

Abstract
P/Q-type calcium channels are high-voltage-gated calcium channels contributing to vesicle release at synaptic terminals. A number of neurological diseases have been attributed to malfunctioning of P/Q channels, including ataxia, migraine and Alzheimer's disease. To date, only two specific P/Q-type blockers are known: both are peptides deriving from the spider venom of Agelenopsis aperta, ω-agatoxins. Other peptidic calcium channel blockers with activity at P/Q channels are available, albeit with less selectivity. A number of low molecular weight compounds modulate P/Q-type currents with different characteristics, and some exhibit a peculiar bidirectional pattern of modulation. Interestingly, there are a number of therapeutics in clinical use, which also show P/Q channel activity. Because selectivity as well as the exact mode of action is different between all P/Q-type channel modulators, the interpretation of clinical and experimental data is complicated and needs a comprehensive understanding of their target profile. The situation is further complicated by the fact that information on potency varies vastly in the literature, which may be the result of different experimental systems, conditions or the splice variants of the P/Q channel. This review attempts to provide a comprehensive overview of the compounds available that affect the P/Q-type channel and should help with the interpretation of results of in vitro experiments and animal models. It also aims to explain some clinical observations by implementing current knowledge about P/Q channel modulation of therapeutically used non-selective drugs. Chances and challenges of the development of P/Q channel-selective molecules are discussed.
Keywords: ion channels, calcium channels, peptide toxins, calcium antagonists, drug discovery, high-throughput screen
Introduction
The P/Q-type calcium channel (also referred to as Cav2.1) is a presynaptic high-voltage-gated calcium channel, which couples neuronal excitation to secretion of neurotransmitter (Ishikawa et al., 2005). The ion-conducting pore is formed by four domains of the α1A subunit, whereas accessory subunits (β, α2δ) modulate channel kinetics and the level of expression. P-type currents were first identified in Purkinje neurons of the cerebellum (Llinás et al., 1989) and are distinguished from Q-type currents identified in cerebellar granule neurons (Randall and Tsien, 1995). Both are characterized by their sensitivity to the venom of Agelenopsis aperta, ω-agatoxin IVA (Mintz et al., 1992a), and are generated by ion channels encoded by the CACNA1A gene. A number of splice variants may explain different phenotypic characteristics of P- and Q-type channels (Bourinet et al., 1999). For convenience and because distinction between these channel subtypes is not always clear, we refer throughout this review to P/Q-type channels. Expression of P/Q-type channels often overlaps with its close analogue, the N-type calcium channel. Yet, the P/Q-type channel is preferably expressed in neurons of the CNS (Bourinet et al., 1999), making it an interesting target for therapeutics addressing neurological disorders.
A number of conditions have been related to P/Q-type channels, some linked by human mutations occurring in familiar inherited diseases (Kisilevsky and Zamponi, 2008). Familiar hemiphlegic migraine is an example of a disorder with altered P/Q-type activity. Here, different mutations in the CACNA1A gene lead to altered calcium influx, possibly causing cortical spreading depression, which is thought to underlie migraine aura (Plomp et al., 2001; van den Maagdenberg et al., 2004). In contrast, decreased P/Q channel activity may lead to absence epilepsy and ataxia (Ophoff et al., 1998). It has recently been shown that amyloid-β (Aβ) oligomers directly increase the recombinant P/Q-type calcium current, and it has been suggested that such modulation can lead to excitotoxic neurodegeneration in Alzheimer's disease (AD; Mezler et al., 2012a). For most of these conditions, there are few or no medications on the market.
In spite of this high, unmet medical need, no specific low molecular weight blockers are known with scaffolds that could serve as structures for lead optimization. This might be due to the fact that the P/Q-type channel is highly homologous to the N-type channel, and that high-throughput assay technology may not successfully deliver specific compounds for lead optimization. Furthermore, development of P/Q-type blockers may be hampered by the fact that peptide tool compounds do not pass the blood–brain barrier, thus do not allow appropriate proof-of-concept studies in animals.
A closer look at the available compound collection may open avenues for drug development, especially when compounds with different biophysical properties are examined for P/Q modulation and may provide clues for a structure–activity relationship. Some proof-of-concept may also come through the interpretation of clinical studies with less specific calcium channel blockers, when taking into account their P/Q-channel activity.
Currently available compounds that carry P/Q-type channel activity can be divided in several groups: (i) Peptidic ion channel blockers deriving from venom of different invertebrate animal species. This group is also host to a subgroup of compounds with two peptides of high selectivity for the P/Q-type channel: the ω-agatoxins. (ii) Low molecular weight compounds that show some efficacy for the P/Q-type channel, but which are not used as therapeutics. (iii) Therapeutics that also affect the P/Q-type channel. Some of those are traditionally named ‘calcium antagonists’ and thought to target the L-type calcium channel. It also comprises some anti-epileptics with P/Q channel activity as well as volatile anaesthetics.
P/Q channels as drug target
Voltage-gated calcium channels (VGCC) are protein complexes that mediate calcium influx in response to membrane depolarization. High threshold VGCC (L-type, P/Q-type, N-type and R-type) are activated by strong depolarization, whereas low threshold calcium channels (T-type) open in response to mild depolarization steps. The topology of the P/Q-type channel is illustrated in Figure 1 (for review, see Pietrobon, 2002). The characteristic of the P/Q-type channel is mainly determined by the α1A subunit, which contains the conducting channel and the voltage sensor. The auxiliary subunits β and α2δ (and sometimes the γ-subunit) occur in most VGCC and influence trafficking or have a regulatory function (Dolphin, 2009). The pore consists of four homologous domains (I–IV), each of which is composed of six transmembrane segments (S1–S6). S4 is thought to be the voltage sensor. The P/Q-type calcium channel is located at axon terminals as well as somatodendritic compartments of central and peripheral neurons, with some preference for the CNS. At presynaptic sites, opening of the channel mediates synaptic vesicle release via an increase in the local calcium concentration. The pore forming subunit of the P/Q-type calcium channel is encoded by the CACNA1A gene, and multiple splice variants exist that are differentially distributed in the CNS.
Figure 1.
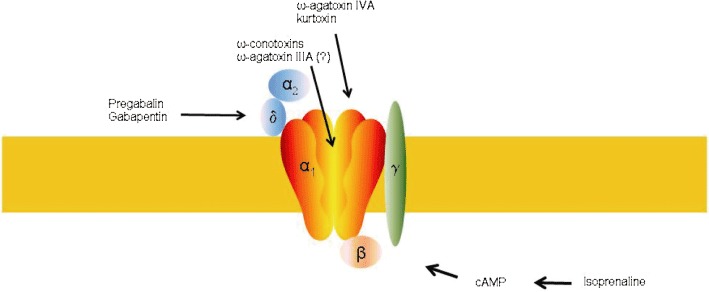
Topology of the P/Q-type channel with potential binding sites of channel modulators. ω-Agatoxin IVA and kurtoxin bind to the outer mouth of the pore forming subunit (linker of the S3–S4 domain), ω-agatoxin IIIA probably to a pore site. ω-Conotoxins bind to the pore region. Pregabalin and gabapentin have been suggested to interact with the α2δ subunit. Isoprenaline probably enhances P/Q currents via second messenger cascades.
Several neurological disorders are caused by mutations in the CACNA1A gene (for review, see Pietrobon, 2010; Rajakulendran et al., 2012). Familiar hemiplegic migraine 1 (FHM1) is a rare, but severe autosomal-dominant subtype of migraine with aura characterized by typical migraine symptoms like unilateral headaches and nausea, but also presents other neurological symptoms such as motor weakness and hemiparesis. Nearly all mutations described in the literature lead to amino acid changes in the α1A subunit, causing a gain-of-function of the P/Q-type calcium channel. A knock-in mouse carrying a FHM1 mutation showed an increased P/Q-type current and a higher susceptibility to cortical spreading depression (van den Maagdenberg et al., 2004; Tottene et al., 2009). The latter is thought to be the pathophysiological correlate of migraine aura. These studies suggest that cortical hyperexcitability may be an underlying cause for the vulnerability of migraine. A key role of cortical spreading depression in migraine pathogenesis has also been derived from human imaging studies (Hadjikhani et al., 2001). Drugs inhibiting cortical spreading depression may thus be candidates for the prophylaxis of migraine. P/Q channel blockade in the CNS will lower neurotransmission and can thus decrease cortical excitability. Several studies have shown that selective P/Q-type channel blockade can prevent spreading depression (Kunkler and Kraig, 2004; Tottene et al., 2011). Taken together, these data encourage the development of P/Q-type channel blockers as a therapeutic strategy for migraine prophylactic treatment.
Mutations in the P/Q-type calcium channel may also lead to a higher susceptibility for epilepsy. Mice with spontaneous mutations in the CACNA1A gene like tottering or learner show patterns of generalized seizures (Fletcher et al., 1996). Mutations in the P/Q-type channel have also been linked to epilepsy in humans (reviewed by Khosravani and Zamponi, 2006), although a robust causal relationship has not been demonstrated. Several types of ataxia have been linked to P/Q-type channel mutations: In episodic ataxia type 2, an acetozolamide-responsive type of generalized ataxia, two mutations have been identified that cause a shift in the open reading frame and result in a truncated α1A subunit. Spinocerebellar ataxia type 6 is a progressive form of ataxia caused by an expansion of the polyglutamate repeat in the C-terminus of the α1A subunit (Zhuchenko et al., 1997). As some of these mutations lead to gain-of-function and others to a lack-of-function of the P/Q-type channel, a single P/Q channel modulator may not be sufficient to treat all P/Q-related disorders.
The P/Q-type calcium channel has also been suggested to contribute to the pathology of AD. It is now an accepted view that Aβ oligomers cause cognitive decline by altering synaptic function in patients with AD. Early studies using non-specific Aβ peptides reported that application of Aβ to neurons causes an increase in calcium currents (He et al., 2002; Rovira et al., 2002). I.c.v. injection of Aβ peptides caused disturbance of synaptic plasticity in rats, which was reversed by calcium antagonists (Freir et al., 2003). Several publications then showed that application of Aβ peptides to neurons increased N and P/Q-type calcium currents (MacManus et al., 2000; Ramsden et al., 2002). We recently tested the effect of Aβ oligomers on recombinantly expressed P/Q-type calcium currents in Xenopus oocytes and showed that the α1A subunit of the channel was specifically modulated, leading to an increased calcium influx. It was speculated that this increase might cause excitotoxicity and lead to synaptic decline in AD (Mezler et al., 2012a). The view that Aβ protein interacts with presynaptic calcium channels in AD patients was supported by the observation that Aβ oligomers co-localize with axon terminals in AD brains (Kokubo et al., 2005; Noguchi et al., 2009). It has also been shown that endogenous Aβ increases the frequency of EPSCs (Abramov et al., 2009), indicating an up-regulation of presynaptic function by amyloid protein.
P/Q-type channels have also been discussed as a drug target for pain (Yakash, 2006; Lewis et al., 2012). Although P/Q-type channels contribute to neurotransmission at nociceptive synapses (Heinke et al., 2004), and efficacy in pain models has been reported (Nebe et al., 1997), N-type channels are likely to be the preferred target for this therapeutic area (for review, see Lewis et al., 2012).
The P/Q-type channel is widely expressed in the CNS. Its general expression in all brain areas – especially in the cerebellum – may be a challenge for drug development, as P/Q blockade in the cerebellum may cause gait and movement disturbances. Indeed, P/Q channel knock-out mice exhibit symptoms of ataxia and dystonia (Jun et al., 1999; Fletcher et al., 2001). Addressing a particular splice variant expressed in the brain region of interest could be a sophisticated approach for drug development to bypass the effects of the cerebellum. Bourinet et al. (1999) identified a number of splice variants with different pharmacological properties. A larger group of splice variants was later identified and exemplifies the diversity of P/Q channel variation (Soong et al., 2002). The challenge would be the identification of a compound with sufficient selectivity for a given splice variant. So far, there is no detailed expression map of the various isoforms available that would support a particular splice variant as drug target. Variant α1A-b shows preferential expression in hippocampal areas (Bourinet et al., 1999) and could be an interesting target for development of compounds against AD. Yet, its full pattern of CNS distribution is not known. A second approach, which is increasingly implemented in drug discovery, is the development of state-dependent therapeutics. These molecules are designed to preferably bind to the inactivated state of the channel and thus are thought to target channels at overactive synapses (and thus only under pathological conditions), while sparing normal synapses. A compound with high level of state-dependency may be favourable, particularly for the indications migraine and epilepsy, where the pathophysiology involves prolonged depolarization of the membrane over seconds or minutes. Other conditions like pain may benefit from use-dependent compounds, which do not block the channel at normal firing patterns, but instead bind to the channel during high-frequency firing. In the pharmaceutical development of ion channel blockers it is now state-of-the-art to strive for a high level of state- or use-dependence in order to increase the therapeutic window. We recently described a high-throughput screening assay with a subsequent electrophysiological secondary screening, which was designed to identify state-dependent P/Q-type channel blockers (Mezler et al., 2012b). Whether these approaches will actually reduce the number and intensity of adverse effects in humans has yet to be shown in clinical trials.
In contrast, some compounds bind to the open state of the channel and thereby delay its deactivation. These drugs lead to a facilitation of calcium influx and may be beneficial in certain types of ataxia, where calcium entry through the P/Q channel is diminished.
Here, we have attempted to give an overview on the available compounds with P/Q channel modulating activity. Only two peptide toxins are selective for P/Q-type channels, the majority of the compounds described are non-selective and often more potent for other targets.
ω-Agatoxins
Spider venoms are a rich source of ion channel blockers. Agatoxins comprise a group of toxins from the American funnel web spider A. aperta that target different classes of ion channels (Adams, 2004). Table 1 summarizes the agatoxins with P/Q channel activity. Two toxins out of this venom screen are specific for P-type channels (i.e. ω-agatoxin IVA and ω-agatoxin IVB). Both peptides share the same specificity and affinity for P-type currents, but seem to exhibit different kinetics (Adams et al., 1993). ω-Agatoxin IVA blocks P-type channels in rat Purkinje neurons with an IC50 of 2–10 nM and only marginally affects other currents (Mintz et al., 1992a, b). ω-Agatoxin IVA blocks Q-type channels less effectively than P-type channels, probably due to different spice variants encoding each subtype (Bourinet et al., 1999). The cloned α1A subunit may reflect the Q-type channel, which would explain the finding that the recombinant α1A is much less sensitive to ω-agatoxin IVA than native P-type currents. Sather et al. (1993), for example, revealed that recombinant α1A channels are 100-fold less sensitive to ω-agatoxin IVA than P-type channels of rat cerebellar Purkinje neurons. Less sensitivity of recombinant α1A channels for ω-agatoxin IVA was also described by other authors (Stea et al., 1994; Bourinet et al., 1999). ω-Agatoxin IVA shifts the activation curve to more positive potentials, indicating that it alters gating of the channel (Winterfield and Swartz, 2000; McDonough et al., 2002). Strong depolarization steps remove the toxin from the channel (Mintz et al., 1992b), indicating that the affinity of the toxin is low for the open state of the channel. In contrast to other calcium channel blockers, ω-agatoxin IVA binds outside of the pore region of the α1A subunit (Winterfield and Swartz, 2000), which may explain its selectivity compared with pore blockers. The ω-agatoxin IVA receptor has been localized to the S3–S4 linker, which is also a binding site for gating modifier molecules on K+ and Na+ channels (Rogers et al., 1996; Li-Smerin et al., 2000). It has been suggested that either the hydrophobic C-terminal part of the peptide (Kim et al., 1995) or charged residues in the mid-part region (Adams et al., 1993) mediate activity. ω-Agatoxin-IVB is the second specific blocker of P-type currents in cerebellar Purkinje neurons with a KD of 3 nM, and no effect on T-type, L-type or N-type calcium channels (Adams et al., 1993). Also, similar to ω-agatoxin IVA, its release from the channel is strongly increased by large depolarizations steps. The only difference between the toxins is the kinetics (block by ω-agatoxin-IVB develops eightfold slower and is also reversed more slowly during washout; Adams et al., 1993). The three-dimensional solution structure of both peptides has been determined by NMR: both are composed of 48 amino acids internally connected by four disulfide bonds (Adams et al., 1993; Kim et al., 1995). In contrast to these specific peptides, ω-agatoxin-IIIA has high affinity to all presynaptic calcium channels (N, P/Q and R) in the low picomolar range (Yan and Adams, 2000). Functionally, it exhibits only a partial block by decreasing single-channel conductance (McDonough et al., 2002). It also blocks L-type channels (Mintz et al., 1991; Ertel et al., 1994).
Table 1.
Reported peptide blockers with P/Q-type channel activity (in alphabetical order)
| Compound | P/Q channel activity | Activity on other channels | Reference | Species |
|---|---|---|---|---|
| Calcicludine | Complete block at 10 nM (native P-type current) to slight block at 100 nM (recombinant P/Q channel) | Block of N and L-type channels at 25–250 nM | Schweitz et al., 1994; Stotz et al., 2000 | Dendroaspis angusticeps |
| DW13.3 | IC50= 4.3 nM | Blocks N-type channels with an IC50 of 14.4 nM, L-type channels with 26.8 nM and R-type channels 96.4 nM | Sutton et al., 1998 | Filistata hibernalis |
| Kurtoxin | 50% inhibition of initial current amplitude (KD= 14 nM), but facilitation of steady-state current | Block of N, L and T-type currents (KD 456, 72 and 49 nM respectively) | Sidach and Mintz, 2002 | Parabuthus transvaalicus |
| Phonetoxin IIA | >70% block at 10 nM | Full block of N-type currents at 3.5 nM, 20% block of R-type currents (17 nM) | Dos Santos et al., 2002 | Phonoetrica nigriventer |
| PnTx3-6 | IC50= 263 nM | IC50 for N-type channel 136 nM, R-type channel 607 nM and L-type channel 122 nM | Vieira et al., 2005 | Phonoetrica nigriventer |
| SNX482 | partical block at 300 nM | R-type complete block at 200 nM (Bourinet et al., 2001); partial block of Na channels at 500 nM | Arroyo et al., 2003 | Hysterocrates gigas |
| ω-agatoxin-IIIA | KD= 9 pM | N-type, R-type (KD= 5–9 pM) | Yan and Adams, 2000 | Agelenopsis aperta |
| ω-agatoxin-IVA | IC50= 2–1000 nM | – | Mintz et al., 1992a, b; Sather et al., 1993; Stea et al., 1994; Bourinet et al., 1999; Hans et al., 1999 | Agelenopsis aperta |
| ω-agatoxin-IVB | KD= 3 nM, complete block at 800 nM | – | Adams et al., 1993 | Agelenopsis aperta |
| ω-conotoxin CVIB | IC50= 23 nM | Blocks N-type channel with an IC50 of 23 nM | Motin et al., 2007 | Conus catus |
| ω-conotoxin MVIIC | IC50 <0.5 µM | Blocks N-type channels with an IC50 of 18 nM | Sather et al., 1993; McDonough et al., 1996 | Conus magus |
| ω-Grammtoxin-SIA | complete block at 50 nM | complete block of N-type current at 500 nM, binding to the drkl K+ channel | McDonough et al., 1997; Takeuchi et al., 2002 | Grammostola spatulata |
| ω-Lsp-IA | Partial block at 10 nM | – (?) | Pluzhnikov et al., 2007 | Geolycosa sp. |
| ω-PnTx3-3 | 79% block at 60 nM | 45% block of L-type current at 80 nM | Leão et al., 2000 | Phonoetrica nigriventer |
References in column 4 report activities on the P/Q-type channel. Activites on other targets are reported in the same references, unless explicitly stated in column 3. The peptide toxins were originally isolated from venom of the species stated in column 5.
Other spider toxins
P/Q blockers have been isolated from a number of spider venoms beyond A. aperta. ω-Grammotoxin SIA was purified from the venom of the tarantula spider Grammostola spatulata (Lampe et al., 1993). It affects both N- and P/Q-type calcium channels (McDonough et al., 1997). Isolated P-type currents in rat cerebellar Purkinje neurons were completely blocked by 50 nM ω-grammotoxin SIA, and this effect seems to be through a modification of channel gating. Resting states are stabilized by the toxin (McDonough et al., 1997). Channel binding has been suggested to occur through a hydrophobic patch of the surface of ω-grammotoxin SIA, but seems not to be restricted to calcium channels (e.g. low affinity binding to K channels; Takeuchi et al., 2002). A peptide homologous to ω-grammotoxin SIA, SNX482, is the 41-amino-acid toxin of the African tarantula Hysterocrates gigas, which has been found to block P/Q channels as well as sodium channels (Arroyo et al., 2003), in addition to its earlier demonstrated effect on R-type currents (Bourinet et al., 2001).
ω-PnTx3-3, a peptide derived from the South American ‘armed’ spider Phoneutrica nigriventer, inhibits most of the isolated P/Q-type current at 60 nM in cerebellar granule neurons, but is also effective for N and L-type currents (Leão et al., 2000). A second toxin was isolated from this spider (i.e. phonetoxin IIA) (Cassola et al., 1998). This toxin is large with 76 amino acids and has some similarity to ω-agatoxin-III family. It irreversibly blocks recombinant P/Q- and N-type currents, and partly inhibits R-type currents (Dos Santos et al., 2002). A third toxin from P. nigriventer, PnTx3-3, blocks L, P/Q, R and N-type channels (Vieira et al., 2005). P/Q currents recombinantly expressed in cell lines were blocked with an IC50 of about 200 nM. A novel 47-amino-acid peptide toxin, ω-Lsp-IA, was recently identified in the venom of a Geolycosa sp.; it attenuates activation kinetics at 10 nM in cerebellar Purkinje neurons and has been suggested to be specific for P/Q-type currents (Pluzhnikov et al., 2007).
DW13.3 is a 74-amino-acid toxin derived from Filistata hibernalis, which blocks all recombinant α1A-E currents in Xenopus oocytes (Sutton et al., 1998), most potently the α1A channel with an IC50 of 4.3 nM. It was also observed to block ω-Agatoxin IVA-sensitive currents in cerebellar Purkinje neurons (saturation at 32–100 nM).
At this point, one may raise the question why particularly arachnids rely on P/Q channel block for prey capture and defence. Do insects have ion channels that are particularly sensitive to P/Q-modulating toxins? Many spiders hunt insects and ω-agatoxin IVA-sensitive currents have indeed been shown to occur in various insect species (Benquet et al., 1999). Furthermore, functional P/Q-like currents can even be recorded in species as low as nematodes (Caenorhabditis elegans; Mathews et al., 2003). It is possible that spiders rely on P/Q blockade to reach a larger spectrum of invertebrate animals.
ω-Conotoxins
Conotoxins are peptidic toxins derived from venomous marine cone snails. Each of the 500 Conus species expresses approximately 100 different conopeptides, so that a pool of more than 50 000 pharmacologically active compounds may exist (Terlau and Olivera, 2004). Most conopeptides target ion channels, some of them with high specificity, and many have been thoroughly used as research tools. ω-Conotoxins target calcium channels and largely derive from fish-hunting cone snails. A derivative of a Conus magus peptide ω-conotoxin MVIIA (an N-type specific inhibitor) is now clinically used under the name Prialt® (ziconotide, SNX-111) as a therapeutic for chronic pain. A number of other conopeptides are in clinical development, including an N-type channel blocker (ω-conotoxin CVID) in phase II (Han et al., 2008). Medicinal chemistry efforts have enabled cyclization of conopeptides to improve bioavailability. Hence, conopeptides may in future be usable for oral drug application, opening further avenues for the development of ion channel-selective therapeutics from peptide blockers (Clark et al., 2005).
Table 1 summarizes reported conopeptides with P/Q channel activity. ω-Conotoxin MVIIC, a peptide identified from a cDNA library from the venom gland of C. magus, inhibits calcium currents in cerebellar Purkinje cells with an IC50 between 1 and 10 µM (Hillyard et al., 1992) and also inhibits P-type currents in hippocampal CA1 pyramidal neurons (Hillyard et al., 1992). It also targets the N-type channel but does not affect the L-type channel. P-type current block by ω-conotoxin MVIIC is slower than for N-type channels and also reverses slowly (McDonough et al., 1996). In contrast to agatoxin, ω-conotoxin MVIIC blocks currents generated by the recombinantly expressed α1A subunit in Xenopus oocytes more potently than the native current (70% block by 5 µM; Stea et al., 1994; IC50 < 0.15 µM). However, the block is rather slow (Sather et al., 1993). ω-Conotoxin MVIIC binds to P-type calcium channels with an affinity of 0.5 nM (estimated by McDonough et al., 1996), but the specific P/Q channel blocker ω-agatoxin-IVA cannot prevent binding of ω-conotoxin MVIIC (McDonough et al., 1996). The high content of basic amino acids residues in ω-conotoxins seems to mediate inhibition (Nadasdi et al., 1995), while a mutation of the tyrosine residue at position 13 disrupts binding of the toxin and may be part of the toxin pharmacophore (Nielsen et al., 1999a). In line with its inhibitory properties on presynaptic calcium channels, ω-conotoxin MVIIC completely prevents synaptic transmission of hippocampal CA3 neurons (Wu and Saggau, 1995).
Another ω-conotoxin exhibiting P/Q channel activity is ω-conotoxin CVIB. It reversibly inhibits both N and P/Q-type calcium channels expressed in Xenopus laevis oocytes with an IC50 of about 23 nM. The R-type current is not affected at 200–500 nM. In dorsal root ganglion cells it blocks isolated P/Q-type as well as N-type currents at 100 nM (Motin et al., 2007). This P/Q-type block (but not the N-type block) is irreversible in these cells.
Other peptide toxins
Table 1 gives an overview of peptides with reported P/Q channel activity. Calcicludine, a 60 amino-acid peptide toxin isolated from the venom of the green mamba Dendroaspis angusticeps, blocks L-type currents recombinantly expressed in HEK293 cells but also exhibits some voltage-dependent block of P/Q- and N-type currents at 100 nM (Stotz et al., 2000). In rat cerebellar Purkinje neurons, it blocks P-type currents more potently with an IC50 of 1–5 nM (Schweitz et al., 1994), with a lower IC50 for L- and N-type channel block (10–100 nM). The peptide binds to olfactory bulb membranes with a KD of 15–36 pM. Kurtoxin, derived from the scorpion venom Parabuthus, is interesting because it reduces high threshold calcium currents in thalamic neurons but enhances P-type currents in Purkinje cells (Sidach and Mintz, 2002).
Low molecular weight calcium channel blockers
In drug development, low molecular weight blockers are usually preferred, as they exhibit several advantages over peptide blockers: First of all, compounds can be selected for tissue penetration, distribution and pharmacokinetics. Peptides usually have extremely low penetration of tissue barriers, which is especially important for CNS indications where the blood–brain barrier often prevents accessibility of the target. Thus, small molecules have a higher potential for improvement of structure–activity relationship. Unfortunately, there is no small molecule known, which is specific to P/Q-type channels. Table 2 provides an overview of the low molecular weight compounds with reported activities for P/Q-type channels. Probably the best-examined small molecule P/Q channel modulator is the cycline-dependent kinase (CDK) inhibitor roscovitine (seliciclib). Roscovitine has been tested for clinical efficacy in a phase II cancer trial (Aldoss et al., 2009). Yan et al. (2002) showed that roscovitine enhanced P/Q-type calcium tail currents with an IC50 of about 20 µM in isolated neostriatal interneurons. This effect was the result of slowed deactivation kinetics. P/Q channel modulation was independent of CDK inhibition. Consequently, roscovitine enhances presynaptic vesicle release in cultured neurons. A subsequent study elaborated on the kinetics of this modulation and found that roscovitine slows deactivation of all recombinantly expressed presynaptic calcium channels (P/Q, N and R) in stably transfected cell lines (Buraei et al., 2006), albeit at high concentrations (EC50 for P/Q: 120 µM). Recently, Buraei and Elmslie (2008) showed that R-roscovitine exhibits both agonist and antagonist effects on all presynaptic calcium channels. Agonist properties were observed at a lower concentration (28 µM) than the antagonistic effect (130 µM). The agonism is specific for the stereoisomer and less pronounced for S-roscovitine and is determined by the residue on the C2 position of the molecule. The antagonism by R-roscovitine was state-dependent, with higher potency at depolarized potentials. Such bidirectional regulation has also been described for two anti-epileptic drugs (benidipine and cilnidipine). Interestingly, a bidirectional modulation of ion channels is brought about by a number of conditions, such as changes in the holding potential (Kass, 1987). It is also not restricted to low molecular weight compounds (Koch et al., 2004; Mezler et al., 2012a) and may simply be observed by changing the expression system (Mezler et al., 2012a). Cho and Meriney (2006) reported a 427% attenuation of the deactivation kinetics of calcium currents by roscovitine in Xenopus motorneurons and consequently increased transmitter release. Apparently, the enhancement of the tail current in such a system predominates, perhaps as the tail current comprises most of the total current during the brief time of an action potential. Such enhanced presynaptic function by roscovitine may cause excitotoxicity, as shown in cultured neurons (Monaco and Vallano, 2005).
Table 2.
Reported LMW blockers with P/Q-type channel activity (in alphabetical order)
| Compound | P/Q channel activity | Activity on other targets | Reference |
|---|---|---|---|
| A-1048400 | IC50= 1.3 µM (inactivated state), 16 µM (hyperpolarized state) | N-type: IC50= 0.8–4.1 µM; T-type channel: IC50= 0.9–2.6 µM | Scott et al., 2012 |
| Amlodipine | IC50= 3–11.5 µM | L-type channel (IC50= 1.2–4.2 µM); N-type (IC50= 0.14–7.9 µM) | Furukawa et al., 1999 |
| Antazoline | IC50= 10 µM | Antagonizes NMDA receptors (IC50 4 µM; Milhaud et al., 2002) | Milhaud et al., 2002 |
| Barnidipine | IC50= 13.1–213 µM | L-type channel (IC50= 1.2–3.1 µM); N-type (IC50= 7.1–1370 µM) | Furukawa et al., 1999 |
| Cilnidipine | IC50= 20.8–58.5 µM | L-type channel (IC50= 5.3–12.7 µM); N-type (IC50= 4.2–39.4 µM) | Furukawa et al., 1999 |
| Diltiazem | IC50= 97 µM; IC50= 169 µM; | L-type channel (IC50= 33.3 µM −40 µM; Diochot et al., 1995; Hockerman et al., 2000) | Ishibashi et al., 1995; Hockerman et al., 2000 |
| Dodecylamine | IC50= 2.1 nM | Blocks L-type channels with an IC50 of 100 nM, N-type channels 1.8 µM, R-type channels 2.0 µM | Beedle and Zamponi, 2000 |
| Eliprodil | IC50= 1.9 µM | Antagonizes NMDA receptors with an IC50 of 670 nM and N and L-type currents with an IC50 of 1.48 µM (Biton et al., 1994) | Biton et al., 1995 |
| Flunarizine | IC50= 1.77–11 µM | hERG (IC50= 5.7 nM) Na channels (use-dependent block at 100 nM; Trepakova et al., 2006), T-type (IC50= 0.1–19); L-type channels (IC50 0.1–11 µM), N-type channel 0.8 µM | Geer et al., 1993; Ye et al., 2011 |
| Fluspirilene | IC50= 6 µM | Nanomolar affinity for D2 receptors (Schotte et al., 1996); N-type: 2 µM; 90% block of T-type current at 1 µM (Enyeart et al., 1992) | Sah and Bean, 1994 |
| Gabapentin | IC50= 98 µM; reduces P/Q-type mediate effect on EPSCs at 20 µM; binds to the α2δ subunit; chronic application modulates P/Q-type inactivation kinetics >0.3 µM; attenuates P/Q-mediated noradrenalin release | L-type (partial block at 100 µM); reduces N-type mediate effect on epscs at 20 µM | Dooley et al., 2002; Fink et al., 2002; Kang et al., 2002; Sutton et al., 2002; Oka et al., 2003a,b; Cunningham et al., 2004 |
| Halothane | Partial block at 0.59 mM | Na channels (IC50= 0.75 mM; Rehberg et al., 1996); enhancement of GABAA receptor-mediated chloride currents (Jones et al., 1992); N-type, R-type, L-type: Partial block at 0.59 mM | Kamatchi et al., 1999; Kameyama et al., 1999 |
| Isoflurane | Partial block at 0.7 mM | Na channels (IC50= 0.85 mM; Rehberg et al., 1996); enhancement of GABAA receptor-mediated chloride currents (Jones et al., 1992); N-type, R-type, L-type: Partial block at 0.7 mM | Kamatchi et al., 1999 |
| Isoproterenol | Enhancement of P/Q currents at 15 µM | β-adrenoreceptor (Kd= 1.7 × 10−7; Brown et al., 1976) | Huang et al., 1996; 1998 |
| Lamotrigine | Partial block at 30 µM | Partial block of N-type channels at 30 µM; partial blockade of sodium channels at 1 µM (Stefani et al., 1997) | Stefani et al., 1996a |
| Levetiracetam | Partial block at 100 µM | Partial block of N-type at 100 µM; binds to SV2A (Lynch et al., 2004) | Pisani et al., 2004 |
| LY393615 | IC50= 4 µM | N-type (IC50= 1.9 µM); R-type (IC50= 5.2 µM) | O'Neill et al., 2001 |
| Memantine | 100 µM memantine abolishes P/Q-type channel mediated glutamate release | Blocks NMDA receptor currents (IC50= 0.47–0.93 µM; Bresink et al., 1996); 100 µM memantine abolishes N-type channel mediated glutamate release | Lu et al., 2010 |
| Neuromed 2 | IC50= 4.5 µM | N-type (IC50 0.12–0.16 µM); T-type (81 nM); L-type (IC50 133 µM) | Yamamoto and Takahara, 2009 |
| Neuromed 5 | IC50= 1.659 µM | N-type (13–60 nM); T-type (0.27–0.579 µM); L-type (144 µM) | Yamamoto and Takahara, 2009 |
| Nicardipine | IC50= 21.1–85 µM | L-type (IC50= 9.6–24 µM); N-type (IC50= 59.9 µM-7.6 mM) | Furukawa et al., 1999 |
| Nimodipine | IC50= 200–500 nM (little activity on P/Q detected by Furukawa et al., 1999) | L-type channel (IC50= 1 µM); | Diochot et al., 1995; Mansvelder et al., 1996 |
| NMED-160 (Neuromed 1) | IC50= 0.12–0.65 µM | N-type (IC50= 0.04–0.2 µM); L-type (IC50= 0.3–0.5 µM) | Yamamoto and Takahara, 2009 |
| R-roscovitine | Decreases step current and enhances tail currents (EC50= 120 µM) | Decreases step current and enhances tail currents of N-type and R-type calcium channels with EC50 of 54 µM; inhibits L-type current, K-currents and Na currents without tail enhancement; Inhibitor of cdc2, cdk2, cdk2 and cdk5 (IC50= 0.2–0.7 µM; Meijer et al., 1997) | Buraei et al., 2006; Buraei and Elmslie, 2008 |
| TROX-1 | IC50= 0.4 µM | N and R-type channels (IC50= 0.4 µM) | Abbadie et al., 2010 |
| Verapamil | Full block at 50 µM; IC50= 62 µM | L-type channel (IC50= 4 µM); N-type channel (full block at 40 µM) | Diochot et al., 1995; Ishibashi et al., 1995; Dobrev et al., 1999 |
| α-Eudesmol | IC50= 3.6 µM | N-type current: IC50= 6.6 µM; | Asakura et al., 1999; Horak et al., 2009 |
References in column 4 report activities on the P/Q-type channel. Activites on other targets are reported in the same references, unless explicitly stated in column 3.
A second compound causing current enhancement of P/Q-type calcium currents is the β-adrenoceptor agonist isoprenaline. The effect on P/Q is mediated by a cAMP cascade (Huang et al., 1996). It causes an increase in the excitatory postsynaptic potential in rat amygdale slices, which can be blocked by ω-agatoxin IVA (Huang et al., 1996). A direct enhancement of ω-agatoxin-sensitive calcium currents by 15 µM isoprenaline was subsequently shown (Huang et al., 1998).
Interestingly, a number of NMDA receptor antagonists have P/Q channel activity: Eliprodil has been reported to block P-type currents in cerebellar Purkinje neurons (Biton et al., 1995). The IC50 for P-type channel block was 1.94 µM, which is in the range of the IC50 for N and L-type channels. The block was not state-dependent. The NMDA receptor antagonist antazoline reversibly blocks P/Q-type channels with an IC50 of 10 µM. This block was state-dependent (Milhaud et al., 2002). It has been suggested that such block may contribute to the neuroprotective properties of imidazolines. In this respect, it should be mentioned that the clinically used NMDA receptor blocker memantine also inhibits P/Q-type/N-type currents (Lu et al., 2010). It is assumed that the therapeutic effect of memantine is mediated by a partial block of NMDA-receptor currents, thereby inhibiting excitotoxicity (Rogawski and Wenk, 2003). One may speculate that a presynaptic calcium channel block could contribute to a common silencing of overactive synapses. In this respect, a state-dependent block of presynaptic calcium channels may leave normal transmission unaltered, while buffering tonic glutamatergic transmission.
Dodecylamine is another low molecular weight blocker with activity for P/Q. It inhibits recombinantly-expressed P/Q-type currents with an IC50 of 2.1 µM, but it is not specific (Beedle and Zamponi, 2000). The block is use-dependent and restricted to the open state.
It should be mentioned that ethanol, although at very high concentrations, inhibits P/Q-type currents (Solem et al., 1997). Although the principal pharmacological effect of ethanol is likely to be on other systems in the CNS, it would be worth examining whether alcohol-induced ataxia could be a result of P/Q channel blockade. The possibility that ataxia is caused by P/Q-channel loss-of-function in several genetic models has been well described in the literature.
Calcium channel blockers in development
The development of calcium channel blockers, particularly of the N-type, has been recently inspired by the FDA approval of the peptidic calcium channel blocker ziconotide (Piralt®). This peptide is a synthetic form of ω-conotoxin MVIIA derived from C. magus and blocks N-type calcium channels on nociceptive A-δ and C nerve fibre endings in lamina I and II of the spinal cord dorsal horn. Ziconotide is efficacious in opioid-resistant pain as well as other severe pain states (Pexton et al., 2011). There does not seem to be development of tolerance (Webster et al., 2009), underlining its advantage over opioids especially for non-cancer patients. However, there are multiple issues regarding adverse effects and the route of administration: First of all, ziconotide is a water-soluble and polar molecule with high molecular weight and thus has limited tissue penetration. Systemic administration inhibits noradrenaline release at sympathetic neurons and therefore exhibits systemic adverse effects like blood pressure changes. It has little effect on parasympathetic nerves (Wermeling, 2005). Thus, intrathecal administration is mandatory. Yet, even with this application route, there are common central side effects like memory impairment, dizziness or speech disorders, leading to dropout rates of up to 39% in clinical studies (Ellis et al., 2008; Webster et al., 2009). Recently, a number of companies tried to overcome these hurdles by the development of state-dependent, low molecular weight compounds with improved pharmacokinetic and side effect profiles. Small molecules would be accessible to chemical optimization processes for improving structure–property relationships (SPR), facilitating oral availability and tissue distribution. A recent trend is the development of state-dependent channel blockers that are designed to only inhibit voltage-gated ion channels at inactivated state. It is thought that these molecules prevent excessive neurotransmission of cells under pain conditions, while leaving normal synaptic function unaltered. Thus, there is the hope for small molecule calcium channel blockers that can be applied systemically with a low side effect profile. These improvements, however, were accompanied by lack of selectivity, especially against the P/Q-type channel (Yamamoto and Takahara, 2009). As a result, most small molecule ‘N-type channel blockers’ in pharmaceutical development are mixed N-P/Q-type blockers (see Table 2 for an overview). Neuromed Pharmaceuticals recently disclosed their compound NMED-160, which blocks N-type and P/Q-type channels in the low nanomolar range. NMED-160 is the only small molecule molecular weight blocker in clinical trials [Neuromed, Merck give up on new pain drug. Philadelphia Business Journal. August 2007. Available from: http://wwwizjournalscom/philadelphia/stories/2007/08/06/daily17html (Last access April 2011)]. Channel block by NMED-160 seems to be use-dependent (McNaughton et al., 2008), which should provide an advantage over non-state-dependent peptides. A number of compounds have been developed fairly recently, some of which have been shown to affect P/Q-type channels (Neuromed 2–7; reviewed by Yamamoto and Takahara, 2009). Merck developed a substituted N-triazole oxindole (TROX-1), which is orally available and showed efficacy in a number of pain models (Abbadie et al., 2010). TROX-1 is potent and state-dependent (IC50= 400 nM). However, in dorsal root ganglion cells, it blocks all CaV2 channels, including the P/Q-type channel, and it also inhibits recombinantly expressed P/Q channels with a potency similar to that for the N-type channel. Elli Lilly published an N-type blocker (LY393615) with an IC50 for N-type channels of 1.9 µM (recombinantly expressed in HEK293 cells), which blocks P/Q channels with similar potency (IC50 for P/Q: 4 µM in isolated Purkinje cells; O'Neill et al., 2001). Abbott Laboratories recently published a state-dependent, orally available calcium channel blocker, which does not affect the L-type calcium channel (A-1048400; Scott et al., 2012). The IC50 for the P/Q-type channel is 16.3 µM at a hyperpolarized state and 1.3 µM at an inactivated state. This compound also potently blocks N-type, R-type and T-type channels (IC50 at inactivated state: 0.8, 0.9 and 1.6 µM respectively). Current drug discovery efforts focusing on the discovery of P/Q-type channel blockers for CNS disorders may provide us with more selective, small molecule blockers for P/Q-type channels (Mezler et al., 2012b).
Clinically used therapeutics that block calcium channels
A number of therapeutics modulate P/Q channels, although the therapeutic effect is thought to be mediated by other targets. A precise understanding of the respective target profile is often missing and may be valuable for the development of more selective compounds with fewer adverse effects. Table 3 gives an overview of clinically used compounds with P/Q-type channel activity.
Table 3.
Clinically used compounds with P/Q channel activity (in alphabetical order)
| Compound | Primary indication(s) | Suggested primary target mediating clinical efficacy | Reference |
|---|---|---|---|
| Amlodipine | Hypertension, angina pectoris | L-type channel | Haria and Wagstaff, 1995; Scholz, 1997 |
| Barnidipine | Hypertension | L-type channel | Liau 2005 |
| Diltiazem | Hypertension, angina pectoris, cardiac arrhythmias | L-type channel | McAuley and Schroeder, 1982 |
| Flunarizine | Migraine | VGCC, Na+ channels | Amery, 1983; Ye et al., 2011 |
| Fluspirilene | Schizophrenia | Dopamine D2 receptors | Galizzi et al., 1986 |
| Gabapentin | Epilepsy, neuropathic pain | α2δ subunit of VGCC | Striano and Striano, 2008 |
| Halothane | Inhalation anaesthesia | Multiple modes of action | Krnjević, 1992 |
| Isoflurane | Inhalation anaesthesia | Multiple modes of action | Krnjević, 1992 |
| Isoprenaline | Bradycardia, heart block, asthma | β-adrenoceptor | Ahlquist, 1976 |
| Lamotrigine | Epilepsy | Na+ channels, VGCC | Rogawski and Löscher, 2004; Elger and Schmidt, 2008 |
| Levitiracetam | Epilepsy | VGCC, SV2 | Elger and Schmidt, 2008 |
| Memantine | Alzheimer's disease | NMDA receptor | Rogawski and Wenk, 2003 |
| Mibefradil | Hypertension | T-type channel | Glasser, 1998 |
| Nicardipine | Hypertension | L-type channel | Pepine and Lambert, 1990 |
| Nimodipine | Cerebral vasospasm after subarachnoid haemorrhage | L-type channel | Tomassoni et al., 2008 |
| Verapamil | Cardiac arrhythmias, hypertension, angina pectoris | L-type channel | Rosen et al., 1975; Scholz, 1997 |
Listed are the primary indications as well as targets suggested to mediate the therapeutic effect. References in column 4 report the pharmacological mechanism suggested to mediate the therapeutic effect.
Calcium antagonists
In 1964, Albrecht Fleckenstein showed that verapamil mimics the effect of Ca2+ removal on electrically-stimulated guinea pig papillary muscle (Fleckenstein-Grün, 1994). He created the name ‘calcium antagonists’ to separate the principle as an alternative to β-receptor blockade and confirmed the idea of calcium channel blockade by voltage-clamp analysis. Shortly afterwards, Bayer AG developed a highly potent calcium channel blocker, Bay a 1040, which was later named nifedipine. In the following years, a large number of calcium antagonists with distinct properties were identified by the pharmaceutical industry, belonging to different classes: benzothiazepines and phenylalkylamines. Some, like verapamil, have inotropic, chronotropic and dromotropic effects besides their vasodilatator properties, whereas nifedipine was largely a vasodilator. Calcium antagonists are used clinically for the treatment of hypertension, coronary heart disease and cardiac arrhythmia. Their principal mode of action is the inhibition of L-type calcium channels in smooth muscle cells (including those of coronary arteries), leading to a block of excitation–contraction coupling and a relaxation of the vasculature. They also inhibit L-type channel-mediated calcium influx into cardiomyocytes and thus inhibit the cardiac action potential. The cardiac pacemaker activity may be brought about by the inhibition of calcium channels (including the T-type channel) in the sinoatrial node as well as the atrioventricular node. After identification and cloning of other VGCC, many therapeutically used calcium antagonists have been evaluated for efficacy on these channels, and it has become clear that many calcium antagonists are not selective for the L-type channel (Fujii et al., 1997; Furukawa et al., 1997).
For example, verapamil, nicardipine and nimodipine block ω-conotoxin GVIA-insensitive and ω-agatoxin IVA-sensitive currents in dorsal root ganglion cells, indicating N- and P/Q-blockade (Diochot et al., 1995). Effective concentrations were in the micromolar range and several-fold higher than for L-type block. Verapamil and diltiazem block P-type currents in cerebellar Purkinje neurons (Ishibashi et al., 1995), and diltiazem may shift the P-type inactivation curve. It was later shown that verapamil blocks P-type currents as well as other high voltage-gated calcium currents in rat striatal slices. Diltiazem also blocks P-type currents in this system (Dobrev et al., 1999). Diltiazem also blocks P/Q-type channels recombinantly expressed in HEK293 cells, although it is fivefold less potent than on L-type channels (Hockerman et al., 2000). Interestingly, when P/Q-type channels containing a nine-amino acid sequence specific for the dihydropyridine binding site were expressed, diltiazem reached the same potency at P/Q-type channels as at L-type channels. Mansvelder et al. (1996) published a study showing that ω-conotoxin GVIA and ω-Agatoxin IVA-sensitive currents are blocked in rat melanotropic cells by the two dihydropyridines nimodipine and nitrendipine with an IC50 of 200–500 nM. This indicates that both nimodipine and nitrendipine affect N- and P/Q-type currents with appreciable potencies.
A comparative study with 10 dihydropyridines was performed on different calcium channels recombinantly expressed in Xenopus oocytes (Furukawa et al., 1999). Nifedipine, nilvadipine, barnidipine, nimodipine, nitrendipine, amlodipine, nicardipine, benidipine, felodipine and cilnidipine all showed appreciable block of L-type channels at 10 µM. Of these, amlodipine, benidipine, cilnidipine, nicardipine and barnidipine also blocked P/Q- and N-type calcium channels. The P/Q channel block by amlodipine, nicardipine and barnidipine was voltage-dependent. Amlodipine was most potent with an IC50 of 3 µM at depolarized states (vs. 11.5 µM at hyperpolarized state). Similar potencies were observed for benidipine, cilnidipine and barnidipine. Some calcium antagonists, like cilnidipine, exhibited a similar block of L-, P/Q- and N-type currents at 10 µM. In contrast to the two studies on native currents described above (Diochot et al., 1995; Mansvelder et al., 1996), nimodipine had only a minor and nitrendipine no effect on P/Q-type channels. An explanation for this contrasting result could be the use of recombinant versus native test systems.
Taken together, these data indicate that several classes of therapeutically used ‘calcium antagonists’ are not specific to the L-type channel of smooth muscle, but also affect other calcium channels including the P/Q-type channel. Further studies need to examine whether their activity on N- and P/Q-type calcium channels explains some of the clinical findings, especially the efficacy in some neurological disorders discussed below.
We recently reported that Aβ globulomer, an oligomeric peptide with a toxic epitope found in AD patients, increases P/Q-type calcium currents recombinantly expressed in Xenopus oocytes (Mezler et al., 2012a). A similar increase in P/Q-type channel activity has also been observed by Ramsden et al. (2002) and MacManus et al. (2000) in cultured neurons, albeit with less relevant Aβ preparations. It has been suggested Aβ oligomers enhance calcium channel flux through P/Q-type channels. Tonically overactive P/Q-type channels at central synapses may cause excessive glutamate release in affected brain regions, leading to excitotoxic cell death (Mezler et al., 2012a). Such neuronal decline should be prevented by P/Q-type channel block. The neuroprotective effect of P/Q-type channel blockers has been thoroughly described in the literature (e.g. Small et al., 1995; Asakura et al., 1997). A few reports state that nimodepine is protective against AD (Tollefson, 1990; Grobe-Einsler and Traber, 1992), although the effect is minimal. Some efficacy of nimodipine in dementia trials was also stated in a Cochrane Review (López-Arrieta and Birks, 2002). Clinical improvement of cognitive decline was observed after treatment with nicardipine (Amenta et al., 2009). In a nucleus basalis lesion model in rats, verapamil was efficacious in the behavioural outcome (Popovićet al., 1997).
P/Q block by verapamil may also explain why a particular type of stroke caused by a mutation in the gene for the P/Q-type calcium channel responds to treatment with verapamil (case report: Knierim et al., 2011). This type of recurrent stroke is associated with seizures and may be prevented by P/Q channel inhibition because of a down-regulation of neuronal firing.
A number of calcium antagonists that are not classically related to the L-type channel block, also show P/Q channel activity. Mibefradil is considered to be a selective T-type calcium channel blocker and is used clinically for the treatment of hypertension. It also has been shown to exhibit some activity for the N-type and P/Q-type channel (Viana et al., 1997).
Flunarizine is a mixed sodium and calcium channel blocker clinically used for the treatment of migraine. Flunarizine blocks P-type currents in neocortical slices, thereby preventing potassium-stimulated calcium influx with an IC50 of 11 µM (Geer et al., 1993). In cultured cortical neurons, the calcium channel block was more potent with an IC50 of 1.77 µM, which is similar to the potency at the sodium channel (IC50 0.94 µM; Ye et al., 2011). Certain types of familiar migraines are caused by mutations in the CACNA1A subunit of the P/Q-type channel. Expression of these mutants in transgenic mice leads to enhanced P/Q channel activity and consequently facilitation of cortical spreading depression (Tottene et al., 2009), which is thought to underlie migraine aura. The phenomena of spreading depression can be blocked by ω-agatoxin IVA (Kunkler and Kraig, 2004). It has been shown that flunarizine enhances the threshold for cortical spreading depression (Wauquier et al., 1985), and one may speculate its preventive effect in migraine can at least in part be attributed to block of P/Q-type channels in the brain. Familiar migraines caused by P/Q mutations also seem to respond to verapamil (Yu and Horowitz, 2003), as do other migraine types (Solomon et al., 1983; Markley et al., 1984).
In summary, there is evidence that certain clinically used calcium antagonists show therapeutic benefit for some neurological diseases that might be linked to P/Q-type calcium channels. If the clinical effects can be attributed to their shared efficacy on P/Q channels, an improvement in P/Q channel specificity as well as target availability may improve their efficacy for these diseases and may reduce their side effects. As all the substances discussed above affect multiple targets, none of these structures may serve as a real lead compound for the development of selective P/Q-type channel blockers.
Antiepileptic drugs
Epileptic seizures are generally caused by a shift in the excitation/inhibition balance in cortical networks towards excitation. This involves enhanced neurotransmission at glutamatergic synapses, which is at least in part mediated by P/Q-type channels (Qian and Noebels, 2001). There is increasing evidence that VGCC, including P/Q-type channels, contribute to idiopathic generalized epilepsies (Zamponi et al., 2010). Mutations in P/Q-type calcium channels have also been linked to absence seizures. Some anti-epileptic drugs interact with the α2δ accessory subunit of VGCC (Vohora et al., 2010). For some, a direct P/Q-type current modulation has been shown. Levetiracetam has been demonstrated to inhibit high-voltage-gated calcium currents in hippocampal pyramidal neurons in slices (Niespodziany et al., 2001). It has also recently been shown to block excitatory postsynaptic potentials in granule cells in slices specifically by blocking the P/Q-type calcium current (Lee et al., 2009). Levetiracetam reduces N- and P/Q-type currents in isolated neocortical neurons without affecting sodium currents at 100 µM, a concentration that attenuates the paroxysmal depolarization shift in a neocortical slice model of epilepsy (Pisani et al., 2004). Lamotrigine also inhibits high-threshold voltage-gated calcium currents with an IC50 of 12.3 µM in isolated rat pyramidal neurons (Stefani et al., 1996a), which is attributed to N- and P-type blockade. Older antiepileptic drugs like carbamazepin and oxcarbamazepine are also active on high threshold calcium currents (Stefani et al., 1995; Zhu et al., 2002), and it has been suggested that carbamazepine has P/Q-activity (Zhu et al., 2002). Some anti-epileptics like felbamate also affect high VGCCs, but not the P/Q-type calcium channel (Stefani et al., 1996b), whereas other anti-epileptics have little effect on high-threshold VGCC (phenytoin; Stefani et al., 1997). Valproate does not seem to have any effect on presynaptic calcium channels, even at very high concentrations (up to 1.5 mM; Sitges et al., 2007).
Gabapentin and pregabalin comprise a class of molecules that bind to the α2δ accessory subunit of VGCC with nanomolar affinities (Suman-Chauhan et al., 1993; Gee et al., 1996). It is thus not surprising that there are multiple studies showing a modulation of VGCC by those drugs. Some authors consider them to be selective for VGCC (reviewed by Sills, 2006). Both, gabapentin and pregabalin, at µM concentrations, attenuate neurotransmitter release in cortical slices by inhibition of P/Q-type channels (Dooley et al., 2002). In cortical synaptosomes, gabapentin also blocks the ω-agatoxin IVA-sensitive increase in potassium-induced calcium levels in the µM range (Fink et al., 2002). In addition, it blocks P/Q-type (and N-type) channels in rat cerebrocortical slices with an IC50 (for P/Q) of 98 µM (Oka et al., 2003a). In another study, Oka et al. (2003b) analysed the effect of gabapentin on depolarization-evoked NOS activity in primary cortical neurons. High concentrations of gabapentin (100 µM) reduced depolarization-induced NOS activity by blockade of P/Q-type and L-type (not N-type) calcium channels. Gabapentin reduced presynaptic vesicle release at low µM concentrations, preferably acting via P/Q-type channel block (Cunningham et al., 2004). Gabapentin also reduced EPSCs and IPSCs in spinal cord, with an IC50 of 23 nM, by reducing P/Q-type calcium currents (Bayer et al., 2004). Whole-cell recordings from dorsal root ganglion cells revealed that gabapentin blocks all N-, P/Q- and L-type channels (Sutton et al., 2002), although the P/Q block appears to be the smaller part. Kang et al. (2002), when recording from P/Q channels recombinantly expressed in Xenopus oocytes, found that chronic but not acute treatment with gabapentin induced a dose-dependent decrease in P/Q-type current inactivation. Inactivation kinetics were modified at concentrations starting at 300 nM. In this respect, it should be noted that the α2δ subunit has been shown to modulate calcium channel kinetics (Qin et al., 1998).
As described above, both familiar migraines as well as Aβ pathology involve a gain-of-function in the P/Q-type current. Thus, it may not come as a surprise that some anti-epileptics are efficacious in these conditions. Piracetam (Croisile et al., 1993) and levetiracetam (Cumbo and Ligori, 2010) are also efficacious in AD patients. Treament with levetiracetam has been correlated with improved cognitive performance, whereas piacetam seems to slow down cognitive decline. Similarly, levetiracetam appears to be beneficial in patients with migraine (Pizza et al., 2011). A number of studies also report efficacy of gabapentin as a prophylactic in migraine (e.g. Di Trapani et al., 2000; Mathew et al., 2001), which may be explained by the ability of the calcium channel blocker gabapentin to prevent cortical spreading depression (Hoffmann et al., 2010).
Mood stabilizers
Calcium channel blockers have been suggested to be beneficial in the treatment of bipolar disorder (Levy and Janicak, 2000). While for some drugs of this class the efficacy has been questioned, data for nimodipine have appeared promising (Pazzaglia et al., 1998). This could be attributed to a better brain penetration by nimodipine as opposed to other calcium channel blockers. It is also possible that nimodipine exhibits different modulatory effects on calcium channels. Whether block of P/Q-type calcium channels contributes to the mood stabilizing properties is unclear. Some data suggest that modulation of presynaptic calcium influx may positively influence bipolar disorder. For example, Chen et al. (2010) implicate presynaptic glutamate release in the pathophysiology of bipolar disorder. Patients with bipolar disorder have been shown to have an increased neurotransmission in the anterior cingulate cortex (Eastwood and Harrison, 2010). Animal studies are needed to clarify whether a specific block of P/Q-type channels could ameliorate symptoms of bipolar disorder.
Anaesthetics
Volatile anaesthetics inhibit P/Q-type currents in the higher µM range, although the block is not specific for this channel isoform. Both isoflurane and halothane increase the rate of inactivation in P/Q-type currents recombinantly expressed in Xenopus oocytes (Kamatchi et al., 1999). Isoflurane also inhibits P/Q-type, N-type and L-type calcium currents in dorsal root ganglion cells (Kameyama et al., 1999) and probably in hippocampal pyramidal cells (Study, 1994). Volatile anaesthetics including halothane prevent glutamate release evoked in synaptosomes via a presynaptic mechanism (Schlame and Hemmings, 1995).
It is generally thought that volatile anaesthetics affect multiple targets in the brain with low potency, including GABAA receptors and VGCC. Thus, it is likely that P/Q channel block has – if at all – a minor contribution to the general anaesthetic state.
Antipsychotics
A number of dopamine receptor antagonists exhibit P-type channel activity, unrelated to a specific structure (Sah and Bean, 1994). Diphenylbutylpiperidines have long been known to exhibit calcium channel activity (Gould et al., 1983) and compromise the activity of the most potent calcium channel blockers (Sah and Bean, 1994). They bind to calcium channels with affinities in the low nM range (Gould et al., 1983). Of all these compounds, fluspirilene is the most potent P-type current blocker, with an IC50 of 6 µM. This block is not specific to P-type channels but also affects N-, L- and T-type channels (Sah and Bean, 1994). At a higher concentration (30 µM), most neuroleptic drugs, including chlorpromazine and haloperidol, have considerable activity on P-type currents. However, these concentrations may not be relevant for antipsychotic activity. As some neuroleptics show the ability to act as a calmodulin antagonist, the effect of fluspirilene was studied in the presence of the calcium chelator BAPTA, but this did not change its potency. Its effect was also not mediated by neurotransmitter activation of G-proteins (that are known to modulate P-type channels). However, the block of P-type currents by fluspirilene was voltage- and frequency dependent (Sah and Bean, 1994). Hence, the binding site of fluspirilene seems to be different from that of ω-agatoxin-IVA and also different from that of the pore blocker Cd2+.
Herbal medication
Some plant extracts that are used as prophylactics for migraine (Diener et al., 2004; Lipton et al., 2004) also inhibit P/Q-type channels. α-Eudesmol (Eucalyptus williamsiania) and petasins (Petasites hybridus) state-dependently inhibit recombinant P/Q-type currents (Horak et al., 2009). In rat cerebellar Purkinje cells, eudesmol (Juniperus virginiana) inhibits P/Q-type currents with an IC50 of 3.6 µM (Asakura et al., 1999). However, whether the ability of these substances to affect P/Q-type currents is therapeutically relevant is questionable.
Is there a path for the development of a selective P/Q-type channel blocker?
One of the challenges in drug discovery is the development of a lead compound with sufficient selectivity for the target. In this respect, VGCCs may be at the extreme end of the spectrum, as selectivity for some types appears to be extremely difficult to obtain. Consequently, there are few fully selective calcium channel blockers. For the development of selective P/Q-type channel blockers, sparse information on structure–activity relationships and perhaps limited high-throughput electrophysiogical methods may have hampered the development of the appropriate lead molecule in the past. The pharmaceutical industry has initiated drug discovery programmes for low molecular weight N-type channel blockers, which was inspired by the approval of the selective peptide N-type channel blocker ziconotide for chronic pain. Yet, no small lead molecule with appreciable selectivity for a presynaptic calcium channel has been forwarded into clinical trials.
Recently, some progress has been made by the pharmaceutical industry to separate N- and P/Q-type blockade from L-type channel activity. Abbott Laboratories, for example, recently presented a small lead molecule – A-1048400 – with high potency for the N-, P/Q- and T-type channel, but which is largely devoid of L-type channel activity (Scott et al., 2012). Separation from L-type channel activity was also reached by Neuromed Pharmaceuticals (see Table 2). Anecdotal reports also indicate separation of N and P/Q channel activity. Neuromed have described a number of compounds that show some selectivity for the N-type versus the P/Q-type channel (e.g. Neuromed 5). Beedle and Zamponi (2000) report that a small molecule – dodecylamine – is largely selective for P/Q-type channels. However, these reports do not provide a clear structure–activity relationship for a development path for small P/Q channel blockers.
An alternative would be a development programme based on one of the two selective peptide P/Q-type channel blockers, ω-agatoxin IVA and ω-agatoxin IVB – in analogy to the N-type blocker ziconotide (Schmidtko et al., 2010). For a number of reasons, ω-agatoxins themselves may not be suitable for CNS therapeutics: firstly, their pharmacokinetic properties are not suitable for oral administration and sufficient brain availability is still an illusion. The latter would be required for the treatment of potentially P/Q channel-related disorders like migraine or AD. Medicinal chemistry approaches have recently succeeded in modifying peptide toxins by cyclization to improve their biophysical properties (for review, see Craik and Adams, 2007). Yet, good bioavailability of peptide toxins is still a challenge, and these molecules do not penetrate the blood–brain barrier. Secondly, administration of ω-agatoxin may have strong adverse effects, as it blocks the channel largely irreversibly (unblock occurs only at large depolarizations; Adams et al., 1993). However, structural information from the binding domain of ω-agatoxin IVA and ω-agatoxin IVB may be a basis for the development of selective and more appropriate peptide analogues. The C-terminal domain of ω-agatoxin IVA has been identified as the active peptide part for P/Q channel blockade (Kim et al., 1995). Some information on active parts of the peptide has also been obtained for ω-agatoxin IVB (Adams et al., 1993). Narrowing down an amino acid sequence to the minimal active motif may provide a basis for the development of peptidomimetics with appropriate pharmacokinetic properties. For ω-conotoxins, channel activity has been to some degree attributed to two conserved amino acid residues Tyr13 and Lys2 (Sato et al., 1993; Kim et al., 1994), together with other residues in loop 2 and 4 (for overview, see Lewis et al., 2012). Lys10 and Arg22, as well as a number of positively charged residues in loops 2 and 4, seem to influence subtype selectivity for the P/Q-type channel (Haack et al., 1993; Nielsen et al., 1999b). There have been encouraging reports on N-type peptide mimetics showing that the development of a pharmacophore in drug discovery based on toxin peptide information is principally possible. Baell et al. (2004) generated peptide mimetics for N-type channel blockers based on the peptide information from ω-conotoxin GVIA. One analogue, compound 4a, mimics three side chains of this peptide and potently blocks N-type calcium channels in the micromolar range. It also retains some selectivity over the P/Q-type channel (approx. 20-fold). Parke-Davis (now Pfizer) designed a small-molecule N-type channel blocker mimicking three residues of ω-conotoxin MIIA (Menzler et al., 2000). Further development also resulted in the generation of an orally-available small-molecule blocker with improved physicochemical properties (e.g. Hu et al., 2000).
A different approach to achieving a selective P/Q-type channel blocker is to address the binding site for selective P/Q channel blockers at the channel. For ω-agatoxin IVA, at least (Winterfield and Swartz, 2000), and possibly kurtoxin (Sidach and Mintz, 2002), this site has been localized and is thought to be the S3-S4 linker at the outer mouth of the pore. A displacement assay using a selective ω-agatoxin-based radioligand could be implemented into a high throughput screening. However, this requires the selective small molecules to be actually available in synthetic compound libraries.
Conclusions
The aim of this review was to provide an overview of the vast number of compounds that modulate the P/Q-type channel. Currently, there are only two selective molecules available, which are peptide blockers derived from spider venom. All other compounds discussed here are nonselective, and their activity on other targets is often higher than that on P/Q-type channels. Yet, the knowledge of the distinct profile of each of those compounds is necessary to interpret and design experiments, and perhaps to analyse clinical studies. Knowledge about the spectrum of targets of each of the classical calcium antagonists may also challenge the view that all of the observed effects in animal models and clinical trials are mediated by L-type channel blockade.
Currently, there is not sufficient information on structure–activity relationships available for a focused development of P/Q channel blockers. Recent advances in high-throughput electrophysiological techniques may facilitate screening for small molecules with higher selectivity. Perhaps one may draw hope from peptide chemistry efforts to create P/Q-type specific peptide mimetics with improved pharmacokinetic profiles.
The development of P/Q-type selective tool compounds and lead molecules with sufficient bioavailability and brain penetration will clearly remain a challenge.
Acknowledgments
This review article was supported by Abbott.
Glossary
- Aβ
amyloid-β
- AD
Alzheimer's disease
- CDK
cycline-dependent kinase
- LMW
low molecular weight
- VGCC
voltage-gated calcium channel
Statement of conflicts of interest
None.
References
- Abbadie C, McManus OB, Sun SY, Bugianesi RM, Dai G, Haedo RJ, et al. Analgesic effects of a substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. J Pharmacol Exp Ther. 2010;334:545–555. doi: 10.1124/jpet.110.166363. [DOI] [PubMed] [Google Scholar]
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Adams ME. Agatoxins: ion channel specific toxins from the American funnel web spider, Agelenopsis aperta. Toxicon. 2004;43:509–525. doi: 10.1016/j.toxicon.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Adams ME, Mintz IM, Reily MD, Thanabal V, Bean BP. Structure and properties of omega-agatoxin IVB, a new antagonist of P-type calcium channels. Mol Pharmacol. 1993;44:681–688. [PubMed] [Google Scholar]
- Ahlquist RP. Present state of alpha- and beta-adrenergic drugs I. The adrenergic receptor. Am Heart J. 1976;92:661–664. doi: 10.1016/s0002-8703(76)80086-5. [DOI] [PubMed] [Google Scholar]
- Aldoss IT, Tashi T, Ganti AK. Seliciclib in malignancies. Expert Opin Investig Drugs. 2009;18:1957–1965. doi: 10.1517/13543780903418445. [DOI] [PubMed] [Google Scholar]
- Amenta F, Lanari A, Mignini F, Silvestrelli G, Traini E, Tomassoni D. Nicardipine use in cerebrovascular disease: a review of controlled clinical studies. J Neurol Sci. 2009;283:219–223. doi: 10.1016/j.jns.2009.02.335. [DOI] [PubMed] [Google Scholar]
- Amery WK. Flunarizine, a calcium channel blocker: a new prophylactic drug in migraine. Headache. 1983;23:70–74. doi: 10.1111/j.1526-4610.1983.hed2302070.x. [DOI] [PubMed] [Google Scholar]
- Arroyo G, Aldea M, Fuentealba J, Albillos A, García AG. SNX482 selectively blocks P/Q Ca2+ channels and delays the inactivation of Na+ channels of chromaffin cells. Eur J Pharmacol. 2003;475:11–18. doi: 10.1016/s0014-2999(03)02084-3. [DOI] [PubMed] [Google Scholar]
- Asakura K, Matsuo Y, Kanemasa T, Ninomiya M. P/Q-type Ca2+ channel blocker omega-agatoxin IVA protects against brain injury after focal ischemia in rats. Brain Res. 1997;776:140–145. doi: 10.1016/s0006-8993(97)00975-x. [DOI] [PubMed] [Google Scholar]
- Asakura K, Kanemasa T, Minagawa K, Kagawa K, Ninomiya M. The nonpeptide alpha-eudexp6l from Juniperus virginiana Linn. (Cupressaceae) inhibits omega-agatoxin IVA-sensitive Ca2+ currents and synaptosomal 45Ca2+ uptake. Brain Res. 1999;823:169–176. doi: 10.1016/s0006-8993(99)01165-8. [DOI] [PubMed] [Google Scholar]
- Baell JB, Duggan PJ, Forsyth SA, Lewis RJ, Lok YP, Schroeder CI. Synthesis and biological evaluation of nonpeptide mimetics of omega-conotoxin GVIA. Bioorg Med Chem. 2004;12:4025–4037. doi: 10.1016/j.bmc.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Bayer K, Ahmadi S, Zeilhofer HU. Gabapentin may inhibit synaptic transmission in the mouse spinal cord dorsal horn through a preferential block of P/Q-type Ca2+ channels. Neuropharmacology. 2004;46:743–749. doi: 10.1016/j.neuropharm.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Beedle AM, Zamponi GW. Block of voltage-dependent calcium channels by aliphatic monoamines. Biophys J. 2000;79:260–270. doi: 10.1016/S0006-3495(00)76288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benquet P, Guen JL, Dayanithi G, Pichon Y, Tiaho F. omega-AgaIVA-sensitive (P/Q-type) and -resistant (R-type) high-voltage-activated Ba(2+) currents in embryonic cockroach brain neurons. J Neurophysiol. 1999;82:2284–2293. doi: 10.1152/jn.1999.82.5.2284. [DOI] [PubMed] [Google Scholar]
- Biton B, Granger P, Carreau A, Depoortere H, Scatton B, Avenet P. The NMDA receptor antagonist eliprodil (SL 82.0715) blocks voltage-operated Ca2+ channels in rat cultured cortical neurons. Eur J Pharmacol. 1994;257:297–301. doi: 10.1016/0014-2999(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Biton B, Granger P, Depoortere H, Scatton B, Avenet P. Block of P-type Ca2+ channels by the NMDA receptor antagonist eliprodil in acutely dissociated rat Purkinje cells. Eur J Pharmacol. 1995;294:91–100. doi: 10.1016/0014-2999(95)00511-0. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, et al. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Stotz SC, Spaetgens RL, Dayanithi G, Lemos J, Nargeot J, et al. Interaction of SNX482 with domains III and IV inhibits activation gating of alpha(1E) (Ca(V)2.3) calcium channels. Biophys J. 2001;81:79–88. doi: 10.1016/S0006-3495(01)75681-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresink I, Benke TA, Collett VJ, Seal AJ, Parsons CG, Henley JM, et al. Effects of memantine on recombinant rat NMDA receptors expressed in HEK 293 cells. Br J Pharmacol. 1996;119:195–204. doi: 10.1111/j.1476-5381.1996.tb15971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM, Fedak SA, Woodard CJ, Aurbach GD. Beta-Adrenergic receptor interactions. Direct comparison of receptor interaction and biological activity. J Biol Chem. 1976;251:1239–1246. [PubMed] [Google Scholar]
- Buraei Z, Elmslie KS. The separation of antagonist from agonist effects of trisubstituted purines on CaV2.2 (N-type) channels. J Neurochem. 2008;105:1450–1461. doi: 10.1111/j.1471-4159.2008.05248.x. [DOI] [PubMed] [Google Scholar]
- Buraei Z, Schofield G, Elmslie KS. Roscovitine differentially affects CaV2 and Kv channels by binding to the open state. Neuropharmacology. 2006;52:883–894. doi: 10.1016/j.neuropharm.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Cassola AC, Jaffe H, Fales HM, Afeche SC, Magnoli F, Cipolla-Neto J. ω-Phonetoxin-IIA: a calcium channel blocker from the spider Phoneutria nigriventer. Pflugers Arch. 1998;436:545–552. doi: 10.1007/s004240050670. [DOI] [PubMed] [Google Scholar]
- Chen G, Henter ID, Manji HK. Presynaptic glutamatergic dysfunction in bipolar disorder. Biol Psychiatry. 2010;67:1007–1009. doi: 10.1016/j.biopsych.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Meriney SD. The effects of presynaptic calcium channel modulation by roscovitine on transmitter release at the adult frog neuromuscular junction. Eur J Neurosci. 2006;23:3200–3208. doi: 10.1111/j.1460-9568.2006.04849.x. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Fischer H, Dempster L, Daly NL, Rosengren KJ, Nevin ST, et al. Engineering stable peptide toxins by means of backbone cyclization: stabilization of the alpha-conotoxin MII. Proc Natl Acad Sci U S A. 2005;102:13767–13772. doi: 10.1073/pnas.0504613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik DJ, Adams DJ. Chemical modification of conotoxins to improve stability and activity. ACS Chem Biol. 2007;2:457–468. doi: 10.1021/cb700091j. [DOI] [PubMed] [Google Scholar]
- Croisile B, Trillet M, Fondarai J, Laurent B, Mauguière F, Billardon M. Long-term and high-dose piracetam treatment of Alzheimer's disease. Neurology. 1993;43:301–305. doi: 10.1212/wnl.43.2.301. [DOI] [PubMed] [Google Scholar]
- Cumbo E, Ligori LD. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer's disease. Epilepsy Behav. 2010;17:461–466. doi: 10.1016/j.yebeh.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Cunningham MO, Woodhall GL, Thompson SE, Dooley DJ, Jones RS. Dual effects of abapentin and pregabalin on glutamate release at rat entorhinal synapses in vitro. Eur J Neurosci. 2004;20:1566–1576. doi: 10.1111/j.1460-9568.2004.03625.x. [DOI] [PubMed] [Google Scholar]
- Di Trapani G, Mei D, Marra C, Mazza S, Capuano A. Gabapentin in the prophylaxis of migraine: a double-blind randomized placebo-controlled study. Clin Ter. 2000;151:145–148. [PubMed] [Google Scholar]
- Diener HC, Rahlfs VW, Danesch U. The first placebo-controlled trial of a special butterbur root extract for the prevention of migraine: reanalysis of efficacy criteria. Eur Neurol. 2004;51:89–97. doi: 10.1159/000076535. [DOI] [PubMed] [Google Scholar]
- Diochot S, Richard S, Baldy-Moulinier M, Nargeot J, Valmier J. Dihydropyridines, phenylalkylamines and benzothiazepines block N-, P/Q- and R-type calcium currents. Pflugers Arch. 1995;431:10–19. doi: 10.1007/BF00374372. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Milde AS, Andreas K, Ravens U. The effects of verapamil and diltiazem on N-, P- and Q-type calcium channels mediating dopamine release in rat striatum. Br J Pharmacol. 1999;127:576–582. doi: 10.1038/sj.bjp.0702574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Calcium channel diversity: multiple roles of calcium channel subunits. Curr Opin Neurobiol. 2009;19:1–8. doi: 10.1016/j.conb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Dooley DJ, Donovan CM, Meder WP, Whetzel SZ. Preferential action of gabapentin and pregabalin at P/Q-type voltage-sensitive calcium channels: inhibition of K+-evoked [3H]-norepinephrine release from rat neocortical slices. Synapse. 2002;45:171–190. doi: 10.1002/syn.10094. [DOI] [PubMed] [Google Scholar]
- Dos Santos RG, Van Renterghem C, Martin-Moutot N, Mansuelle P, Cordeiro MN, Diniz CR, et al. Phoneutria nigriventer omega-phonetoxin IIA blocks the Cav2 family of calcium channels and interacts with omega-conotoxin-binding sites. J Biol Chem. 2002;277:13856–13862. doi: 10.1074/jbc.M112348200. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Harrison PJ. Markers of glutamate synaptic transmission and plasticity are increased in the anterior cingulate cortex in bipolar disorder. Biol Psychiatry. 2010;67:1010–1016. doi: 10.1016/j.biopsych.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elger CE, Schmidt D. Modern management of epilepsy: a practical approach. Epilepsy Behav. 2008;12:501–539. doi: 10.1016/j.yebeh.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Ellis DJ, Dissanayake S, McGuire D, Charapata SG, Staats PS, Wallace MS, et al. Continuous intrathecal infusion of ziconotide for treatment of chronic malignant and nonmalignant pain over 12 months: a prospective, open-label study. Neuromodulation. 2008;11:40–49. doi: 10.1111/j.1525-1403.2007.00141.x. [DOI] [PubMed] [Google Scholar]
- Enyeart JJ, Biagi BA, Mlinar B. Preferential block of T-type calcium channels by neuroleptics in neural crest-derived rat and human C cell lines. Mol Pharmacol. 1992;42:364–372. [PubMed] [Google Scholar]
- Ertel EA, Smith MM, Leibowitz MD, Cohen CJ. Isolation of myocardial L-type calcium channel gating currents with the spider toxin omega-Aga-IIIA. J Gen Physiol. 1994;103:731–753. doi: 10.1085/jgp.103.5.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink K, Dooley DJ, Meder WP, Suman-Chauhan N, Duffy S, Clusmann H, et al. Inhibition of neuronal Ca(2+) influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology. 2002;42:229–236. doi: 10.1016/s0028-3908(01)00172-1. [DOI] [PubMed] [Google Scholar]
- Fleckenstein-Grün G. Historical development of calcium antagonism- in memoriam Albrecht Fleckenstein. High Blood Press. 1994;3:284–290. [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Tottene A, Lennon VA, Wilson SM, Dubel SJ, Paylor R, et al. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001;15:1288–1290. doi: 10.1096/fj.00-0562fje. [DOI] [PubMed] [Google Scholar]
- Freir DB, Costello DA, Herron CE. A beta 25-35-induced depression of long-term potentiation in area CA1 in vivo and in vitro is attenuated by verapamil. J Neurophysiol. 2003;89:3061–3069. doi: 10.1152/jn.00992.2002. [DOI] [PubMed] [Google Scholar]
- Fujii S, Kameyama K, Hosono M, Hayashi Y, Kitamura K. Effect of cilnidipine, a novel dihydropyridine Ca++-channel antagonist, on N-type Ca++ channel in rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1997;280:1184–1191. [PubMed] [Google Scholar]
- Furukawa T, Nukada T, Suzuki K, Fujita Y, Mori Y, Nishimura M, et al. Voltage and pH dependent block of cloned N-type Ca2+ channels by amlodipine. Br J Pharmacol. 1997;121:1136–1140. doi: 10.1038/sj.bjp.0701226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Yamakawa T, Midera T, Sagawa T, Mori Y, Nukada T. Selectivities of dihydropyridine derivatives in blocking Ca(2+) channel subtypes expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1999;291:464–473. [PubMed] [Google Scholar]
- Galizzi JP, Fosset M, Romey G, Laduron P, Lazdunski M. Neuroleptics of the diphenylbutylpiperidine series are potent calcium channel inhibitors. Proc Natl Acad Sci U S A. 1986;83:7513–7517. doi: 10.1073/pnas.83.19.7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel nticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- Geer JJ, Dooley DJ, Adams ME. K(+)-stimulated 45Ca2+ flux into rat neocortical mini-slices is blocked by omega-Aga-IVA and the dual Na+/Ca2+ channel blockers lidoflazine and flunarizine. Neurosci Lett. 1993;158:97–100. doi: 10.1016/0304-3940(93)90621-q. [DOI] [PubMed] [Google Scholar]
- Glasser SP. The relevance of T-type calcium antagonists: a profile of mibefradil. J Clin Pharmacol. 1998;38:659–669. doi: 10.1002/j.1552-4604.1998.tb04804.x. [DOI] [PubMed] [Google Scholar]
- Gould RJ, Murphy KM, Reynolds IJ, Snyder SH. Antischizophrenic drugs of the diphenylbutylpiperidine type act as calcium channel antagonists. Proc Natl Acad Sci U S A. 1983;80:5122–5125. doi: 10.1073/pnas.80.16.5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobe-Einsler R, Traber J. Clinical results with nimodipine in Alzheimer disease. Clin Neuropharmacol. 1992;15(Suppl. 1):416A–417A. doi: 10.1097/00002826-199201001-00217. [DOI] [PubMed] [Google Scholar]
- Haack JA, Kinser P, Yoshikami D, Olivera BM. Biotinylated derivatives of omega-conotoxins GVIA and MVIID: probes for neuronal calcium channels. Neuropharmacology. 1993;32:1151–1159. doi: 10.1016/0028-3908(93)90009-r. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TS, Teichert RW, Olivera BM, Bulaj G. Conus venoms – a rich source of peptide-based therapeutics. Curr Pharm Des. 2008;14:2462–2479. doi: 10.2174/138161208785777469. [DOI] [PubMed] [Google Scholar]
- Hans M, Urrutia A, Deal C, Brust PF, Staudermann K, Ellis SB, et al. Structural elements in domain IV that influence biophysical and pharmacological properties of human α1A–containing high-voltage-activated calcium channels. Biophys J. 1999;76:1384–1400. doi: 10.1016/S0006-3495(99)77300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haria M, Wagstaff AJ. Amlodipine. A reappraisal of its pharmacological properties and therapeutic use in cardiovascular disease. Drugs. 1995;50:560–586. doi: 10.2165/00003495-199550030-00009. [DOI] [PubMed] [Google Scholar]
- He LM, Chen LY, Lou XL, Qu AL, Zhou Z, Xu T. Evaluation of beta-amyloid peptide 25-35 on calcium homeostasis in cultured rat dorsal root ganglion neurons. Brain Res. 2002;939:65–75. doi: 10.1016/s0006-8993(02)02549-0. [DOI] [PubMed] [Google Scholar]
- Heinke B, Balzer E, Sandkühler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci. 2004;19:103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, Ramachandran J, et al. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 1992;9:69–77. doi: 10.1016/0896-6273(92)90221-x. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Dilmac N, Scheuer T, Catterall WA. Molecular determinants ofdiltiazem block in domains IIIS6 and IVS6 of L-type Ca(2+) channels. Mol Pharmacol. 2000;58:1264–1270. doi: 10.1124/mol.58.6.1264. [DOI] [PubMed] [Google Scholar]
- Hoffmann U, Dileköz E, Kudo C, Ayata C. Gabapentin suppresses cortical spreading depression susceptibility. J Cereb Blood Flow Metab. 2010;30:1588–1592. doi: 10.1038/jcbfm.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak S, Koschak A, Stuppner H, Striessnig J. Use-dependent block of voltage-gated Cav2.1 Ca2+ channels by petasins and eudesmol isomers. J Pharmacol Exp Ther. 2009;330:220–226. doi: 10.1124/jpet.109.151183. [DOI] [PubMed] [Google Scholar]
- Hu LY, Ryder TR, Rafferty MF, Taylor CP, Feng MR, et al. The discovery of [1-(4-dimethylamino-benzyl)-piperidin-4-yl]-[4-(3,3-dimethylbutyl)-phen yl]-(3-methyl-but-2-enyl)-amine, an N-type Ca+2 channel blocker with oral activity for analgesia. Bioorg Med Chem. 2000;8:1203–1212. doi: 10.1016/s0968-0896(00)00077-8. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Wang SJ, Gean PW. Selective enhancement of P-type calcium currents by isoproterenol in the rat amygdala. J Neurosci. 1998;18:2276–2282. doi: 10.1523/JNEUROSCI.18-06-02276.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi H, Yatani A, Akaike N. Block of P-type Ca2+ channels in freshly dissociated rat cerebellar Purkinje neurons by diltiazem and verapamil. Brain Res. 1995;695:88–91. doi: 10.1016/0006-8993(95)00815-8. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Kaneko M, Shin HS, Takahashi T. Presynaptic N-type and P/Q-type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J Physiol. 2005;568:199–209. doi: 10.1113/jphysiol.2005.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Brooks PA, Harrison NL. Enhancement of gamma-aminobutyric acid-activated Cl- currents in cultured rat hippocampal neurones by three volatile anaesthetics. J Physiol. 1992;449:279–293. doi: 10.1113/jphysiol.1992.sp019086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun K, Piedras-Rentería ES, Smith SM, Wheeler DB, Lee SB, Lee TG, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci U S A. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamatchi GL, Chan CK, Snutch T, Durieux ME, Lynch C., 3rd Volatile anesthetic inhibition of neuronal Ca channel currents expressed in Xenopus oocytes. Brain Res. 1999;831:85–96. doi: 10.1016/s0006-8993(99)01401-8. [DOI] [PubMed] [Google Scholar]
- Kameyama K, Aono K, Kitamura K. Isoflurane inhibits neuronal Ca2+ channels through enhancement of current inactivation. Br J Anaesth. 1999;82:402–411. doi: 10.1093/bja/82.3.402. [DOI] [PubMed] [Google Scholar]
- Kang MG, Felix R, Campbell KP. Long-term regulation of voltage-gated Ca(2+) channels by gabapentin. FEBS Lett. 2002;528:177–182. doi: 10.1016/s0014-5793(02)03295-7. [DOI] [PubMed] [Google Scholar]
- Kass RS. Voltage-dependent modulation of cardiac calcium channel current by optical isomers of Bay K 8644: implications for channel gating. Circ Res. 1987;61:I1–I5. [PubMed] [Google Scholar]
- Khosravani H, Zamponi GW. Voltage-gated calcium channels and idiopathic generalized epilepsies. Physiol Rev. 2006;86:941–966. doi: 10.1152/physrev.00002.2006. [DOI] [PubMed] [Google Scholar]
- Kim JI, Takahashi M, Ogura A, Kohno T, Kudo Y, Sato K. Hydroxyl group of Tyr13 is essential for the activity of omega-conotoxin GVIA, a peptide toxin for N-type calcium channel. J Biol Chem. 1994;269:23876–23878. [PubMed] [Google Scholar]
- Kim JI, Konishi S, Iwai H, Kohno T, Gouda H, Shimada I, et al. Three-dimensional solution structure of the calcium channel antagonist omega-agatoxin IVA: consensus molecular folding of calcium channel blockers. J Mol Biol. 1995;250:659–671. doi: 10.1006/jmbi.1995.0406. [DOI] [PubMed] [Google Scholar]
- Kisilevsky AE, Zamponi GW. Presynaptic calcium channels: structure, regulators, and blockers. Handb Exp Pharmacol. 2008;184:45–75. doi: 10.1007/978-3-540-74805-2_3. [DOI] [PubMed] [Google Scholar]
- Knierim E, Leisle L, Wagner C, Weschke B, Lucke B, Bohner G, et al. Recurrent stroke due to a novel voltage sensor mutation in Cav2.1 responds to verapamil. Stroke. 2011;42:e14–e17. doi: 10.1161/STROKEAHA.110.600023. [DOI] [PubMed] [Google Scholar]
- Koch ED, Olivera BM, Terlau H, Conti F. The binding of kappa-Conotoxin PVIIA and fast C-type inactivation of Shaker K+ channels are mutually exclusive. Biophys J. 2004;86:191–209. doi: 10.1016/S0006-3495(04)74096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo H, Kayed R, Glabe CG, Yamaguchi H. Soluble Abeta oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer's disease brain. Brain Res. 2005;1031:222–228. doi: 10.1016/j.brainres.2004.10.041. [DOI] [PubMed] [Google Scholar]
- Krnjević K. Cellular and synaptic actions of general anaesthetics. Gen Pharmacol. 1992;23:965–975. doi: 10.1016/0306-3623(92)90274-n. [DOI] [PubMed] [Google Scholar]
- Kunkler PE, Kraig RP. P/Q Ca2+ channel blockade stops spreading depression and related pyramidal neuronal Ca2+ rise in hippocampal organ culture. Hippocampus. 2004;14:356–367. doi: 10.1002/hipo.10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe RA, Defeo PA, Davison MD, Young J, Herman JL, Spreen RC, et al. Isolation and pharmacological characterization of omega-grammotoxin SIA, a novel peptide inhibitor of neuronal voltage-sensitive calcium channel responses. Mol Pharmacol. 1993;44:451–460. [PubMed] [Google Scholar]
- Leão RM, Cruz JS, Diniz CR, Cordeiro MN, Beirão PS. Inhibition of neuronal high-voltage activated calcium channels by the omega-phoneutria nigriventer Tx3-3 peptide toxin. Neuropharmacology. 2000;39:1756–1767. doi: 10.1016/s0028-3908(99)00267-1. [DOI] [PubMed] [Google Scholar]
- Lee CY, Chen CC, Liou HH. Levetiracetam inhibits glutamate transmission through presynaptic P/Q-type calcium channels on the granule cells of the dentate gyrus. Br J Pharmacol. 2009;158:1753–1762. doi: 10.1111/j.1476-5381.2009.00463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy NA, Janicak PG. Calcium channel antagonists for the treatment of bipolar disorder. Bipolar Disord. 2000;2:108–119. doi: 10.1034/j.1399-5618.2000.020204.x. [DOI] [PubMed] [Google Scholar]
- Lewis RJ, Dutertre S, Vetter I, Christie MJ. Conus venom Peptide pharmacology. Pharmacol Rev. 2012;64:259–298. doi: 10.1124/pr.111.005322. [DOI] [PubMed] [Google Scholar]
- Liau CS. Barnidipine: a new calcium channel blocker for hypertension treatment. Expert Rev Cardiovasc Ther. 2005;3:207–213. doi: 10.1586/14779072.3.2.207. [DOI] [PubMed] [Google Scholar]
- Lipton RB, Göbel H, Einhäupl KM, Wilks K, Mauskop A. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004;63:2240–2244. doi: 10.1212/01.wnl.0000147290.68260.11. [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y, Hackos DH, Swartz KJ. alpha-helical structural elements within the voltage-sensing domains of a K(+) channel. J Gen Physiol. 2000;115:33–50. doi: 10.1085/jgp.115.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Lin JW, Cherksey B. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc Natl Acad Sci U S A. 1989;86:1689–1693. doi: 10.1073/pnas.86.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Arrieta JM, Birks J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. 2002;(3):CD000147. doi: 10.1002/14651858.CD000147. [DOI] [PubMed] [Google Scholar]
- Lu CW, Lin TY, Wang SJ. Memantine depresses glutamate release through inhibition of voltage-dependent Ca2+ entry and protein kinase C in rat cerebralcortex nerve terminals: an NMDA receptor-independent mechanism. Neurochem Int. 2010;57:168–176. doi: 10.1016/j.neuint.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101:9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–710. doi: 10.1016/s0896-6273(04)00085-6. [DOI] [PubMed] [Google Scholar]
- MacManus A, Ramsden M, Murray M, Henderson Z, Pearson HA, Campbell VA. Enhancement of (45)Ca(2+) influx and voltage-dependent Ca(2+) channel activity by beta-amyloid-(1-40) in rat cortical synaptosomes and cultured cortical neurons. Modulation by the proinflammatory cytokine interleukin-1beta. J Biol Chem. 2000;275:4713–4718. doi: 10.1074/jbc.275.7.4713. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Stoof JC, Kits KS. Dihydropyridine block of omega-agatoxin IVA- and omega-conotoxin GVIA-sensitive Ca2+ channels in rat pituitary melanotropic cells. Eur J Pharmacol. 1996;311:293–304. doi: 10.1016/0014-2999(96)00432-3. [DOI] [PubMed] [Google Scholar]
- Markley HG, Cheronis JC, Piepho RW. Verapamil in prophylactic therapy of migraine. Neurology. 1984;34:973–976. doi: 10.1212/wnl.34.7.973. [DOI] [PubMed] [Google Scholar]
- Mathew NT, Rapoport A, Saper J, Magnus L, Klapper J, Ramadan N, et al. Efficacy of gabapentin in migraine prophylaxis. Headache. 2001;41:119–128. doi: 10.1046/j.1526-4610.2001.111006119.x. [DOI] [PubMed] [Google Scholar]
- Mathews EA, García E, Santi CM, Mullen GP, Thacker C, Moerman DG, et al. Critical residues of the Caenorhabditis elegans unc-2 voltage-gated calcium channel that affect behavioral and physiological properties. J Neurosci. 2003;23:6537–6545. doi: 10.1523/JNEUROSCI.23-16-06537.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley BJ, Schroeder JS. The use of diltiazem hydrochloride in cardiovascular disorders. Pharmacotherapy. 1982;2:121–133. doi: 10.1002/j.1875-9114.1982.tb04518.x. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP. Inhibition of calcium channels in rat central and peripheral neurons by omega-conotoxin MVIIC. J Neurosci. 1996;16:2612–2623. doi: 10.1523/JNEUROSCI.16-08-02612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough SI, Lampe RA, Keith RA, Bean BP. Voltage-dependent inhibition of N- and P-type calcium channels by the peptide toxin omega-grammotoxin-SIA. Mol Pharmacol. 1997;52:1095–1104. doi: 10.1124/mol.52.6.1095. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Boland LM, Mintz IM, Bean BP. Interactions among toxins that inhibit N-type and P-type calcium channels. J Gen Physiol. 2002;119:313–328. doi: 10.1085/jgp.20028560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton NCL, Horridge E, Gleave R, Beswick PJ, Chen YH, Powell AJ, et al. Piperazine amide calcium canne blockers such as NMED-160 block Cav2.2, Cav3.2 and Cav1.2 human recombinant calcium channels in both a tonic and use-dependent manner. FENS Abstr. 2008 4, 124.24. [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Menzler S, Bikker JA, Suman-Chauhan N, Horwell DC. Design and biological evaluation of non-peptide analogues of omega-conotoxin MVIIA. Bioorg Med Chem Lett. 2000;10:345–347. doi: 10.1016/s0960-894x(99)00699-x. [DOI] [PubMed] [Google Scholar]
- Mezler M, Barghorn S, Schoemaker H, Gross G, Nimmrich V. Aβ oligomer directly modulates P/Q-type calcium currents in Xenopus oocytes. Br J Pharmacol. 2012a;165:1572–1583. doi: 10.1111/j.1476-5381.2011.01646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezler M, Hermann D, Swensen AM, Draguhn A, Terstappen GC, Gross G, et al. Development and validation of a fluorescence-based HTS assay for the identification of P/Q-type calcium channel blockers. Comb Chem High Throughput Screen. 2012b;15:372–385. doi: 10.2174/138620712800194503. [DOI] [PubMed] [Google Scholar]
- Milhaud D, Fagni L, Bockaert J, Lafon-Cazal M. Inhibition of voltage-gated Ca2+ channels by antazoline. Neuroreport. 2002;13:1711–1714. doi: 10.1097/00001756-200210070-00004. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Adams ME, Bean BP. Inhibition of N- and L-type Ca2+ channels by the spider venom toxin omega-Aga-IIIA. Proc Natl Acad Sci U S A. 1991;88:6628–6631. doi: 10.1073/pnas.88.15.6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 1992a;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992b;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- Monaco EA, 3rd, Vallano ML. Roscovitine triggers excitotoxicity in cultured granule neurons by enhancing glutamate release. Mol Pharmacol. 2005;68:1331–1342. doi: 10.1124/mol.105.012732. [DOI] [PubMed] [Google Scholar]
- Motin L, Yasuda T, Schroeder CI, Lewis RJ, Adams DJ. Omega-conotoxin CVIB differentially inhibits native and recombinant N- and P/Q-type calcium channels. Eur J Neurosci. 2007;25:435–444. doi: 10.1111/j.1460-9568.2006.05299.x. [DOI] [PubMed] [Google Scholar]
- Nadasdi L, Yamashiro D, Chung D, Tarczy-Hornoch K, Adriaenssens P, Ramachandran J. Structure-activity analysis of a Conus peptide blocker of N-type neuronal calcium channels. Biochemistry. 1995;34:8076–8081. doi: 10.1021/bi00025a013. [DOI] [PubMed] [Google Scholar]
- Nebe J, Vanegas H, Neugebauer V, Schaible HG. Omega-agatoxin IVA, a P-type calcium channel antagonist, reduces nociceptive processing in spinal cord neurons with input from the inflamed but not from the normal knee joint – an electrophysiological study in the rat in vivo. Eur J Neurosci. 1997;9:2193–2201. doi: 10.1111/j.1460-9568.1997.tb01386.x. [DOI] [PubMed] [Google Scholar]
- Nielsen KJ, Adams DA, Alewood PF, Lewis RJ, Thomas L, Schroeder T. Effects of chirality at Tyr13 on the structure-activity relationships of omega-conotoxins from Conus magus. Biochemistry. 1999a;38:6741–6751. doi: 10.1021/bi982980u. [DOI] [PubMed] [Google Scholar]
- Nielsen KJ, Adams D, Thomas L, Bond T, Alewood PF, Craik DJ, et al. Structure-activity relationships of omega-conotoxins MVIIA, MVIIC and 14 loop splice hybrids at N and P/Q-type calcium channels. J Mol Biol. 1999b;289:1405–1421. doi: 10.1006/jmbi.1999.2817. [DOI] [PubMed] [Google Scholar]
- Niespodziany I, Klitgaard H, Margineanu DG. Levetiracetam inhibits the high-voltage-activated Ca(2+) current in pyramidal neurones of rat hippocampal slices. Neurosci Lett. 2001;306:5–8. doi: 10.1016/s0304-3940(01)01884-5. [DOI] [PubMed] [Google Scholar]
- Noguchi A, Matsumura S, Dezawa M, Tada M, Yanazawa M, Ito A, et al. Isolation and characterization of patient-derived, toxic, high mass amyloid beta-protein (Abeta) assembly from Alzheimer disease brains. J Biol Chem. 2009;284:32895–32905. doi: 10.1074/jbc.M109.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Itoh Y, Wada M, Yamamoto A, Fujita T. A comparison of Ca2+ channel blocking mode between gabapentin and verapamil: implication for protection against hypoxic injury in rat cerebrocortical slices. Br J Pharmacol. 2003a;139:435–443. doi: 10.1038/sj.bjp.0705246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Itoh Y, Wada M, Yamamoto A, Fujita T. Gabapentin blocks L-type and P/Q-type Ca2+ channels involved in depolarization-stimulated nitric oxide synthase activity in primary cultures of neurons from mouse cerebral cortex. Pharm Res. 2003b;20:897–899. doi: 10.1023/a:1024078704020. [DOI] [PubMed] [Google Scholar]
- O'Neill MJ, Hicks CA, Ward MA, Osborne DJ, Wishart G, Mathews KS, et al. LY393615, a novel neuronal Ca(2+) and Na(+) channel blocker with neuroprotective effects in models of in vitro and in vivo cerebral ischemia. Brain Res. 2001;888:138–149. doi: 10.1016/s0006-8993(00)03043-2. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Frants RR, Ferrari MD. P/Q-type Ca2+ channel defects in migraine, ataxia and epilepsy. Trends Pharmacol Sci. 1998;19:121–127. doi: 10.1016/s0165-6147(98)01182-1. [DOI] [PubMed] [Google Scholar]
- Pazzaglia PJ, Post RM, Ketter TA, Callahan AM, Marangell LB, Frye MA, et al. Nimodipine monotherapy and carbamazepine augmentation in patients with refractory recurrent affective illness. J Clin Psychopharmacol. 1998;18:404–413. doi: 10.1097/00004714-199810000-00009. [DOI] [PubMed] [Google Scholar]
- Pepine CJ, Lambert CR. Cardiovascular effects of nicardipine. Angiology. 1990;41:978–986. [PubMed] [Google Scholar]
- Pexton T, Moeller-Bertram T, Schilling JM, Wallace MS. Targeting voltage-gated calcium channels for the treatment of neuropathic pain: a review of drug development. Expert Opin Investig Drugs. 2011;20:1277–1284. doi: 10.1517/13543784.2011.600686. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Calcium channels and channelopathies of the central nervous system. Mol Neurobiol. 2002;25:31–50. doi: 10.1385/MN:25:1:031. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Cav2.1 channelopathies. Eur J Physiol. 2010;460:375–393. doi: 10.1007/s00424-010-0802-8. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Martella G, De Persis C, Costa C, Pisani F, et al. Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia. 2004;45:719–728. doi: 10.1111/j.0013-9580.2004.02204.x. [DOI] [PubMed] [Google Scholar]
- Pizza V, Busillo V, Agresta A, Bisogno A, Capasso A. Elderly patients with migraine: an open-label study on prophylaxis therapy with levetiracetam. Cent Nerv Syst Agents Med Chem. 2011;11:31–34. doi: 10.2174/187152411794961086. [DOI] [PubMed] [Google Scholar]
- Plomp JJ, van den Maagdenberg AM, Molenaar PC, Frants RR, Ferrari MD. Mutant. P/Q-type calcium channel electrophysiology and migraine. Curr Opin Investig Drugs. 2001;2:1250–1260. [PubMed] [Google Scholar]
- Pluzhnikov K, Vassilevski A, Korolkova Y, Fisyunov A, Iegorova O, Krishtal O, et al. omega-Lsp-IA, a novel modulator of P-type Ca2+ channels. Toxicon. 2007;50:993–1004. doi: 10.1016/j.toxicon.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Popović M, Popović N, Jovanova-Nesić K, Bokonjić D, Dobrić S, Rosić N. Open field behavior in nucleus basalis magnocellularis-lesioned rats treated with physostigmine and verapamil. Int J Neurosci. 1997;91:181–188. doi: 10.3109/00207459708986375. [DOI] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Presynaptic Ca2+ channels and neurotransmitter release at the terminal of a mouse cortical neuron. J Neurosci. 2001;21:3721–3728. doi: 10.1523/JNEUROSCI.21-11-03721.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Olcese R, Stefani E, Birnbaumer L. Modulation of human neuronal alpha 1E-type calcium channel by alpha 2 delta-subunit. Am J Physiol. 1998;274:C1324–C1331. doi: 10.1152/ajpcell.1998.274.5.C1324. [DOI] [PubMed] [Google Scholar]
- Rajakulendran S, Kaski D, Hanna MG. Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol. 2012;8:86–96. doi: 10.1038/nrneurol.2011.228. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Henderson Z, Pearson HA. Modulation of Ca2+ channel currents in primary cultures of rat cortical neurones by amyloid beta protein (1-40) is dependent on solubility status. Brain Res. 2002;956:254–261. doi: 10.1016/s0006-8993(02)03547-3. [DOI] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehberg B, Xiao YH, Duch DS. Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anesthetics. Anesthesiology. 1996;84:1223–1233. doi: 10.1097/00000542-199605000-00025. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer's disease. CNS Drug Rev. 2003;9:275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Rosen MR, Wit AL, Hoffman BF. Electrophysiology and pharmacology of cardiac arrhythmias. VI. Cardiac effects of verapamil. Am Heart J. 1975;89:665–673. doi: 10.1016/0002-8703(75)90514-1. [DOI] [PubMed] [Google Scholar]
- Rovira C, Arbez N, Mariani J. Abeta(25-35) and Abeta(1-40) act on different calcium channels in CA1 hippocampal neurons. Biochem Biophys Res Commun. 2002;296:1317–1321. doi: 10.1016/s0006-291x(02)02072-7. [DOI] [PubMed] [Google Scholar]
- Sah DW, Bean BP. Inhibition of P-type and N-type calcium channels by dopamine receptor antagonists. Mol Pharmacol. 1994;45:84–92. [PubMed] [Google Scholar]
- Sather WA, Tanabe T, Zhang JF, Mori Y, Adams ME, Tsien RW. Distinctive biophysical and pharmacological properties of class A (BI) calcium channel alpha 1 subunits. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-t. [DOI] [PubMed] [Google Scholar]
- Sato K, Park NG, Kohno T, Maeda T, Kim JI, Kato R, et al. Role of basic residues for the binding of omega-conotoxin GVIA to N-type calcium channels. Biochem Biophys Res Commun. 1993;194:1292–1296. doi: 10.1006/bbrc.1993.1964. [DOI] [PubMed] [Google Scholar]
- Schlame M, Hemmings HC., Jr Inhibition by volatile anesthetics of endogenous glutamate release from synaptosomes by a presynaptic mechanism. Anesthesiology. 1995;82:1406–1416. doi: 10.1097/00000542-199506000-00012. [DOI] [PubMed] [Google Scholar]
- Schmidtko A, Lötsch J, Freynhagen R, Geisslinger G. Ziconotide for treatment of severe chronic pain. Lancet. 2010;375:1569–1577. doi: 10.1016/S0140-6736(10)60354-6. [DOI] [PubMed] [Google Scholar]
- Scholz H. Pharmacological aspects of calcium channel blockers. Cardiovasc Drugs Ther. 1997;10(Suppl. 3):869–872. doi: 10.1007/BF00051613. [DOI] [PubMed] [Google Scholar]
- Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology. 1996;124:57–73. doi: 10.1007/BF02245606. [DOI] [PubMed] [Google Scholar]
- Schweitz H, Heurteaux C, Bois P, Moinier D, Romey G, Lazdunski M. Calcicludine, a venom peptide of the Kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high affinity for L-type channels in cerebellar granule neurons. Proc Natl Acad Sci U S A. 1994;91:878–882. doi: 10.1073/pnas.91.3.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott VE, Vortherms TA, Niforatos W, Swensen AM, Neelands T, Milicic I, et al. A-1048400 is a novel, orally active, state-dependent neuronal calcium channel blocker that produces dose-dependent antinociception without altering hemodynamic function in rats. Biochem Pharmacol. 2012;83:406–418. doi: 10.1016/j.bcp.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Sidach SS, Mintz IM. Kurtoxin, a gating modifier of neuronal high- and low-threshold ca channels. J Neurosci. 2002;22:2023–2034. doi: 10.1523/JNEUROSCI.22-06-02023.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sills GJ. The mechanisms of action of gabapentin and pregabalin. Curr Opin Pharmacol. 2006;6:108–113. doi: 10.1016/j.coph.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Sitges M, Guarneros A, Nekrassov V. Effects of carbamazepine, phenytoin, valproic acid, oxcarbazepine, lamotrigine, topiramate and vinpocetine on the presynaptic Ca2+ channel-mediated release of [3H]glutamate: comparison with the Na+ channel-mediated release. Neuropharmacology. 2007;53:854–862. doi: 10.1016/j.neuropharm.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Small DL, Monette R, Mealing G, Buchan AM, Morley P. Neuroprotective effects of omega-Aga-IVA against in vitro ischaemia in the rat hippocampal slice. Neuroreport. 1995;6:1617–1620. doi: 10.1097/00001756-199508000-00007. [DOI] [PubMed] [Google Scholar]
- Solem M, McMahon T, Messing RO. Protein kinase A regulates regulates inhibition of N- and P/Q-type calcium channels by ethanol in PC12 cells. J Pharmacol Exp Ther. 1997;282:1487–1495. [PubMed] [Google Scholar]
- Solomon GD, Steel JG, Spaccavento LJ. Verapamil prophylaxis of migraine. A double-blind, placebo-controlled study. JAMA. 1983;250:2500–2502. [PubMed] [Google Scholar]
- Soong TW, DeMaria CD, Alvania RS, Zweifel LS, Liang MC, Mittman S, et al. Systematic identification of splice variants in human P/Q-type channel alpha1(2.1) subunits: implications for current density and Ca2+-dependent inactivation. J Neurosci. 2002;22:10142–10152. doi: 10.1523/JNEUROSCI.22-23-10142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, et al. Localization and functional properties of a rat brain alpha 1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc Natl Acad Sci U S A. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani A, Pisani A, De Murtas M, Mercuri NB, Marciani MG, Calabresi P. Action of GP 47779, the active metabolite of oxcarbazepine, on the corticostriatal system. II. Modulation of high-voltage-activated calcium currents. Epilepsia. 1995;36:997–1002. doi: 10.1111/j.1528-1157.1995.tb00958.x. [DOI] [PubMed] [Google Scholar]
- Stefani A, Spadoni F, Siniscalchi A, Bernardi G. Lamotrigine inhibits Ca2+ currents in cortical neurons: functional implications. Eur J Pharmacol. 1996a;307:113–116. doi: 10.1016/0014-2999(96)00265-8. [DOI] [PubMed] [Google Scholar]
- Stefani A, Calabresi P, Pisani A, Mercuri NB, Siniscalchi A, Bernardi G. Felbamate inhibits dihydropyridine-sensitive calcium channels in central neurons. J Pharmacol Exp Ther. 1996b;277:121–127. [PubMed] [Google Scholar]
- Stefani A, Spadoni F, Bernardi G. Differential inhibition by riluzole, lamotrigine, and phenytoin of sodium and calcium currents in cortical neurons: implications for neuroprotective strategies. Exp Neurol. 1997;147:115–122. doi: 10.1006/exnr.1997.6554. [DOI] [PubMed] [Google Scholar]
- Stotz SC, Spaetgens RL, Zamponi GW. Block of voltage-dependent calcium channel by the green mamba toxin calcicludine. J Membr Biol. 2000;174:157–165. doi: 10.1007/s002320001040. [DOI] [PubMed] [Google Scholar]
- Striano P, Striano S. Gabapentin: a Ca2+ channel alpha 2-delta ligand far beyond epilepsy therapy. Drugs Today. 2008;44:353–368. doi: 10.1358/dot.2008.44.5.1186403. [DOI] [PubMed] [Google Scholar]
- Study RE. Isoflurane inhibits multiple voltage-gated calcium currents in hippocampal pyramidal neurons. Anesthesiology. 1994;81:104–116. doi: 10.1097/00000542-199407000-00016. [DOI] [PubMed] [Google Scholar]
- Suman-Chauhan N, Webdale L, Hill DR, Woodruff GN. Characterisation of [3H]gabapentin binding to a novel site in rat brain: homogenate binding studies. Eur J Pharmacol. 1993;244:293–301. doi: 10.1016/0922-4106(93)90155-3. [DOI] [PubMed] [Google Scholar]
- Sutton KG, Siok C, Stea A, Zamponi GW, Heck SD, Volkmann RA, et al. Inhibition of neuronal calcium channels by a novel peptide spider toxin, DW13.3. Mol Pharmacol. 1998;54:407–418. doi: 10.1124/mol.54.2.407. [DOI] [PubMed] [Google Scholar]
- Sutton KG, Martin DJ, Pinnock RD, Lee K, Scott RH. Gabapentin inhibits high-threshold calcium channel currents in cultured rat dorsal root ganglion neurones. Br J Pharmacol. 2002;135:257–265. doi: 10.1038/sj.bjp.0704439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Park E, Lee C, Kim J, Takahashi H, Swartz K, et al. Solution structure of omega-grammotoxin SIA, a gating modifier of P/Q and N-type Ca(2+) channel. J Mol Biol. 2002;321:517–526. doi: 10.1016/s0022-2836(02)00595-8. [DOI] [PubMed] [Google Scholar]
- Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- Tollefson GD. Short-term effects of the calcium channel blocker nimodipine (Bay-e-9736) in the management of primary degenerative dementia. Biol Psychiatry. 1990;27:1133–1142. doi: 10.1016/0006-3223(90)90050-c. [DOI] [PubMed] [Google Scholar]
- Tomassoni D, Lanari A, Silvestrelli G, Traini E, Amenta F. Nimodipine and its use in cerebrovascular disease: evidence from recent preclinical and controlled clinical studies. Clin Exp Hypertens. 2008;30:744–766. doi: 10.1080/10641960802580232. [DOI] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, et al. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron. 2009;61:762–773. doi: 10.1016/j.neuron.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Tottene A, Urbani A, Pietrobon D. Role of different voltage-gated Ca2+ channels in cortical spreading depression: specific requirement of P/Q-type Ca2+ channels. Channels (Austin) 2011;5:110–114. doi: 10.4161/chan.5.2.14149. [DOI] [PubMed] [Google Scholar]
- Trepakova ES, Dech SJ, Salata JJ. Flunarizine is a highly potent inhibitor of cardiac hERG potassium current. J Cardiovasc Pharmacol. 2006;47:211–220. doi: 10.1097/01.fjc.0000200810.18575.80. [DOI] [PubMed] [Google Scholar]
- Viana F, Van den Bosch L, Missiaen L, Vandenberghe W, Droogmans G, Nilius B, et al. Mibefradil (Ro 40-5967) blocks multiple types of voltage-gated calcium channels in cultured rat spinal motoneurones. Cell Calcium. 1997;22:299–311. doi: 10.1016/s0143-4160(97)90068-3. [DOI] [PubMed] [Google Scholar]
- Vieira LB, Kushmerick C, Hildebrand ME, Garcia E, Stea A, Cordeiro MN, et al. Inhibition of high voltage-activated calcium channels by spider toxin PnTx3-6. J Pharmacol Exp Ther. 2005;314:1370–1377. doi: 10.1124/jpet.105.087023. [DOI] [PubMed] [Google Scholar]
- Vohora D, Saraogi P, Yazdani MA, Bhowmik M, Khanam R, Pillai KK. Recent advances in adjunctive therapy for epilepsy: focus on sodium channel blockers as third-generation antiepileptic drugs. Drugs Today (Barc) 2010;46:265–277. doi: 10.1358/dot.2010.46.4.1445795. [DOI] [PubMed] [Google Scholar]
- Wauquier A, Ashton D, Marrannes R. The effects of flunarizine in experimental models related to the pathogenesis of migraine. Cephalalgia. 1985;5(Suppl. 2):119–123. doi: 10.1177/03331024850050S222. [DOI] [PubMed] [Google Scholar]
- Webster LR, Fisher R, Charapata S, Wallace MS. Long-term intrathecal ziconotide for chronic pain: an open-label study. J Pain Symptom Manage. 2009;37:363–372. doi: 10.1016/j.jpainsymman.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Wermeling DP. Ziconotide, an intrathecally administered N-type calcium channel antagonist for the treatment of chronic pain. Pharmacotherapy. 2005;25:1084–1094. doi: 10.1592/phco.2005.25.8.1084. [DOI] [PubMed] [Google Scholar]
- Winterfield JR, Swartz KJ. A hot spot for the interaction of gating modifier toxins with voltage-dependent ion channels. J Gen Physiol. 2000;116:637–644. doi: 10.1085/jgp.116.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Block of multiple presynaptic calcium channel types by omega-conotoxin-MVIIC at hippocampal CA3 to CA1 synapses. J Neurophysiol. 1995;73:1965–1972. doi: 10.1152/jn.1995.73.5.1965. [DOI] [PubMed] [Google Scholar]
- Yakash TL. Calcium channels as therapeutic targets in neuropathic pain. J Pain. 2006;7:S13–S30. doi: 10.1016/j.jpain.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Takahara A. Recent updates of N-type calcium channel blockers with therapeutic potential for neuropathic pain and stroke. Curr Top Med Chem. 2009;9:377–395. doi: 10.2174/156802609788317838. [DOI] [PubMed] [Google Scholar]
- Yan L, Adams ME. The spider toxin omega-Aga IIIA defines a high affinity site on neuronal high voltage-activated calcium channels. J Biol Chem. 2000;275:21309–21316. doi: 10.1074/jbc.M000212200. [DOI] [PubMed] [Google Scholar]
- Yan Z, Chi P, Bibb JA, Ryan TA, Greengard P. Roscovitine: a novel regulator of P/Q-type calcium channels and transmitter release in central neurons. J Physiol. 2002;540:761–770. doi: 10.1113/jphysiol.2001.013376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Yan LY, Xue LJ, Wang Q, Zhou ZK, Xiao H, et al. Flunarizine blocks voltage-gated Na(+) and Ca(2+) currents in cultured rat cortical neurons: a possible locus of action in the prevention of migraine. Neurosci Lett. 2011;487:394–399. doi: 10.1016/j.neulet.2010.10.064. [DOI] [PubMed] [Google Scholar]
- Yu W, Horowitz SH. Treatment of sporadic hemiplegic migraine with calcium-channel blocker verapamil. Neurology. 2003;60:120–121. doi: 10.1212/01.wnl.0000042051.16284.70. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Lory P, Perez-Reyes E. Role of voltage-gated calcium channels in epilepsy. Pflugers Arch. 2010;460:395–403. doi: 10.1007/s00424-009-0772-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Okada M, Murakami T, Kawata Y, Kamata A, Kaneko S. Interaction between carbamazepine, zonisamide and voltage-sensitive Ca2+ channel on acetylcholine release in rat frontal cortex. Epilepsy Res. 2002;49:49–60. doi: 10.1016/s0920-1211(02)00015-3. [DOI] [PubMed] [Google Scholar]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium cannel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]