

Abstract
BACKGROUND AND PURPOSE
The purpose of the study was to investigate renal endothelium-dependent vasodilatation in a model of severe hypertension associated with kidney injury.
EXPERIMENTAL APPROACH
Changes in perfusion pressure were measured in isolated, perfused kidneys taken from 18-week-old Wistar–Kyoto rat (WKY), spontaneously hypertensive rats (SHR) and SHR treated for 2 weeks with Nω-nitro-L-arginine methyl ester in the drinking water (L-NAME-treated SHR, 6 mg·kg−1·day−1).
KEY RESULTS
Acetylcholine caused similar dose-dependent renal dilatation in the three groups. In vitro administration of indomethacin did not alter the vasodilatation, while the addition of Nw-nitro-L-arginine (L-NA) produced a differential inhibition of the vasodilatation, (inhibition in WKY > SHR > L-NAME-treated SHR). Further addition of ODQ, an inhibitor of soluble guanylyl cyclase, abolished the responses to sodium nitroprusside but did not affect the vasodilatation to acetylcholine. However, the addition of TRAM-34 (or charybdotoxin) inhibitors of Ca2+-activated K+ channels of intermediate conductance (KCa3.1), blocked the vasodilatation to acetylcholine, while apamin, an inhibitor of Ca2+-activated K+ channels of small conductance (KCa2.3), was ineffective. Dilatation induced by an opener of KCa3.1/KCa2.3 channels, NS-309, was also blocked by TRAM-34, but not by apamin. The magnitude and duration of NS-309-induced vasodilatation and the renal expression of mRNA for KCa3.1, but not KCa2.3, channels followed the same ranking order (WKY < SHR < L-NAME-treated SHR).
CONCLUSIONS AND IMPLICATIONS
In SHR kidneys, an EDHF-mediated response, involving activation of KCa3.1 channels, contributed to the mechanism of endothelium-dependent vasodilatation. In kidneys from L-NAME-treated SHR, up-regulation of this pathway fully compensated for the decrease in NO availability.
Keywords: SHR, L-NAME chronic treatment, EDHF-mediated responses, KCa3.1, KCa2.3, perfused rat kidney
Introduction
Hypertension is a major pathological factor in the development of cardiovascular disease and in the progression towards end-stage renal disease, the incidence of both being increased during aging (Zhou and Frohlich, 2007). The aging spontaneously hypertensive rat (SHR) is an animal model exhibiting end-stage renal disease that shows some similarities with those observed in clinical hypertension (i.e. elevated renal vascular resistance, impaired renal functions and increased renal fibrosis). However, this interesting model is difficult to manage since SHR require careful husbandry and up to the age of 12–16 months when these renal dysfunctions become manifest (Zhou and Frohlich, 2007). However, SHR treated with a moderate dose of a NOS inhibitor show impaired kidney function and altered renal structures similar to those observed in untreated, ageing SHR, and hence, represents an accelerated model of hypertension associated with kidney damage (Zhou and Frohlich, 2007). This experimental model of severe hypertension and nephrosclerosis is also associated with cardiac fibrosis and arterial stiffening (Ono et al., 2001; Terata et al., 2003; Zhou and Frohlich, 2007; Vayssettes-Courchay et al., 2008; 2011; Isabelle et al., 2012).
Endothelial cells control vascular tone not only by releasing NO but also by generating other vasoactive mediators such as metabolites of arachidonic acid. In addition, they also elicit endothelium-dependent hyperpolarizations (EDHF-mediated responses; Félétou et al., 2011). In the renal vasculature, endothelium-dependent vasodilatation is an important regulatory mechanism and disruption of these processes, including the activity of various populations of potassium channels, can profoundly alter haemodynamic resistance, renal blood flow and, hence, affect glomerular filtration pressure and renal function (Benter et al., 2005; Sorensen et al., 2012). In hypertension, this endothelial dysfunction is characterized by an alteration of the balance between endothelial vasoconstrictor and vasodilator responses (Büssemaker et al., 2003; Félétou and Vanhoutte, 2006a; Michel et al., 2008). The purpose of the present work was, in the accelerated model of hypertension associated with kidney damage (i.e. SHR treated with a moderate dose of a NOS inhibitor), to study renal function and to characterise endothelial function. In isolated perfused kidneys from these SHR, the role of the various endothelial mechanisms involved in acetylcholine-induced vasodilatation was assessed, with a particular emphasis on EDHF-mediated responses.
Methods
All studies reported were performed in accordance with the European Community Guidelines for the use of experimental animals and were approved by the ethical committee on Animal Experiments of the Servier Research Institute. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 123 animals were used in the experiments described here.
Experimental procedures
Male spontaneously hypertensive rats (SHR) were purchased from Janvier (Le Genest St Isle, France). All animals were fed a standard laboratory diet and water ad libitum and were maintained in a humidity- and temperature-controlled room. Four groups of 4-month-old Wistar–Kyoto rat (WKY) and SHR were treated for 2 weeks. Group 1: control WKY rats drinking regular water; group 2: L-NAME-treated WKY rats drinking water containing 50 mg·L−1 of Nω-nitro-L-arginine methyl ester (L-NAME); group 3: control SHR drinking regular water; group 4: L-NAME-treated SHR drinking water containing 50 mg·L−1 of L-NAME (approximately 6 mg·kg−1·day−1).
In WKY rats, this moderate dose of L-NAME administered for this period of time did not significantly affect arterial blood pressure (systolic blood pressure: 140 ± 6 mm Hg, n= 10) or body weight and did not influence either the renal parameters measured or the vascular reactivity of the isolated perfused kidney. Therefore, for the sake of clarity, the data from this group of animals will not be presented (see also Figures S1 and S2).
Urine volume and protein determination
At the end of the 2 week treatment period, animals were placed in individual metabolic cages, with access to regular water or L-NAME containing water ad libitum, depending on the groups, but without food, for 24 h urine collection. The collected urine from each animal was centrifuged at 2500× g and at 4°C for 15 min, and then stored at −80°C until biochemical analysis (auto analyser Pentra 400 ABX). Urinary excretion of nitrite was measured by a colorimetric assay (Cayman Chemicals, Spibio, France).
Blood pressure measurement and blood sampling
Animals were anaesthetized with sodium pentobarbital (55 mg·kg−1, i.p.) and then placed on a homoeothermic blanket to maintain rectal temperature at 37°C. After a tracheotomy, a catheter was inserted into the right carotid artery for blood pressure measurement and blood sampling. Systolic blood pressure and heart rate were recorded through a pressure transducer (P10EZ, Statham, France) connected to a data acquisition system (Biopac 02, BIOPAC Systems Inc., CEROM, Paris, France). At the end of the blood pressure measurement, blood samples were collected into two tubes containing lithium-heparin and EDTA respectively. Then the left kidney was cannulated for perfusion pressure experiments, and the right kidney was used for histological analysis.
Iron-nitroxyl-haemoglobin (HbNO) measurement
Blood samples were collected in heparinised (50 U·mL−1) tubes and sodium dithionite (20 mg·mL−1) was added to prevent excessive oxygenation. Red blood cells were obtained by centrifugation (2500×g, 10 min), and HbNO levels were assayed by electron spin resonance and recorded in a finger Dewar refrigerated with liquid nitrogen and with a Magnettech MS200. System settings were the following: BO field (3340 G), BO sweep (300 G), Sweep-time (30 s), Pass (4), Modulation (7000), MW attenuation (4 db). Spectra were analysed on the Analysis 2.02 software provided with the instrument.
RT-qPCR analysis
Freshly collected aortae and kidneys were snap-frozen in liquid nitrogen and the latter homogenized on ice in 300 µL of RLT lysis buffer for mRNA extraction (RNeasy® mini kit; Qiagen, Hilden, Germany). Samples with a RQI >7 (RNA quality integrity) were converted to cDNA (1 µg of starting material) with the Superscript™ III first-strand cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA). Expression of KCa3.1 and KCa2.3 protein was quantified by quantitative RT-qPCR using an iCycler iQ Detection System (Bio-Rad Laboratories, Hercules, CA). Each reaction was performed using the IQ™ SYBR® Green supermix (containing 3 mM MgCl2 and the iTaq DNA polymerase at 25 units·mL−1; Bio-Rad), 2 µL of DNA samples and 150 nM of each specific primer in a final volume of 25 µL. Samples were denatured for 300 s at 95°C and amplified for 40 cycles as follows: denaturation for 20 s at 95°C and annealing for 1 min at the specific oligonucleotide optimal temperature (Table S1). Each real-time PCR run included cDNAs in duplicate in parallel with serial dilutions of a cDNA mix tested for each primer pair to generate a linear standard curve with the mean cycle thresholds. This curve was then used to estimate the relative quantity of the relevant mRNA in each sample. The values obtained with our samples were normalized to the geometric mean of the values obtained with three internal controls: hypoxanthine-guanine phosphoribosyltransferase (HPRT), β-actin and GAPDH. The specificity of PCR products was confirmed by a melting curve analysis. All the primers were designed with the Beacon Designer Software (Premier Biosoft, Palo Alto, CA, USA).
Isolated, perfused rat kidney
Isolated, perfused kidneys were prepared as previously described (Collis and Vanhoutte, 1977). Briefly, after anaesthesia, the left kidney was exposed by midline ventral laparotomy. The left renal artery was cannulated via an incision made in the aorta. Then the kidney was perfused at constant flow by a peristaltic pump (Gilson Minipuls 2), with a warmed (37°C) and oxygenated (95% O2–5% CO2) Tyrode solution of the following composition (mM): NaCl 137; KCl 2.7; CaCl2 1.8; MgCl2 1.1; NaHCO3 12.0; NaHPO4 0.42; Na2-Ca-EDTA 0.026 and glucose 5.6. The perfused kidney was removed from the surrounding adipose tissue and placed in a perfusion chamber pre-warmed at 37°C. The perfusate was not re-circulated. Flow rate was adjusted to optimum flow rate corresponding to 5 mL·min−1 per g of kidney weight (Collis and Vanhoutte, 1977; Heuzé-Joubert et al., 1992). The changes in renal vascular resistance were recorded as changes in perfusion pressure measured downstream from the pump via a pressure transducer (P10EZ, Statham, France), which was connected to a data acquisition system IOX2 (EMKA Technologies, Paris, France). Once the perfusion pressure reached its steady state, after a 60 min equilibration period, experiments were started.
Pharmacological agents were administered in three different ways: (i) 20 µL bolus could be injected into the perfusion circuit (doses were expressed in moles); (ii) the inhibitors or antagonists could be infused into the perfusion circuit by an infusion pump placed upstream to the perfusion pump; or (iii) compounds could be directly added into the perfusing Tyrode solution (concentrations were expressed in mol·L−1).
Experimental design
Part 1
Under the present experimental conditions, papaverine (10 nmol) did not affect basal perfusion pressure, indicating that the perfused rat kidney lacks intrinsic tone. A concentration–response curve to noradrenaline (0.001–10 nmol) was performed. The maximal perfusion pressure achieved (basal perfusion pressure + the increase in perfusion pressure produced by the highest dose of noradrenaline tested) was not significantly different between the various groups tested. However, kidneys from L-NAME-treated SHR were slightly but significantly more sensitive to the α-adrenoceptor agonist than kidneys from other groups of rats (Figure S1). In order to study endothelium-dependent and -independent vasodilator compounds, a stable level of tension was produced by the infusion of the metabolically stable α-adrenoceptor agonist, methoxamine. The rate of infusion was slightly different between groups and between kidneys from different animals within the same group in order to achieve the same stable level of perfusion pressure (70% to 80% of the maximal perfusion pressure), either in control conditions or in the presence of the various pharmacological agents studied. Then vasodilator substances were injected as a bolus into the circuit at intervals of at least 10 min: acetylcholine (from 1 pmol to 10 nmol), sodium nitroprusside (from 10 pmol to 10 nmol) and forskolin (6 nmol).
Part 2
In the vasodilator responses to acetylcholine, the contributions of cyclooxygenase derivatives and/or NO were studied. The experiments were performed in the presence of the cyclooxygenase inhibitor, indomethacin (3 µmol·L−1) added to the Tyrode solution or/and of the NOS inhibitor, Nω-nitro-L-arginine (L-NA) perfused at 100 µmol·L−1, 20 min before the initiation of the perfusion with methoxamine (Heuzé-Joubert et al., 1992).
Part 3
To assess the role of calcium-activated potassium channels involved in the EDHF-mediated vasodilator responses, a third series of experiments was performed in the presence of indomethacin and L-NA combined. The selective small-conductance (KCa2.3) channel blocker, apamin (0.5 µmol·L−1) and the selective intermediate-conductance (KCa3.1) channel blocker, TRAM-34 (0.5, 1 and 5 µmol·L−1) were perfused 30 min after the stabilization of the methoxamine-induced constriction. In some experiments, charybdotoxin (10 nmol·L−1), a non-selective blocker of KCa3.1 channels was also studied. All channel nomenclature follows Alexander et al., (2011).
Data analysis
Perfusion pressure was measured in mmHg and changes in renal vascular pressure were expressed as the difference from basal perfusion pressure. Values are expressed as mean ± SEM, and n indicates the number of animals from which the kidneys were taken. Vasodilator responses to each agonist were measured at the peak of maximum change and were expressed as a percentage of the constriction induced by methoxamine, just before the injection of agonist. As NS-309 induced a long-lasting vasodilatation in kidneys from the SHR–L-NAME group, the area under each individual curve (AUC) expressed in arbitrary units was also used to compute the vasodilator responses induced by NS 309 alone or in the presence of potassium channel inhibitors. In order to evaluate significant differences between groups, two-sided one- and two-way anova, followed by Bonferroni complementary tests, were performed. In all cases, P < 0.05 was considered to be significant.
Materials
Acetylcholine chloride, forskolin (from Coleus forskohli), indomethacin, L-NA, L-NAME, methoxamine hydrochloride, NS-309 (6–7-dichloro-1H-indole-2,3-dione 3-oxime), ODQ (1H-(1,2,4)-oxadiazolo(4,2-a)quinoxalin-1-one) and TRAM-34 (1-[(2-chlorophenyl) diphenylmethyl]-1H-pyrazole) were obtained from Sigma-Aldrich (St Quentin-Fallavier, France); sodium nitroprusside was purchased from Prolabo (France), apamin and charybdotoxin from Latoxan (Valence, France). Forskolin, indomethacin, NS-309, ODQ and TRAM-34 were prepared as stock solutions in DMSO and subsequently diluted in physiological salt solution. All other drugs were dissolved in deionized water.
Results
Physiological and biochemical parameters
The systolic blood pressure was significantly higher in SHR than in WKY and in L-NAME-treated SHR than in SHR (without changes in heart rate), whilst the body weight was significantly lower in SHR than in WKY, and in L-NAME-treated SHR than in SHR. Blood HbNO was significantly less in L-NAME-treated SHR than in the two other groups, and urine levels of nitrite/nitrate were significantly less in SHR and L-NAME-treated SHR than in WKY (Table 1).
Table 1.
Physiological and biochemical parameters in normotensive WKY, and SHR with or without treatment with L-NAME
| WKY (untreated) | SHR (untreated) | SHR (L-NAME) | |
|---|---|---|---|
| SBP (mmHg) | 123 ± 5 (11) | 212 ± 10 (21)* | 246 ± 6 (22)*$ |
| Heart rate (beats·min−1) | 387 ± 9 (11) | 380 ± 10 (21) | 383 ± 8 (22) |
| Body weight (g) | 391 ± 9 (30) | 333 ± 7 (33)* | 282 ± 8 (43)*$ |
| Kidney weight (g) | 1.46 ± 0.03 (30) | 1.32 ± 0.02 (33)* | 1.10 ± 0.01 (43)*$ |
| Kidney/body weight (103) | 3.63 ± 0.13 (30) | 4.03 ± 0.09 (33) | 4.16 ± 0.16 (43)* |
| Micro-albuminuria (mg·24 h−1) | <0.01 (9) | 0.09 ± 0.01 (12)* | 2.7 ± 0.68 (13)*$ |
| Urinary proteins (mg·24 h−1) | 19.6 ± 2.6 (15) | 11.7 ± 0.8 (15)* | 91.1 ± 7.8 (13)*$ |
| Creatinine (mL·min−1·kg−1) | 0.61 ± 0.05 (8) | 0.38 ± 0.02 (11)* | 0.17 ± 0.02 (10)*$ |
| HbNO (arbitrary units) | 2171 ± 365 (6) | 1582 ± 255 (6) | 399 ± 75 (7)*$ |
| Nitrite/nitrate (µmol·24 h−1) | 4.02 ± 0.81 (10) | 1.38 ± 0.22 (14)* | 0.66 ± 0.18 (17)* |
Values are shown as mean ± SEM with the number of animal from which kidneys were taken, shown in brackets.
P < 0.05 significantly different from WKY,
P < 0.05, significantly different from untreated SHR; one-way anova, followed by a Bonferroni post hoc test, three multiple comparisons.
SBP, systolic blood pressure; HbNO, iron nitroxyl-haemoglobin.
The weight of the kidneys was significantly lower in L-NAME-treated SHR than in the other groups and a profound renal dysfunction was observed, characterized by an increased micro-albuminuria and urinary protein excretion and a decreased creatinine clearance (Table 1). In SHR, the renal histological sections did not show any major structural alterations when compared with normotensive WKY of the same age, while, in L-NAME-treated SHR, major changes were observed, including tubulopathies and, in small to medium-size arteries, wall hyperplasia and fibrinoid necrosis (Figure S2).
Basal perfusion pressure of isolated perfused kidneys
The basal perfusion pressures were comparable in WKY and SHR: 62 ± 2 (n= 19) and 65 ± 5 mmHg (n= 11), respectively, but significantly enhanced in those of L-NAME-treated SHR, 90 ± 6 mmHg (n= 19) (P < 0.05 vs. SHR, one-way anova).
Vasodilator responses
Infusion of methoxamine (2–5 µM), an α1-adrenoceptor agonist, increased the renal perfusion pressure in WKY, SHR and L-NAME-treated SHR to 144 ± 8 (n= 12), 165 ± 8 (n= 20) and 147 ± 6 mmHg (n= 33) respectively. In all the groups, the increase in perfusion pressure in responses to methoxamine remained stable during the duration of the experiments. The level of perfusion achieved (basal pressure plus methoxamine-induced pressure) was similar in each group of rats.
In WKY kidneys, acetylcholine (1 pmol to 10 nmol) caused a dose-dependent dilatation with a maximal amplitude of 54.8 ± 7.7 mmHg (n= 9). In SHR, the dilator responses to acetylcholine were slightly but significantly increased when compared with those observed in WKY (Figure 1A). In L-NAME-treated SHR, the dilator responses to acetylcholine were similar to those observed in WKY and SHR (Figure 1A). In contrast, when compared with the two other groups, the dilatation induced by the endothelium-independent NO donor, sodium nitroprusside (10 pmol to 10 nmol) was significantly increased in the kidneys of L-NAME-treated SHR (Figure 1B). Forskolin (6 nmol), an adenylate cyclase activator, produced similar dilatation in the three groups [WKY: 67.0 ± 4.7% (n= 6); SHR: 68.6 ± 8.4% (n= 8); L-NAME-treated SHR: 70.8 ± 4.9% (n= 8)].
Figure 1.
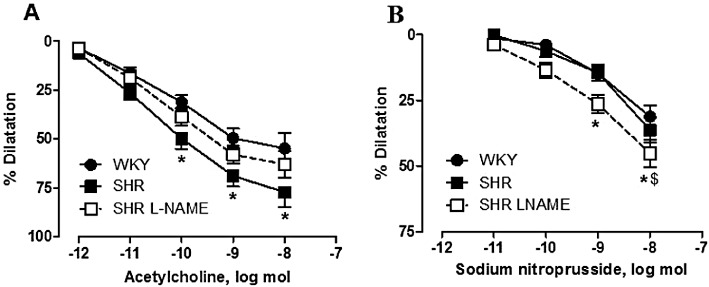
Bolus injections of increasing doses of (A) acetylcholine and (B) sodium nitroprusside during vasoconstrictions caused by methoxamine in isolated perfused kidneys from WKY, SHR and SHR treated with L-NAME (SHR L-NAME). The results are shown as % dilatation of the increase in perfusion pressure induced by methoxamine, and values are expressed as means ± SEM. (n= 9–10). *P < 0.05, significantly different from the WKY group; $P < 0.05, significant difference between the SHR and L-NAME-treated SHR groups; two-way anova, followed by a Bonferroni post hoc test, 15 and 12 multiple comparisons for acetylcholine and sodium nitroprusside experiments, respectively.
Acetylcholine-induced vasodilatation: NO and prostacyclin
Indomethacin (3 µmol·L−1), an inhibitor of cyclooxygenases, did not significantly modify either the basal tone or the amplitude of the constriction induced by methoxamine (WKY: 151 ± 15 mmHg, SHR: 161 ± 6 mmHg, n= 7; L-NAME-treated SHR: 144 ± 18 mmHg, n= 6). The presence of indomethacin did not affect the dilator responses to acetylcholine or sodium nitroprusside (Figure 2).
Figure 2.
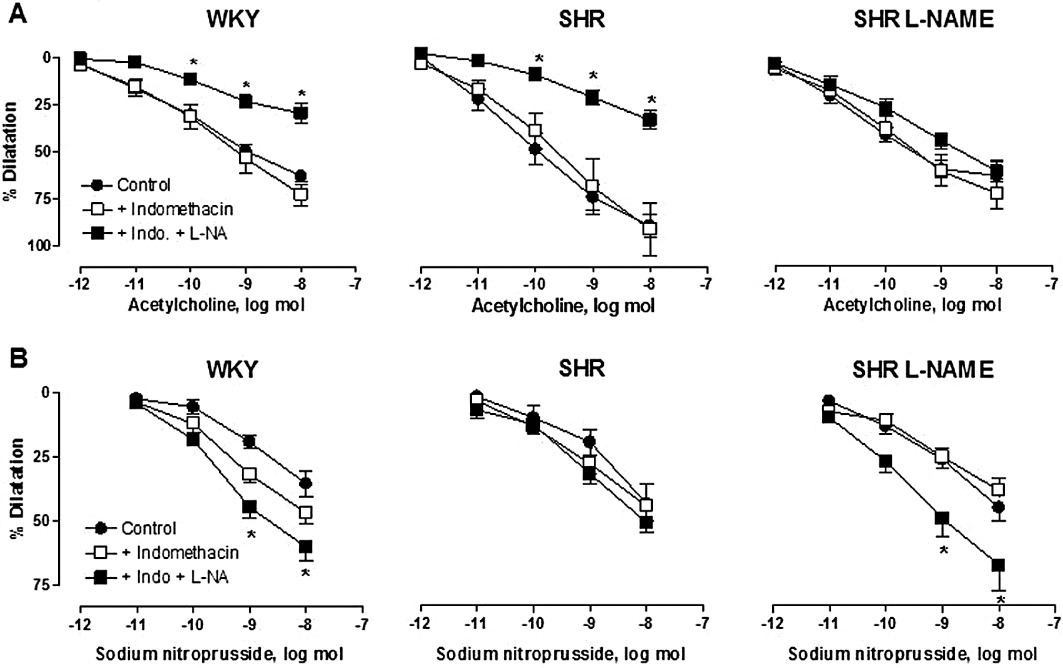
Effects of indomethacin (INDO) and L-NA on the dilatation due to acetylcholine (A) and sodium nitroprusside (B) of isolated perfused kidney from WKY, SHR and SHR treated with L-NAME (SHR L-NAME). The results are shown as % dilatation of the increase in perfusion pressure induced by methoxamine, and values are expressed as means ± SEM. (n= 5–10). *P < 0.05, significant difference between the indomethacin and the indomethacin + L-NA groups; two-way anova followed by a Bonferroni post hoc test, 15 and 12 multiple comparisons for acetylcholine and sodium nitroprusside experiments respectively.
In the presence of indomethacin, the additional infusion of another NOS inhibitor, L-NA (100 µmol·L−1), induced only a small increase in the basal perfusion pressure of WKY and L-NAME-treated SHR kidneys (7.5 ± 1.5 mmHg, n= 13, and 10.7 ± 1.5 mmHg, n= 16, respectively), but a larger increase in that of SHR (33.7 ± 7.1 mmHg, n= 14). In the presence of the combination of indomethacin plus L-NA, the perfusion pressure reached after methoxamine perfusion was 136 ± 10 mmHg (n= 12), 171 ± 10 mmHg (n= 12) and 157 ± 8 mmHg (n= 22) for WKY, SHR and L-NAME-treated SHR respectively (P= NS).
L-NA produced a substantial inhibition of the dilatation to acetylcholine in WKY and SHR kidneys, but only a modest inhibition in those from L-NAME-treated SHR (Figure 2A). Thus, the response to acetylcholine at the highest dose studied of 10 nmol was significantly diminished by 59% and 61% in WKY and SHR kidneys, respectively; while in the L-NAME-treated SHR, the response was only diminished by 16%. In contrast, the responses to sodium nitroprusside (10 nmol) were increased by 22% and 12% in WKY and SHR, respectively, and by 43% in the L-NAME-treated SHR groups (Figure 2B).
In L-NAME-treated SHR kidneys, in the presence of indomethacin plus L-NA, the dilator responses to acetylcholine were not impaired any further by the additional presence of the inhibitor of soluble guanylyl cyclase, ODQ (3 µmol·L−1). In contrast, this concentration of ODQ abolished sodium nitroprusside-induced vasodilatation (Figure 3).
Figure 3.
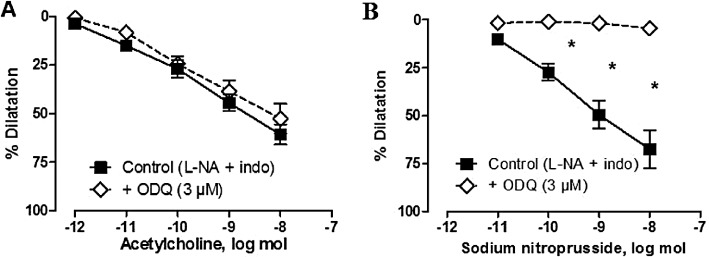
Effects of ODQ on the dilatation evoked by acetylcholine and sodium nitroprusside in isolated perfused kidneys from L-NAME-treated SHR (in the presence of L-NA plus indomethacin). The results are shown as % dilatation of the increase in perfusion pressure induced by methoxamine and values are expressed as mean ± SEM (n= 5–8). *P < 0.05, significantly different from control; two-way anova, followed by a Bonferroni post hoc test, five and four multiple comparisons for acetylcholine and sodium nitroprusside experiments respectively.
Acetylcholine-induced vasodilatation: potassium channels
In the presence of indomethacin and L-NA, TRAM-34 (0.5 and 1 µM), a potent and selective inhibitor of KCa3.1 channels did not affect the renal vasoconstriction to methoxamine. However, at the higher concentration of 5 µM, it significantly reduced the constrictions to methoxamine in kidneys from the three groups of rats [WKY: from 152 ± 14 to 122 ± 12 mmHg (n= 6); SHR: from 192 ± 3 to 159 ± 25 mmHg (n= 7) and L-NAME-treated SHR: from 168 ± 7 to 123 ± 11 mmHg (n= 6)].
TRAM-34 (1 and 5 µM) inhibited the response to acetylcholine in both WKY and SHR kidneys (Figure 4) without altering those to sodium nitroprusside (10 nmol) and forskolin (6 nmol) (Table 2) In kidneys from L-NAME-treated SHR, TRAM-34 (0.5, 1 and 5 µmol·L−1) caused a significant and dose-dependent inhibition of the vasodilator responses to acetylcholine (Figure 4 and Table 2). Similarly, in L-NAME-treated SHR kidneys, charybdotoxin (10 nmol·L−1) [a non-selective inhibitor of KCa3.1 channels and of large conductance calcium-activated potassium channels (KCa1.1) and some voltage-activated potassium channels (KV)], significantly inhibited the vasodilator responses to 10 nmol acetylcholine (59.6 ± 4.1%, n= 9 and 27.1 ± 7.2%, n= 3; in control condition or in the presence of charybdotoxin, respectively: P < 0.05), without altering the vasodilatation to sodium nitroprusside or forskolin (Table 2).
Figure 4.
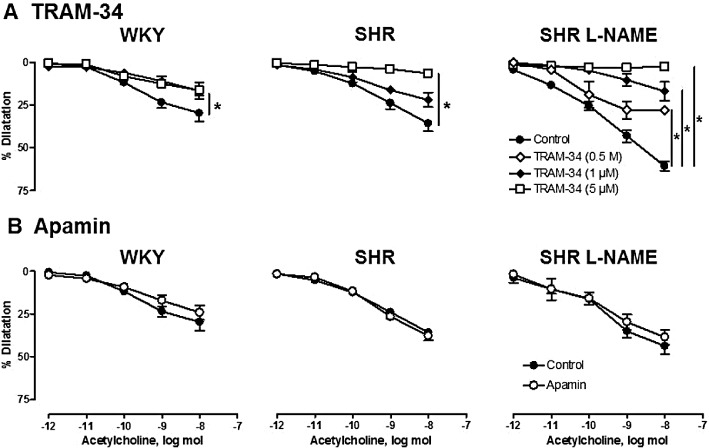
Effects of TRAM-34 (0.5, 1 and 5 µmol·L−1, top) and apamin (0.5 µmol·L−1, bottom) on the dilatation due to acetylcholine in isolated perfused kidneys from WKY, SHR and L-NAME-treated SHR (n= 3–7). The experiments were performed in the presence of indomethacin (3 µM) and L-NA (100 µM). The results are shown as % dilatation of the increase in perfusion pressure induced by methoxamine and values are expressed as means ± SEM (n≥ 3). *P < 0.05, significantly different from control; two-way anova, followed by a Bonferroni post hoc test, 10–15 and 5 multiple comparisons for TRAM-34 and apamin experiments respectively.
Table 2.
Maximal decreases in perfusion pressure caused by sodium nitroprusside (SNP: 10 nmol) and forskolin (6 nmol) in kidneys from WKY, SHR and L-NAME-treated SHR (in the presence of indomethacin and L-NA). Effects of the potassium channel blockers: TRAM-34, apamin and charybdotoxin
| WKY (untreated) | SHR (untreated) | SHR (L-NAME) | ||
|---|---|---|---|---|
| Control | SNP | 60.3 ± 5.4 (5) | 51.0 ± 3.7 (6) | 64.6 ± 4.4 (14) |
| Forskolin | 62.1 ± 6.9 (6) | 57.3 ± 3.2 (7) | 67.0 ± 4.6 (14) | |
| TRAM-34 (0.5 µmol·L−1) | SNP | – | – | 53.2 ± 5.7 (5) |
| Forskolin | 56.3 ± 4.7 (5) | |||
| TRAM-34 (1 µmol·L−1) | SNP | 64.9 ± 5.3 (4) | 42.9 ± 1.5 (3) | 58.6 ± 6.3 (5) |
| Forskolin | 45.3 ± 6.4 (5) | 49.2 ± 5.2 (3) | 58.9 ± 7.0 (5) | |
| TRAM-34 (5 µmol·L−1) | SNP | 66.8 ± 4.6 (5) | 58.4 ± 4.7 (5) | 46.3 ± 5.3 (5) |
| Forskolin | 65.9 ± 7.7 (6) | 55.3 ± 6.7 (5) | 54.0 ± 2.9 (5) | |
| Apamin (0.5 µmol·L−1) | SNP | 46.6 ± 9.1 (3) | 48.4 ± 4.3 (3) | 52.1 ± 6.8 (4) |
| Forskolin | 55.5 ± 6.9 (3) | 59.4 ± 3.8 (3) | 52.4 ± 4.5 (6) | |
| Apamin (0.5 µmol·L−1) + TRAM-34 (5 µmol·L−1) | SNP | – | – | 43.4 ± 7.8 (5) |
| Forskolin | 71.8 ± 9.4 (5) | |||
| Charybdotoxin (10 nmol·L−1) | SNP | – | – | 65.5 ± 9.8 (3) |
| Forskolin | 68.2 ± 9.8 (3) |
Values are expressed in mmHg and shown as mean ± SEM with the number of animal from which kidneys were taken, shown in brackets.
In contrast, the perfusion of apamin (0.5 µmol·L−1), a selective inhibitor of KCa2.3 channels did not modify the renal vasoconstriction to methoxamine, did not affect the vasodilator responses to acetylcholine, sodium nitroprusside or forskolin and did not influence the inhibitory effect of TRAM-34 (Figure 4, Table 2).
NS-309-induced vasodilatation
In the presence of indomethacin and L-NA, and in the kidneys of the three groups of rats, the bolus administration of NS-309, a mixed activator of KCa3.1 and KCa2.3 channels (10 nmol), caused vasodilatation. The amplitude and the time course of these responses were larger in SHR than in WKY and larger in L-NAME-treated SHR than in SHR (Figure 5 and Table 3).
Figure 5.
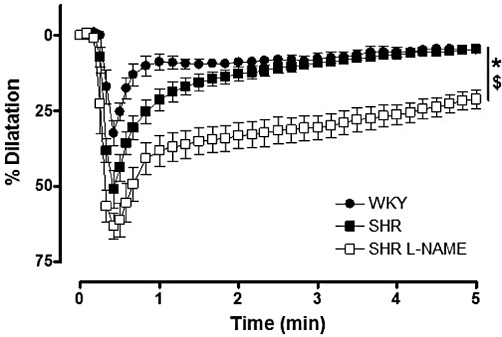
Bolus injection of NS 309 (10 nmol) in isolated perfused kidneys from normotensive WKY, SHR and L-NAME-treated SHR (n= 5–7). The experiments were performed in the presence of indomethacin plus L-NA. The results are shown as % dilatation of the increase in perfusion pressure induced by methoxamine and values are expressed as mean ± SEM. *P < 0.05, AUC significantly different from control; $P < 0.05, significant difference between SHR and L-NAME-treated SHR; one-way anova, followed by a Bonferroni post hoc test, three multiple comparisons.
Table 3.
Effects of TRAM-34 and apamin on NS-309 (10 nmol)-induced vasodilatation (in the presence of indomethacin + L-NA)
| WKY (untreated) | SHR (untreated) | SHR (L-NAME) | |
|---|---|---|---|
| Control | 40.9 ± 7.6 (5) | 65.0 ± 7.5 (7) | 161.2 ± 14.7 (7) |
| TRAM-34 (0.5 µmol·L−1) | 96.4 ± 15.4 (5)* | ||
| TRAM-34 (1 µmol·L−1) | 43.8 ± 13.8 (5) | 41.7 ± 8.5 (3) | 48.6 ± 10.9 (3)* |
| TRAM-34 (5 µmol·L−1) | 48.1 ± 11.9 (5) | 18.4 ± 5.2 (5)* | 38.6 ± 6.1 (3)* |
| Apamin (0.5 µmol·L−1) | 24.3 ± 11.4 (2) | 67.6 ± 5.6 (3) | 164.4 ± 29.1 (6) |
| Apamin + TRAM-34 (5 µmol·L−1) | 42.5 ± 8.3 (6)* |
Values are expressed as AUC (arbitrary units) and are shown as mean ± SEM with the number of animal from which kidneys were taken in brackets.
P < 0.05, significantly different from control; one-way anova.
TRAM-34 (1 and 5 µM) did not affect the vasodilator responses to NS 309 in WKY kidneys but produced a significant concentration-dependent but partial inhibition in kidneys from both SHR and SHR L-NAME (Figure 6). Thus, in the presence of TRAM-34 (5 µM), the residual responses to NS 309 observed in SHR and L-NAME-treated SHR were similar to the response observed in WKY kidneys. In the latter group, charybdotoxin (10 nmol·L−1) also significantly inhibited the vasodilator responses to NS-309 (63.5 ± 4.1%, n= 7 and 30.9 ± 7.1%, n= 3; in control and charybdotoxin-treated kidneys, P < 0.05). In contrast, in isolated kidneys from the three groups of rats, the presence of apamin (0.5 µM), did not affect the vasodilatation to NS 309 (Figure 6). In addition, the combination of apamin (0.5 µM) plus TRAM-34 (5 µM) was no more effective in inhibiting NS 309-induced vasodilatation than the presence of TRAM-34 alone (Table 3).
Figure 6.
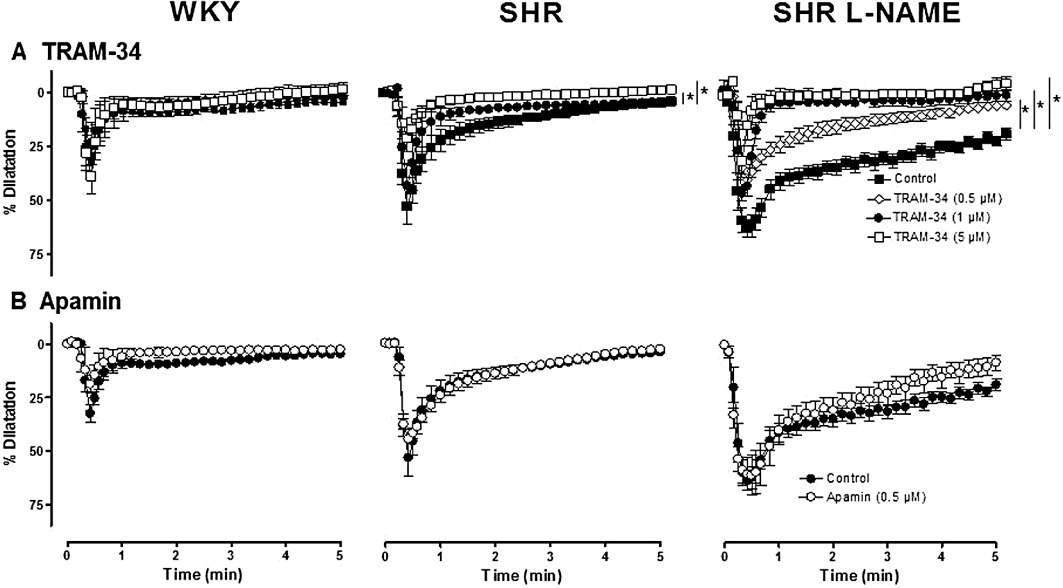
Effects of TRAM-34 (0.5, 1 and 5 µM) on the dilatation due to NS 309 (10 nmol) in isolated perfused kidneys from WKY, SHR and L-NAME-treated SHR (n= 3–7). The experiments were performed in the presence of indomethacin plus L-NA. The results are shown as % dilatation of the increase in perfusion pressure induced by methoxamine, and values are expressed as mean ± SEM. *P < 0.05, AUC significantly different from control; one-way anova followed by a Bonferroni post hoc test, 2–3 and 1 multiple comparisons for TRAM-34 and apamin experiments respectively.
KCa2.3 and KCa3.1 mRNA expression
The expression of mRNA for KCa2.3 protein was not significantly different in kidneys of WKY, SHR and L-NAME-treated SHR. However, that for KCa3.1 protein was significantly enhanced in L-NAME-treated SHR kidneys (Figure 7). In contrast, in aorta of the three groups of rats, the mRNA expression for these two potassium channel proteins was not significantly different (data not shown).
Figure 7.
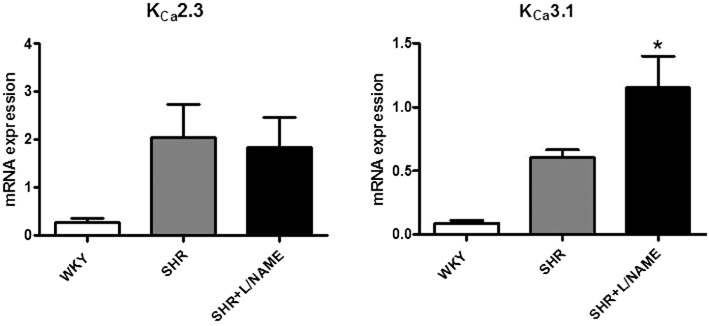
Quantitative RT-PCR for mRNA for KCa2.3 and KCa3.1 protein in kidneys of WKY, SHR and L-NAME-treated SHR (n= 8–13). The values are normalized to the geometric mean of the values obtained with three internal controls: HPRT, β-actin and GAPDH. Values are expressed as mean ± SEM. *P < 0.05, significant difference between L-NAME-treated SHR and WKY kidneys; one-way anova-1 followed by a Bonferroni post hoc test, three multiple comparisons.
Discussion
The present study confirmed that chronic administration of a moderate dose of L-NAME over a 2 week period in 4-month-old SHR induced severe hypertension and pronounced renal injury, characterised by increased proteinuria and reduced creatinine clearance. Interestingly, in L-NAME-treated SHR, the marked structural alterations in the renal microvasculature were associated with only minor changes in acetylcholine-induced renal vasodilatation, most likely due to the development of a compensatory mechanism involving the activation of the intermediate-conductance calcium-activated potassium channels (KCa3.1).
NO is a major vasodilator and a regulator of renal haemodynamics and blood pressure (Baylis and Qiu, 1996). The dose of L-NAME chosen in this study (6 mg·kg−1·day−1) was moderate and did not produce any significant alterations in the normotensive WKY strain. In this strain, the L-NAME doses producing significant changes in blood pressure are above 10 mg·kg−1·day−1 and, in order to induce substantial long-lasting changes in blood pressure, a dose of 100 mg·kg−1·day−1 is routinely used (Arnal et al., 1992). In contrast to the WKY, in the SHR, this same low dose of NOS inhibitor was sufficient to produce a substantial further increase in systolic blood pressure (Ono et al., 1995; Benter et al., 2005; Hilgers and Webb, 2007; Vayssettes-Courchay et al., 2011; Isabelle et al., 2012). In control SHR, the circulating iron-nitrosyl-haemoglobin, an index of blood NO levels, and the concentration of urinary nitrite/nitrate were lower than in corresponding WKY, confirming an overall reduction of NO bioavailability in this genetic model of hypertension (Benter et al., 2005). These two indices of NO generation were further reduced in L-NAME-treated SHR, confirming NOS inhibition and a further reduction in NO bioavailability. Additionally, in the L-NAME-treated SHR, the inhibition of NO generation was associated with profound renal injuries as shown by structural alterations in renal arteries of small and medium size as well as tubular injuries. These alterations are similar to those observed in ageing SHR (Ono et al., 1995; 2001; Hilgers and Webb, 2007; Zhou and Frohlich, 2007) and are comparable to those attributed to a reduced NO availability in human cardiovascular pathologies (Taddei et al., 1993; Panza et al., 1995; Virdis et al., 2010).
Hypertension is associated with endothelial dysfunction, which is generally characterised by impaired endothelial dilator responses, observed both in vivo and in vitro, either in isolated arteries or in perfused organs. However, these endothelial dysfunctions present different characteristics depending on the vascular bed, the animal model used and/or the age of the animals (see Félétou and Vanhoutte, 2006a). In the aorta or in the mesenteric vascular bed of SHR, the impaired endothelium-dependent dilatation is associated with the generation of endothelium-derived and cyclooxygenase-derived contracting factors with no or little alteration in the production of NO (Lüscher et al., 1990; Fujii et al., 1992; Mantelli et al., 1995; Hutri-Kahonen et al., 1997; Félétou et al., 2009) whereas, in the carotid artery, the attenuated endothelium-dependent relaxations to acetylcholine are probably related to a smooth muscle dysfunction in the responsiveness to NO, with no involvement of endothelium-derived contracting factors (Hongo et al., 1988; Lüscher et al., 1988). In perfused kidneys of young SHR, when compared with those of normotensive WKY rats, the endothelium-dependent dilatation to acetylcholine or to bradykinin is minimally inhibited (Hayakawa et al., 1993), unaffected (Burton et al., 1991) or even enhanced (Cachofeiro and Nasjletti, 1991, Chamorro et al., 2004; Simonet et al., 2009). In the present study, we observed a significant enhancement of acetylcholine-induced decrease in perfusion pressure, which could not be attributed to any change in smooth muscle sensitivity towards NO, as the endothelium-independent responses to the NO donor, sodium nitroprusside were unaffected.
Chronic L-NAME treatment also inhibited the renal NOS. In the kidneys of L-NAME-treated SHR, the vasodilator response to sodium nitroprusside, a donor of NO, was significantly enhanced, a result which is in agreement with previous reports showing that the inhibition of NOS increases the sensitivity of soluble guanylyl cyclase to NO (Moncada et al., 1991; Fulton et al., 1992; Desai et al., 2006). Furthermore, both basal and acetylcholine-induced NO release were inhibited. Indeed, in isolated kidneys of L-NAME-treated SHR, the in vitro administration of a different NOS inhibitor, L-NA, produced a reduced intrinsic vasoconstriction and induced virtually no inhibition of acetylcholine-induced vasodilatation.
In WKY and SHR kidneys, the endothelium-dependent vasodilatation to acetylcholine involves NO because the acute administration of L-NA produced a significant but partial inhibition of the responses. Indomethacin, an inhibitor of cyclooxygenase, was without effect, ruling out prostaglandins, and especially prostacyclin, as a component of this mechanism. In many peripheral arteries, EDHF-mediated responses are associated with the opening of endothelial calcium-activated potassium channels of small and/or intermediate conductance (KCa2.3 and KCa3.1; Eichler et al., 2003; Félétou and Vanhoutte, 2006b; Grgic et al., 2009a). Apamin, a toxin from bee venom, is a very selective inhibitor of KCa2.3;channels while charybdotoxin, a scorpion toxin, is a non-selective inhibitor of KCa3.1, KCa1.1 and some KV channels. The clotrimazole derivative TRAM-34 is a selective KCa3.1 channel blocker with more than a 1000-fold selectivity ratio compared with KCa2.3 and KCa1.1 channels (Castle, 1999; Wulff et al., 2000). In isolated kidneys from WKY, the COX-NOS-independent relaxations to acetylcholine were unaffected by the additional presence of apamin but were virtually abolished by TRAM-34 alone.These data are in agreement with earlier reports using kidneys from normotensive rats and showing that EDHF-mediated responses were attenuated by charybdotoxin, whilst apamin had no effect (Fulton et al., 1992; Rapacon et al., 1996; Mieyal et al., 1998). Moreover, our data are in accord with results recently obtained in vivo, in an elegant study involving renal haemodynamics in anaesthetized rats (Edgley et al., 2008). Thus, in isolated and perfused rat kidneys, the EDHF-mediated responses elicited by acetylcholine involve predominantly the activation of calcium-activated potassium channels and especially KCa3.1 channels. Furthermore, the TRAM-34-sensitive response was greater in kidneys from SHR than in WKY, indicating an up-regulation of this pathway in the kidneys of the hypertensive strain.
In L-NAME-treated SHR kidneys, the vasodilatation to acetylcholine was virtually unaffected. Again, indomethacin did not modify the responses to acetylcholine, ruling out prostacyclin as a component of this vasodilatation. Moreover, in the presence of the cyclooxygenase inhibitor and after acute administration of L-NA, no further inhibition of the responses to acetylcholine was observed. Furthermore, the addition of both L-NA and the selective inhibitor of soluble guanylyl cyclase, ODQ, used at a concentration that totally blocked the vasodilator responses to sodium nitroprusside, did not affect the vasodilatation to acetylcholine Thus, a large component of this response is NO- and prostacyclin-independent and therefore could be attributed to a compensatory EDHF-like mechanism. These data are in agreement with previous results in normotensive rats, showing that, after chronic, but not acute treatment, with L-NAME, a proper hypotensive response to acetylcholine is restored and that compensatory mechanism(s) develop in resistant vascular beds, including the mesenteric (Ruiz-Marcos et al., 2001; Hilgers et al., 2010) and the renal circulatory beds (Bryant et al., 1995; Vargas et al., 1996; Maeso et al., 1999; Strøbæk et al., 2004; Desai et al., 2006). Very similar observations have been reported in mice with genetic deletion of eNOS (Brandes et al., 2000; Ding et al., 2000; Huang et al., 2000) or in patients with essential hypertension or primary hyperparathyroidism (Taddei et al., 2006; Sainsbury et al., 2007; Virdis et al., 2010). However, the mechanisms underlying the compensatory effect of EDHF-mediated responses, when the endothelial production of NO is impaired, have not been fully characterized.
In L-NAME-treated SHR, the EDHF-mediated response is significantly and markedly enhanced when compared with control WKY or SHR. The predominant role of KCa3.1 has been demonstrated by its sensitivity to TRAM-34 and further confirmed by the inhibition elicited by charybdotoxin. In contrast to TRAM-34, this toxin did not totally abolish the vasodilator effect of acetylcholine, possibly because TRAM-34 can also block non-selective cationic channels involved in calcium homeostasis (Schilling and Eder, 2007), which could be associated to EDHF-mediated responses (Félétou and Vanhoutte, 2006b).
NS-309 has been described as a potent opener of both KCa3.1 and KCa2.3 channels, with a preferential selectivity for the former (Strøbæk et al., 2004; Leuranguer et al., 2008). In kidneys from WKY rats, NS 309 produced a transient vasodilatation that was not affected by the presence of either TRAM-34 or apamin. This vasodilatation in WKY and, in the kidneys of SHR and L-NAME-treated SHR, the residual vasodilatation to NS-309, which was insensitive to blockers of KCa3.1 channels, may involve a non-selective effect of NS 309, the inhibition of L-type voltage-operated calcium channels (Morimura et al., 2006). Nevertheless, in kidneys of SHR and L-NAME-treated SHR, NS-309 induced dilatation inhibited by either TRAM-34 or charybdotoxin but not by apamin alone, indicating the predominant activation of KCa3.1 channels. In L-NAME-treated SHR, the amplitude of the dilator responses to NS-309 was increased and the duration was markedly prolonged. Because the combination of TRAM-34 plus apamin was not more effective than TRAM-34 alone, the involvement of KCa2.3 channels in this dilator response could again be excluded. Again, the TRAM-34-sensitive response was larger in SHR than in WKY kidneys and, significantly and markedly enhanced in those from L-NAME-treated SHR, compared with kidneys from control SHR. Altogether, these findings suggest that an enhanced contribution of KCa3.1 channel activation compensated for the decrease in NO bioavailability. In SHR kidneys, a minor up-regulation of the TRAM-34-sensitive response (to both acetylcholine and NS 309) was associated with a moderate decrease in NO bioavailability, whereas in kidneys from L-NAME-treated SHR, a pronounced up-regulation of the TRAM-34-sensitive response was associated with a major decrease in NO bio-availability. Interestingly, in the kidneys of these rats, these functional responses were accompanied by similar changes in expression of the mRNA for KCa3.1, but not in that of KCa2.3 channels (which do not contribute to the vasodilator response). Furthermore, this up-regulation of KCa3.1 channels was observed in the kidney but not in the aorta, a blood vessel that does not exhibit EDHF-mediated responses (Félétou and Vanhoutte, 2006b). Unfortunately, the present quantitative RT-PCR experiments performed on the whole kidney does not allow us to determine whether or not the KCa3.1 channel overexpression involves endothelial and/or other cell subtypes, for instance renal fibroblasts (Grgic et al., 2009b).
In various pathological rat models of hypertension (SHR, SHR-SP and angiotensin-II infused) and type-II diabetes (Zucker and cafeteria diet-induced obesity), a maintenance or an up-regulation of the activity of KCa3.1 channels, in some instance associated with an increased expression of KCa3.1 protein has been reported; whilst the reverse was observed for the KCa2.3 channel (Burnham et al., 2006; Hilgers and Webb, 2007; Giachini et al., 2009; Chadha et al., 2010; Weston et al., 2010), suggesting that KCa3.1 channels are the last line of defence to maintain endothelium-dependent vasodilatation. Expression of KCa3.1 protein is negatively regulated by the repressor element-1 silencing transcription factor (REST) (Cheong et al., 2005; Tharp et al., 2008). In SHR-SP rats, it has been suggested that up-regulation of KCa3.1 channels via a decreased expression of REST, compensates for the decreased NO bioavailability (Giachini et al., 2009). This regulating mechanism may occur in other models including the renal resistance vessels of L-NAME-treated SHR rats studied in the present work.
The physiopathological role of EDHF-mediated responses in the kidney is basically unknown. In rats with low NO activity, EDHF-mediated responses might compensate for the NO deficiency and act as a dilator system in attenuating endothelial dysfunction. However, the EDHF contribution, in contrast to the NO contribution, is positively related to the development of proteinuria (Gschwend et al., 2002), suggesting that, although EDHF-mediated responses seem to attenuate endothelial dysfunction in the kidney, they could be involved in the detrimental mechanisms associated with these renal alterations. Indeed, the KCa3.1 channel can also play a pivotal role in promoting mitogenesis, not only in endothelial and vascular smooth muscle cells (Köhler et al., 2003; Grgic et al., 2005), but also in renal fibroblasts (Grgic et al., 2009b). In a model of balloon catheter injury and in a model of renal fibrosis, TRAM-34 treatment prevented restenosis in the former and reduced the chronic tubulointerstitial damage in the latter (Köhler et al., 2003; Grgic et al., 2009b).
Conclusions and perspectives
In conclusion, this study confirmed that, in SHR, chronic L-NAME treatment increased blood pressure and markedly altered renal structure and function. In these animals, the renal COX-NOS-independent vasodilatation in response to either acetylcholine or to NS-309, a preferential KCa3.1 channel opener, was increased and was associated with an enhanced expression of mRNA for KCa3.1 protein, suggesting that these EDHF-mediated responses involve an up-regulation of the KCa3.1 channels. The pathophysiological significance of the compensatory contribution of KCa3.1 in the context of renal failure remains to be clarified. This would require a chronic in vivo study. However, NS 309 remains a poorly selective tool that, besides activating both KCa3.1 and KCa2.3 channels, interacts with other targets for instance, hERG (KV11.1) and L-type Cav channels and adenosine A2A receptors. Furthermore, the pharmacokinetic properties of this compound are not compatible with chronic in vivo administration (Félétou et al., 2011). Performing experiments with the more selective and recently described compound SKA-31 (not commercially available when our studies were initiated) would be more appropriate, although this compound is not orally active (Sankaranarayanan et al., 2009; Damkjaer et al., 2012). Such experiments would answer this fundamental question: ‘Does the compensating renal vasodilator response by an EDHF mechanism influence kidney function or delay functional and/or structural alterations in the kidneys?’
Acknowledgments
The authors are grateful to Léa Guillaume and Olympe Beugnot for technical assistance and Catherine de Montrion, Monique Naze, Christelle Le Clanche and Jean-François Boivin for biochemical and histological analysis.
Glossary
- EDHF
endothelium-derived hyperpolarizing factor
- HbNO
iron-nitroxyl-haemoglobin
- L-NA
Nω-nitro-L-arginine
- L-NAME
Nω-nitro-L-arginine methyl ester
- NS-309
6–7-dichloro-1 H-indole-2,3-dione 3-oxime
- ODQ
1H-(1,2,4)-oxadiazolo(4,2-a)quinoxalin-1-one
- SHR
spontaneously hypertensive rat
- TRAM-34
1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole
- WKY
rat, Wistar–Kyoto rat
Conflict of interests
All the authors were employees of Servier at the time of the experiments described here. None of the authors has a conflict of interest with the material presented in the submitted manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article:
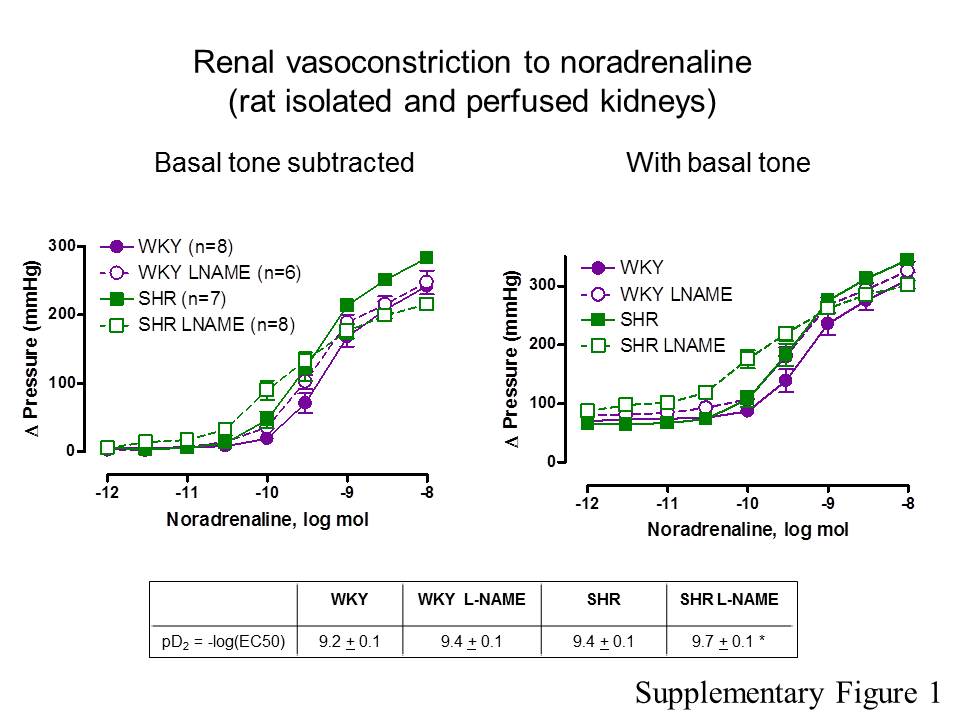
Figure S1 Noradrenaline-induced increases inperfusion pressure in rat isolated and perfused kidneys. Themaximal perfusion pressure achieved (basal tone + the increase inperfusion pressure produced by the highest dose of noradrenalinetested) is not significantly different. However, kidneys fromL-NAME-treated SHR were significantly more sensitive to theα-adrenoceptor agonist than kidneys from other groups ofrats. pD2 values are shown in the inset. *P <0.05, significant difference between the L-NAME-treated SHR and theother groups; one-way ANOVA, followed by a Bonferroni posthoc test, six multiple comparisons.
{kind=link}
Figure S2 Renal histology in WKY,L-NAME-treated WKY, SHR and L-NAME-treated SHR. Black arrows in thesection from kidneys of L-NAME-treated SHR show tubulopathy,arterial wall hyperplasia, fibrinoid necrosis and red blood cellinfiltration. A, arteriole; G, glomerulus; T, tubule. A 100 μmscale is shown on each section.
{kind=link}
Table S1 RT-qPCR: Primer sequences and annealing temperature
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnal JF, Warin L, Michel JB. Determinants of aortic cyclic guanosine monophosphate in hypertension induced by chronic inhibition of nitric oxide synthase. J Clin Invest. 1992;90:647–652. doi: 10.1172/JCI115906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylis C, Qiu C. Importance of nitric oxide in the control of renal hemodynamics. Kidney Int. 1996;49:1727–1731. doi: 10.1038/ki.1996.256. [DOI] [PubMed] [Google Scholar]
- Benter IF, Francis I, Cojocel C, Juggi JS, Yousif MH, Canatan H. Contribution of cytochrome metabolites of arachidonic acid to hypertension and end-organ damage in SHR treated with L-NAME. Auton Autacoid Pharmacol. 2005;25:143–154. doi: 10.1111/j.1474-8673.2005.00343.x. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Schmitz-Winnenthal F-H, Félétou M, Gödecke A, Huang P-L, Vanhoutte PM, et al. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild type and endothelial NO synthase knock-out mice. Proc Natl Acad Sci U S A. 2000;97:9747–9752. doi: 10.1073/pnas.97.17.9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CE, Allcock GH, Warner TD. Comparison of effects of chronic and acute administration of NG-nitro-L-arginine methyl ester to the rat on inhibition of nitric oxide-mediated responses. Br J Pharmacol. 1995;114:1673–1679. doi: 10.1111/j.1476-5381.1995.tb14956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham MP, Johnson IT, Weston AH. Impaired small-conductance Ca2+-activated K+ channel-dependent EDHF responses in Type II diabetic ZDF rats. Br J Pharmacol. 2006;148:434–441. doi: 10.1038/sj.bjp.0706748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GA, Haylor J, de Jonge A. Renal sensitivity to endothelium-derived-relaxing-factor-mediated vasodilatation in the spontaneously hypertensive rat. Clin Sci (Lond) 1991;80:435–441. doi: 10.1042/cs0800435. [DOI] [PubMed] [Google Scholar]
- Büssemaker E, Popp R, Fisslthaler B, Larson CM, Fleming I, Busse R, et al. Aged spontaneously hypertensive rats exhibit a selective loss of EDHF-mediated relaxation in the renal artery. Hypertension. 2003;42:562–568. doi: 10.1161/01.HYP.0000088852.28814.E2. [DOI] [PubMed] [Google Scholar]
- Cachofeiro V, Nasjletti A. Increased vascular responsiveness to bradykinin in kidneys of SHR. Effect of N omega-nitro-L-arginine. Hypertension. 1991;15:683–688. doi: 10.1161/01.hyp.18.5.683. [DOI] [PubMed] [Google Scholar]
- Castle NA. Recent advances in the biology of small conductance calcium-activated potassium channels. Perspect Drug Discov. 1999;15/16:131–154. [Google Scholar]
- Chadha PS, Haddock RE, Howitt L, Morris MJ, Murphy TV, Grayson TH, et al. Obesity upregulates IKCa and myoendothelial gap junctions to maintain endothelial vasodilator function. J Pharmacol Exp Ther. 2010;335:284–293. doi: 10.1124/jpet.110.167593. [DOI] [PubMed] [Google Scholar]
- Chamorro V, Moreno JM, Wangensteen R, Sainz J, Rodriguez-Gomez I, Osuna A, et al. Effects of deoxycorticosterone on renal vascular reactivity and flow-pressure curve in spontaneously hypertensive rats. J Physiol Pharmacol. 2004;55:17–26. [PubMed] [Google Scholar]
- Cheong A, Bingham AJ, Li J, Kumar B, Sukumar P, Munsch C, et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol Cell. 2005;20:45–52. doi: 10.1016/j.molcel.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Collis MG, Vanhoutte PM. Vascular reactivity of isolated perfused kidneys from males and female spontaneously hypertensive rats. Circ Res. 1977;41:759–767. doi: 10.1161/01.res.41.6.759. [DOI] [PubMed] [Google Scholar]
- Damkjaer M, Nielsen G, Bodendiek S, Staehr M, Gramsbergen JB, de Wit C, et al. Pharmacological activation of KCa3.1/KCa2.3 channels produces endothelial hyperpolarization and lowers blood pressure in conscious dogs. Br J Pharmacol. 2012;165:223–234. doi: 10.1111/j.1476-5381.2011.01546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai KM, Gopalakrishnan V, Hiebert LM, McNeill JR, Wilson TW. EDHF-mediated rapid restoration of hypotensive response to acetylcholine after chronic, but not acute, nitric oxide synthase inhibition in rats. Eur J Pharmacol. 2006;546:120–126. doi: 10.1016/j.ejphar.2006.06.072. [DOI] [PubMed] [Google Scholar]
- Ding H, Kubes P, Triggle C. Potassium and acetylcholine-induced vasorelaxation in mice lacking endothelial nitric oxide synthase. Br J Pharmacol. 2000;129:1194–1200. doi: 10.1038/sj.bjp.0703144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgley AJ, Tare M, Evans RG, Skordilis C, Parkington HC. In vivo regulation of endothelium-dependent vasodilation in the rat renal circulation and the effect of streptozotocin-induced diabetes. Am J Physiol Regul Integr Comp Physiol. 2008;295:R829–R839. doi: 10.1152/ajpregu.00861.2007. [DOI] [PubMed] [Google Scholar]
- Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, et al. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br J Pharmacol. 2003;138:594–601. doi: 10.1038/sj.bjp.0705075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félétou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder. Am J Physiol. 2006a;291:985–1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- Félétou M, Vanhoutte PM. EDHF, the Complete Story. Boca Raton, FL: Taylor & Francis CRC Press; 2006b. [Google Scholar]
- Félétou M, Verbeuren TJ, Vanhoutte PM. Endothelium-dependent contractions in SHR: a tale of prostanoid TP and IP receptors. Br J Pharmacol. 2009;156:563–574. doi: 10.1111/j.1476-5381.2008.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félétou M, Köhler R, Vanhoutte PM. Nitric oxide: orchestrator of endothelium-dependent responses. Ann Med. 2011 doi: 10.3109/07853890.2011.585658. doi: 10.3109/07853890.2011.585658 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Fujii K, Tominaga M, Ohmori S, Kobayashi K, Koga T, Takata Y, et al. Decreased endothelium-dependent hyperpolarization to acetylcholine in smooth muscle of the mesenteric artery of spontaneously hypertensive rats. Circ Res. 1992;70:660–669. doi: 10.1161/01.res.70.4.660. [DOI] [PubMed] [Google Scholar]
- Fulton D, McGiff JC, Quilley J. Contribution of NO and cytochrome P450 to the vasodilator effect of bradykinin in the rat kidney. Br J Pharmacol. 1992;107:722–725. doi: 10.1111/j.1476-5381.1992.tb14513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachini FR, Carneiro FS, Lima VV, Carneiro ZN, Dorrance A, Webb RC, et al. Upregulation of intermediate calcium-activated potassium channels counterbalance the impaired endothelium-dependent vasodilation in stroke-prone spontaneously hypertensive rats. Transl Res. 2009;154:183–193. doi: 10.1016/j.trsl.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J, et al. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2005;25:704–709. doi: 10.1161/01.ATV.0000156399.12787.5c. [DOI] [PubMed] [Google Scholar]
- Grgic I, Kaistha BP, Hoyer J, Köhler R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses – relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009a;157:509–526. doi: 10.1111/j.1476-5381.2009.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, Sautter J, et al. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc Natl Acad Sci U S A. 2009b;106:14518–14523. doi: 10.1073/pnas.0903458106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwend S, Buikema H, Navis G, Henning RH, De Zeeuw D, Van Dokkum RPE. Endothelial dilatory function predicts individual susceptibility to renal damage in the 5/6 nephrectomised rat. J Am Soc Nephrol. 2002;13:292–2915. doi: 10.1097/01.asn.0000036865.22253.d4. [DOI] [PubMed] [Google Scholar]
- Hayakawa H, Hirata Y, Suzuki E, Sugimoto T, Matsuoka H, Kikuchi K, et al. Mechanisms for altered endothelium-dependent vasorelaxation in isolated kidneys from experimental hypertensive rats. Am J Physiol. 1993;264:H1535–H1541. doi: 10.1152/ajpheart.1993.264.5.H1535. [DOI] [PubMed] [Google Scholar]
- Heuzé-Joubert I, Mennecier P, Simonet S, Laubie M, Verbeuren TJ. Effect of vasodilators, including nitric oxide, on the release of GMPc and AMPc in isolated perfused rat kidney. Eur J Pharmacol. 1992;220:161–171. doi: 10.1016/0014-2999(92)90744-o. [DOI] [PubMed] [Google Scholar]
- Hilgers RH, Webb RC. Reduced expression of SKCa and IKCa channel proteins in rat small mesenteric arteries during angiotensin II-induced hypertension. Am J Physiol. 2007;292:H2275–H2284. doi: 10.1152/ajpheart.00949.2006. [DOI] [PubMed] [Google Scholar]
- Hilgers RH, Janssen GM, Fazzi GE, De Mey JG. Twenty-four-hour exposure to altered blood flow modifies endothelial Ca2+-activated K+ channels in rat mesenteric arteries. J Pharmacol Exp Ther. 2010;333:210–217. doi: 10.1124/jpet.109.161448. [DOI] [PubMed] [Google Scholar]
- Hongo K, Nakagomi T, Kassell NF, Sasaki T, Lehman M, Vollmer DG, et al. Effects of aging and hypertension on endothelium-dependent vascular relaxation in rat carotid artery. Stroke. 1988;19:892–897. doi: 10.1161/01.str.19.7.892. [DOI] [PubMed] [Google Scholar]
- Huang A, Sun D, Smith CJ, Connetta JA, Shesely EG, Koller A, et al. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am J Physiol. 2000;278:H762–H768. doi: 10.1152/ajpheart.2000.278.3.H762. [DOI] [PubMed] [Google Scholar]
- Hutri-Kahonen N, Kahonen M, Tolvanen JP, Wu X, Sallinen K, Porsti I. Ramipril therapy improves arterial dilation in experimental hypertension. Cardiovasc Res. 1997;33:188–195. doi: 10.1016/s0008-6363(96)00197-6. [DOI] [PubMed] [Google Scholar]
- Isabelle M, Simonet S, Ragonnet C, Sansilvestri-Morel P, Clavreul N, Vayssettes-Courchay C, et al. Chronic reduction of nitric oxide level in adult spontaneously hypertensive rats (SHR) induces an aortic stiffness similar to old SHR. J Vasc Res. 2012;49:309–318. doi: 10.1159/000337470. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, et al. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;2:1119–1125. doi: 10.1161/01.CIR.0000086464.04719.DD. [DOI] [PubMed] [Google Scholar]
- Leuranguer V, Gluais P, Vanhoutte PM, Verbeuren TJ, Félétou M. Openers of calcium-activated potassium channels and endothelium-dependent hyperpolarizations in the guinea pig carotid artery. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:101–109. doi: 10.1007/s00210-008-0267-x. [DOI] [PubMed] [Google Scholar]
- Lüscher TF, Diederich D, Weber E, Vanhoutte PM, Buhler FR. Endothelium-dependent responses in carotid and renal arteries of normotensive and hypertensive rats. Hypertension. 1988;11:573–578. doi: 10.1161/01.hyp.11.6.573. [DOI] [PubMed] [Google Scholar]
- Lüscher TF, Aarhus LL, Vanhoutte PM. Indomethacin improves the impaired endothelium-dependent relaxations in small mesenteric arteries of the spontaneously hypertensive rats. Am J Hypertens. 1990;3:55–58. doi: 10.1093/ajh/3.1.55. [DOI] [PubMed] [Google Scholar]
- Maeso R, Navarro-Cid J, Rodrigo E, Ruilope LM, Cachofeiro V, Lahera V. Effects of antihypertensive therapy on factors mediating endothelium-dependent relaxations in rats treated chronically with L-NAME. J Hypertens. 1999;17:221–227. doi: 10.1097/00004872-199917020-00006. [DOI] [PubMed] [Google Scholar]
- Mantelli L, Amerini S, Ledda F. Role of nitric oxide and endothelium-derived hyperpolarizing factor in vasorelaxant effect of acetylcholine as influenced by aging and hypertension. J Cardiovasc Pharmacol. 1995;25:595–602. doi: 10.1097/00005344-199504000-00013. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel FS, Man GS, Man RY, Vanhoutte PM. Hypertension and the absence of EDHF-mediated responses favour endothelium-dependent contractions in renal arteries of the rat. Br J Pharmacol. 2008;155:217–226. doi: 10.1038/bjp.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieyal P, Fulton D, McGiff JC, Quilley J. NO-independent vasodilation to acetylcholine in the rat isolated kidney utilizes a charybdotoxin-sensitive, intermediate-conductance Ca2+-activated K+ channel. J Pharmacol Exp Ther. 1998;285:659–664. [PubMed] [Google Scholar]
- Moncada S, Rees D, Schulz R, Palmer MJ. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc Natl Acad Sci U S A. 1991;88:2166–2170. doi: 10.1073/pnas.88.6.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura K, Yamamura H, Ohya S, Imaizumi Y. Voltage-dependent Ca2+-channel block by openers of intermediate and small conductance Ca2+-activated K+ channels in urinary bladder smooth muscle cells. J Pharmacol Sci. 2006;100:237–241. doi: 10.1254/jphs.sc0060011. [DOI] [PubMed] [Google Scholar]
- Ono H, Ono Y, Frohlich ED. Nitric oxide synthase inhibition in spontaneously hypertensive rats. Hypertension. 1995;26:249–255. doi: 10.1161/01.hyp.26.2.249. [DOI] [PubMed] [Google Scholar]
- Ono H, Ono Y, Takanohashi A, Matsuoka H, Frohlich ED. Apoptosis and glomerular injury after prolonged nitric oxide synthase inhibition in spontaneously hypertensive rats. Hypertension. 2001;38:1300–1306. doi: 10.1161/hy1201.096118. [DOI] [PubMed] [Google Scholar]
- Panza JA, Garcia CE, Kilcoyne CM, Quyyumi A, Cannon RO., 3rd Impaired endothelium-dependent vasodilation in patients with essential hypertension: evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation. 1995;91:1732–1738. doi: 10.1161/01.cir.91.6.1732. [DOI] [PubMed] [Google Scholar]
- Rapacon M, Mieyal P, McGiff JC, Quilley J. Contribution of calcium-activated potassium channels to the vasodilator effect of bradykinin in the isolated, perfused kidney of the rat. Br J Pharmacol. 1996;118:1504–1508. doi: 10.1111/j.1476-5381.1996.tb15566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Marcos FM, Ortiz MC, Fortepiani LA, Nadal FJ, Atucha NM, Garcia-Estan J. Mechanism of the increase pressor response to vasopressors in the mesenteric bed of nitric oxide-deficient hypertensive rats. Eur J Pharmacol. 2001;412:273–279. doi: 10.1016/s0014-2999(00)00795-0. [DOI] [PubMed] [Google Scholar]
- Sainsbury CAR, Coleman J, Brady AJ, Connell JMC, Hillier C, Petrie JR. Endothelium-dependent relaxation to inhibition of nitric oxide synthesis, but sensitive to blockade of calcium-activated potassium channels in essential hypertension. J Hum Hypertens. 2007;21:808–814. doi: 10.1038/sj.jhh.1002226. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, et al. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol. 2009;75:281–295. doi: 10.1124/mol.108.051425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T, Eder C. TRAM-34 inhibits nonselective cation channels. Pflugers Arch. 2007;454:559–563. doi: 10.1007/s00424-007-0232-4. [DOI] [PubMed] [Google Scholar]
- Simonet S, Bousquenaud M, Isabelle M, Vayssettes-Courchay C, Félétou M, Verbeuren TJ. Endothelial dysfunction in isolated perfused kidneys of L-NAME-treated spontaneous hypertensive rats. Arch Cardiovasc Dis. 2009;102:S31–S32. [Google Scholar]
- Sorensen CM, Braunstein TH, Holstein-Rathlou NH, Salomonsson M. Role of vascular potassium channels in the regulation of renal hemodynamics. Am J Physiol Renal Physiol. 2012;302:F505–518. doi: 10.1152/ajprenal.00052.2011. [DOI] [PubMed] [Google Scholar]
- Strøbæk D, Teuber L, Jørgensen TD, Ahring PK, Kjær K, Hansen RS, et al. Activation of human IK and SK Ca2+-activated K+ channels by NS 309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime) Biochim Biophys Acta. 2004;1665:1–5. doi: 10.1016/j.bbamem.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Mattei P, Salvetti A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension. 1993;21:929–933. doi: 10.1161/01.hyp.21.6.929. [DOI] [PubMed] [Google Scholar]
- Taddei S, Versari D, Cipriano A, Ghiadoni L, Galetta F, Franzoni F, et al. Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpolarizing factor in essential hypertensive patients. J Am Coll Cardiol. 2006;48:508–515. doi: 10.1016/j.jacc.2006.04.074. [DOI] [PubMed] [Google Scholar]
- Terata Y, Saito T, Fujiwara Y, Hasegawa H, Miura H, Watanabe H, et al. Pitavastatin Inhibits Upregulation of Intermediate Conductance Calcium-Activated Potassium Channels and Coronary Arteriolar Remodeling Induced by Long-Term Blockade of Nitric Oxide Synthesis. Pharmacology. 2003;68:169–176. doi: 10.1159/000070455. [DOI] [PubMed] [Google Scholar]
- Tharp DL, Wamhoff BR, Wulff H, Raman G, Cheong A, Bowles DK. Local delivery of the KCa3.1 blocker, TRAM-34, prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis. Arterioscler Thromb Vasc Biol. 2008;28:1084–1089. doi: 10.1161/ATVBAHA.107.155796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas F, Osuna A, Fernández-Rivas A. Vascular reactivity and flow-pressure curve in isolated kidneys from rats with N-nitro-L-arginine methyl ester-induced hypertension. J Hypertens. 1996;14:373–379. doi: 10.1097/00004872-199603000-00015. [DOI] [PubMed] [Google Scholar]
- Vayssettes-Courchay C, Isabelle M, Ragonnet C, Butin S, Verbeuren TJ. NOS inhibition in the SHR causes simultaneous alterations of the baroreflex, aortic distensibility and left ventricular function [Abstract] Hypertension. 2008;52:4. [Google Scholar]
- Vayssettes-Courchay C, Ragonnet C, Isabelle M, Verbeuren TJ. Aortic stiffness in vivo in hypertensive rat via echo-tracking: analysis of the pulsatile distension waveform. Am J Physiol. 2011;301:H382–H390. doi: 10.1152/ajpheart.00094.2011. [DOI] [PubMed] [Google Scholar]
- Virdis A, Cetani F, Giannarelli C, Banti C, Ghiadoni L, Ambrogini E, et al. The sulfaphenazole-sensitive pathway acts as a compensatory mechanism for impaired nitric oxide availability in patients with primary hyperparathyroidism. Effect of surgical treatment. J Clin Endocrinol Metab. 2010;95:920–927. doi: 10.1210/jc.2009-1669. [DOI] [PubMed] [Google Scholar]
- Weston AH, Porter EL, Harno E, Edwards G. Impairment of endothelial SKCa channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol. 2010;160:836–843. doi: 10.1111/j.1476-5381.2010.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Frohlich ED. Analogy of cardiac and renal complications in essential hypertension and aged SHR or L-NAME/SHR. Med Chem. 2007;3:61–65. doi: 10.2174/157340607779317634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.