewed in Schiffrin (2005).
The work by Opocensky et al. (2006) has shown that in hypertensive rats podocyte injury preceded proteinuria. The selective ETA antagonist atrasentan, but not the mixed ETA/B antagonist bosentan, prevented podocyte injury and substantially reduced proteinuria despite the fact that animals were still markedly hypertensive (Opocensky et al., 2006). Importantly, treatment was begun after hypertension had been established, and the beneficial effects on podocyte injury were seen at an early stage of renal injury, that is in the absence of established glomerulosclerosis. Podocyte injury prior to the development of glomerulosclerosis was also observed in Dahl hypertensive rats, and this was sensitive to mineralocorticoid receptor blockade (Nagase et al., 2006). Importantly, unlike ET or mineralocorticoid receptor blockade, antihypertensive treatment with hydralazine had no effect on podocyte injury, again indicating that the renoprotective effects are specific and largely pressure independent (Nagase et al., 2006). Interestingly, Boffa et al. (2001) have shown reversal of vascular fibrosis and collagen deposition in a model of NO-deficient hypertension following ET receptor blockade.
Diabetic nephropathy
DN is now the commonest cause of end-stage renal failure. A number of studies have assessed the effects of different ET antagonists in models of DN with the majority using streptozocin to induce diabetes mellitus. Early studies with mixed ETA/B antagonists showed a fall in proteinuria and, where measured, BP (Benigni et al., 1998a; Hocher et al., 2001) There was also evidence of reduced glomerulosclerosis (Hocher et al., 2001). An important comparator in such studies is the use of ACE inhibitors, which are the mainstay of treatment for DN. When assessing proteinuria (or albuminuria) none of the studies show that ET receptor antagonists are superior to ACE inhibitors. Some studies show equivalent efficacy (Gagliardini et al., 2009; Watson et al., 2010), whereas others show that ACE inhibitors are better (Zoja et al., 2011). The combination of ACE inhibition with ET receptor antagonism may have additional advantages. In uninephrectomized streptozocin-induced diabetic rats treatment with lisinopril and avosentan normalized the proteinuria, reduced interstitial and glomerular scarring, prevented the loss of podocytes and maintained expression of nephrin (Gagliardini et al., 2009). Here combination therapy was more effective than either drug alone.
ET receptor antagonists have also been shown to have a number of effects on markers of glomerular injury and inflammation in models of DN. Treatment with the ETA receptor antagonist atrasentan, in streptozocin-induced diabetes, reduced albumin permeability in isolated glomeruli. In addition, treatment reduced expression of intercellular adhesion molecule 1 and MCP-1, whilst preserving expression of nephrin (Saleh et al., 2010). Furthermore, the selective ETA receptor antagonist sitaxentan reduced renal Mϕ infiltration and expression of pro-inflammatory cytokines in different models of DN (Sasser et al., 2007; Zoja et al., 2011). These BP-independent, anti-inflammatory and podocyte protective effects of ET receptor antagonism may provide additional longer-term benefits in DN.
Glomerulonephritis
Glomerular disease outside of diabetes and hypertension has been less extensively studied with respect to ET receptor blockade. In particular, there are very few studies in inflammatory glomerulonephritis. The major studies of note are outlined in Table 1. Whether ET receptor antagonists are able to alter Mϕ infiltration and function, podocyte biology and mesangial cell responses in glomerulonephritis has not been assessed.
Table 1.
Models of glomerulonephritis and the effects of ET receptor antagonism
| Model | Selective ETA or mixed ETA/B antagonism | Effects | Reference |
|---|---|---|---|
| Immune complex GN (rat) | Mixed ETA/B | Reduction in proteinuria and glomerular injury | (Gomez-Garre et al., 1996) |
| Anti-Thy1 nephritis (characterized by acute mesangiolysis followed by mesangial proliferation; rat) | Selective ETA | Reduced glomerular cell proliferation | (Fukuda et al., 1996) |
| No assessment of inflammation, injury or proteinuria | |||
| Membranous nephropathy (rat) | ACE inhibitor and ETA receptor antagonist: alone and in combination | Reduced proteinuria | (Benigni et al., 1998b) |
| Lower serum creatinine | |||
| Less glomerular and tubulointerstitial injury | |||
| Combination therapy superior to either intervention alone | |||
| Heyman nephritis (rat) | ACE inhibitor, ARB, ETA receptor antagonist: alone and in combination | Similar BP reduction | (Amann et al., 2001) |
| ACE inhibitor superior in reducing proteinuria and glomerulosclerosis | |||
| Additional effects of adding ETA antagonist to either ACE inhibitor or ARB |
ARB, angiotensin receptor blocker.
Clinical studies
Most of the clinical studies in CKD relating to ET receptor antagonism have focussed on changes in BP and proteinuria and have shown reductions in both. These have been reviewed elsewhere (Dhaun et al., 2006; Barton, 2008) and suggest that both selective ETA and mixed ETA/B antagonist approaches may be of benefit in clinical CKD. There is only one study that relates to renal inflammation. This suggests that urinary ET-1, which is a measure of renal ET-1 synthesis (Benigni et al., 1991; Dhaun et al., 2009), is increased in those with active renal inflammation (Dhaun et al., 2009). Interestingly, urinary ET-1 levels fell following successful treatment of the inflammation. These data, however, were limited to those with lupus nephritis. It would be interesting to see if the same holds true for those patients with other forms of renal inflammatory disease such as small vessel vasculitis or interstitial nephritis. Furthermore, whether urinary ET-1 may be a useful biomarker of disease relapse in these conditions remains unclear.
Most clinical studies of ET receptor antagonists have restricted themselves to diabetic renal disease using BP and proteinuria as their end points. Future studies in patients with non-diabetic renal disease are now needed. Furthermore, the impact of ET receptor antagonists on clinical end points other than BP and proteinuria would be of great interest – some potential candidates include changes in urinary biomarkers (such as neutrophil gelatinase-associated lipocalin and kidney injury molecule-1), histological renal injury and novel cardiovascular risk factors (such as arterial stiffness, endothelial dysfunction, serum urate and asymmetric dimethylarginine).
Conclusion
The role of ET-1 in regulating BP and renal haemodynamics is well established. The paracrine effects of ET-1 that are likely to be important in the development and progression of CKD are less clearly understood. Renal ET-1 production is increased in most causes of renal injury. Glomerular endothelial cells are probably the principal source of renal-derived ET-1 and this may affect a number of local cell types, including podocytes, mesangial cells, other glomerular endothelial cells and inflammatory cells. Currently available data suggest that the majority of the pathological effects of ET-1, at least within the kidney, are mediated via the ETA receptor. On podoytes, ET-1 causes alterations in the actin cytoskeleton, foot process effacement and loss of proteins such as nephrin that maintain the slit diaphragm. This contributes to the development of proteinuria, a powerful marker of renal disease progression. ET-1 activates mesangial cells to release pro-inflammatory and profibrotic cytokines; it stimulates cell proliferation and increases production of matrix proteins that can lead to glomerular sclerosis. ET-1 is also chemoattractant to leucocytes including Mϕs, which infiltrate the glomerulus or interstitium and may further contribute to renal inflammation. More studies – both preclinical and clinical – are needed to further define the effects of ET-1 on these different renal cell types and to establish whether ET receptor antagonism may be of benefit in inhibiting inflammation and maintaining glomerular architecture.
Glossary
- Ang
II, angiotensin II
- BP
blood pressure
- CD
collecting duct
- CKD
chronic kidney disease
- DC
dendritic cell
- DN
diabetic nephropathy
- ET-1
enothelin-1
- ETA
GEnC, glomerular endothelial cell
- GFR
glomerular filtration rate
- Mϕ
macrophage
- MCP-1
macrophage chemotactic protein-1
- MIP-1β
macrophage inflammatory protein-1β
- RBF
renal blood flow
- TLR
toll-like receptor
Conflicts of interest
DCK has no conflict of interest. ND and DJW have received research grants from Pfizer. DJW has acted as a consultant to Pfizer.
References
- Abassi Z, Francis B, Wessale J, Ovcharenko E, Winaver J, Hoffman A. Effects of endothelin receptors ET(A) and ET(B) blockade on renal haemodynamics in normal rats and in rats with experimental congestive heart failure. Clin Sci. 2002;103:245–248. doi: 10.1042/CS103S245S. [DOI] [PubMed] [Google Scholar]
- Achmad TH, Rao GS. Chemotaxis of human blood monocytes toward endothelin-1 and the influence of calcium channel blockers. Biochem Biophys Res Commun. 1992;189:994–1000. doi: 10.1016/0006-291x(92)92302-e. [DOI] [PubMed] [Google Scholar]
- Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, et al. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest. 2004;114:504–511. doi: 10.1172/JCI21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann K, Simonaviciene A, Medwedewa T, Koch A, Orth S, Gross ML, et al. Blood pressure-independent additive effects of pharmacologic blockade of the renin-angiotensin and endothelin systems on progression in a low-renin model of renal damage. J Am Soc Nephrol. 2001;12:2572–2584. doi: 10.1681/ASN.V12122572. [DOI] [PubMed] [Google Scholar]
- Amiri F, Paradis P, Reudelhuber TL, Schiffrin EL. Vascular inflammation in absence of blood pressure elevation in transgenic murine model overexpressing endothelin-1 in endothelial cells. J Hypertens. 2008;26:1102–1109. doi: 10.1097/HJH.0b013e3282fc2184. [DOI] [PubMed] [Google Scholar]
- Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–732. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- Arinami T, Ishikawa M, Inoue A, Yanagisawa M, Masaki T, Yoshida MC, et al. Chromosomal assignments of the human endothelin family genes: the endothelin-1 gene (EDN1) to 6p23-p24, the endothelin-2 gene (EDN2) to 1p34, and the endothelin-3 gene (EDN3) to 20q13.2-q13.3. Am J Hum Genet. 1991;48:990–996. [PMC free article] [PubMed] [Google Scholar]
- Attina T, Camidge R, Newby DE, Webb DJ. Endothelin antagonism in pulmonary hypertension, heart failure, and beyond. Heart. 2005;91:825–831. doi: 10.1136/hrt.2004.053991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon CR, Cary NR, Davenport AP. Endothelin peptide and receptors in human atherosclerotic coronary artery and aorta. Circulation Res. 1996;79:794–801. doi: 10.1161/01.res.79.4.794. [DOI] [PubMed] [Google Scholar]
- Barton M. Reversal of proteinuric renal disease and the emerging role of endothelin. Nat Clin Pract Nephrol. 2008;4:490–501. doi: 10.1038/ncpneph0891. [DOI] [PubMed] [Google Scholar]
- Benigni A, Remuzzi G. Endothelin antagonists. Lancet. 1999;353:133–138. doi: 10.1016/S0140-6736(98)09423-9. [DOI] [PubMed] [Google Scholar]
- Benigni A, Perico N, Gaspari F, Zoja C, Bellizzi M, Gabanelli M, et al. Increased renal endothelin production in rats with reduced renal mass. Am J Physiol. 1991;260:331–339. doi: 10.1152/ajprenal.1991.260.3.F331. [DOI] [PubMed] [Google Scholar]
- Benigni A, Zoja C, Corna D, Orisio S, Longaretti L, Bertani T, et al. A specific endothelin subtype A receptor antagonist protects against injury in renal disease progression. Kidney Int. 1993;44:440–444. doi: 10.1038/ki.1993.263. [DOI] [PubMed] [Google Scholar]
- Benigni A, Colosio V, Brena C, Bruzzi I, Bertani T, Remuzzi G. Unselective inhibition of endothelin receptors reduces renal dysfunction in experimental diabetes. Diabetes. 1998a;47:450–456. doi: 10.2337/diabetes.47.3.450. [DOI] [PubMed] [Google Scholar]
- Benigni A, Corna D, Maffi R, Benedetti G, Zoja C, Remuzzi G. Renoprotective effect of contemporary blocking of angiotensin II and endothelin-1 in rats with membranous nephropathy. Kidney Int. 1998b;54:353–359. doi: 10.1046/j.1523-1755.1998.00011.x. [DOI] [PubMed] [Google Scholar]
- Blank U, Essig M, Scandiuzzi L, Benhamou M, Kanamaru Y. Mast cells and inflammatory kidney disease. Immunol Rev. 2007;217:79–95. doi: 10.1111/j.1600-065X.2007.00503.x. [DOI] [PubMed] [Google Scholar]
- Boesen EI, Krishnan KR, Pollock JS, Pollock DM. ETA activation mediates angiotensin II-induced infiltration of renal cortical T cells. J Am Soc Nephrol. 2011;22:2187–2192. doi: 10.1681/ASN.2010020193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffa JJ, Tharaux PL, Dussaule JC, Chatziantoniou C. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension. 2001;37:490–496. doi: 10.1161/01.hyp.37.2.490. [DOI] [PubMed] [Google Scholar]
- Bohle A, Wehrmann M, Bogenschutz O, Batz C, Vogl W, Schmitt H, et al. The long-term prognosis of the primary glomerulonephritides. A morphological and clinical analysis of 1747 cases. Pathol Res Pract. 1992;188:908–924. doi: 10.1016/s0344-0338(11)80252-9. [DOI] [PubMed] [Google Scholar]
- Callera GE, Montezano AC, Touyz RM, Zorn TM, Carvalho MH, Fortes ZB, et al. ETA receptor mediates altered leukocyte-endothelial cell interaction and adhesion molecules expression in DOCA-salt rats. Hypertension. 2004;43:872–879. doi: 10.1161/01.HYP.0000117296.30296.14. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Turner DR, Ogg CS, Chantler C, Williams DG. The long-term prognosis of patients with focal segmental glomerulosclerosis. Clin Nephrol. 1978;10:213–218. [PubMed] [Google Scholar]
- Chou SY, Porush JG. Renal actions of endothelin-1 and endothelin-3: interactions with the prostaglandin system and nitric oxide. Am J Kidney Dis. 1995;26:116–123. doi: 10.1016/0272-6386(95)90164-7. [DOI] [PubMed] [Google Scholar]
- Collino F, Bussolati B, Gerbaudo E, Marozio L, Pelissetto S, Benedetto C, et al. Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am J Physiol. 2008;294:1185–1194. doi: 10.1152/ajprenal.00442.2007. [DOI] [PubMed] [Google Scholar]
- Cui P, Tani K, Kitamura H, Okumura Y, Yano M, Inui D, et al. A novel bioactive 31-amino acid endothelin-1 is a potent chemotactic peptide for human neutrophils and monocytes. J Leukoc Biol. 2001;70:306–312. [PubMed] [Google Scholar]
- Davenport AP, Nunez DJ, Brown MJ. Binding sites for 125I-labelled endothelin-1 in the kidneys: differential distribution in rat, pig and man demonstrated by using quantitative autoradiography. Clin Sci. 1989;77:129–131. doi: 10.1042/cs0770129. [DOI] [PubMed] [Google Scholar]
- Denton KM, Shweta A, Finkelstein L, Flower RL, Evans RG. Effect of endothelin-1 on regional kidney blood flow and renal arteriole calibre in rabbits. Clin Exp Pharmacol Physiol. 2004;31:494–501. doi: 10.1111/j.1440-1681.2004.04036.x. [DOI] [PubMed] [Google Scholar]
- DeNucci G, Thomas R, D'Orleans-Juste P, Antunes E, Walder C, Warner TD, et al. Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin and endothelium-derived relaxing factor. Proc Natl Acad Sci U S A. 1988;85:9797–9800. doi: 10.1073/pnas.85.24.9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaun N, Goddard J, Webb DJ. The endothelin system and its antagonism in chronic kidney disease. J Am Soc Nephrol. 2006;17:943–955. doi: 10.1681/ASN.2005121256. [DOI] [PubMed] [Google Scholar]
- Dhaun N, Pollock DM, Goddard J, Webb DJ. Selective and mixed endothelin receptor antagonism in cardiovascular disease. Trends Pharmacol Sci. 2007;28:573–579. doi: 10.1016/j.tips.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Dhaun N, Lilitkarntakul P, Macintyre IM, Muilwijk E, Johnston NR, Kluth DC, et al. Urinary endothelin-1 in chronic kidney disease and as a marker of disease activity in lupus nephritis. Am J Physiol. 2009;296:1477–1483. doi: 10.1152/ajprenal.90713.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaun N, MacIntyre IM, Kerr D, Melville V, Johnston NR, Haughie S, et al. Selective endothelin-A receptor antagonism reduces proteinuria, blood pressure, and arterial stiffness in chronic proteinuric kidney disease. Hypertension. 2011;57:772–779. doi: 10.1161/HYPERTENSIONAHA.110.167486. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Stewart DJ, Cernacek P, Gosselin G. Human pulmonary circulation is an important site for both clearance and production of endothelin-1. Circulation. 1996;94:1278–1284. doi: 10.1161/01.cir.94.7.1578. [DOI] [PubMed] [Google Scholar]
- Ehrenreich H, Anderson RW, Fox CH, Rieckmann P, Hoffman GS, Travis WD, et al. Endothelins, peptides with potent vasoactive properties, are produced by human macrophages. J Exp Med. 1990;172:1741–1748. doi: 10.1084/jem.172.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid H, de Bold ML, Chen JH, de Bold AJ. Epicardial mesothelial cells synthesize and release endothelin. J Cardiovasc Pharmacol. 1994;24:715–720. doi: 10.1097/00005344-199424050-00005. [DOI] [PubMed] [Google Scholar]
- Evans RG, Madden AC, Oliver JJ, Lewis TV. Effects of ET(A)- and ET(B)-receptor antagonists on regional kidney blood flow, and responses to intravenous endothelin-1, in anaesthetized rabbits. J Hypertens. 2001;19:1789–1799. doi: 10.1097/00004872-200110000-00013. [DOI] [PubMed] [Google Scholar]
- Fligny C, Barton M, Tharaux PL. Endothelin and podocyte injury in chronic kidney disease. Contrib Nephrol. 2011;172:120–138. doi: 10.1159/000328692. [DOI] [PubMed] [Google Scholar]
- Freed MI, Wilson DE, Thompson KA, Harris RZ, Ilson BE, Jorkasky DK. Pharmacokinetics and pharmacodynamics of SB 209670, an endothelin receptor antagonist: effects on the regulation of renal vascular tone. Clin Pharmacol Ther. 1999;65:473–482. doi: 10.1016/S0009-9236(99)70066-4. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Yanagida T, Okuda S, Tamaki K, Ando T, Fujishima M. Role of endothelin as a mitogen in experimental glomerulonephritis in rats. Kidney Int. 1996;49:1320–1329. doi: 10.1038/ki.1996.188. [DOI] [PubMed] [Google Scholar]
- Gagliardini E, Corna D, Zoja C, Sangalli F, Carrara F, Rossi M, et al. Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am J Physiol. 2009;297:1448–1456. doi: 10.1152/ajprenal.00340.2009. [DOI] [PubMed] [Google Scholar]
- Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J Clin Invest. 2000;105:925–933. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasic S, Wagner OF, Vierhapper H, Nowotny P, Waldhausl W. Regional haemodynamic effects and clearance of endothelin-1 in humans: renal and peripheral tissues may contribute to overall disposal of the peptide. J Cardiovasc Pharmacol. 1992;19:176–180. doi: 10.1097/00005344-199202000-00004. [DOI] [PubMed] [Google Scholar]
- Ge Y, Stricklett PK, Hughes AK, Yanagisawa M, Kohan DE. Collecting duct-specific knockout of the endothelin A receptor alters renal vasopressin responsiveness, but not sodium excretion or blood pressure. Am J Physiol. 2005;289:F692–F698. doi: 10.1152/ajprenal.00100.2005. [DOI] [PubMed] [Google Scholar]
- Ge Y, Bagnall A, Stricklett PK, Strait K, Webb DJ, Kotelevtsev Y, et al. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am J Physiol. 2006;291:F1274–F1280. doi: 10.1152/ajprenal.00190.2006. [DOI] [PubMed] [Google Scholar]
- Ge Y, Bagnall A, Stricklett PK, Webb D, Kotelevtsev Y, Kohan DE. Combined knockout of collecting duct endothelin A and B receptors causes hypertension and sodium retention. Am J Physiol. 2008;295:1635–1640. doi: 10.1152/ajprenal.90279.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard J, Eckhart C, Johnston NR, Cumming AD, Rankin AJ, Webb DJ. Endothelin A receptor antagonism and angiotensin-converting enzyme inhibition are synergistic via an endothelin B receptor-mediated and nitric oxide-dependent mechanism. J Am Soc Nephrol. 2004a;15:2601–2610. doi: 10.1097/01.ASN.0000141313.84470.4B. [DOI] [PubMed] [Google Scholar]
- Goddard J, Johnston NR, Hand MF, Cumming AD, Rabelink TJ, Rankin AJ, et al. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: a comparison of selective and combined endothelin receptor blockade. Circulation. 2004b;109:1186–1193. doi: 10.1161/01.CIR.0000118499.69469.51. [DOI] [PubMed] [Google Scholar]
- Gomez-Garre D, Largo R, Liu XH, Gutierrez S, Lopez-Armada MJ, Palacios I, et al. An orally active ETA/ETB receptor antagonist ameliorates proteinuria and glomerular lesions in rats with proliferative nephritis. Kidney Int. 1996;50:962–972. doi: 10.1038/ki.1996.397. [DOI] [PubMed] [Google Scholar]
- Greka A, Mundel P. Cell biology and pathology of podocytes. Ann Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JK, Cantley LG. Cellular maintenance and repair of the kidney. Ann Rev Physiol. 2010;72:357–376. doi: 10.1146/annurev.physiol.010908.163245. [DOI] [PubMed] [Google Scholar]
- Gurbanov K, Rubinstein I, Hoffman A, Abassi Z, Better OS, Winaver J. Differential regulation of renal regional blood flow by endothelin-1. Am J Physiol. 1996;271:1166–1172. doi: 10.1152/ajprenal.1996.271.6.F1166. [DOI] [PubMed] [Google Scholar]
- Guruli G, Pflug BR, Pecher S, Makarenkova V, Shurin MR, Nelson JB. Function and survival of dendritic cells depend on endothelin-1 and endothelin receptor autocrine loops. Blood. 2004;104:2107–2115. doi: 10.1182/blood-2003-10-3559. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Webb DJ. Contribution of endogenous generation of endothelin-1 to basal vascular tone. Lancet. 1994;344:852–854. doi: 10.1016/s0140-6736(94)92827-4. [DOI] [PubMed] [Google Scholar]
- Herman WH, Emancipator SN, Rhoten RL, Simonson MS. Vascular and glomerular expression of endothelin-1 in normal human kidney. Am J Physiol. 1998;275:8–17. doi: 10.1152/ajprenal.1998.275.1.F8. [DOI] [PubMed] [Google Scholar]
- Hocher B, Thone-Reineke C, Rohmeiss P, Schmager F, Slowinski T, Burst V, et al. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J Clin Invest. 1997;99:1380–1389. doi: 10.1172/JCI119297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocher B, Schwarz A, Fagan KA, Thone-Reineke C, El-Hag K, Kusserow H, et al. Pulmonary fibrosis and chronic lung inflammation in ET-1 transgenic mice. Am J Respir Cell Mol Biol. 2000;23:19–26. doi: 10.1165/ajrcmb.23.1.4030. [DOI] [PubMed] [Google Scholar]
- Hocher B, Schwarz A, Reinbacher D, Jacobi J, Lun A, Priem F, et al. Effects of endothelin receptor antagonists on the progression of diabetic nephropathy. Nephron. 2001;87:161–169. doi: 10.1159/000045906. [DOI] [PubMed] [Google Scholar]
- Ibsen H, Olsen MH, Wachtell K, Borch-Johnsen K, Lindholm LH, Mogensen CE, et al. Reduction in albuminuria translates to reduction in cardiovascular events in hypertensive patients: losartan intervention for endpoint reduction in hypertension study. Hypertension. 2005;45:198–202. doi: 10.1161/01.HYP.0000154082.72286.2a. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Kohno M, Horio T, Yasunari K, Yokokawa K, Kano H, et al. Effect of thrombin and PDGF on endothelin production in cultured mesangial cells derived from spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1995;22:197–198. doi: 10.1111/j.1440-1681.1995.tb02879.x. [DOI] [PubMed] [Google Scholar]
- Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, et al. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci U S A. 1989;86:2863–2867. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inscho EW, Imig JD, Cook AK, Pollock DM. ET(A) and ET(B) receptors differentially modulate afferent and efferent arteriolar responses to endothelin. Br J Pharmacol. 2005;146:1019–1026. doi: 10.1038/sj.bjp.0706412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa K, Yoshizumi M, Tsuchiya K, Houchi H, Minakuchi K, Izawa Y, et al. Dual effects of endothelin-1 (1-31): induction of mesangial cell migration and facilitation of monocyte recruitment through monocyte chemoattractant protein-1 production by mesangial cells. Hypertens Res. 2004;27:433–440. doi: 10.1291/hypres.27.433. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Homma T, Matsuda Y, Kon V. Endothelin receptor subtype B mediates autoinduction of endothelin-1 in rat mesangial cells. J Biol Chem. 1995;270:6997–7003. doi: 10.1074/jbc.270.12.6997. [DOI] [PubMed] [Google Scholar]
- Jaffer FE, Knauss TC, Poptic E, Abboud HE. Endothelin stimulates PDGF secretion in cultured human mesangial cells. Kidney Int. 1990;38:1193–1198. doi: 10.1038/ki.1990.333. [DOI] [PubMed] [Google Scholar]
- Juergens UR, Racke K, Uen S, Haag S, Lamyel F, Stober M, et al. Inflammatory responses after endothelin B (ETB) receptor activation in human monocytes: new evidence for beneficial anti-inflammatory potency of ETB-receptor antagonism. Pulm Pharmacol Ther. 2008;21:533–539. doi: 10.1016/j.pupt.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Karet FE, Davenport AP. Localization of endothelin peptides in human kidney. Kidney Int. 1996;49:382–387. doi: 10.1038/ki.1996.56. [DOI] [PubMed] [Google Scholar]
- Kaw S, Hecker M, Vane JR. The two-step conversion of big endothelin 1 to endothelin 1 and degradation of endothelin 1 by subcellular fractions from human polymorphonuclear leukocytes. Proc Natl Acad Sci U S A. 1992;89:6886–6890. doi: 10.1073/pnas.89.15.6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KJ, Dominguez JH. Rapid progression of diabetic nephropathy is linked to inflammation and episodes of acute renal failure. Am J Nephrol. 2010;32:469–475. doi: 10.1159/000320749. [DOI] [PubMed] [Google Scholar]
- Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- Kluth DC. Pro-resolution properties of macrophages in renal injury. Kidney Int. 2007;72:234–236. doi: 10.1038/sj.ki.5002332. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Sakamoto Y, Shibasaki M, Kimura I, Matsuo H. Human alveolar macrophages synthesize endothelins by thrombin. J Immunol. 1997;158:5442–5447. [PubMed] [Google Scholar]
- Kohan DE. Endothelins in the normal and diseased kidney. Am J Kid Dis. 1997;29:2–26. doi: 10.1016/s0272-6386(97)90004-4. [DOI] [PubMed] [Google Scholar]
- Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuc R, Davenport AP. Comparison of endothelin-A and endothelin-B receptor distribution visualized by radioligand binding versus immunocytochemical localization using subtype selective antisera. J Cardiovasc Pharmacol. 2004;44:224–226. doi: 10.1097/01.fjc.0000166260.35099.d5. [DOI] [PubMed] [Google Scholar]
- Lehrke I, Waldherr R, Ritz E, Wagner J. Renal endothelin-1 and endothelin receptor type B expression in glomerular diseases with proteinuria. J Am Soc Nephrol. 2001;12:2321–2329. doi: 10.1681/ASN.V12112321. [DOI] [PubMed] [Google Scholar]
- Mallick NP, Short CD, Hunt LP. How far since Ellis? The Manchester study of glomerular disease. Nephron. 1987;46:113–124. doi: 10.1159/000184325. [DOI] [PubMed] [Google Scholar]
- Mathieson PW. The podocyte as a target for therapies – new and old. Nat Rev Nephrol. 2012;8:52–56. doi: 10.1038/nrneph.2011.171. [DOI] [PubMed] [Google Scholar]
- Maurer M, Wedemeyer J, Metz M, Piliponsky AM, Weller K, Chatterjea D, et al. Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature. 2004;432:512–516. doi: 10.1038/nature03085. [DOI] [PubMed] [Google Scholar]
- Mencarelli M, Pecorelli A, Carbotti P, Valacchi G, Grasso G, Muscettola M. Endothelin receptor A expression in human inflammatory cells. Regul Pept. 2009;158:1–5. doi: 10.1016/j.regpep.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Mishra R, Leahy P, Simonson MS. Gene expression profile of endothelin-1-induced growth in glomerular mesangial cells. Am J Physiol. 2003;285:1109–1115. doi: 10.1152/ajpcell.00105.2003. [DOI] [PubMed] [Google Scholar]
- Montanari A, Biggi A, Carra N, Fasoli E, Calzolari M, Corsini F, et al. Endothelin-A blockade attenuates systemic and renal hemodynamic effects of L-NAME in humans. Hypertension. 2000;35:518–523. doi: 10.1161/01.hyp.35.1.518. [DOI] [PubMed] [Google Scholar]
- Montanari A, Carra N, Perinotto P, Iori V, Fasoli E, Biggi A, et al. Renal hemodynamic control by endothelin and nitric oxide under angiotensin II blockade in man. Hypertension. 2002;39:715–720. doi: 10.1161/hy0202.104399. [DOI] [PubMed] [Google Scholar]
- Morigi M, Buelli S, Angioletti S, Zanchi C, Longaretti L, Zoja C, et al. In response to protein load podocytes reorganize cytoskeleton and modulate endothelin-1 gene: implication for permselective dysfunction of chronic nephropathies. Am J Pathol. 2005;166:1309–1320. doi: 10.1016/S0002-9440(10)62350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigi M, Buelli S, Zanchi C, Longaretti L, Macconi D, Benigni A, et al. Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol. 2006;169:1965–1975. doi: 10.2353/ajpath.2006.051331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;47:1084–1093. doi: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- Nakano D, Pollock DM. Contribution of endothelin A receptors in endothelin 1-dependent natriuresis in female rats. Hypertension. 2009;53:324–330. doi: 10.1161/HYPERTENSIONAHA.108.123687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odoux C, Crestani B, Lebrun G, Rolland C, Aubin P, Seta N, et al. Endothelin-1 secretion by alveolar macrophages in systemic sclerosis. Am J Respir Crit Care Med. 1997;156:1429–1435. doi: 10.1164/ajrccm.156.5.96-11004. [DOI] [PubMed] [Google Scholar]
- Opocensky M, Kramer HJ, Backer A, Vernerova Z, Eis V, Cervenka L, et al. Late-onset endothelin-A receptor blockade reduces podocyte injury in homozygous Ren-2 rats despite severe hypertension. Hypertension. 2006;48:965–971. doi: 10.1161/01.HYP.0000245117.57524.d6. [DOI] [PubMed] [Google Scholar]
- Orisio S, Benigni A, Bruzzi I, Corna D, Perico N, Zoja C, et al. Renal endothelin gene expression is increased in remnant kidney and correlates with disease progression. Kidney Int. 1993;43:354–358. doi: 10.1038/ki.1993.53. [DOI] [PubMed] [Google Scholar]
- Orth SR, Amann K, Gehlen F, Unger L, Wagner J, Raschack M, et al. Adult human mesangial cells (HMCs) express endothelin-B-receptors which mediate endothelin-1-induced cell growth. J Cardiovasc Pharmacol. 2000;36:232–237. doi: 10.1097/00005344-200036051-00069. [DOI] [PubMed] [Google Scholar]
- Ortmann J, Amann K, Brandes RP, Kretzler M, Munter K, Parekh N, et al. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension. 2004;44:974–981. doi: 10.1161/01.HYP.0000149249.09147.b4. [DOI] [PubMed] [Google Scholar]
- Patrignani P, Del Maschio A, Bazzoni G, Daffonchio L, Hernandez A, Modica R, et al. Inactivation of endothelin by polymorphonuclear leukocyte-derived lytic enzymes. Blood. 1991;78:2715–2720. [PubMed] [Google Scholar]
- Pernow J, Franco-Cereceda A, Matran R, Lundberg JM. Effect of endothelin-1 on regional vascular resistance in the pig. J Cardiovasc Pharmacol. 1989;13:205–206. doi: 10.1097/00005344-198900135-00058. [DOI] [PubMed] [Google Scholar]
- Plato CF, Pollock DM, Garvin JL. Endothelin inhibits thick ascending limb chloride flux via ET(B) receptor-mediated NO release. Am J Physiol. 2000;279:326–333. doi: 10.1152/ajprenal.2000.279.2.F326. [DOI] [PubMed] [Google Scholar]
- Pollock DM. Contrasting pharmacological ETB receptor blockade with genetic ETB deficiency in renal responses to big ET-1. Physiol Genom. 2001;6:39–43. doi: 10.1152/physiolgenomics.2001.6.1.39. [DOI] [PubMed] [Google Scholar]
- Rabelink TJ, Kaasjager KAH, Boer P, Stroes EG, Braam B, Koomans HA. Effects of endothelin-1 on renal function in humans. Implications for physiology and pathophysiology. Kidney Int. 1994;46:376–381. doi: 10.1038/ki.1994.284. [DOI] [PubMed] [Google Scholar]
- Radford MG, Jr, Donadio JV, Jr, Bergstralh EJ, Grande JP. Predicting renal outcome in IgA nephropathy. J Am Soc Nephrol. 1997;8:199–207. doi: 10.1681/ASN.V82199. [DOI] [PubMed] [Google Scholar]
- Rebibou JM, He CJ, Delarue F, Peraldi MN, Adida C, Rondeau E, et al. Functional endothelin 1 receptors on human glomerular podocytes and mesangial cells. Nephrol Dial Transplant. 1992;7:288–292. doi: 10.1093/oxfordjournals.ndt.a092130. [DOI] [PubMed] [Google Scholar]
- Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339:1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- Rubinstein I, Gurbanov K, Hoffman A, Better OS, Winaver J. Differential effect of endothelin-1 on renal regional blood flow: role of nitric oxide. J Cardiovasc Pharmacol. 1995;26:S208–S210. [PubMed] [Google Scholar]
- Ruggenenti P, Perna A, Remuzzi G. Retarding progression of chronic renal disease: the neglected issue of residual proteinuria. Kidney Int. 2003;63:2254–2261. doi: 10.1046/j.1523-1755.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Sasaki S, Hirata Y, Imai T, Ando K, Ida T, et al. Production of endothelin-1 by rat cultured mesangial cells. Biochem Biophys Res Commun. 1990;169:462–468. doi: 10.1016/0006-291x(90)90354-p. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, et al. Cloning of a cDNA encoding a non-isopeptide selective subtype of the endothelin receptor. Nature. 1990;348:732–735. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM. Endothelin-1 increases glomerular permeability and inflammation independent of blood pressure in the rat. Hypertension. 2010;56:942–949. doi: 10.1161/HYPERTENSIONAHA.110.156570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM. Endothelin receptor A-specific stimulation of glomerular inflammation and injury in a streptozotocin-induced rat model of diabetes. Diabetologia. 2011;54:979–988. doi: 10.1007/s00125-010-2021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasser JM, Sullivan JC, Hobbs JL, Yamamoto T, Pollock DM, Carmines PK, et al. Endothelin A receptor blockade reduces diabetic renal injury via an anti-inflammatory mechanism. J Am Soc Nephrol. 2007;18:143–154. doi: 10.1681/ASN.2006030208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffrin EL. Vascular endothelin in hypertension. Vascul Pharmacol. 2005;43:19–29. doi: 10.1016/j.vph.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Schmetterer L, Dallinger S, Bobr B, Selenko N, Eichler H-G, Woltz M. Systemic and renal effects of an ETA receptor subtype-specific antagonist in healthy subjects. Br J Pharmacol. 1998;124:930–934. doi: 10.1038/sj.bjp.0701923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MP, Boesen EI, Pollock DM. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu Rev Pharmacol Toxicol. 2007;47:731–759. doi: 10.1146/annurev.pharmtox.47.120505.105134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson MS, Ismail-Beigi F. Endothelin-1 increases collagen accumulation in renal mesangial cells by stimulating a chemokine and cytokine autocrine signaling loop. J Biol Chem. 2011;286:11003–11008. doi: 10.1074/jbc.M110.190793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson MS, Wann S, Mene P, Dubyak GR, Kester M, Nakazato Y, et al. Endothelin stimulates phospholipase C, Na+/H+ exchange, c-fos expression, and mitogenesis in rat mesangial cells. J Clin Invest. 1989;83:708–712. doi: 10.1172/JCI113935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen SS, Madsen JK, Pedersen EB. Systemic and renal effects of intravenous infusion of endothelin-1 in healthy human volunteers. Am J Physiol. 1994;266:411–418. doi: 10.1152/ajprenal.1994.266.3.F411. [DOI] [PubMed] [Google Scholar]
- Spath M, Pavenstadt H, Muller C, Petersen J, Wanner C, Schollmeyer P. Regulation of phosphoinositide hydrolysis and cytosolic free calcium induced by endothelin in human glomerular epithelial cells. Nephrol Dial Transplant. 1995;10:1299–1304. [PubMed] [Google Scholar]
- Speciale L, Roda K, Saresella M, Taramelli D, Ferrante P. Different endothelins stimulate cytokine production by peritoneal macrophages and microglial cell line. Immunology. 1998;93:109–114. doi: 10.1046/j.1365-2567.1998.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirig R, Potapova I, Shaw-Boden J, Tsui J, Rieben R, Shaw SG. TLR2 and TLR4 agonists induce production of the vasoactive peptide endothelin-1 by human dendritic cells. Mol Immunol. 2009;46:3178–3182. doi: 10.1016/j.molimm.2009.05.179. [DOI] [PubMed] [Google Scholar]
- Takeda M, Iwasaki S, Hellings SE, Yoshida H, Homma T, Kon V. Divergent expression of Eta and Etb receptors in response to cyclosporine in mesangial cells. Am J Pathol. 1994;144:473–479. [PMC free article] [PubMed] [Google Scholar]
- Tostes RC, Touyz RM, He G, Chen X, Schiffrin EL. Contribution of endothelin-1 to renal activator protein-1 activation and macrophage infiltration in aldosterone-induced hypertension. Clin Sci. 2002;103:25–30. doi: 10.1042/CS103S025S. [DOI] [PubMed] [Google Scholar]
- Watson AM, Li J, Schumacher C, de Gasparo M, Feng B, Thomas MC, et al. The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia. 2010;53:192–203. doi: 10.1007/s00125-009-1540-3. [DOI] [PubMed] [Google Scholar]
- Webb DJ, Monge JC, Rabelink TJ, Yanagisawa M. Endothelin: new discoveries and rapid progress in the clinic. Trend Pharmacol Sci. 1998;19:5–8. doi: 10.1016/s0165-6147(97)01144-9. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Saleem MA. Nephrin-signature molecule of the glomerular podocyte? J Pathol. 2010;220:328–337. doi: 10.1002/path.2661. [DOI] [PubMed] [Google Scholar]
- Wesson DE, Simoni J, Green DF. Reduced extracellular pH increases endothelin-1 secretion by human renal microvascular endothelial cells. J Clin Invest. 1998;101:578–583. doi: 10.1172/JCI854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SH, Simari RD, Lerman A. The effect of endothelin-1 on nuclear factor kappa B in macrophages. Biochem Biophys Res Commun. 2001;286:968–972. doi: 10.1006/bbrc.2001.5485. [DOI] [PubMed] [Google Scholar]
- Wright CD, Cody WL, Dunbar JB, Jr, Doherty AM, Hingorani GP, Rapundalo ST. Characterization of endothelins as chemoattractants for human neutrophils. Life Sci. 1994;55:1633–1641. doi: 10.1016/0024-3205(94)00330-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Hirohama T, Uemura H. Endothelin B receptor-like immunoreactivity in podocytes of the rat kidney. Arch Histol Cytol. 2002;65:245–250. doi: 10.1679/aohc.65.245. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Inoue A, Ishikawa T, Kasuya Y, Kimura S, Kumagaye S, et al. Primary structure, synthesis, and biological activity of rat endothelin, an endothelium-derived vasoconstrictor peptide. Proc Natl Acad Sci U S A. 1988a;85:6964–6967. doi: 10.1073/pnas.85.18.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988b;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Ishizaki Y, Sasaki T, Murota S. Effect of carbon dioxide and oxygen on endothelin production by cultured porcine cerebral endothelial cells. Stroke. 1991;22:378–383. doi: 10.1161/01.str.22.3.378. [DOI] [PubMed] [Google Scholar]
- Zoja C, Cattaneo S, Fiordaliso F, Lionetti V, Zambelli V, Salio M, et al. Distinct cardiac and renal effects of ETA receptor antagonist and ACE inhibitor in experimental type 2 diabetes. Am J Physiol. 2011;301:1114–1123. doi: 10.1152/ajprenal.00122.2011. [DOI] [PubMed] [Google Scholar]
- Zouki C, Baron C, Fournier A, Filep JG. Endothelin-1 enhances neutrophil adhesion to human coronary artery endothelial cells: role of ET(A) receptors and platelet-activating factor. Br J Pharmacol. 1999;127:969–979. doi: 10.1038/sj.bjp.0702593. [DOI] [PMC free article] [PubMed] [Google Scholar]