

Abstract
BACKGROUND AND PURPOSE
Hydrogen sulfide (H2S), generated by enzymes such as cystathionine-γ-lyase (CSE) from L-cysteine, facilitates pain signals by activating the Cav3.2 T-type Ca2+ channels. Here, we assessed the involvement of the CSE/H2S/Cav3.2 pathway in cystitis-related bladder pain.
EXPERIMENTAL APPROACH
Cystitis was induced by i.p. administration of cyclophosphamide in mice. Bladder pain-like nociceptive behaviour was observed and referred hyperalgesia was evaluated using von Frey filaments. Phosphorylation of ERK in the spinal dorsal horn was determined immunohistochemically following intravesical administration of NaHS, an H2S donor.
KEY RESULTS
Cyclophosphamide caused cystitis-related symptoms including increased bladder weight, accompanied by nociceptive changes (bladder pain-like nociceptive behaviour and referred hyperalgesia). Pretreatment with DL-propargylglycine, an inhibitor of CSE, abolished the nociceptive changes and partly prevented the increased bladder weight. CSE protein in the bladder was markedly up-regulated during development of cystitis. Mibefradil or NNC 55–0396, blockers of T-type Ca2+ channels, administered after the symptoms of cystitis appeared, reversed the nociceptive changes. Further, silencing of Cav3.2 protein by repeated intrathecal administration of mouse Cav3.2-targeting antisense oligodeoxynucleotides also significantly attenuated the nociceptive changes, but not the increased bladder weight. Finally, the number of cells staining positive for phospho-ERK was increased in the superficial layer of the L6 spinal cord after intravesical administration of NaHS, an effect inhibited by NNC 55–0396.
CONCLUSION AND IMPLICATIONS
Endogenous H2S, generated by up-regulated CSE, caused bladder pain and referred hyperalgesia through the activation of Cav3.2 channels, one of the T-type Ca2+ channels, in mice with cyclophosphamide-induced cystitis.
Keywords: hydrogen sulfide, T-type calcium channel, bladder pain, cystitis, cystathionine-γ-lyase, phosphorylation of ERK, Cav3.2
Introduction
Hydrogen sulfide (H2S), a toxic gas occurring in volcanic zones and hot spring areas, is synthesized from L-cysteine by several enzymes including cystathionine-γ-lyase (CSE) and cystathionine-β-syntase in mammals and plays various roles as a gasotransmitter in health and disease (Wang, 2002; Fiorucci et al., 2006). The functions of H2S in sensory neurons include the regulation of the secretion of HCO3- in the duodenum (Ise et al., 2011) and Cl- in the colon (Schicho et al., 2006), contraction of bladder smooth muscle (Patacchini et al., 2004) and induction of neurogenic inflammation in the airways (Trevisani et al., 2005), lungs (Bhatia et al., 2006) and pancreas (Bhatia et al., 2005). We have demonstrated that H2S activates the Cav3.2 T-type Ca2+ channels expressed in sensory neurons, leading to the facilitation of somatic pain processing (Kawabata et al., 2007; Maeda et al., 2009; Takahashi et al., 2010; channel nomenclature follows Alexander et al., 2011). Furthermore, H2S also plays an important role in the processing of visceral pain, because exogenously administered NaHS, a H2S donor compound, evokes colonic or pancreatic pain signals and endogenous H2S generated by CSE contributes to pancreatitis-related referred hyperalgesia (Matsunami et al., 2009; Nishimura et al., 2009).
The most common visceral pain in the field of urology is bladder pain, a symptom of lower urinary tract diseases such as cystitis, cystolithiasis and bladder cancer, which impairs the quality of life in patients. Interstitial cystitis or painful bladder syndrome is an idiopathic disease, presenting with bladder pain and urinary frequency or urgency (Kelada and Jones, 2007). The bladder pain accompanying interstitial cystitis is regional, chronic and spread over the lower pelvic and suprapubic area and mimics neuropathic pain (Theoharides et al., 2008). Interestingly, there is evidence that NaHS administered intravesically produces changes in urodynamic parameters after chemical disruption of the urothelial barrier in rats, although the mechanisms underlying the effect of NaHS in vivo have yet to be analysed (Streng et al., 2008). It is still open to question whether endogenously synthesized H2S is involved in the bladder pain accompanying cystitis. Therefore, the present study investigated the possible involvement of endogenous H2S and the Cav3.2 T-type Ca2+ channels, one of molecular targets for H2S (Maeda et al., 2009; Takahashi et al., 2010), in the processing of bladder pain in mice with cyclophosphamide-induced cystitis, an animal model of human interstitial cystitis (Wantuch et al., 2007).
Methods
Animals
All animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85-23, revised 1996) and were approved by the Committee for the Care and Use of Laboratory Animals of Kinki University. The ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010) have been followed. Female ddY mice weighing 18–25 g (total = 421) were purchased from Japan SLC Inc. (Shizuoka, Japan) or Kiwa Laboratory Animals Co., Ltd. (Wakayama, Japan). The animals were housed in a temperature-controlled room under a 12-h day per night cycle at about 24°C and had free access to food and water.
Induction of cyclophosphamide-induced cystitis and assessment of bladder pain-like nociceptive behaviour and referred hyperalgesia (nociceptive changes)
Cystitis was induced in mice by the i.p. administration of cyclophosphamide at 300 mg·kg−1, and bladder pain-like nociceptive behaviour was defined as described previously (Olivar and Laird, 1999) with minor modifications (Miki et al., 2011). Mice were placed on a raised wire mesh under a clear plastic box (23.5 × 16.6 × 12.4 cm), and acclimated to the experimental environment for 1 h. Bladder pain-like nociceptive behaviour was observed from 3.5 to 4 h after the administration of cyclophosphamide; that is the number of times the animal licked the external urethral opening was measured for 30 min. Immediately after the observation of nociceptive behaviour, the region between the anus and urethral opening was stimulated mechanically using four distinct von Frey filaments (Laird et al., 2002) with strengths of 0.008, 0.07, 0.4 and 1.0 g, in ascending order of strength, for the determination of referred hyperalgesia. The mechanical stimulation with each filament was applied 10 times at intervals of 5–10 s. To prevent ‘wind-up’ effects or desensitization, repeated stimulation of the same point was avoided. The scoring of nociceptive behaviour was as follows: 0 = no response; 1 = licking or biting of the external urethral opening and/or the surrounding area, leaving the position, bending of the trunk, raising the upper half of the body and thrashing limbs; 2 = jumping. Data are expressed as the total score for responses to 10 challenges with each filament.
Evaluation of the severity of cyclophosphamide-induced cystitis in mice
Immediately after the von Frey test, the mice were killed by cervical dislocation, and the bladder was removed and weighed to estimate the bladder oedema. In some experiments, the bladder was fixed, embedded in paraffin, sectioned, and stained with haematoxylin and eosin for morphological analysis.
Silencing of Cav3.2 by intrathecal administration of antisense (AS) oligodeoxynucleotides (ODNs)
Knockdown of Cav3.2 channels was achieved by repeated intrathecal administration of the mixture of two distinct AS-ODNs. AS-ODNs targeting mouse Cav3.2 protein or scrambled (SC) ODNs were synthesized by Sigma-Aldrich Japan (Ishikari, Japan). The sequences were as follows: AS-ODN-Cav3.2 (No. 1, TGA AGT GGT AAT GGT GGT GAT GGT GGT; No.2, GAG TGA TGA TGG ACA GGA ACG AGA CCG); SC-ODN-Cav3.2 (No. 1, TAA GTG GTG GTA TGA GGG TGT TTG GGA; No.2, GGG AAA GAC CAC GGG TAA TGG TAG GAC). A mixture of two types of AS-ODN-Cav3.2 or SC-ODN-Cav3.2 solution (No.1 and 2; 1 µg·µL−1 each) in a volume of 5 µL was administered intrathecal, once a day for 3 d in mice.
Western blot analysis
The mice were killed by cervical dislocation 4–4.5 h after the i.p. administration of cyclophosphamide, and the bladder was excised and frozen in liquid nitrogen. In the knockdown experiments, the bilateral dorsal root ganglia (DRG) at the L1-L6 spinal levels were also collected. The tissue samples were homogenized in a RIPA buffer (PBS, 1% Igepal CA-630, 0.5% sodium deoxycholate and 0.1% SDS), containing 0.1 mg·mL−1 PMSF, 0.15 U·mL−1 aprotinin and 1 mM sodium orthovanadate. After the addition of 2-mercaptoethanol and bromophenol blue, the supernatant was denatured at 95–100°C for 5 min, and the proteins were separated by electrophoresis on a 12.5% (for the detection of CSE) or 7.5% [for Cav3.2 and transient receptor potential ankyrin 1 (TRPA1)] SDS-polyacrylamide gel (Wako Pure Chemicals, Osaka, Japan) and transferred onto a PVDF membrane (Immobilon-P, Millipore, Billerica, MA, USA). The membrane was blocked with a blocking solution containing 5% skim milk, 137 mM NaCl, 0.1% Tween 20 and 20 mM Tris-HCl (pH 7.6). After a wash, the membrane was incubated overnight at 4°C with an anti-CSE rabbit polyclonal antibody (1:1000), an anti-Cav3.2 rabbit polyclonal antibody (1:200) (Sigma-Aldrich Japan), an anti-TRPA1 rabbit polyclonal antibody (1:1000) (Novus Biologicals, Littleton, CO, USA) or an anti-GAPDH rabbit polyclonal antibody (1:3000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After another wash, the membrane was incubated with a horseradish peroxidase-conjugated anti-rabbit antibody (1:1000) (Cell Signaling Technology, Beverly, MA, USA). Immunolabelled proteins (CSE, 44 kDa; Cav3.2, 230 kDa; TRPA1, 110 kDa; GAPDH, 37 kDa) were visualized with enhanced chemiluminescence detection reagent (Amersham Biosciences, Little Chalfont, UK). The resulting films were scanned and quantified using densitometric software (Win ROOF, Mitani Corporation, Fukui, Japan).
Immunohistochemical analysis of phosphorylation of ERK in the spinal cord following intravesical administration of a donor for H2S in mice
The mice received indomethacin (i.p., 10 mg·kg−1) 30 min before catheterization of the bladder via the urethra, in order to reduce the background staining of phospho-ERK (p-ERK) because of endogenous prostanoid formation in response to mechanical stimuli with catheterization and/or intravesical administration, as described previously (Miki et al., 2011). Under anaesthesia with urethane (1.5 g·kg−1, i.p.) the urine was drained off through the catheter by applying light pressure to the abdomen, and the mouse was allowed to rest for 25 min. The animal then received intravesical administration of NaHS, a donor for H2S, (50 nmol per mouse; 50 µL of 1 mM solution) and 5 min later, was transcardially perfused with 20 mL of 1% paraformaldehyde in a 0.2 M phosphate buffer (pH 7.4) and subsequently with 140 mL of 4% paraformaldehyde in the same buffer for fixation. The spinal cord was removed, postfixed in the latter solution, and cryoprotected in a phosphate-buffered 30% sucrose solution overnight at 4°C. The L6 spinal cord segment was serially cut into 30 µm sections using a freezing microtome. After incubation for 20 min in a 0.3% hydrogen peroxide solution containing methanol, the sections were incubated in 1% normal goat serum for 30 min, and reacted overnight at 4°C with a rabbit polyclonal antibody against human phospho-p44/42 MAPK (Thr202/Tyr204) (Cell Signaling Technology) at a dilution of 1:500. After being washed in PBS, the sections were incubated in biotinylated anti-goat IgG for 1 h and reacted with an avidin-biotin complex (VECSTATIN Elite ABC kit, Vector, Burlingame, CA, USA) for 30 min at 4°C, and finally developed with a 0.05 M Tris-HCl buffer (pH 7.6) containing 0.05% 3,3′-diaminobenzidine-tetra HCl, 0.4% nickel ammonium sulphate, and 0.035% hydrogen peroxide for 10 min at 24°C. The sections were then rinsed three times with the Tris-HCl buffer for 10 min. Cells staining positive for p-ERK were counted in five sections of the L6 spinal cord from each mouse, and the cell number was determined bilaterally in four distinct regions, the medial dorsal horn (MDH), lateral dorsal horn (LDH), dorsal commissure (DCM) and sacral parasympathetic nucleus (Birder and de Groat, 1992; Miki et al., 2011).
Assay of H2S content in the mouse bladder tissue
Tissue H2S content was determined as described previously (Li et al., 2005) with minor modifications. The mice were killed by cervical dislocation, and the bladder was removed, weighed and homogenized in an ice-cold 100 mM potassium phosphate buffer including 10% trichloroacetic acid and 1% zinc acetate. After addition of 2.1 mM N,N-dimethyl-p-phenylenediamine sulfate and 3.15 mM FeCl3, the mixture was incubated at room temperature for 20 min and centrifuged. The absorbance of the filtered supernatant was measured at a wave-length of 670 nm. The concentration of H2S in each sample was calculated against a calibration curve of Na2S solution (15–240 µM).
Statistical analysis
Data are presented as means with SEM. Statistical significance for parametric data was analysed by Student's t-test for comparisons between two groups and by an analysis of variance followed by Tukey's test for multiple comparisons. For non-parametric analyses, the Kruskal–Wallis H-test followed by a least significant difference-type test was employed for multiple comparisons. Significance was set at a level of P < 0.05.
Materials
Cyclophosphamide, DL-propargylglycine, mibefradil, NNC 55–0396, and verapamil were purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in saline. HC-030031 was purchased from Tocris Bioscience (Bristol, UK) and dissolved in 0.5% methylcellulose. AP18 was purchased from Enzo Life Sciences Inc. (Farmingdale, NY, USA) and dissolved in PBS containing 0.46% Tween 80 and 7.5% DMSO. NaHS was obtained from Kishida Chemical Co, Ltd (Osaka, Japan) and dissolved in distilled water. Indomethacin was purchased from Wako Pure Chemicals and dissolved in 4% sodium hydrogen carbonate.
Results
Characterization of cyclophosphamide-induced cystitis accompanied by bladder pain and referred hyperalgesia in mice
As reported previously (Olivar and Laird, 1999; Miki et al., 2011), i.p. administration of cyclophosphamide (300 mg·kg−1) induced cystitis characterized by mucosal oedema and a partial rupture of urothelial cells (Figure 1A), and by a marked increase in bladder weight, used as an indicator of oedema (Figure 1D). Cyclophosphamide also provoked bladder pain-like nociceptive behaviour from 3.5 to 4 h and referred hyperalgesia in the region between the anus and urethral opening at 4 h post-administration (Figure 1B and C). Cyclophosphamide-induced cystitis is believed to be produced by accumulation of acrolein, a hepatic metabolite of cyclophosphamide, in the bladder (Fraiser and Kehrer, 1992), which exhibits cytotoxicity including DNA damage (Liu et al., 2010) and also stimulates TRPA1 channels (Bautista et al., 2006). To determine the contribution of TRPA1 channels to the pathogenesis of cyclophosphamide-induced cystitis, we examined the effect of pretreatment with a TRPA1 channel blocker on cyclophosphamide-induced cystitis. Pretreatment with AP18, a TRPA1 channel blocker, given i.p. at 10 mg·kg−1, partly prevented the cyclophosphamide-induced increase in bladder weight (Figure 1D), but failed to attenuate the nociceptive changes (bladder pain-like nociceptive behaviour or referred hyperalgesia) (Figure 1B and C). Note that AP18 at the same dose (equivalent to 47.7 µmol·kg−1) completely inhibited the mechanical hyperalgesia following intraplantar administration of allyl isothiocyanate, a TRPA1 channel agonist, in mice (Okubo et al., 2012).
Figure 1.
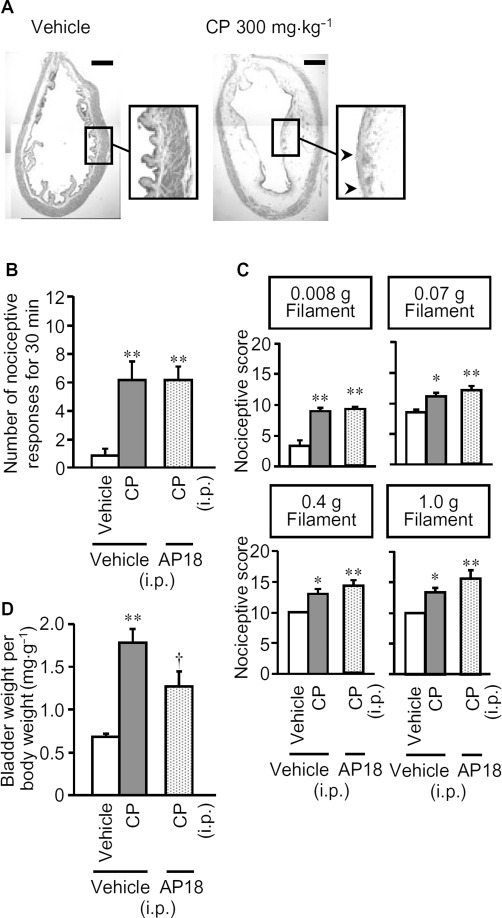
Characterization of the cyclophosphamide-induced cystitis accompanied by bladder pain-like nociceptive behaviour and referred hyperalgesia in mice. Cyclophosphamide (CP; 300 mg·kg−1) or vehicle was administered i.p. to mice. (A) Morphological characteristics of bladder tissue from mice with cyclophosphamide-induced cystitis, stained with haematoxylin and eosin. Arrow heads show the rupture of urothelial cells. Scale bars indicate 1 mm. Nociceptive behaviour (B) was observed 3.5–4 h after cyclophosphamide, and referred hyperalgesia was evaluated 4 h after cyclophosphamide (C). After the nociceptive test, the mice were killed and the bladder weight was measured as an indicator of bladder oedema (D). AP18, an inhibitor of TRPA1 channels (10 mg·kg−1) or vehicle was given i.p. to mice 30 min before cyclophosphamide. Data show means with SEM for six mice. *P < 0.05, **P < 0.01 versus vehicle + vehicle. †P < 0.05 versus vehicle + CP.
Involvement of CSE, an H2S-synthesizing enzyme, in cyclophosphamide-induced cystitis and bladder pain and referred hyperalgesia in mice
To investigate the role of endogenous H2S synthesized by CSE in the development of cyclophosphamide-induced cystitis accompanied by bladder pain, we examined the effect of DL-propargylglycine (PPG), a CSE inhibitor. PPG at 100 mg·kg−1 administered i.p. 60 min before cyclophosphamide markedly inhibited the cyclophosphamide-induced nociceptive changes, i.e., bladder pain-like nociceptive behaviour and referred hyperalgesia (Figure 2A and B), and reduced the increase in bladder weight (Figure 2C). In agreement with the results from inhibition experiments, Western blot analysis revealed a marked up-regulation of CSE at protein levels in the bladder tissue of mice with cyclophosphamide-induced cystitis, which was not affected by pretreatment with the TRPA1 blocker, AP18 (Figure 3). Surprisingly, combined pretreatment with AP18 (10 mg·kg−1) and PPG (100 mg·kg−1) did not affect the increased bladder weight (Figure 2D), although either compound, given alone, did decrease bladder weight (see Figures 1D and 2C).
Figure 2.
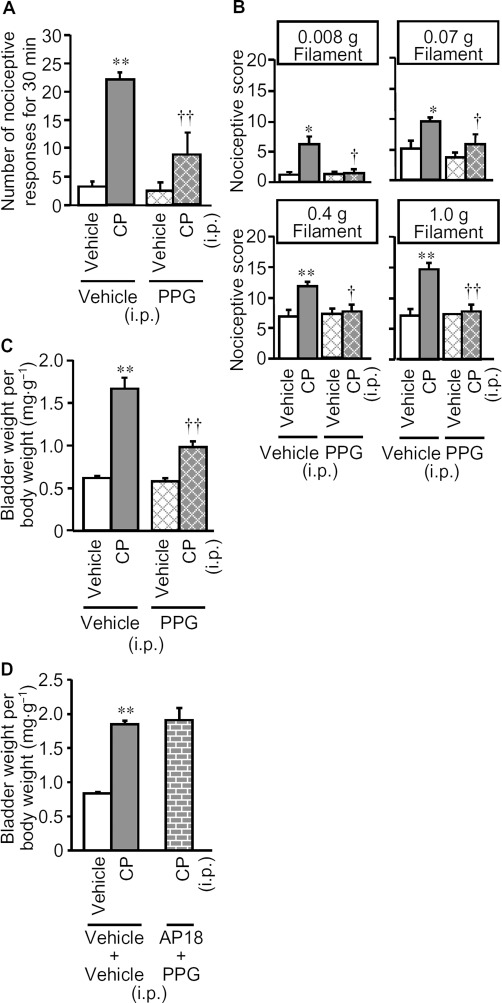
Effect of PPG, a CSE inhibitor, on cyclophosphamide-induced bladder pain-like nociceptive behaviour, referred hyperalgesia and increases in bladder weight. (A, B, C) PPG (100 mg·kg−1) or vehicle was administered i.p. to mice 60 min before cyclophosphamide (CP; 300 mg·kg−1) or vehicle. Nociceptive behaviour (A) was observed 3.5–4 h after cyclophosphamide and referred hyperalgesia was evaluated 4 h after cyclophosphamide (B). After the nociceptive tests, the mice were killed and the bladder weight was measured as an indicator of bladder oedema (C). (D) Effects of pretreatment with PPG in combination with AP18, a TRPA1 channel blocker, on cyclophosphamide-induced increases in bladder weight. PPG (100 mg·kg−1) and AP18 (10 mg·kg−1) were administered i.p. to mice 60 min and 30 min, respectively, before cyclophosphamide (300 mg·kg−1) or vehicle. Data show the mean with SEM for 7–8 (A, B and C) or 5–6 (D) mice. *P < 0.05, **P < 0.01 versus vehicle + vehicle. †P < 0.05, ††P < 0.01 versus vehicle + CP.
Figure 3.
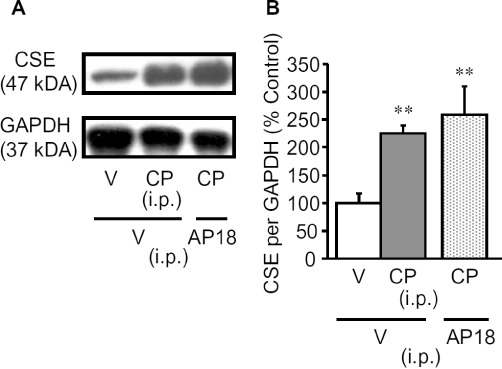
Enhanced expression of CSE protein in the bladder of mice with cyclophosphamide-induced cystitis. (A) Typical photographs of Western blots for CSE in the bladder. (B) CSE protein levels in the bladder quantified by densitometry. The mice were killed for excision of the bladder 4 h after i.p. cyclophosphamide (CP; 300 mg·kg−1) or vehicle. AP18 (10 mg·kg−1) or vehicle (V) was administered i.p. to mice 30 min before cyclophosphamide. Data show the mean with SEM for four to six experiments. **P < 0.01 versus vehicle + vehicle.
Inhibitors of T-type, but not L-type, Ca2+ channels or TRPA1 channels reverse the cyclophosphamide-induced nociceptive changes in mice
T-type Ca2+ channels are known to mediate the facilitation of somatic, pancreatic and colonic nociception by H2S (Kawabata et al., 2007; Maeda et al., 2009; Matsunami et al., 2009; Nishimura et al., 2009; Takahashi et al., 2010), while the functions of L-type Ca2+ channels and TRPA1 channels could also be modulated by H2S (Garcia-Bereguiain et al., 2008; Streng et al., 2008; Sun et al., 2008). To clarify the mechanisms by which CSE-derived H2S participated in the cystitis-related bladder pain, we examined the effects of the T-type Ca2+ channel blockers mibefradil and NNC 55–0396, the L-type Ca2+ channel blocker verapamil and the TRPA1 channel blocker AP18, given i.p. after the symptoms of cystitis appeared. Mibefradil (10 mg·kg−1) or NNC 55–0396 (20 mg·kg−1) given i.p. 3 h after cyclophosphamide, completely suppressed the cystitis-related nociceptive changes (Figure 4A and B), while neither reversed the already increased bladder weight (Figure 4C). In contrast, verapamil (5 mg·kg−1 i.p.) with the same schedule, exerted no inhibitory effects on the nociceptive changes or on the increased bladder weight (Figure 5). Although pretreatment with AP18 (10 mg·kg−1) partly prevented the increased bladder weight without affecting nociceptive changes (see Figure 1), post-treatment with this compound, i.e., after the symptoms of cystitis appeared, failed to modify the nociceptive changes or the increased bladder weight (Figure 5). HC-030031, another TRPA1 channel blocker, (300 mg·kg−1) given in the same manner, also failed to significantly reduce the nociceptive changes induced by cyclophosphamide (Figure 5).
Figure 4.
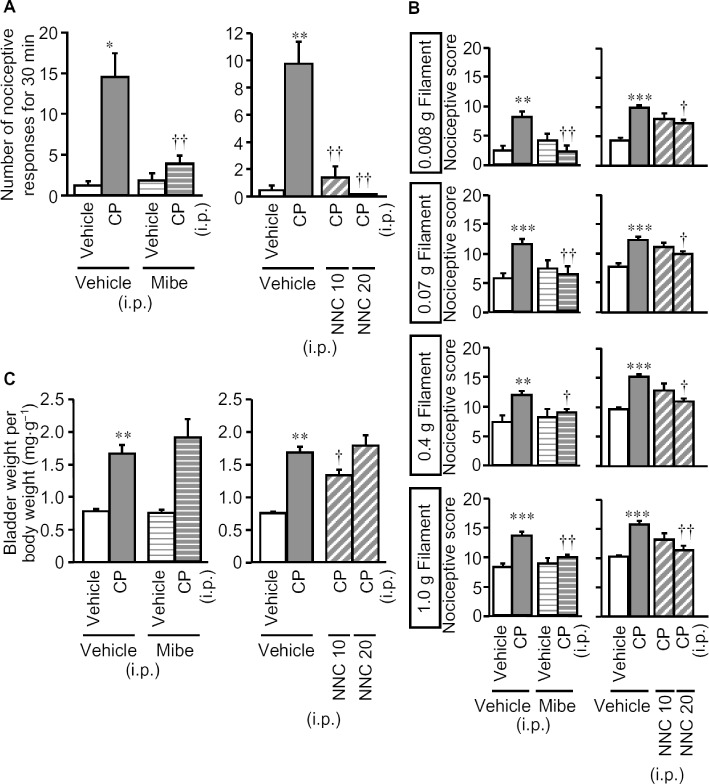
Effect of mibefradil and NNC 55–0396, T-type Ca2+ channel blockers, on cyclophosphamide-induced nociceptive changes and increases in bladder weight. Mibefradil (Mibe; 10 mg·kg−1), NNC 55–0396 (NNC; 10 or 20 mg·kg−1) or vehicle was administered i.p. to mice 3 h after cyclophosphamide (CP; 300 mg·kg−1, i.p). Nociceptive behaviour (A) was observed 3.5–4 h after cyclophosphamide, and referred hyperalgesia was evaluated 4 h after cyclophosphamide (B). After the nociceptive tests, the mice were killed and the bladder weight was measured as an indicator of bladder oedema (C). Data show the mean with SEM for 7–11 (left panels in A, B and C) or 5–13 (right panels in A, B and C) mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle + vehicle. †P < 0.05, ††P < 0.01 versus vehicle + CP.
Figure 5.
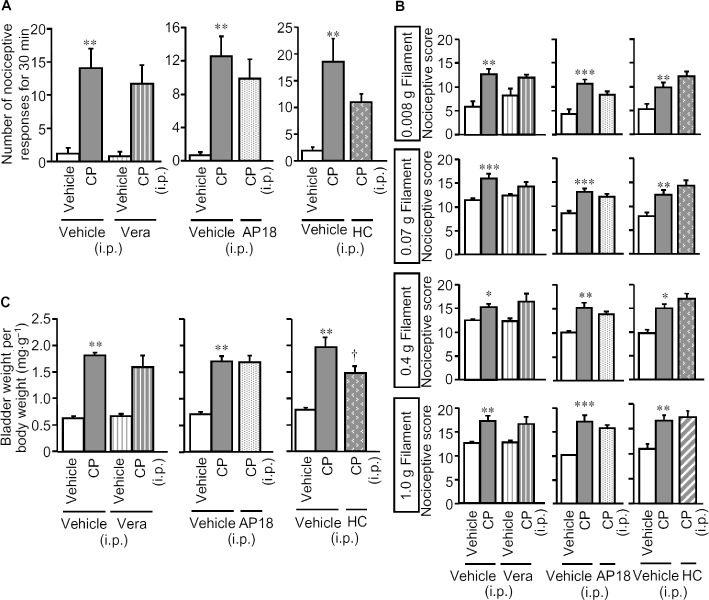
Lack of effects of post-treatment with verapamil, an L-type Ca2+ channel blocker, or AP18 and HC-030031, TRPA1 channel blockers, on cyclophosphamide-induced bladder pain-like nociceptive behaviour, referred hyperalgesia and increases in bladder weight. Verapamil (Vera; 5 mg·kg−1), AP18 (10 mg·kg−1), HC-030031 (300 mg·kg−1) or vehicle was administered i.p. to mice 3 h after the i.p. administration of cyclophosphamide (CP; 300 mg·kg−1) or vehicle. Nociceptive behaviour (A) was observed 3.5–4 h after cyclophosphamide, and referred hyperalgesia was evaluated 4 h after cyclophosphamide (B). After the nociceptive tests, the mice were killed and the bladder weight was measured as an indicator of bladder oedema (C). Data show the mean with SEM for five to six (left panels in A, B and C), six (centre panels in A, B and C) or 9–10 (right panels in A, B and C) mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle + vehicle. †P < 0.05 versus vehicle + CP.
Silencing of Cav3.2 protein suppresses the cyclophosphamide-induced nociceptive changes
Among the three isoforms T-type Ca2+ channels (Cav3.1, Cav3.2 and Cav3.3), the Cav3.2 channels are considered to be the most important for pain signalling (Bourinet et al., 2005; Choi et al., 2007; Messinger et al., 2009) and mediate H2S-induced facilitation of somatic pain processing (Maeda et al., 2009; Takahashi et al., 2010). Hence, we examined the effect of knocking down Cav3.2 channels with AS-ODN-Cav3.2 on the cyclophosphamide-induced bladder pain in mice. First, we confirmed that repeated intrathecal administration of AS-ODN-Cav3.2 to the mice greatly decreased the level of Cav3.2 protein, but not TRPA1 protein, in the DRG to 20% of the levels in control mice treated with SC-ODN, indicating successful and specific silencing of Cav3.2 protein in the sensory neurons (Figure 6A and B). The knockdown of Cav3.2 channels significantly suppressed the cyclophosphamide-induced nociceptive changes (Figure 6C and D), although the increase in bladder weight was unaffected (Figure 6E).
Figure 6.
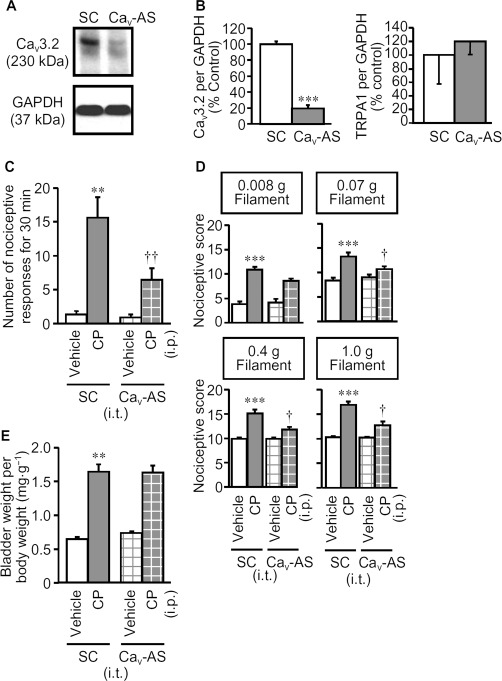
Effect of knockdown of Cav3.2 protein by repeated intrathecal administration of AS-ODN-Cav3.2 on cyclophosphamide-induced nociceptive changes and increases in bladder weight. AS-ODN-Cav3.2 (Cav-AS) or SC-ODN (SC) (10 µg per mouse in a volume of 5 µL) was administrated i.t. once a day for 3 d. (A) Typical photographs of Western blots for Cav3.2 protein in the DRG of the mice treated with Cav-AS or SC. (B) Cav-AS-induced decrease in expression levels of Cav3.2 protein, but not TRPA1 protein, quantified by densitometry. Cyclophosphamide (CP; 300 mg·kg−1) or vehicle was administered i.p. to mice treated with Cav-AS or SC. Nociceptive behaviour (C) was observed 3.5–4 h after cyclophosphamide, and referred hyperalgesia was evaluated 4 h after cyclophosphamide (D). After the nociceptive tests, the mice were killed and the bladder weight was measured as an indicator of bladder oedema (E). Data show the mean with SEM for four (A and B) or 11–12 (C, D and E) mice. **P < 0.01, ***P < 0.001 versus SC + vehicle. †P < 0.05 versus SC + CP.
Expression levels of Cav3.2 protein in the DRG and H2S levels in the bladder tissue following administration of cyclophosphamide in mice
Expression levels of Cav3.2 protein in the DRG tended to increase 4 h after systemic administration of cyclophosphamide in mice. Quantification of the Western blots showed that the expression level for Cav3.2 protein was 192% higher in cyclophosphamide-injected mice than in vehicle-treated animals. However, we could not detect a significant increase in bladder H2S levels after cyclophosphamide treatment: the corresponding H2S levels were 123 ± 25.5 % (n = 3) in the cyclophosphamide-treated mice and 100 ± 25.5 % in the vehicle-treated mice (n = 3).
Intravesical administration of an H2S donor facilitates phosphorylation of ERK in the spinal cord through T-type Ca2+ channels
To ask whether H2S in the bladder activated sensory neurons via T-type Ca2+ channels, we measured p-ERK in the four distinct regions of the L6 spinal cord (Figure 7A) after intravesical administration of NaHS, an H2S donor, in anesthetized mice. As reported previously (Miki et al., 2011), intravesical administration of distilled water itself caused the phosphorylation of ERK in the spinal cord at the L6 level, an effect attenuated by pretreatment with indomethacin (Figure 7B). In mice pretreated with indomethacin, intravesical administration of NaHS (50 nmol per mouse) caused a prompt increase of p-ERK in the superficial layers of the spinal cord (Figure 7C). The number of p-ERK-positive cells in the four regions (Figure 7A) was significantly increased only in MDH and LDH by the intravesical administration of NaHS (Figure 7D). In addition, pretreatment with the T-type Ca2+ channel blocker NNC 55–0396, (i.p. 20 mg·kg−1) completely inhibited the phosphorylation of ERK in the superficial layers of the L6 spinal cord (Figure 8A) and the increased number of p-ERK-positive cells in the spinal MDH and LDH regions (Figure 8B) following intravesical administration of NaHS.
Figure 7.
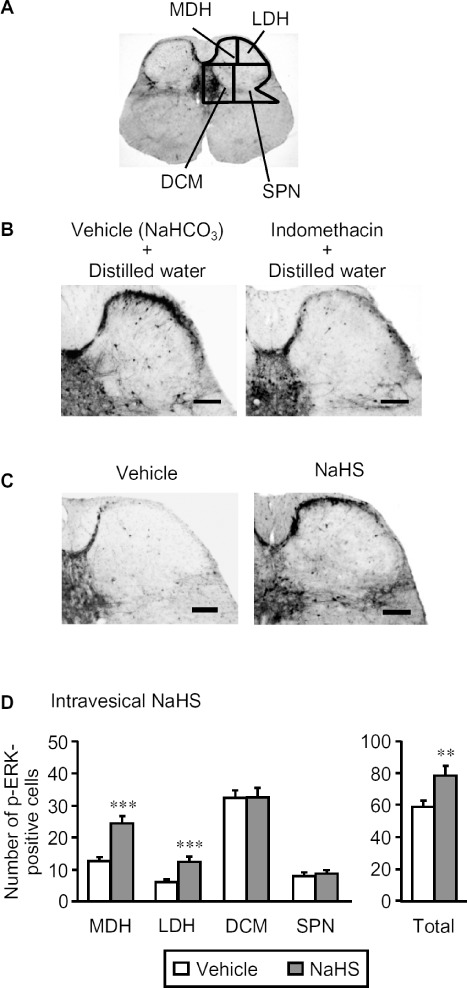
Effect of intravesical NaHS on the number of p-ERK-positive cells in the L6 spinal cord of the mice. The mice were given indomethacin (i.p., 10 mg·kg−1) 55 min before challenge with NaHS. For immunostaining of p-ERK, the spinal cord was transcardially perfused and fixed 5 min after the intravesical administration of NaHS (50 nmol per mouse; 50 µL of 1mM solution). (A) Drawings of four separated regions of the L6 spinal cord in mice. (B) Typical microphotographs for the immunostaining of p-ERK 5 min after distilled water (vehicle) in the mice pretreated with indomethacin or vehicle (NaHCO3). (C) Typical microphotographs for the immunostaining of p-ERK caused by intravesical NaHS in mice pretreated with indomethacin. Scale bars indicate 100 µm. (D) The number of p-ERK-positive cells in the four regions of the bilateral spinal cord following intravesical NaHS. Data indicate the mean with the SEM for 35–40 (seven to eight mice) slices. **P < 0.01, ***P < 0.001 versus vehicle. SPN, sacral parasympathetic nucleus.
Figure 8.
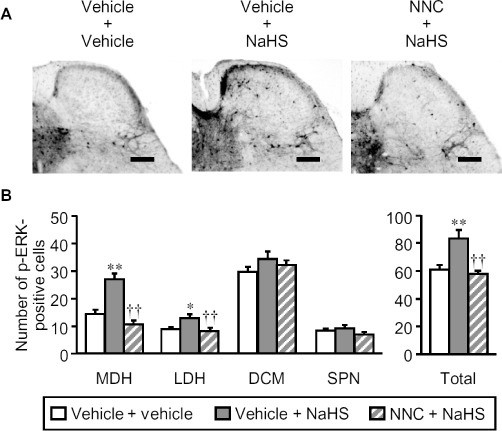
Effect of NNC 55–0396, a selective T-type Ca2+ channel blocker, on the phosphorylation of ERK in the spinal cord of mice after intravesical NaHS. The mice were given indomethacin (i.p., 10 mg·kg−1) 55 min before challenge with NaHS. NNC 55–0396 (NNC; 20 mg·kg−1) or vehicle was administered i.p. 30 min before the intravesical administration of NaHS (50 nmol per mouse; 50 µL of 1mM) or vehicle. For immunostaining of p-ERK, the spinal cord was transcardially perfused and fixed 5 min after the intravesical administration of NaHS. (A) Typical microphotographs showing the effect of NNC on the phosphorylation of spinal ERK after intravesical NaHS in mice. Scale bars indicate 100 µm. (B) Effect of NNC on the intravesical NaHS-induced increase in the number of p-ERK-positive cells in the four regions of the bilateral spinal cord. Data indicate the mean with the SEM for 30–35 (six to seven mice) slices. *P < 0.05, **P < 0.01 versus vehicle + vehicle. ††P < 0.01 versus vehicle + NaHS. SPN, sacral parasympathetic nucleus.
Pretreatement with T-type Ca2+ channel inhibitors prevents the cyclophosphamide-induced cystitis and nociceptive changes in mice
Finally, to study the involvement of T-type Ca2+ channels in the development of cystitis, we determined the preventive effects of pretreatment with mibefradil and NNC 55–0396, on the cyclophosphamide-induced increase in bladder oedema and nociceptive changes in mice. A single pretreatment with mibefradil (i.p. 10 mg·kg−1) significantly reduced the increased bladder weight (Figure 9C), but not the nociceptive changes (Figure 9A and B), whereas the same dose of mibefradil, given twice (one injection before and one 3 h after cyclophosphamide treatment), significantly prevented all cystitis symptoms including the nociceptive changes (Figure 9). On the other hand, a single pretreatment with NNC 55–0396 (20 mg·kg−1) significantly reduced both the increased bladder weight and the nociceptive changes (Figure 9).
Figure 9.
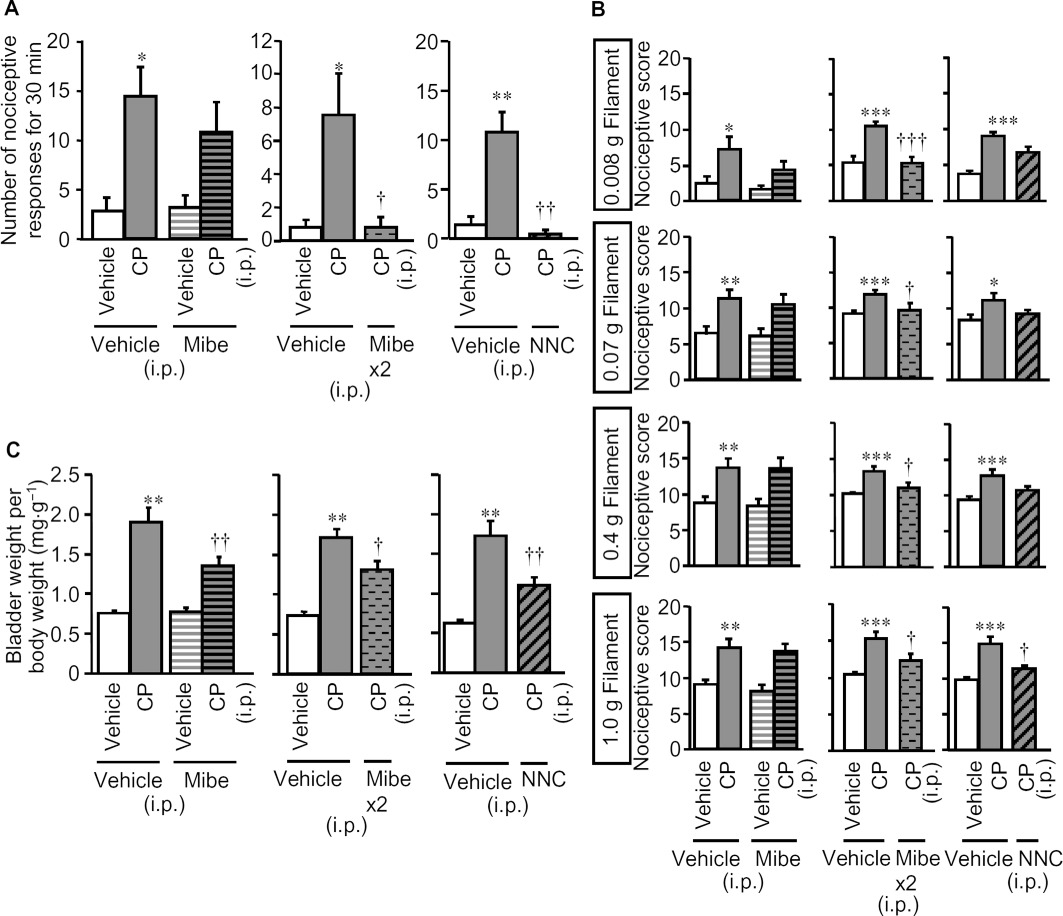
Preventive effects of pretreatment with mibefradil and NNC 55–0396, T-type Ca2+ channel blockers, on cyclophosphamide-induced bladder pain-like nociceptive behaviour, referred hyperalgesia and increases in bladder weight in mice. Mibefradil (Mibe) was given i.p. once 30 min before cyclophosphamide (CP; i.p., 300 mg·kg−1) (left panels in A, B and C), or (Mibe x2) 30 min before and 3 h after i.p. cyclophosphamide (centre panels in A, B and C). NNC 55–0396 (20 mg·kg−1) was administered i.p. once 30 min before cyclophosphamide (right panels in A, B and C). Nociceptive behaviour (A) was observed 3.5–4 h after cyclophosphamide and referred hyperalgesia was evaluated 4 h after cyclophosphamide (B). After the nociceptive tests, the mice were killed and the bladder weight was measured as an indicator of bladder oedema (C). Data show the mean with SEM for seven to eight (left panels in A, B and C), 10–14 (centre panels in A, B and C) and eight (right panels in A, B and C) mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle + vehicle. †P < 0.05, ††P < 0.01, †††P < 0.001 versus vehicle + CP.
Discussion and conclusions
Prevention of the cyclophosphamide-induced increase in the bladder weight and the nociceptive changes (bladder pain and referred hyperalgesia) by inhibition of CSE, an H2S-forming enzyme, and the cyclophosphamide-induced up-regulation of CSE in the bladder suggested the involvement of CSE-derived H2S in the pathogenesis of this cystitis model. Our data from the experiments using the inhibitors of T-type Ca2+ channels and the AS-ODN-Cav3.2 for silencing of Cav3.2 protein strongly imply a critical role of the Cav3.2 T-type Ca2+ channels, known to be one of molecular targets for H2S, in the cystitis-related nociceptive changes. The notion that H2S in the bladder tissue causes excitation of nociceptors via T-type Ca2+ channels, is supported by our results showing that phosphorylation of spinal ERK following intravesical administration of the H2S donor was reversed by blockade of T-type Ca2+ channels. Together, the present study suggests that the CSE/H2S/Cav3.2 channel pathway was involved in the bladder pain accompanying cyclophosphamide-induced cystitis, a possible model for interstitial cystitis.
Cyclophosphamide-induced cystitis in mice was characterized by nociceptive changes (bladder pain and referred hyperalgesia) and increased bladder weight, and by the histological profile including mucosal oedema and a partial rupture of urothelial cells, but little or no inflammatory cell infiltration (see Figure 1), as reported previously (Olivar and Laird, 1999; Boucher et al., 2000; Wang et al., 2008; Miki et al., 2011). These features are similar to those of human interstitial cystitis (Wantuch et al., 2007). Our findings that pretreatment with AP18, a TRPA1 channel inhibitor, partly prevented the increase in bladder weight, but not the nociceptive changes in bladder following cyclophosphamide (see Figure 1), imply that the pathogenesis of this cystitis model would involve both TRPA1 channel-dependent and -independent actions of acrolein, a major toxic metabolite of cyclophosphamide (Fraiser and Kehrer, 1992). Considering that the cyclophosphamide-induced upregulation of CSE in the bladder was not inhibited by pretreatment with the TRPA1 channel blocker (see Figure 3), CSE-derived H2S appears to be fully capable of causing the nociceptive changes, independently of TRPA1 channels. Although the pathogenesis of human interstitial cystitis is still open to question, it would be interesting to explore the clinical impact of pharmacological intervention in the CSE/H2S/ T-type Ca2+ channel pathway in patients with interstitial cystitis in future. Nonetheless, it was surprising that combined pretreatment with AP18 and PPG did not affect the cyclophosphamide-induced bladder oedema (see Figure 2D), while each of them exhibited significant inhibitory effects (see Figures 1D and 2C). These findings remain to be analysed further.
Our evidence for involvement of CSE-derived H2S in the nociceptive changes is in agreement with our previous reports concerning lipopolysaccharide-induced inflammatory hyperalgesia (Kawabata et al., 2007), neuropathic pain (Takahashi et al., 2010) and pancreatitis-related pain (Nishimura et al., 2009). Particularly, considering the up-regulation of CSE protein in pancreatic tissue during cerulein-induced pancreatitis accompanied by pancreatic pain (Nishimura et al., 2009), the present study indicates a key role of endogenous H2S generated by CSE in visceral inflammatory pain. The tendency towards up-regulation of Cav3.2 protein expression in DRG may also contribute to the endogenous H2S-dependent nociceptive changes in the mice treated with cyclophosphamide. It is open to question if the enhanced T-type Ca2+ channel activity involves cellular or molecular events such as the altered membrane translocation and phosphorylation of Cav3.2 channels. It is noteworthy that pretreatment with PPG, a CSE inhibitor, partly prevented the increased bladder tissue weight caused by cyclophosphamide (see Figure 2C), indicating a contribution of endogenous H2S to the development of oedema in the bladder tissue. Further, the preventive effects of pretreatment with T-type Ca2+ channel blockers on the increased bladder weight and nociceptive changes (see Figure 9) suggest that the pro-inflammatory effect of endogenous H2S might be, at least in part, mediated by activation of T-type Ca2+ channels. In contrast, because the knockdown of Cav3.2 protein did not reduce the cyclophosphamide-induced increase in the bladder weight (see Figure 6E), the possibility cannot be ruled out that T-type Ca2+ channels other than Cav3.2 might contribute to the cyclophosphamide-induced bladder inflammation. Together, several inconsistencies, as described earlier, may preclude a definitive conclusion regarding the involvement of the H2S and Cav3.2 channel signalling in the formation of bladder oedema.
The pharmacological profile of cyclophosphamide-induced nociceptive changes and phosphorylation of spinal ERK following intravesical NaHS (see Figures 4, 5 and 7) and the data from Cav3.2 protein knockdown experiments (see Figure 6) clearly demonstrate that T-type Ca2+ channels are the primary target for endogenous and exogenous H2S. H2S should be capable of activating TRPA1 channels, because NaHS in a concentration range of 1–10 mM causes cytosolic Ca2+ mobilization in TRPA1-transfected cells, but not untransfected cells (Streng et al., 2008). The lack of effect of post-treatment with AP18 or HC-030031 on the nociceptive changes after cyclophosphamide in the present study (see Figure 5) implied that the concentration of H2S generated in the bladder tissue after cyclophosphamide might not be high enough to activate TRPA1 channels, or that activation of TRPA1 channels by H2S, if any, might not contribute critically to the bladder pain, but to the development of detrusor overactivity during cystitis, as reported elsewhere (Streng et al., 2008). Although there is conflicting evidence that H2S facilitates and inhibits the functions of L-type Ca2+ channels (Garcia-Bereguiain et al., 2008; Sun et al., 2008), the lack of effect of verapamil on the bladder pain/referred hyperalgesia in mice treated with cyclophosphamide (see Figure 5) makes the involvement of L-type Ca2+ channels much less likely.
The levels of p-ERK in the spinal cord are now often used as an immediate marker for visceral pain (Galan et al., 2003; Million et al., 2006). We have reported that the administration of NaHS into the colon and pancreatic duct evoked prompt phosphorylation of ERK in the superficial layer of the spinal dorsal horn at T13-L1 and L5-S1 levels and at T9 level, respectively, in mice and/or rats (Matsunami et al., 2009; Fukushima et al., 2010). The present finding that intravesical NaHS caused prompt phosphorylation of ERK in the superficial layers (MDH and LDH) of the L6 spinal cord (see Figure 7) is consistent with the previous studies determining immediate phosphorylation of ERK and delayed expression of Fos in the spinal cord following intravesical acetic acid in rats (Birder and de Groat, 1992; Cruz et al., 1996; 2007). Clearly, it would appear, from the inhibitory effect of NNC 55–0396 (see Figure 7) that the phosphorylation of spinal ERK following intravesical NaHS results from excitation of nociceptors via T-type Ca2+ channels, considering. Thus, exogenously applied H2S activates T-type Ca2+ channels in the bladder, leading to neuronal excitation. However, it has yet to be demonstrated whether endogenous levels of H2S in the bladder are capable of activating T-type Ca2+ channels during the development of cyclophosphamide-induced cystitis.
In conclusion, in the cyclophosphamide-induced cystitis mouse model, endogenous H2S, generated by upregulated CSE, following progression of the disease, facilitated the excitation of sensory neurons through the activation of Cav3.2 T-type Ca2+ channels, leading to bladder pain. The present study thus suggests that the treatment of pain by targeting CSE or the Cav3.2 T-type Ca2+ channels would contribute usefully to the design of a new therapeutic strategy for patients suffering from interstitial cystitis.
Acknowledgments
This work was supported in part by the ‘Antiaging Center Project’ for Private Universities from Ministry of Education, Culture, Sports, Science and Technology, 2008–2012, and by a Grant-in-Aid for Research Activity Start-up.
Glossary
- AS
antisense
- CSE
cystathionine-γ-lyase
- DCM
dorsal commissure
- DRG
dorsal root ganglia
- LDH
lateral dorsal horn
- MDH
medial dorsal horn
- ODNs
oligodeoxynucleotides
- p-ERK
phospho-ERK
- PPG
DL-propargylglycine, SC, scrambled
- TRPA1
transient receptor potential ankyrin 1
Conflicts of interest
The authors have no conflicts of interest to report.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Wong FL, Fu D, Lau HY, Moochhala SM, Moore PK. Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 2005;19:623–625. doi: 10.1096/fj.04-3023fje. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Zhi L, Zhang H, Ng SW, Moore PK. Role of substance P in hydrogen sulfide-induced pulmonary inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2006;291:L896–L904. doi: 10.1152/ajplung.00053.2006. [DOI] [PubMed] [Google Scholar]
- Birder LA, de Groat WC. Increased c-fos expression in spinal neurons after irritation of the lower urinary tract in the rat. J Neurosci. 1992;12:4878–4889. doi: 10.1523/JNEUROSCI.12-12-04878.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher M, Meen M, Codron JP, Coudore F, Kemeny JL, Eschalier A. Cyclophosphamide-induced cystitis in freely-moving conscious rats: behavioral approach to a new model of visceral pain. J Urol. 2000;164:203–208. [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315–324. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Na HS, Kim J, Lee J, Lee S, Kim D, et al. Attenuated pain responses in mice lacking Ca(V)3.2 T-type channels. Genes Brain Behav. 2007;6:425–431. doi: 10.1111/j.1601-183X.2006.00268.x. [DOI] [PubMed] [Google Scholar]
- Cruz CD, Ferreira D, McMahon SB, Cruz F. The activation of the ERK pathway contributes to the spinal c-fos expression observed after noxious bladder stimulation. Somatosens Mot Res. 2007;24:15–20. doi: 10.1080/08990220601143265. [DOI] [PubMed] [Google Scholar]
- Cruz F, Avelino A, Coimbra A. Desensitization follows excitation of bladder primary afferents by intravesical capsaicin, as shown by c-fos activation in the rat spinal cord. Pain. 1996;64:553–557. doi: 10.1016/0304-3959(95)00157-3. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Fraiser L, Kehrer JP. Murine strain differences in metabolism and bladder toxicity of cyclophosphamide. Toxicology. 1992;75:257–272. doi: 10.1016/0300-483x(92)90007-2. [DOI] [PubMed] [Google Scholar]
- Fukushima O, Nishimura S, Matsunami M, Aoki Y, Nishikawa H, Ishikura H, et al. Phosphorylation of ERK in the spinal dorsal horn following pancreatic pronociceptive stimuli with proteinase-activated receptor-2 agonists and hydrogen sulfide in rats: evidence for involvement of distinct mechanisms. J Neurosci Res. 2010;88:3198–3205. doi: 10.1002/jnr.22480. [DOI] [PubMed] [Google Scholar]
- Galan A, Cervero F, Laird JM. Extracellular signaling-regulated kinase-1 and -2 (ERK 1/2) mediate referred hyperalgesia in a murine model of visceral pain. Brain Res Mol Brain Res. 2003;116:126–134. doi: 10.1016/s0169-328x(03)00284-5. [DOI] [PubMed] [Google Scholar]
- Garcia-Bereguiain MA, Samhan-Arias AK, Martin-Romero FJ, Gutierrez-Merino C. Hydrogen sulfide raises cytosolic calcium in neurons through activation of L-type Ca2+ channels. Antioxid Redox Signal. 2008;10:31–42. doi: 10.1089/ars.2007.1656. [DOI] [PubMed] [Google Scholar]
- Ise F, Takasuka H, Hayashi S, Takahashi K, Koyama M, Aihara E, et al. Stimulation of duodenal HCO(3) secretion by hydrogen sulphide in rats: relation to prostaglandins, nitric oxide and sensory neurones. Acta Physiol (Oxf) 2011;201:117–126. doi: 10.1111/j.1748-1716.2010.02152.x. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Ishiki T, Nagasawa K, Yoshida S, Maeda Y, Takahashi T, et al. Hydrogen sulfide as a novel nociceptive messenger. Pain. 2007;132:74–81. doi: 10.1016/j.pain.2007.01.026. [DOI] [PubMed] [Google Scholar]
- Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275:223–229. doi: 10.1007/s00404-006-0247-7. [DOI] [PubMed] [Google Scholar]
- Laird JM, Souslova V, Wood JN, Cervero F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J Neurosci. 2002;22:8352–8356. doi: 10.1523/JNEUROSCI.22-19-08352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19:1196–1198. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- Liu XY, Zhu MX, Xie JP. Mutagenicity of acrolein and acrolein-induced DNA adducts. Toxicol Mech Methods. 2010;20:36–44. doi: 10.3109/15376510903530845. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Aoki Y, Sekiguchi F, Matsunami M, Takahashi T, Nishikawa H, et al. Hyperalgesia induced by spinal and peripheral hydrogen sulfide: evidence for involvement of Cav3.2 T-type calcium channels. Pain. 2009;142:127–132. doi: 10.1016/j.pain.2008.12.021. [DOI] [PubMed] [Google Scholar]
- Matsunami M, Tarui T, Mitani K, Nagasawa K, Fukushima O, Okubo K, et al. Luminal hydrogen sulfide plays a pronociceptive role in mouse colon. Gut. 2009;58:751–761. doi: 10.1136/gut.2007.144543. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, et al. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain. 2009;145:184–195. doi: 10.1016/j.pain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Matsunami M, Nakamura S, Okada H, Matsuya H, Kawabata A. ONO-8130, a selective prostanoid EP1 receptor antagonist, relieves bladder pain in mice with cyclophosphamide-induced cystitis. Pain. 2011;152:1373–1381. doi: 10.1016/j.pain.2011.02.019. [DOI] [PubMed] [Google Scholar]
- Million M, Wang L, Wang Y, Adelson DW, Yuan PQ, Maillot C, et al. CRF2 receptor activation prevents colorectal distension induced visceral pain and spinal ERK1/2 phosphorylation in rats. Gut. 2006;55:172–181. doi: 10.1136/gut.2004.051391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Fukushima O, Ishikura H, Takahashi T, Matsunami M, Tsujiuchi T, et al. Hydrogen sulfide as a novel mediator for pancreatic pain in rodents. Gut. 2009;58:762–770. doi: 10.1136/gut.2008.151910. [DOI] [PubMed] [Google Scholar]
- Okubo K, Matsumura M, Kawaishi Y, Aoki Y, Matsunami M, Okawa Y, et al. Hydrogen sulfide-induced mechanical hyperalgesia and allodynia require activation of both Ca(v) 3.2 and TRPA1 channels in mice. Br J Pharmacol. 2012;166:1738–1743. doi: 10.1111/j.1476-5381.2012.01886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivar T, Laird JM. Cyclophosphamide cystitis in mice: behavioural characterisation and correlation with bladder inflammation. Eur J Pain. 1999;3:141–149. doi: 10.1053/eujp.1998.0105. [DOI] [PubMed] [Google Scholar]
- Patacchini R, Santicioli P, Giuliani S, Maggi CA. Hydrogen sulfide (H2S) stimulates capsaicin-sensitive primary afferent neurons in the rat urinary bladder. Br J Pharmacol. 2004;142:31–34. doi: 10.1038/sj.bjp.0705764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schicho R, Krueger D, Zeller F, Von Weyhern CW, Frieling T, Kimura H, et al. Hydrogen sulfide is a novel prosecretory neuromodulator in the Guinea-pig and human colon. Gastroenterology. 2006;131:1542–1552. doi: 10.1053/j.gastro.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Streng T, Axelsson HE, Hedlund P, Andersson DA, Jordt SE, Bevan S, et al. Distribution and function of the hydrogen sulfide-sensitive TRPA1 ion channel in rat urinary bladder. Eur Urol. 2008;53:391–399. doi: 10.1016/j.eururo.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Sun YG, Cao YX, Wang WW, Ma SF, Yao T, Zhu YC. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res. 2008;79:632–641. doi: 10.1093/cvr/cvn140. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Aoki Y, Okubo K, Maeda Y, Sekiguchi F, Mitani K, et al. Upregulation of Ca(v)3.2 T-type calcium channels targeted by endogenous hydrogen sulfide contributes to maintenance of neuropathic pain. Pain. 2010;150:183–191. doi: 10.1016/j.pain.2010.04.022. [DOI] [PubMed] [Google Scholar]
- Theoharides TC, Whitmore K, Stanford E, Moldwin R, O'Leary MP. Interstitial cystitis: bladder pain and beyond. Expert Opin Pharmacother. 2008;9:2979–2994. doi: 10.1517/14656560802519845. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Patacchini R, Nicoletti P, Gatti R, Gazzieri D, Lissi N, et al. Hydrogen sulfide causes vanilloid receptor 1-mediated neurogenic inflammation in the airways. Br J Pharmacol. 2005;145:1123–1131. doi: 10.1038/sj.bjp.0706277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- Wang ZY, Wang P, Merriam FV, Bjorling DE. Lack of TRPV1 inhibits cystitis-induced increased mechanical sensitivity in mice. Pain. 2008;139:158–167. doi: 10.1016/j.pain.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Wantuch C, Piesla M, Leventhal L. Pharmacological validation of a model of cystitis pain in the mouse. Neurosci Lett. 2007;421:250–252. doi: 10.1016/j.neulet.2007.05.043. [DOI] [PubMed] [Google Scholar]