

Abstract
BACKGROUND AND PURPOSE
cGMP is involved in the regulation of many cellular processes including cardiac and smooth muscle contractility, aldosterone synthesis and inhibition of platelet activation. Intracellular effects cGMP are mediated by cGMP-dependent PKs, cGMP-regulated PDEs and cGMP-gated ion channels. PKG inhibitors are widely used to discriminate PKG-specific effects. They can be divided into cyclic nucleotide-binding site inhibitors such as Rp-phosphorothioate analogues (Rp-cGMPS), ATP-binding site inhibitors such as KT5823, and substrate binding site inhibitors represented by the recently described DT-oligopeptides. As it has been shown that Rp-cGMPS and KT5823 have numerous non-specific effects, we analysed the pharmacological properties of the oligopeptide (D)-DT-2 described as a highly specific, membrane–permeable, PKG inhibitor.
EXPERIMENTAL APPROACH
Specificity and potency of (D)-DT-2 to inhibit PKG activity was evaluated using biochemical assays in vitro and by substrate phosphorylation analysis in various cell types including human platelets, rat mesangial cells and rat neonatal cardiomyocytes.
KEY RESULTS
Despite potent inhibition of PKGI in vitro, (D)-DT-2 lost specificity for PKG in cell homogenates and particularly in living cells, as demonstrated by phosphorylation of different substrates. Instead, (D)-DT-2 modulated activity of other kinases including ERK, p38, PKB and PKC, thereby inducing unpredicted and often opposing functional effects.
CONCLUSIONS AND IMPLICATIONS
We conclude that DT-oligopeptides, as other inhibitors, cannot be used to specifically inhibit PKG in intact cells. Therefore, no specific pharmacological PKG inhibitors are available, and reliable studies of PKG signalling can only be made by using RNA knockdown or genetic deletion methods.
Keywords: cAMP, cGMP PKA, PKG inhibitors
Introduction
cGMP is synthesized from GTP by two classes of GCs which include transmembrane particulate GCs, which serve as receptors for natriuretic peptides and the soluble form (sGC), which serves as the cytosolic receptor for NO (Lohmann and Walter, 2005). cGMP is involved in the regulation of many diverse cellular processes such as cardiac and smooth muscle cell contractility, aldosterone synthesis, inhibition of platelet activation, immune cell response, neuronal excitability and synaptic plasticity (Lohmann and Walter, 2005; Hofmann et al., 2006; 2009; Walter and Gambaryan, 2009). Intracellular effects of cGMP are mediated by a number of effectors, including cGMP-gated channels, cGMP-regulated PDEs (PDE2 and 3) and cGMP-dependent PKs (PKGs; nomenclature follows Alexander et al., 2011). Most cells contain at least one of the three known PKG subtypes (PKG Iα, PKG Iβ, PKG II) (Hofmann et al., 2009). However, very often, the same cells express cGMP-regulated PDEs, whose activation/inhibition by cGMP can promote or inhibit PKA activity as well. For example, in aldosterone producing cells of the adrenal zona glomerulosa, PKG II activity is involved in the regulation of basal aldosterone production (Gambaryan et al., 2003), whereas activation of PDE2 by cGMP inhibits cAMP-induced aldosterone production (MacFarland et al., 1991; Nikolaev et al., 2005). In cardiac myocytes, PKG I, PDE2 and PDE3 are localized in different compartments and can play even opposing roles in regulation of cardiac contractility (Castro et al., 2010; Mika et al., 2012). In renin-secreting juxtaglomerular cells, cGMP signalling is even more complicated. In these cells, PKG I, PKG II, PDE3 and PDE2 are expressed (Gambaryan et al., 1996; 1998; Castrop et al., 2010). Depending on experimental conditions (in vivo experiments, isolated glomeruli or isolated juxtaglomerular cells) and the site of cGMP synthesis (activation of particulate versus soluble GC), cGMP can stimulate or inhibit renin release by activation of PKG or PDEs (Kurtz, 2011). Several approaches (knockout animal models, overexpression of active and inactive PKGs, PKG-specific activators/inhibitors) have been used to characterize PKG-specific effects and distinguish them from other cGMP mediators (Smolenski et al., 1998b; Hofmann et al., 2006; 2009).
Currently used PKG inhibitors can be divided into three classes: cyclic nucleotide-binding site inhibitors such as Rp-phosphorothioate analogues (Rp-cGMPS), ATP-binding site inhibitors such as KT5823 and substrate-binding site inhibitors represented by the recently described DT-oligopeptides. Although in our previous experiments, Rp-cGMPS compounds (Rp-8-pCPT-cGMPS, Rp-8-Br-PET-cGMPS) were potent inhibitors of PKG activity in mesangial cells and platelets (Burkhardt et al., 2000), we later recognized that they also display PKG-independent unspecific effects on platelets (Gambaryan et al., 2004). In smooth muscle cells, Rp-8-Br-PET-cGMPS stimulates PKG I instead of inhibiting its activity (Valtcheva et al., 2009). KT5823, a widely used cell-permeable PKG inhibitor, does not inhibit basal or stimulated PKG activity in platelets, mesangial and smooth muscle cells, but may potently inhibit other PKs (Wyatt et al., 1991; Komalavilas and Lincoln, 1996; Burkhardt et al., 2000; Bain et al., 2003). DT-oligopeptides, which bind to the substrate-binding site of PKG catalytic domain and inhibit PKG activity by competing with the substrates, were introduced as ‘highly specific, membrane permeable peptide blockers of PKG’ (Nickl et al., 2010). Here, we show that the oligopeptide (D)-DT-2 was a specific PKG I inhibitor only for purified PKG Iα/β enzymes but did not inhibit PKG activity in intact cells.
Methods
In vitro PK assay
PKA c-subunit (PKAc) and PKG type Iα were purified from bovine heart and bovine lung, respectively, as described earlier (Kaczmarek et al., 1980; Walter et al., 1980). Human PKG Iβ and PKG II were expressed in Sf9 cells and purified by affinity chromatography (Pohler et al., 1995). The activity of the purified kinases was measured by the phosphocellulose method (Roskoski, 1983). Briefly, PK activity was assayed at 30°C in a total volume of 100 µL containing 20 mM Tris/HCl buffer pH 7.4, 10 mM MgCl2, 5 mM β-mercaptoethanol, 0.01% (w/v) bovine serum albumin, 2 nM PK, 20 µM Vasptide and 5 µM cGMP (for PKG) or 20 µM Kemptide (for PKA) as described (Butt et al., 1994), and indicated concentrations of (D)-DT-2. The reaction was started by the addition of 50 µM [32γ]-ATP (1000 cpm pmol−1) and terminated after 5 min by the addition of 0.1 M EDTA. Phosphorylated peptide was bound to P81-paper (Whatman, Rothenburg, Germany), washed with 75 mM phosphoric acid and quantified by liquid scintillation counting. The half maximal inhibitory concentration (IC50) for PKG Iα and PKG Iβ was determined graphically by plotting kinase activity versus varying (D)-DT-2 concentrations at a constant substrate level and represented the average of three independent experiments.
Preparation of human washed platelets
Our studies with human platelets were approved and recently (24 September 2008) reconfirmed by the local ethics committee of the University of Würzburg (Studies no. 67/92 and 114/04). Blood was obtained from healthy volunteers according to our institutional guidelines and the Declaration of Helsinki.
Human platelets were prepared and used as previously reported (Gambaryan et al., 2010) with small modifications. Blood was collected into 1/7 volume of ACD solution (12 mM citric acid, 15 mM sodium citrate, 25 mM d-glucose and 2 µM EGTA, final concentrations). PRP was obtained by 5 min centrifugation at 330×g. To reduce leukocyte contamination, PRP was diluted 1:1 with PBS and centrifuged at 240 g for 10 min. Subsequently, the supernatant was centrifuged for 10 min at 430×g, then the pelleted platelets were washed once in CGS buffer (composition: 120 mM sodium chloride, 12.9 mM trisodium citrate, 30 mM d-glucose, pH 6.5), and resuspended in HEPES buffer (150 mM sodium chloride, 5 mM potassium chloride, 1 mM magnesium chloride, 10 mM d-glucose, 10 mM HEPES, pH 7.4). After 15 min rest in a 37°C water bath, washed platelets were used for experiments.
Animals
All animal care and experimental procedures complied with the guidelines of the Federal German Government and were approved by the Ethical Committee of the University of Würzburg. The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). A total of 40 animals were used in the experiments described here.
Experiments on rat mesangial cells
Rat mesangial cells (RMC) were isolated from whole kidneys of 1 day old rats (Sprague Dawley; University of Göttingen animal facility) and cultured using a sieving technique as previously described (Burkhardt et al., 2000). Cells were cultured in RPMI1640 containing 20% fetal calf serum, 2 mM L-glutamine, 0.1 mM sodium pyruvate, 5 mM HEPES, pH 7.2, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, 0.1% non-essential amino acids and 0.1% growth supplements (5 µg·mL−1 insulin, 5 µg·mL−1 transferrin, 5 ng·mL−1 sodium selenite). For experiments, cells at passage 5 to 12 at a density of approximately 5 × 105 cells per well (6 well plate) were incubated in RPMI without FCS at 37°C for 3 h. (D)-DT-2 was added 30 min prior to stimulating with ANP, the NO-donor SNP or thrombin for 5 min at indicated concentrations. After washing with PBS, cells were lysed by adding SDS gel loading buffer. For experiments in permeabilized cells, saponin (0.001%) was added to the cells 5 min prior to (D)-DT-2. After 10 min, PKG or PKA was stimulated by 5 µM (final concentration) of cGMP or cAMP, respectively.
Experiments on neonatal rat ventricular myocytes
Neonatal rat ventricular myocytes (NRVM) were isolated from 0-1 day old Sprague Dawley rats (University of Göttingen) and cultured as previously described (Grebe et al., 2011). Twenty-four hours after isolation, cells were washed with serum-free media and pre-incubated with 20 µM (D)-DT-2 for 30 min. Next, cells were stimulated with indicated chemicals and processed for Western blot analysis.
FRET experiments on human embryonic kidney (HEK) 293 cells
HEK293a cells were cultured in DMEM medium containing 10% FCS, seeded on 24 mm glass coverslips and transfected with the plasmids for A-kinase activity reporter (AKAR3, kindly provided by Jin Zhang, Baltimore, USA), rat GC-A receptor for ANP and wild-type or dominant-negative (K405A) PKGIβ (Smolenski et al., 2000). Twenty-four hours after transfection, the cells were used for FRET measurements performed exactly as described by Borner et al., (2011). The cells were stimulated with 100 nM ANP, and the amplitudes of FRET responses were analysed.
Confocal microscopy
Confocal microscopy was performed using Zeiss LSM710 microscope (Carl Zeiss MicroImaging, Jena, Germany) equipped with the Plan-Apochromat ×63/1.40 oil-immersion objective. Images were acquired and analysed using ZEN 2010 software (Zeiss). To determine approximate cell volume, 3D stacks of RMC and NRCM transfected with a GFP plasmid were acquired and analysed using the ZEN 2010 software.
Experiments with human washed platelets and RMC lysate
Human washed platelets or RMC were lysed in the buffer (10 mM HEPES, 150 mM NaCl, 2 mM EGTA, 1% Triton ×100, pH 7.4) with protease inhibitors (1 mM PMSF, 10 µg·mL−1 pepstatin A, 20 mg·mL−1 leupeptin) and frozen in liquid nitrogen. In addition, 10 µL of 10× concentrated reaction buffer (100 mM HEPES, 50 mM MgCl2, 5 mM EGTA, 10 mM dithiothreitol 1 mM ATP, pH 7.4) was added to the 90 µL of cell lysate. The samples were pre-incubated with (D)-DT-2 (20 µM) for 10 min then stimulated with 2 µM of cGMP or cAMP for 2 min and stopped with 100 µL of 2× SDS gel loading buffer.
Western blot analysis
Washed platelets were pre-incubated for the indicated time/concentration with (D)-DT-2, stimulated with the NO donor DEA-NO, thrombin or collagen and stopped by adding 2× SDS gel loading buffer. RMC were pre-incubated with (D)-DT-2 for 30 min and stimulated with ANP, DEA-NO or thrombin for 10 min. NRVM were pre-incubated with (D)-DT-2 or ERK inhibitor U0126 for 30 min and stimulated with ANP or SNP. An equal volume of 2× SDS gel loading buffer was directly added to platelet suspensions, cells were washed with PBS and lysed in 2× SDS gel loading buffer (500 µL per confluent well of 6 well plate). For Western blotting, cell lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and the membranes were incubated with appropriate primary antibodies [PKG I antibody described by Gambaryan et al., (1996), phospho PDE5-Ser92 and total PDE5 (generous gift from Dr S. Rybalkin, University of Washington, Seattle, USA), phospho-VASP-Ser239, PKG Iα, PKG Iβ, phospho–p38 (Thr180/Tyr182) MAPK antibody, PKC substrate MARCKS phospho-Ser159/163 antibody, phospho-ERK (Thr202/Tyr20), total p38, total ERK phospho-PKB-Ser473 and total PKB] overnight at 4°C. For calculation of PKG Iα/β concentrations in cells, defined amounts of purified kinase and cells were loaded on the same gel. Cell volumes were calculated from confocal microscopic images. For visualization of the signal, goat anti-rabbit or anti-mouse IgG conjugated with horseradish peroxidase was used as secondary antibodies, followed by ECL detection. Blots were scanned using SilverFast software and analysed densitometrically by NIH Image J software for uncalibrated optical density.
Platelet aggregation and calcium measurements
Platelet aggregation was measured using an Apact (LabiTec, Ahrensburg, Germany) aggregometer. Washed human platelets (3 × 108·mL−1) were pre-incubated with 10 µM of (D)-DT-2 for 10 min. Platelet aggregation was induced by addition of 0.01 U·mL−1 thrombin or 5 µg·mL−1 collagen. Aggregation was measured with continuous stirring at 1000 rpm and 37°C.
Calcium transients were determined with the fluorescence indicator Fura-2. Platelets were loaded with Fura-2-AM in PRP for 45 min at 37°C. Excess dye and plasma were removed by centrifugation. The pelleted platelets were then resuspended in HEPES buffer and diluted to a density of 2 × 108 platelets·mL−1. The measurement was carried out with a Perkin-Elmer (Waltham, MA, USA) LS50 luminometer. Fura-2 fluorescence was measured at 340 nm excitation. 1 mM Ca++ was added immediately before the experiment.
FACS analysis of platelet integrin αIIbβ3 activation
FACS analysis was performed on a Becton Dickinson FACScalibur using CELLQuest software, version 3.1f (Becton Dickinson, Heidelberg, Germany). Washed platelets were pre-incubated with (D)-DT-2 for 10 min and stimulated with thrombin (0.01 U·mL−1) or collagen for 1 min; incubations were then simultaneously terminated for Western blot and FACS analysis. For FACS analysis, after agonist stimulation, washed platelets were labelled for 5 min at room temperature with FITC-conjugated antibody against activated αIIbβ3 integrins, PAC-1-FITC (BD-Bioscience Heidelberg, Germany). Stimulation was stopped by diluting platelets (1:10) in PBS/5.5 mM glucose/0.5%, and platelets were analysed by flow cytometry.
Data analysis
All experiments were performed at least three times and summary data are expressed as means ± SEM. Differences between groups were analysed by anova followed by Bonferroni's test, and Student's t-test was used when appropriate. P < 0.05 was considered statistically significant.
Materials
(D)-DT-2 was from Biolog (Bremen, Germany); atrial natriuretic peptide (ANP), sodium nitroprusside (SNP), saponin, phospho–p38 (Thr180/Tyr182) MAPK antibody were from Sigma (Deisenhofen, Germany); thrombin was from Roche (Mannheim, Germany); DEA-NO was from Alexis Biochemicals (Lörrach, Germany); collagen was from Nycomed (Linz, Austria); tat peptide was from Antibodies-online (Aachen, Germany); U0126 was from Calbiochem (Schwalbach, Germany); phospho-VASP-Ser239, PKG Iα, PKG Iβ antibodies were from Nanotools (Teningen, Germany); PKC substrate MARCKS phospho-Ser159/163 antibody was from Epitomics (Hamburg, Germany); phospho-ERK (Thr202/Tyr20), total p38, total ERK, phospho-PKB-Ser473, total PKB antibodies were from Cell Signaling (Frankfurt am Main, Germany)
Results
In vitro characterization of (D)-DT-2 effects on PKG (Iα, Iβ, II) and PKA activity
The oligopeptides DT-2 (Dostmann et al., 2000) and (D)-DT-2 (Nickl et al., 2010) have been characterized in vitro as highly specific inhibitors (IC50= 12.0 ± 0.8 nM) for 2 nM of PKG Iα (selectivity of more than 15 000-fold compared to PKA). Using in vitro PK assays, we tested the commercially available oligopeptide (D)-DT-2 for its ability to inhibit all known PKG isoforms (Iα, Iβ and II) and the catalytic subunit of PKA. (D)-DT-2 concentration-dependently and equally inhibited PKG Iα and PKG Iβ, (Figure 1A, B), but did not inhibit PKG II (Figure 1C) or PKA (Figure 1D), even at high concentrations. Calculated IC50 values for 2 nM of PKG Iα and PKG Iβ were 9 ± 2 nM and 7.5 ± 1.8 nM, respectively, indicating that (D)-DT-2 concentrations should be at least three times higher than the kinase concentration to reach half maximal inhibition.
Figure 1.
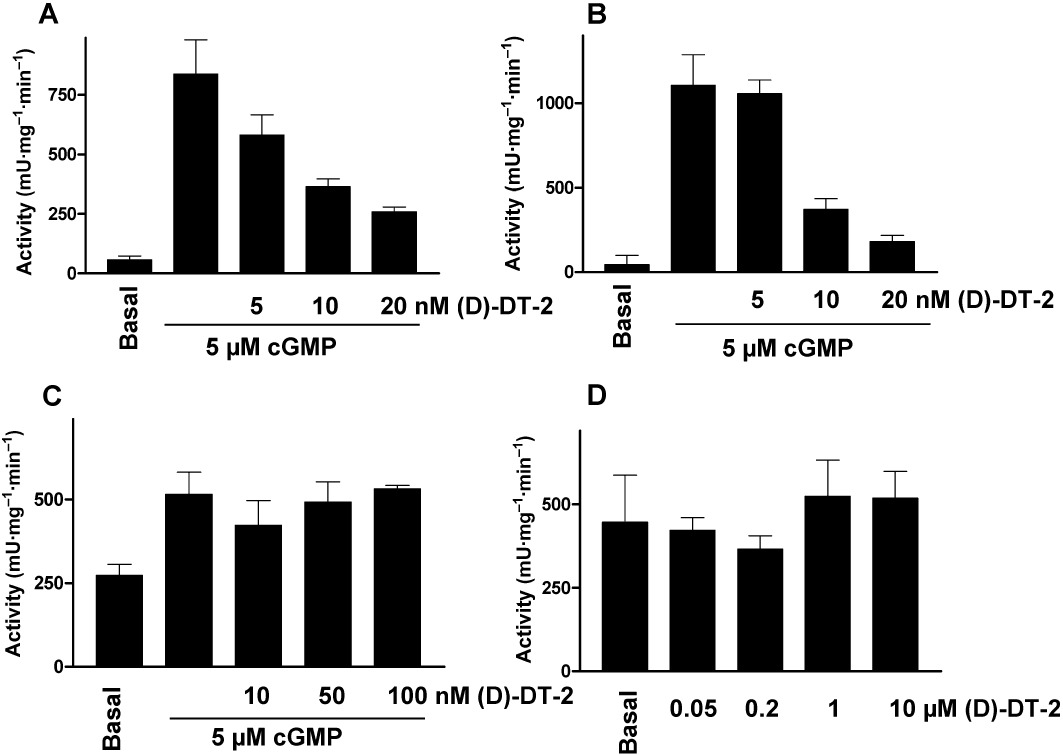
In vitro, (D)-DT-2 selectively inhibits PKG Iα and PKG Iβ but not PKG II or PKA. Indicated concentrations of (D)-DT-2 were added to purified (A) PKG Iα, (B) PKG Iβ, (C) PKG II and (D) PKAc (2 nM each). PKGs were stimulated by addition of 5 µM cGMP (PKAc is catalytically active). Kinase activity was determined by phosphorylation of Vasptide (for PKGs) or Kemptide (for PKAc), both used at 20 µM.
In human platelets, only PKG Iβ is expressed at a concentration of 7.3 ± 0.8 µM (Eigenthaler et al., 1992). In RMC and in NRVM, no PKG Iβ was detected, while the concentrations of PKG Iα were 0.36 ± 0.03 µM for RMC and 0.30 ± 0.02 µM for NRVM, as calculated by Western blotting according to the standard concentrations of the purified kinase (Figure 2A) and by measuring cell volume by confocal microscopy (Figure 2B, C). Therefore, we used micromolar concentrations of (D)-DT-2 in all other experiments. Next, we tested whether the selectivity of (D)-DT-2 for PKG Iα/β was still present in cell homogenates. We used whole cell homogenates from human platelets, which express PKG Iβ, and RMC, which express PKG Iα, to assess PKG and PKA activity by phosphorylation of the well-known PKG and PKA substrate vasodilator-stimulated phosphoprotein (VASP). Phosphorylation of VASP at Ser157, the preferred PKA site, alters the apparent molecular mass of VASP on SDS/PAGE from 46 kDa to 50 kDa, whereas phosphorylation on Ser239, the preferred site for PKG, can be analysed by phospho-Ser239-specific monoclonal VASP antibodies, which can be used as a marker of PKG activity in vitro and in intact cells (Smolenski et al., 1998a). However, both kinases (PKA and PKG) can phosphorylate VASP at Ser239 and Ser157. Unexpectedly, in contrast to purified kinases (Figure 1), in both cases (homogenates from platelets and RMC), (D)-DT-2 lost its specificity and potently and concentration-dependently inhibited both PKG and PKA activities (Figure 3).
Figure 2.

Quantitative characterization of PKG expression in RMC and NRVM. Indicated concentrations of purified PKG Iα/β and cells were loaded on the gel and processed for Western blotting. Blots were scanned using SilverFast software and analysed densitometrically by NIH Image J software for uncalibrated optical density. Cell volumes calculated from confocal 3D stacks (representative microscopy images are shown in B and C. Scale bars, 10 µM) were 6.3 pL for RMC and 3.5 pL for NRCM. In both cell types, only PKG Iα is expressed in concentrations of 0.36 ± 0.03 µM for RMC and 0.30 ± 0.02 µM for NRVM.
Figure 3.
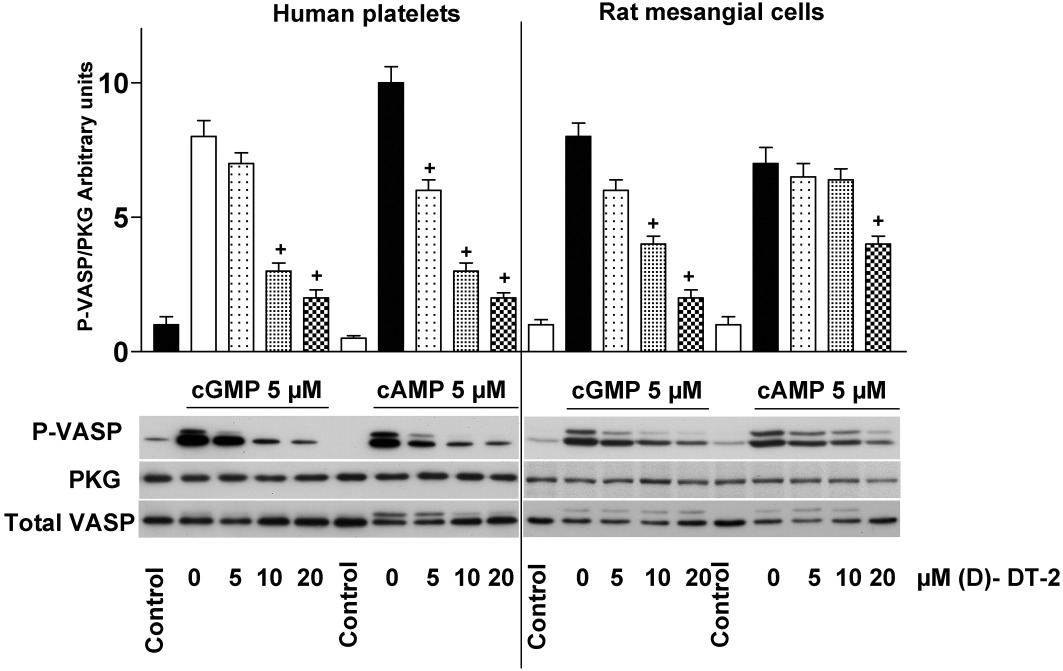
(D)-DT-2 loses its selectivity against PKG and PKA in cell homogenates. Lysates of human platelets or RMC were pre-incubated (10 min) with indicated concentrations of (D)-DT-2. PKG and PKA were stimulated by cGMP or cAMP, respectively (for 5 min, 5 µM each), Samples were processed for Western blotting. PKG activity was evaluated by VASP-Ser239 phosphorylation (P-VASP). The apparent shift of the VASP signal from 46 kDa to 50 kDa is due to phosphorylation at Ser157. PKG I and total VASP expression are served as loading control. Blots were scanned using SilverFast software and analysed densitometrically by NIH Image J software for uncalibrated optical density. The intensity of P-VASP signal was normalized to the total VASP signal, and then this ratio for each sample was expressed relative to the control which was designated as unity. Results are mean ± SEM, n= 5, +P < 0.05 (Student's t-test). In contrast to in vitro assays (Figure 1), (D)-DT-2 inhibits both PKA and PKG activation in the whole cell lysate.
Experiments on intact washed human platelets
In intact human platelets, PKG Iβ concentration has been calculated as 7.3 µM (Eigenthaler et al., 1992); therefore, we used very high (up to 100 µM) concentrations of (D)-DT-2 to evaluate the inhibition of PKG in intact platelets (Figure 4A). As fluorescein-labelled DT-2 was translocated into smooth muscle cells within 30 min (Dostmann et al., 2000), we also incubated our cells with (D)-DT-2 for up to 30 min (Figure 4B). In both cases, (D)-DT-2 failed to inhibit DEA-NO-stimulated PKG activity assessed by phosphorylation of the established PKG substrates VASP and PDE5. Next, we tested whether (D)-DT-2 could inhibit basal (unstimulated) PKG activity and its effect on several other PKs including MAP kinases (p38 and ERK), PKC and PKB, which are important markers of platelet activation. Platelets were stimulated by thrombin or collagen, which do not activate PKG, and platelet activation was assessed with the PAC-1 antibody which binds only to activated integrin αIIbβ3. Kinase activities were evaluated by phospho-specific antibodies which recognize activated forms of kinases (p38, ERK, PKB), or by phosphorylation of the established kinase substrates (VASP for PKG and MARKS for PKC). (D)-DT-2 had no effect on the basal PKG activity, but concentration-dependently inhibited thrombin-stimulated activation of p38, ERK, PKB and PKC (Figure 5A). Inhibition of these kinases correlates with the inhibition of integrin activation, aggregation (Figure 5B) and calcium mobilization (Figure 5C). In collagen-stimulated platelets, (D)-DT-2 had an opposite effect on platelet activation. Starting from 5 µM, (D)-DT-2 enhanced PKB and PKC activity and PAC-1 binding (Figure 5A), which corresponds to an increased platelet aggregation (Figure 5D). However, (D)-DT-2 had no effect on collagen-induced calcium mobilization (Figure 5E).
Figure 4.
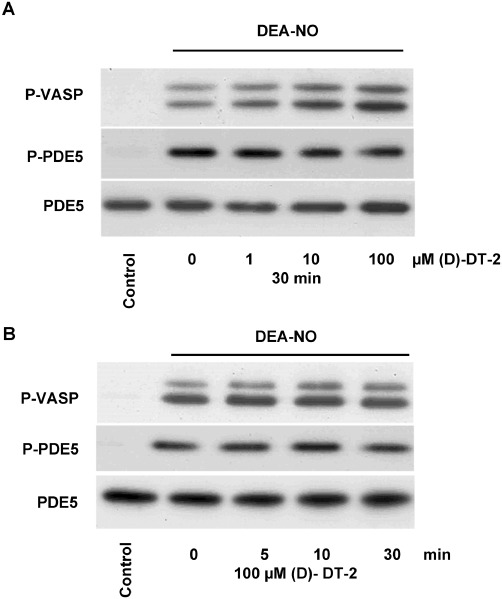
(D)-DT-2 does not inhibit PKG activity in intact human platelets. 100 µL of washed human platelets (2 × 108 cells·mL−1) were pre-incubated with indicated concentrations of (D)-DT-2 for 30 min (A), or with 100 µM of (D)-DT-2 for indicated time points (B). PKG was activated by DEA-NO (1 µM, 1 min). Samples were processed for Western blotting and PKG activity was evaluated by VASP-Ser239 (P-VASP) and PDE5-Ser92 (PDE5) phosphorylation. PDE5 expression served as a loading control. The results are representative of four independent experiments.
Figure 5.
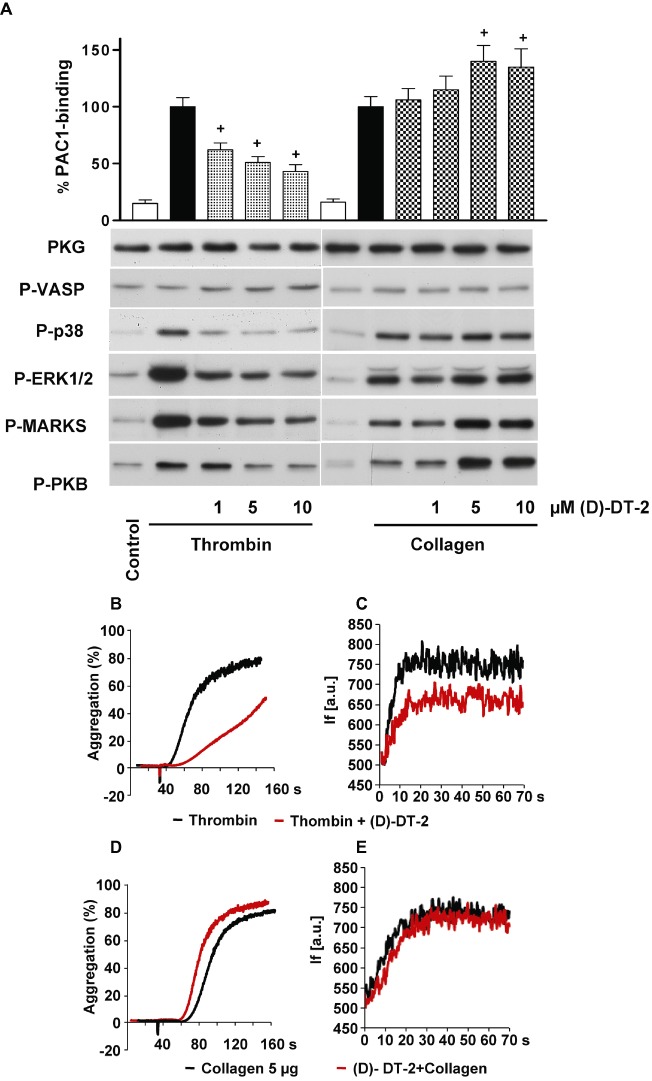
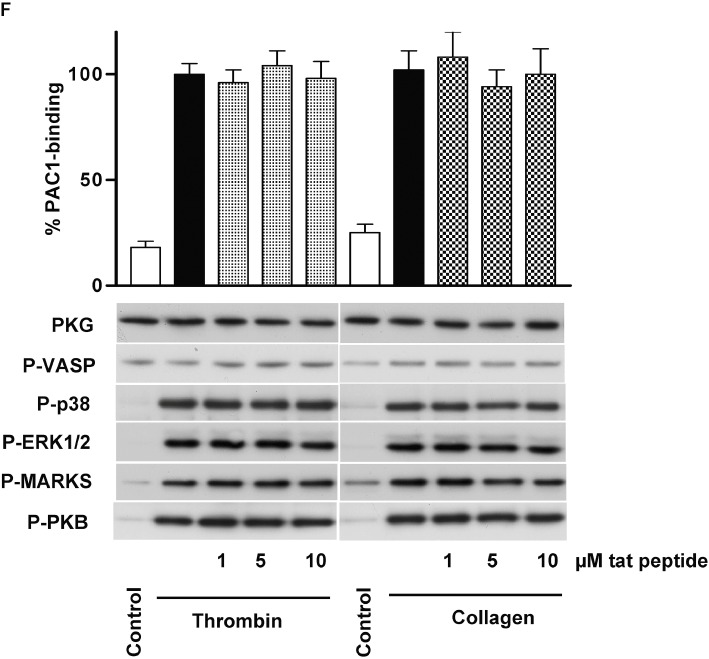
(D)-DT-2 PKG-independently inhibits thrombin-stimulated and enhances collagen-stimulated platelet activation. (A) Human washed platelets were pre-incubated with indicated concentrations of (D)-DT-2 for 10 min and then stimulated with thrombin (0.01 U·mL−1, 1 min) or collagen (5 µg·mL−1, 1 min). 20 µL of platelet suspension was used for FACS analysis of integrin αIIbβ3 activation (PAC-1 binding). The rest of the samples were processed for Western blotting. (D)-DT-2 does not inhibit basal PKG activity in both cases (P-VASP panel); however, it strongly and concentration-dependently inhibits thrombin-stimulated platelet activation assessed by integrin activation (PAC-1 binding). In addition, (D)-DT-2 inhibits MAP kinases (P-p38 and P-ERK1/2 panels), PKC (inhibition of established PKC substrate, P-MARKS panel) and PKB (P-PKB panel). In collagen-stimulated platelets, (D)-DT-2 starting from 5 µM significantly enhances integrin activation which corresponds to increased activation of PKC and PKB. These data correspond to (D)-DT-2 effects on platelet aggregation (B) and calcium mobilization (C) induced by thrombin, or collagen (D, E). Pre-incubation of (D)-DT-2 (10 µM, 10 min) strongly inhibits thrombin-induced platelet aggregation (B) and calcium mobilization (C) and slightly enhances collagen-induced aggregation (D), without affecting calcium mobilization (E). (F) shows the same experiment as in (A) except that (D)-DT-2 was replaced by the tat peptide. All results are representative of at least three independent experiments. Data on PAC-1 binding (A, F) are means ± SEM, n= 4, +P < 0.05 (anova followed by Bonferroni's test).
In additional experiments performed with the tat peptide, which was added to the (D)-DT-2 sequence to make it cell permeable (Nickl et al., 2010), we could show that (D)-DT-2 effects on platelets were not mediated by the tat-peptide, but were directly connected with (D)-DT-2 (Figure 5F). Our data clearly indicated that (D)-DT-2 did not inhibit PKG activity in intact platelets but, unexpectedly, did inhibit (in the case of thrombin stimulation) or enhance (after collagen stimulation) the activation of several other PKs (p38, ERK, PKC, PKB).
Experiments on RMC and NRVM
To test the ability of (D)-DT-2 to inhibit PKG Iα in intact cells, we used RMC and NRVM which only express PKG Iα at concentrations of 0.36 µM and 0.30 µM, respectively. In these cells, PKG is activated by stimulation of two different receptors: membrane-bound GC-A (by ANP) and soluble GC (by SNP). Activation of both receptors similarly stimulated PKG activity (Figure 6A, B). Pre-incubation with (D)-DT-2 (20 µM, for 30 min) had no effect on PKG activity (as evident from P-VASP blot). However, it slightly potentiated basal and thrombin-induced ERK activation in RMC Figure 6A), whereas in NRVM, it strongly, and comparable to the specific ERK inhibitor (U0126), inhibited ERK activation (P-ERK blot). Next, we tested whether the lack of PKG inhibition in RMC originates from a limited diffusion of the peptide across the cell membrane. RMC were permeabilized by 0.001% saponin, and PKG or PKA were activated by cGMP or cAMP, respectively. In intact cells, cGMP and cAMP added to the media did not stimulate PKG or PKA, while in permeabilized cells, both kinases were strongly activated by the cyclic nucleotides (Figure 6C). Pre-incubation with (D)-DT-2 (20 µM, 30 min), in contrast to the cell lysates (compare with Figure 3), did not inhibit PKG or PKA activity in permeabilized cells (Figure 6C).
Figure 6.
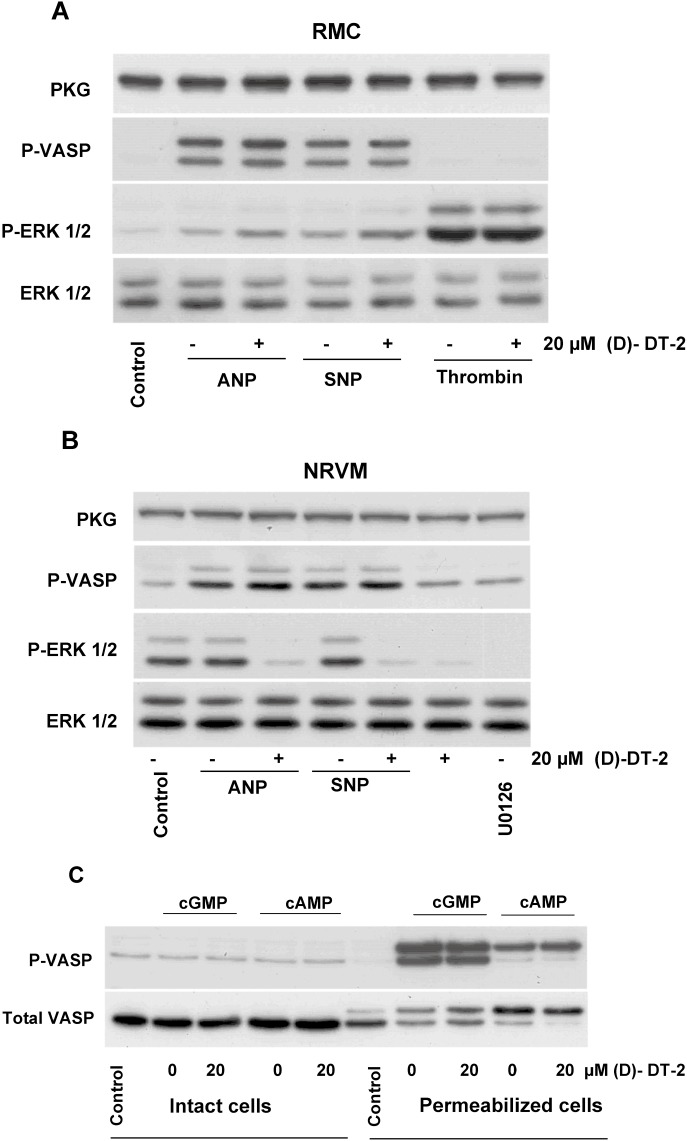
(D)-DT-2 does not inhibit PKG activation in intact and permeabilized RMC and in NRVM. (A) Intact RMC, (B) NRVM and (C) saponin-permeabilized RMC were pre-incubated for 30 min with 20 µM of (D)-DT-2 or ERK inhibitor U0126 (only for NRVM, 1 µM, 5 min) before 5 min stimulation with ANP (10 nM), SNP (5 µM), thrombin (0.1 U·mL−1) (in A), cGMP or cAMP (in C). Cells were washed with PBS and processed for Western blot analysis for VASP and ERK phosphorylation and PKG expression. In intact RMC, (D)-DT-2 does not inhibit PKG activity (P-VASP panel) and slightly enhances basal and thrombin-induced ERK1/2 phosphorylation. In NRVM, (D)-DT-2 also does not inhibit PKG activity (P-VASP panel). However, in contrast to RMC (compared to A), it potently (comparable to the specific ERK inhibitor U0126) inhibits ERK phosphorylation. In permeabilized RMC (C), in contrast to cell lysate (compare with Figure 3), (D)-DT-2 did not inhibit PKG or PKA activated by cGMP or cAMP, respectively. Incubation of non-permeabilized cells with cGMP or cAMP does not activate PKG or PKA. The data shown are representative of four independent experiments using different cell preparations.
FRET experiments in HEK 293 cells
To directly identify possible inhibitory effects of (D)-DT-2 in living cells, we used a FRET-based PKA substrate sensor AKAR3 (Zhang et al., 2005; Allen and Zhang, 2006), which in our experiments, was also phosphorylated by PKG Iβ in transfected HEK293 cells. HEK293 cells do not express detectable amounts of PKG I and GC-A. Therefore, in addition to AKAR3, the cells were co-transfected with GC-A and PKG Iβ. The specificity of AKAR3 phosphorylation by PKG was tested in experiments where the dominant-negative PKG (dnPKG Iβ) was transfected instead of active PKG Iβ. Activation of GC-A by ANP strongly enhanced the FRET signal of AKAR3, without any detectable changes in FRET response in the cells transfected with dnPKG Iβ. Again, pre-incubation with (D)-DT-2 did not inhibit PKG activity measured with the FRET sensors in these cells (Figure 7).
Figure 7.
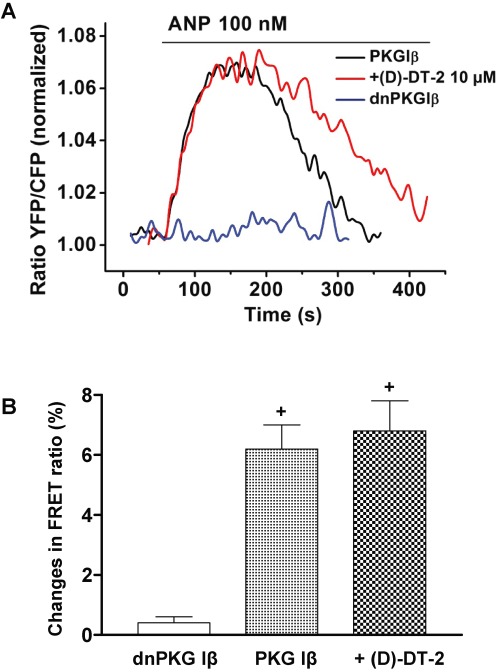
(D)-DT-2 does not inhibit the ANP-induced FRET response of the PKA/PKG activity sensor AKAR3. HEK293 cells transfected with AKAR3, GC-A and active PKG Iβ, or dominant-negative dnPKG Iβ plasmids, were stimulated with 100 nM ANP. FRET responses were recorded from single cells and compared with those measured in cells after 30 min pre-incubation with 10 µM (D)-DT-2. Representative FRET recordings are shown in (A). Quantification of the FRET data is in (B). Data shown are means ± SEM, n= 5, +P < 0.05 (Student's t-test).
Discussion
The main function of PKs is the phosphorylation of target proteins (substrates) by covalent addition of inorganic phosphate to serine, threonine or tyrosine at a distinct consensus substrate sequence. Therefore, inhibition of specific substrate phosphorylation in intact cells should be regarded as the only reliable criteria for PK inhibitors. Unfortunately, in many, if not most, of the studies involving PKG inhibitors, conclusions about the mediation by PKG of the effects of cGMP are drawn from the blockade or abolition of these effects. Such conclusions solely rely on the observed functional effects of the inhibitors used, without prior demonstration of their specificity for PKG and inhibition of PKG substrate phosphorylation. From the updated list of PKG substrates presented in a recent review (Francis et al., 2010), it becomes evident that each cell type contains at least some of the identified PKG substrates, and against some of them, including VASP, LASP, PDE5, IRAG, phospho-specific antibodies are available. Among them, VASP, which is expressed in many tissues and cell types (Gambaryan et al., 2001), is one of the best characterized PKG and PKA substrates (Butt et al., 1994). A peptide, corresponding to the PKG phosphorylation site of VASP (LRKVSKQE, also called Vasptide) is used as a substrate for in vitro determination of purified PKG activity (Butt et al., 1994). The oligopeptides DT-2 (YGRKKRRQRRRPP-LRKKKKKH, sequence of DT-2 is in bold) and (D)-DT-2 are stable against proteolysis (Nickl et al., 2010) and were developed from the peptide library as pseudo-substrates for PKG which can bind to the substrate binding site of the PKG catalytic domain and competitively inhibit PKG activity. High selectivity of DT-2 for PKG over PKA was shown by in vitro assays. However, the selectivity of DT-2 against other Ser/Thr PKs has not been investigated. In addition, the in vivo inhibitory potential of DT compounds was shown only at the functional level (Dostmann et al., 2000; Krieg et al., 2005), without demonstrating inhibition of PKG-specific substrate phosphorylation. We chose (D)-DT-2 as the best characterized PKG inhibitor to analyse PKG-specific effects in both cell lysate and intact cells. We used human platelets, RMC and cardiac myocytes (NRVM) because these cells have been used earlier by us (Burkhardt et al., 2000; Gambaryan et al., 2004) and others (Wyatt et al., 1991; Valtcheva et al., 2009) to demonstrate that the PKG inhibitors KT5823 (competitive binding at ATP-site) and Rp-cGMPS (competitive binding at cGMP binding sites) did not inhibit the kinase but had some PKG-independent functional effects.
(D)-DT-2 is a highly specific inhibitor for purified PKG Iα/β enzymes which does not inhibit PKA in vitro (Figure 1). The calculated IC50 values for PKG Iα/β in these experiments are in agreement with the published data (Dostmann et al., 2000). Binding of (D)-DT-2 to PKG Iα was mapped by photoaffinity cross linking to the catalytic core on residues 356–372, also known as the glycine-rich loop which is essential for ATP binding (Pinkse et al., 2009). This domain is highly conserved in all species and homologous between various PKG isoforms (Uhler, 1993). Nevertheless, PKG II activity is not blocked by (D)-DT-2. Differences in binding affinity are also observed in the highly homologous cyclic nucleotide-binding domains for cGMP that vary from 0.07 µM for PKG II to 0.1 µM for PKG Iα and 0.9 µM for PKG Iβ and is most likely due to structural differences between the isoforms (Osborne et al., 2011). Therefore, it is tempting to speculate that there might be also some structural differences in the catalytic site of all three PKGs that would allow differentiation between substrates. An example of such differences in substrate recognition is a novel PKG substrate, LASP-1. While VASP is phosphorylated by all three PKGs to similar extent, LASP-1 is preferred by PKG Iβ (Butt et al., 2003). As the (D)-DT-2 peptide was designed and screened for PKG Iα inhibition (Dostmann et al., 2000), the observed preferential inhibitory potential for PKG I is possible.
However, in intact human platelets, expressing PKG Iβ, even at very high (up to 100 µM) concentrations and at up to 30 min incubation time, which is sufficient for this peptide to enter the cells (Dostmann et al., 2000), (D)-DT-2 did not inhibit the PKG-mediated phosphorylation of the established PKG substrates VASP and PDE5 (Figure 4). In contrast to intact platelets, in whole cell lysates, (D)-DT-2 dose-dependently inhibited PKG activity. However, even in this case, it cannot be used as PKG-specific inhibitor, because it also inhibits PKA activity. Next, we tested whether (D)-DT-2 inhibited basal PKG activity and/or interacted with other PKs. We used platelet agonists (thrombin, collagen) which do not activate PKG but stimulate the activity of several other PKs. Surprisingly, we found that this ‘highly specific’ PKG inhibitor modulated other kinases such as PKC, PKB and MAP kinases (Figure 5).
We can only speculate about the exact mechanism of (D)-DT-2 mediated inhibition of thrombin-activated platelets or the potentiation of collagen-activated platelets. However, these effects are obviously not connected with the tat peptide, because the tat peptide itself had no effect on platelets used under the same conditions (Figure 5F). Possibly, in platelets, (D)-DT-2 can interact with thrombin receptors, thereby inhibiting thrombin-induced platelet activation, whereas binding to collagen receptors might further potentiate collagen-induced platelet activation. In contrast, experiments on RMC and NRVM, where (D)-DT-2 also PKG-independently and unexpectedly potentiated (RMC, Figure 6A) or inhibited (NRVM, Figure 6B) basal ERK activity, suggested that these effects of (D)-DT-2 might be intracellular and not connected to cell membrane receptors. Importantly, in NRVM, the oligopeptide inhibited basal ERK activity (Figure 6B) potently (comparable to the specific ERK inhibitor U0126) but independently of PKG.
One possible explanation for this cross-inhibition might be the high similarity of the catalytic domain between Ser/Thr kinases. They all contain a common catalytic domain with an ATP-binding site characterized by the Rossmann fold motif GXGXXG and a substrate recognition site. Binding of (D)-DT-2 to PKG Iα was mapped by photoaffinity cross linking to the catalytic core on residues 356–372, which is essential for ATP binding (Pinkse et al., 2009). This domain is highly homologous among Ser/Thr kinases and conserved between all three PKGs (Uhler, 1993). Another important aspect, which was not taken into account by the developers of (D)-DT-2, is the substrate specificity of the kinases. Several Ser/Thr kinases including PKA, PKG, PKC, PKB, Pim1, RSK1, SLK1, DMPK and ZIPK have a conspicuous overlapping substrate specificity with a preference for basic amino acids N-terminally at positions −2 and −3 from the phosphorylated serine or threonine residue and varying amino acids at position −1 (Miller and Blom, 2009). A further disadvantage of the (D)-DT-2 is its competition with other PKG substrates in intact cells. To date, more than 50 PKG substrates have been identified (Francis et al., 2010). They are expressed at various intracellular concentrations ranging from nanomolar amounts up to 25 µM for VASP (Eigenthaler et al., 1992) and 18 µM for PDE5 in human platelets (Wangorsch et al., 2011). Accordingly, (D)-DT-2 as a PKG inhibitor should compete with many endogenous substrates. Therefore, it is not surprising that (D)-DT-2 is not a specific PKG inhibitor in intact cells and can modulate the activity of several other Ser/Thr kinases. All the data reviewed above indicate that the use of peptide-based pseudo-substrates as PK inhibitors for intact cells is probably not the correct approach.
In summary, we showed that the ‘highly specific’ PKG inhibitor (D)-DT-2 was even less specific than Rp-cGMPS or KT5823 because (i) PKG activity was not inhibited in several types of intact cells; (ii) PKG specificity over PKA was lost in whole cell homogenates; and (iii) PKG-independent inhibition/stimulation of other PKs was rather unpredictable. Therefore, replacing PKA by PKG in the title of the recent comprehensive review on PKA inhibitors (Murray, 2008), we can clearly affirm that ‘Pharmacological PKG inhibition: all may not be what it seems’.
Acknowledgments
This study was supported by the DFG (SFB688, GA 1561/1-1) and Ministry of Education and Science of Russian Federation (grant 14.740.11.0918).
Glossary
- ANP
atrial natriuretic peptide
- GC-A receptor
guanylyl cyclase-A receptor
- NRVM
neonatal rat ventricular myocytes
- RMC
rat mesangial cells
- SNP
sodium nitroprusside
- VASP
vasodilator-stimulated phosphoprotein
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (5th Edition) 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 2006;348:716–721. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ, et al. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc. 2011;6:427–438. doi: 10.1038/nprot.2010.198. [DOI] [PubMed] [Google Scholar]
- Burkhardt M, Glazova M, Gambaryan S, Vollkommer T, Butt E, Bader B, et al. KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J Biol Chem. 2000;275:33536–33541. doi: 10.1074/jbc.M005670200. [DOI] [PubMed] [Google Scholar]
- Butt E, Abel K, Krieger M, Palm D, Hoppe V, Hoppe J, et al. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269:14509–14517. [PubMed] [Google Scholar]
- Butt E, Gambaryan S, Gottfert N, Galler A, Marcus K, Meyer HE. Actin binding of human LIM and SH3 protein is regulated by cGMP- and cAMP-dependent protein kinase phosphorylation on serine 146. J Biol Chem. 2003;278:15601–15607. doi: 10.1074/jbc.M209009200. [DOI] [PubMed] [Google Scholar]
- Castro LR, Schittl J, Fischmeister R. Feedback control through cGMP-dependent protein kinase contributes to differential regulation and compartmentation of cGMP in rat cardiac myocytes. Circ Res. 2010;107:1232–1240. doi: 10.1161/CIRCRESAHA.110.226712. [DOI] [PubMed] [Google Scholar]
- Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C. Physiology of kidney renin. Physiol Rev. 2010;90:607–673. doi: 10.1152/physrev.00011.2009. [DOI] [PubMed] [Google Scholar]
- Dostmann WR, Taylor MS, Nickl CK, Brayden JE, Frank R, Tegge WJ. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Ialpha inhibit NO-induced cerebral dilation. Proc Natl Acad Sci U S A. 2000;97:14772–14777. doi: 10.1073/pnas.97.26.14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigenthaler M, Nolte C, Halbrugge M, Walter U. Concentration and regulation of cyclic nucleotides, cyclic-nucleotide-dependent protein kinases and one of their major substrates in human platelets. Estimating the rate of cAMP-regulated and cGMP-regulated protein phosphorylation in intact cells. Eur J Biochem. 1992;205:471–481. doi: 10.1111/j.1432-1033.1992.tb16803.x. [DOI] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaryan S, Hausler C, Markert T, Pohler D, Jarchau T, Walter U, et al. Expression of type II cGMP-dependent protein kinase in rat kidney is regulated by dehydration and correlated with renin gene expression. J Clin Invest. 1996;98:662–670. doi: 10.1172/JCI118837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaryan S, Wagner C, Smolenski A, Walter U, Poller W, Haase W, et al. Endogenous or overexpressed cGMP-dependent protein kinases inhibit cAMP-dependent renin release from rat isolated perfused kidney, microdissected glomeruli, and isolated juxtaglomerular cells. Proc Natl Acad Sci U S A. 1998;95:9003–9008. doi: 10.1073/pnas.95.15.9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaryan S, Hauser W, Kobsar A, Glazova M, Walter U. Distribution, cellular localization, and postnatal development of VASP and Mena expression in mouse tissues. Histochem Cell Biol. 2001;116:535–543. doi: 10.1007/s00418-001-0353-3. [DOI] [PubMed] [Google Scholar]
- Gambaryan S, Butt E, Marcus K, Glazova M, Palmetshofer A, Guillon G, et al. cGMP-dependent protein kinase type II regulates basal level of aldosterone production by zona glomerulosa cells without increasing expression of the steroidogenic acute regulatory protein gene. J Biol Chem. 2003;278:29640–29648. doi: 10.1074/jbc.M302143200. [DOI] [PubMed] [Google Scholar]
- Gambaryan S, Geiger J, Schwarz UR, Butt E, Begonja A, Obergfell A, et al. Potent inhibition of human platelets by cGMP analogs independent of cGMP-dependent protein kinase. Blood. 2004;103:2593–2600. doi: 10.1182/blood-2003-09-3349. [DOI] [PubMed] [Google Scholar]
- Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem. 2010;285:18352–18363. doi: 10.1074/jbc.M109.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebe C, Klingebiel TM, Grau SP, Toischer K, Didie M, Jacobshagen C, et al. Enhanced expression of DYRK1A in cardiomyocytes inhibits acute NFAT activation but does not prevent hypertrophy in vivo. Cardiovasc Res. 2011;90:521–528. doi: 10.1093/cvr/cvr023. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Bernhard D, Lukowski R, Weinmeister P. cGMP regulated protein kinases (cGK) Handb Exp Pharmacol. 2009;191:137–162. doi: 10.1007/978-3-540-68964-5_8. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK, Jennings KR, Strumwasser F, Nairn AC, Walter U, Wilson FD, et al. Microinjection of catalytic subunit of cyclic AMP-dependent protein kinase enhances calcium action potentials of bag cell neurons in cell culture. Proc Natl Acad Sci U S A. 1980;77:7487–7491. doi: 10.1073/pnas.77.12.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisphosphate receptor. Cyclic GMP-dependent protein kinase mediates cAMP and cGMP dependent phosphorylation in the intact rat aorta. J Biol Chem. 1996;271:21933–21938. doi: 10.1074/jbc.271.36.21933. [DOI] [PubMed] [Google Scholar]
- Krieg T, Philipp S, Cui L, Dostmann WR, Downey JM, Cohen MV. Peptide blockers of PKG inhibit ROS generation by acetylcholine and bradykinin in cardiomyocytes but fail to block protection in the whole heart. Am J Physiol Heart Circ Physiol. 2005;288:H1976–H1981. doi: 10.1152/ajpheart.00883.2004. [DOI] [PubMed] [Google Scholar]
- Kurtz A. Renin release: sites, mechanisms, and control. Annu Rev Physiol. 2011;73:377–399. doi: 10.1146/annurev-physiol-012110-142238. [DOI] [PubMed] [Google Scholar]
- Lohmann SM, Walter U. Tracking functions of cGMP-dependent protein kinases (cGK) Front Biosci. 2005;10:1313–1328. doi: 10.2741/1621. [DOI] [PubMed] [Google Scholar]
- MacFarland RT, Zelus BD, Beavo JA. High concentrations of a cGMP-stimulated phosphodiesterase mediate ANP-induced decreases in cAMP and steroidogenesis in adrenal glomerulosa cells. J Biol Chem. 1991;266:136–142. [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika D, Leroy J, Vandecasteele G, Fischmeister R. PDEs create local domains of cAMP signaling. J Mol Cell Cardiol. 2012;52:323–329. doi: 10.1016/j.yjmcc.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Miller ML, Blom N. Kinase-specific prediction of protein phosphorylation sites. Methods Mol Biol. 2009;527:299–310. doi: 10.1007/978-1-60327-834-8_22. [DOI] [PubMed] [Google Scholar]
- Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:re4. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- Nickl CK, Raidas SK, Zhao H, Sausbier M, Ruth P, Tegge W, et al. (D)-Amino acid analogues of DT-2 as highly selective and superior inhibitors of cGMP-dependent protein kinase Ialpha. Biochim Biophys Acta. 2010;1804:524–532. doi: 10.1016/j.bbapap.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Gambaryan S, Engelhardt S, Walter U, Lohse MJ. Real-time monitoring of the PDE2 activity of live cells: hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. J Biol Chem. 2005;280:1716–1719. doi: 10.1074/jbc.C400505200. [DOI] [PubMed] [Google Scholar]
- Osborne BW, Wu J, McFarland CJ, Nickl CK, Sankaran B, Casteel DE, et al. Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure. 2011;19:1317–1327. doi: 10.1016/j.str.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkse MW, Rijkers DT, Dostmann WR, Heck AJ. Mode of action of cGMP-dependent protein kinase-specific inhibitors probed by photoaffinity cross-linking mass spectrometry. J Biol Chem. 2009;284:16354–16368. doi: 10.1074/jbc.M808521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohler D, Butt E, Meissner J, Muller S, Lohse M, Walter U, et al. Expression, purification, and characterization of the cGMP-dependent protein kinases I beta and II using the baculovirus system. FEBS Lett. 1995;374:419–425. doi: 10.1016/0014-5793(95)01168-e. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr Assays of protein kinase. Methods Enzymol. 1983;99:3–6. doi: 10.1016/0076-6879(83)99034-1. [DOI] [PubMed] [Google Scholar]
- Smolenski A, Bachmann C, Reinhard K, Honig-Liedl P, Jarchau T, Hoschuetzky H, et al. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J Biol Chem. 1998a;273:20029–20035. doi: 10.1074/jbc.273.32.20029. [DOI] [PubMed] [Google Scholar]
- Smolenski A, Burkhardt AM, Eigenthaler M, Butt E, Gambaryan S, Lohmann SM, et al. Functional analysis of cGMP-dependent protein kinases I and II as mediators of NO/cGMP effects. Naunyn Schmiedebergs Arch Pharmacol. 1998b;358:134–139. doi: 10.1007/pl00005234. [DOI] [PubMed] [Google Scholar]
- Smolenski A, Poller W, Walter U, Lohmann SM. Regulation of human endothelial cell focal adhesion sites and migration by cGMP-dependent protein kinase I. J Biol Chem. 2000;275:25723–25732. doi: 10.1074/jbc.M909632199. [DOI] [PubMed] [Google Scholar]
- Uhler MD. Cloning and expression of a novel cyclic GMP-dependent protein kinase from mouse brain. J Biol Chem. 1993;268:13586–13591. [PubMed] [Google Scholar]
- Valtcheva N, Nestorov P, Beck A, Russwurm M, Hillenbrand M, Weinmeister P, et al. The commonly used cGMP-dependent protein kinase type I (cGKI) inhibitor Rp-8-Br-PET-cGMPS can activate cGKI in vitro and in intact cells. J Biol Chem. 2009;284:556–562. doi: 10.1074/jbc.M806161200. [DOI] [PubMed] [Google Scholar]
- Walter U, Gambaryan S. cGMP and cGMP-dependent protein kinase in platelets and blood cells. Handb Exp Pharmacol. 2009;191:533–548. doi: 10.1007/978-3-540-68964-5_23. [DOI] [PubMed] [Google Scholar]
- Walter U, Miller P, Wilson F, Menkes D, Greengard P. Immunological distinction between guanosine 3′:5′-monophosphate-dependent and adenosine 3′:5′-monophosphate-dependent protein kinases. J Biol Chem. 1980;255:3757–3762. [PubMed] [Google Scholar]
- Wangorsch G, Butt E, Mark R, Hubertus K, Geiger J, Dandekar T, et al. Time-resolved in silico modeling of fine-tuned cAMP signaling in platelets: feedback loops, titrated phosphorylations and pharmacological modulation. BMC Syst Biol. 2011;5:178. doi: 10.1186/1752-0509-5-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt TA, Pryzwansky KB, Lincoln TM. KT5823 activates human neutrophils and fails to inhibit cGMP-dependent protein kinase phosphorylation of vimentin. Res Commun Chem Pathol Pharmacol. 1991;74:3–14. [PubMed] [Google Scholar]
- Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437:569–573. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]