


Abstract
Transcription factors (TFs) regulate gene expression by binding to short DNA sequence motifs, yet their binding specificities alone cannot explain how certain TFs drive a diversity of biological processes. In order to investigate the factors that control the functions of the pleiotropic TF STAT3, we studied its genome-wide binding patterns in four different cell types: embryonic stem cells, CD4+ T cells, macrophages and AtT-20 cells. We describe for the first time two distinct modes of STAT3 binding. First, a small cell type-independent mode represented by a set of 35 evolutionarily conserved STAT3-binding sites that collectively regulate STAT3’s own functions and cell growth. We show that STAT3 is recruited to sites with E2F1 already pre-bound before STAT3 activation. Second, a series of different transcriptional regulatory modules (TRMs) assemble around STAT3 to drive distinct transcriptional programs in the four cell types. These modules recognize cell type-specific binding sites and are associated with factors particular to each cell type. Our study illustrates the versatility of STAT3 to regulate both universal- and cell type-specific functions by means of distinct TRMs, a mechanism that might be common to other pleiotropic TFs.
INTRODUCTION
The precise spatio-temporal regulation of gene expression programs determines an organism’s development and the interaction with its environment. Transcription factors (TFs) control this process by binding to short DNA sequences (typically 6–8 bp), yet their binding specificities cannot explain the various cell type-specific functions of many TFs. Protein binding microarrays have shown that members of TF families such as homeodomains bind to very similar sequences, which therefore cannot account on their own for the enormous diversity of functional roles of homeodomain TFs during animal development (1,2). Potentially, cell type specificity emerges from the interplay of TF DNA sequence specificity, co-factors and epigenetics (3). However, despite vast efforts to understand the mechanisms that determine cell type-specific TF activity, the exact mechanisms continue to remain frustratingly elusive. A number of studies have shown that key TFs associate locally with co-activators to constitute ‘transcriptional regulatory modules’ (TRMs) that endow the key TF with cell type-specific functions. An important example was provided in embryonic stem cells (ESCs), where TFs assemble around the core heterodimer SOX2-OCT4 and NANOG (4). In hematopoietic progenitor cells, the TRM centers around GATA2, RUNX1 and SCL/TAL1 (5), whereas in developing B cells the TRM clusters around E2A, EBF1 and FOXO1 (6). Finally, in trophectoderm stem cells, the TF core around which the TRM assembles includes SMARCA4, EOMES, TCFAP2A, GATA3 and ETS2 (and possibly STAT3 too) (7). Although experimentally characterized TRMs are very informative as to the co-activators that key TFs need to associate with to perform their biological functions, these TRM models have not yet been able to provide an explanation for how pleiotropic TFs bring about functional specificity in distinct cell types. Examples for the pleiotropic functions of TFs are as follows: (i) the ESC factor SOX2 is also active in neural progenitor cells (8), (ii) the essential hematopoietic factor SCL/TAL1 is also robustly expressed in neural progenitor cells, (iii) the B-cell development factor FOXO1 is known to regulate adipocyte differentiation (9) and (iv) the trophectoderm stem-cell factor GATA3 is crucial at various stages of CD4+ T-cell development (10).
Therefore, a fundamental question in transcriptional regulation is how a given TF can perform highly divergent and at the same time crucial functions across distinct cell types (11). To address this problem, we set out to investigate the mechanisms that enable STAT3 to regulate distinctive gene sets leading to diverse biological outcomes in various cell types. STAT3 has been profiled by ChIP-seq in multiple cell types, including ESCs (4), CD4+ T cells (12,13), macrophages (14) and AtT-20 corticotroph cells (15). Crucially, for the dissection of cell type-specific functions, STAT3 has radically different roles in each one of these cell types: in ESCs, STAT3 maintains pluripotency (16), whereas in CD4+ T cells STAT3 drives the differentiation toward Th17 cells (13,17) and is also required for Th2 cells (18). In macrophages, STAT3 is essential for the initiation of the anti-inflammatory response mediated by IL-10 (19,20), and in AtT-20 corticotroph cells, STAT3 promotes adrenocorticotropic hormone production as part of the hypothalamo–pituitary–adrenal axis in response to stress and inflammation (15,21). Clearly, these diverse functions imply that STAT3 is able to target different enhancers to regulate distinct genes depending on the biological context. Other advantages of using STAT3 as a model to investigate TF functional specificity in the four distinct cellular types described earlier are as follows: (i) STAT3 is an essential regulator in these cell types and cannot be replaced by other factors; (ii) STAT3 is activated upon induction by a cytokine and thus constitutes a natural switch that produces easily distinguishable outcomes and (iii) upon activation, STAT3 initiates a measurable response that is either a developmental program or a response to an environmental stimulus.
Here, we analyze genome-wide STAT3 binding data from ChIP-seq libraries profiled in ESCs, CD4+ T cells, macrophages and AtT-20 cells and show that STAT3 has two modes of binding: (i) a small number of STAT3-binding sites that are common to all four cell types analyzed which regulate a core set of genes that are pre-bound by E2F1 and which encode a self-regulatory loop for STAT3 and (ii) the larger sets of STAT3-binding events that are cell type-specific and therefore responsible for the distinct biological outcomes of STAT3 in the various cell types. Moreover, by integrating data on predicted TF-binding sites, protein–protein interactions and gene expression, we built TRM models that predict the unique associations of STAT3 with distinct sets of co-factors and which explain how STAT3 directs both its cell type-independent and cell type-specific functions. This is the first example of a TF having both cell type-independent and cell type-specific functions mediated by distinct TRMs, a modus operandi that might be shared by other pleiotropic TFs.
MATERIALS AND METHODS
ChIP-seq read mapping and peak calling
ChIP-seq reads were mapped to the mouse genome (mm9) using Bowtie (v0.12.7) (22) with the setting ‘best’ and with reads mapping to more than one genomic location being excluded (−m). Peak discovery was performed using MACS (v1.4.1) (23) using the parameters bandwidth = 200 and genomesize = mm. The m-fold parameter was adjusted until MACS could find ∼1000–2500 high-quality peaks to construct a model. The P-value for each ChIP-seq library was determined by increasing the P-value from 1 × 10−10 until the number of peaks discovered by chance alone (false positives) was close to 1% or the P-value reached 1 × 10−5. Detailed genome mapping statistics and the full list of settings used by MACS for all of the ChIP-seq data used in this article are described in Supplementary Table S1.
Gene Ontology (GO) on the ChIP-seq lists was done using GREAT (v2.0.1) (24) using the whole genome as a background. Statistical analysis was performed using SciPy and data visualization used matplotlib, pycairo, R and glbase (https://bitbucket.org/oaxiom/glbase/wiki/Home/). For evolutionary conservation analysis, all pre-computed phastCons scores were obtained from the UCSC genome browser.
The random backgrounds used for motif enrichment analysis or for comparison were generated by randomly sampling 1 or 10% of the appropriate control ChIP-seq library. Sequence libraries were made unique, allowing only a single read per genomic position prior to sampling random sites. This removed repeats that typically attract a large number of sequence tags and give spurious regions of overlap. For the ‘shared overlap’, ‘any two cell types’ and ‘any three cell types’, the backgrounds were combined in equal proportions relative to the size of the contribution of the cell-type STAT3 binding to the overlap.
Gene expression data
Expression profiles were obtained from the Gene Expression Omnibus for ESCs (GSE27708) (25), IL-21-treated anti-CD3/CD28-activated-CD4+ T cells (GSE19198) (12), IL-6-treated anti-CD3/CD28-activated-naive CD4+ T cells (GSE21671) (13), IL-10-treated peritoneal exudate cells macrophages (GSE31531) (14), IL-6-treated liver cells (GSE21060) (26) and AtT-20 cells (GSE19042) (21). For ESCs, AtT-20 and CD4+ T cells, raw data were processed using the BrainArray custom CDFs (27), while the RNA-seq macrophage data for PEC macrophage RNA-seq, transcripts were quantified using RSEM (28) and annotated to Ensembl genes (release 66).
Motif discovery and identification of TRMs
HOMER (v3.6) (29) was used for de novo motif discovery with default parameters, with ±200 bp around the peak summits of the list of STAT3 binding peaks and a random background sampling of 1% of the appropriate control ChIP-seq library. TRMs were identified by integrating motif enrichment, gene expression and protein–protein interaction information. De novo motifs discovered by HOMER that did not resemble STAT3 or a STAT3 half-site were searched again for enrichment using FIMO (30) to remove motifs with a z-score < 5.0 in all STAT3 binding lists. De novo motifs detected by HOMER were annotated with Tomtom (MEME suite) (matching motifs with a q < 0.05 were kept) (31) using a library of motifs obtained from the JASPAR and UniPROBE databases (32,33). Known motifs were mapped to human genes and pairwise similarities among motifs were computed with Tomtom to determine clusters of TFs with similar binding preferences. The original gene annotations for the motifs were mapped to the human homologs using the Biomart tool from Ensembl. These mapped genes were filtered by expression in the appropriate cell type to reduce redundancy. Based on the distribution of intensities, genes were considered not expressed if the normalized intensity value was <5.2 for ESCs, <4.1 in CD4+ T cells or <3.9 in AtT-20 cells. For the macrophage RNA-seq data, those transcripts with a transcripts-per-million value <1.0 were disregarded. The human protein–protein interaction network was obtained from the BioGRID database (34) and nodes were removed for non-expressed genes in each of the data sets. Additionally, the nodes UBC and SUMO2 were also removed as they connect to 60 and 8% of the entire interactome, respectively. Full details on our method for reconstructing TRMs will be published elsewhere.
ChIP-qPCR of STAT3-bound sites in macrophages
PEC macrophages were purified from pooled 6- to 8-week-old male C57Bl6JcL mice (CLEA Japan) and treated with IL-10 (R&D Systems). Formaldehyde cross-linked chromatin was extracted and subjected to ChIP using antibodies to STAT3 (SantaCruz, sc-482), E2F1 (Millipore, 05-379) and GFP (Santa Cruz, sc-8334), as previously described (14). qPCR was performed on an ABI 7900, using SYBR qPCR mix (TOYOBO) according to the manufacturer’s instructions. Primers used in this study are described in Supplementary Table S2.
RESULTS
A catalogue of STAT3 genomic binding sites in ESCs, CD4+ T cells, macrophages and AtT-20 cells
To date, the genome-wide binding pattern of STAT3 has been reported by ChIP-seq in ESCs (4), CD4+ T cells (12), Th17 cells (13), macrophages (14) and AtT-20 corticotroph cells (15). Although each publication provides an alignment to the genome and a set of characterized peaks, these were performed at different times where the sequencing reads were aligned to different mouse genome assemblies (mm8 and mm9), using various software tools (ELAND, AceView and bowtie) and a variety of peak discovery algorithms (MACS, ChIPseq and AceView). In order to compare the various ChIP-seq libraries, these and their corresponding control libraries (raw sequence reads) were uniformly reanalyzed, except for the STAT3 ChIP-seq library prepared in Th17 cells as it lacks a paired control library (13). Nevertheless CD4+ T cells contain small numbers of Th17 cells, meaning that the CD4+ T-cell ChIP-seq library will contain many Th17-specific STAT3-binding sites. For each cell type, ChIP-seq library replicates were merged into a single fastq file and bowtie (22) was used to align the reads to the mm9 version of the mouse genome. The peak discovery tool used was MACS (23), which reports the number of peaks discovered by chance alone by reversing the experimental and control libraries. At the default cutoff (P = 1 × 10−5), MACS has a tendency to inflate the number of peaks reported for larger sequence libraries at the expense of increasing the number of false positives. To correct for this bias, we adjusted the MACS P-value cutoff to limit the number of peaks discovered by chance alone (false positives) to ∼1% (Supplementary Figure S1 and Supplementary Table S1). Thus, we report 2651 (ESCs), 5152 (CD4+ T cells), 1724 (macrophages) and 7982 (AtT-20) peaks. Of the original sets of peaks defined in the respective publications, our new peak lists contain 66% of the ESCs peaks, 81% of the CD4+ T-cell peaks, 87% of the macrophage peaks and 100% of the AtT-20 peaks. Although the majority of the peaks that were missed are lower ranked peaks (Supplementary Figure S2), we also report new additional peaks in CD4+ T cells (756), ESCs (245), macrophages (372) and AtT-20 cells (4881). Similarly, most of these new peaks are lower ranked peaks by fold enrichment of the experimental libraries over the control libraries (Supplementary Figure S2).
STAT3 regulates a core set of genes across all four cellular types
STAT3 peaks were defined by extending the summits determined by MACS 200 bp either side of each summit, and overlapping peaks were merged by taking the midpoint between the two summits. Whenever peaks overlapped, they tended to be close (Figure 1A) and only 35 peaks were found to be common to all four libraries (Figure 1B). These 35 peaks overlapping across all four libraries were highly ranked STAT3-binding sites, with one-half of these 35 overlapping peaks being listed in the top 1000 STAT3 peaks in all four libraries (Figure 1C). Moreover, a Monte Carlo simulation of the overlap suggested that the number of overlapping peaks that would be expected among all four libraries by chance alone is zero (expected overlapping sites: 0.002 ± 0.045) (Figure 1B). Therefore, the 35 STAT3-binding events shared by all four ChIP-seq libraries are not random, while most STAT3-binding events are highly specific to each cell type (Supplementary Figures S3 and S4; Supplementary Table S3). Conservation of transcriptional regulation can also be gene-centric, where regulatory TF-binding sites do not overlap but still regulate the same nearby gene. This has been reported for SMAD3 (35) and during evolution for CEBPA (36). However, we did not find robust evidence for systematic gene-centric regulation as the number of gene-centric observations was close to the values expected by chance alone (Supplementary Figure S5).
Figure 1.

STAT3 binds to a small but significant ‘shared overlap’ of binding sites in divergent cell types, but is otherwise strongly cell type specific. (A) Summary of the distances between the overlapping peaks for each pair of STAT3 ChIP-seq libraries. The distance was measured as the number of base pairs between the summits of the two overlapping peaks. (B) Overlap of the binding peaks in three STAT3 libraries culled from mouse ESCs, CD4+ T cells, macrophages and AtT-20 cells. Peaks were considered to overlap if their peak summits were within 200 bp of one another. The overlap was simulated by generating lists of faux ChIP-seq peaks followed by the assessment of their overlap (this was performed 1000 times to generate the number of overlaps expected by chance-value listed in brackets). (C) The ‘shared overlap’ sites are more likely found in the most highly ranked STAT3-binding sites. STAT3-binding sites were ranked by fold enrichment and then the cumulative overlap of STAT3 peaks appearing in all four cell types was plotted for ESCs, CD4+ T cells, macrophages and AtT-20 cells. (D) Genome distributions of STAT3-binding sites orientated with respect to the nearest gene to the STAT3-binding site. Coloured bars describe the distance from the STAT3-binding sites to the nearest TSS, as described in the key. The gray regions denote a random background and represent the expected distribution of peaks were the binding sites randomly distributed across the sequenceable genome.
The distribution of STAT3-binding events relative to gene locations is typical of other TF ChIP-seq libraries, with one-third of STAT3 peaks located within 10 kb of the nearest TSS (Figure 1D and Supplementary Table S4). However, the ‘shared overlap’ of 35 STAT3-binding events across all four cellular types has a very strong bias toward the TSS, with 30 of 35 (∼86%) STAT3 peaks being located within 10 kb of the TSS. As proximity to the TSS has been linked with the control of gene expression (4,37), this suggests that the 35 STAT3 peaks likely regulate the expression of the genes they lie closest to. Many of these genes are essential for STAT3 function, including Stat3 itself, Socs3, Bcl3 and Ptpn1 (Figure 2A). Not surprisingly the shared 35 STAT3-binding events show a much greater degree of evolutionary conservation than any set of cell type-specific STAT3 peaks (Figure 2B). Most of the 35 STAT3-binding events shared across all four cellular types are located within 10 kb of a TSS, although the assignment between TF-binding events and gene regulation is still an open question as no generally applicable model has yet been described. The association by proximity of TF-binding events with the genes nearby shows that typically only 10–20% of such genes are differentially expressed (10,14,38). To investigate the effects of the 35 STAT3-binding events on the expression of the closest genes, we analyzed expression data for the four cellular types analyzed here plus two additional conditions where STAT3 is also activated by a cytokine: naive CD4+ T cells stimulated by IL-6 (13) and liver cells stimulated by IL-6 (for which the STAT3 genome-wide binding profile is not known) (26). Gene expression values of the cytokine-stimulated cells were combined and ranked by fold-change relative to controls, while the expression data from ESCs were inverted (as the withdrawal of LIF causes ESCs to differentiate). Remarkably, about one-half of the genes are up-regulated upon cytokine stimulation in all six conditions (Figure 3B) and strikingly only a few genes are down-regulated. This indicates that a core unit of STAT3 regulation occurs in all these biological contexts and cellular types, including the well-characterized targets of STAT3, Socs3 and Bcl3, which are up-regulated in all six conditions.
Figure 2.

The shared overlap of STAT3-binding sites controls the expression of a core set of genes important for STAT3 function. (A) Genomic views of STAT3 ChIP-seq sequence tag densities for four STAT3-binding sites common to ESCs, CD4+ T cells, macrophages and AtT-20 cells. STAT3 binding is shown around the TSS of Stat3 and Socs3, within the second intron of Bcl3 and ∼6 kb 5′ of the TSS of Ptpn1. (B) Average evolutionary conservation for the different categories of STAT3-binding events. STAT3 peak summits were extended by 1 kb either side and the Euarchontoglires evolutionary conservation scores were annotated as determined by phastCons in the UCSC Genome Browser (http://genome.ucsc.edu/).
Figure 3.

The shared overlap STAT3-binding sites co-regulate a set of key genes important for STAT3 function in multiple cell types. (A) Significantly over-represented terms from the ‘Pathway Commons’ category for the genes associated with the shared overlap. Shown here are the top five terms only. A significant q-value of 0.05 is represented by a solid black line and a q-value of 0.01 by a dotted gray line. (B) The expression of the closest genes within 200 kb of the shared overlap was measured in a series of gene expression microarray data sets which show the activation of STAT3 or the loss of STAT3 activity (LIF withdrawal from ESCs). Expression data are reversed in ESCs for clarity, but is otherwise down-regulated upon removal of LIF whereas all other treatments show up-regulation in response to cytokine stimulation. P-values are from a Wilcoxon test between the treated and untreated conditions: CD4+ T cells treated with IL-21 (P = 5.59 × 10−5) (GSE19198), naïve CD4+ T cells treated with IL-6 (P = 2.45 × 10−3) (GSE21671), ESCs upon withdrawal of LIF from the medium (P = 0.027) (GSE27708), peritoneal macrophages stimulated with IL-10 (P = 2.14 × 10−4) (GSE31529), AtT-20 cells treated with LIF (P = 1.00 × 10−4) (GSE19042) and liver cells stimulated with IL-6 (P = 0.032) (GSE21060). Genes marked in green do not have a corresponding probe on the microarray.
The 35 STAT3-binding events common to all four cell types regulate a diverse set of genes involved in the maintenance of specific cellular activities. GO analysis using GREAT (24) reported an over-representation of signal transduction pathways related to STAT3 (Figure 3A). The promoters of Stat3 and Stat1 are both bound and induced by the recruitment of STAT3 in all four cell types (Figure 3B). Moreover, it is remarkable to find that three genes involved in three different pathways that negatively regulate JAK-STAT signaling are induced by STAT3, suggesting that STAT3 is acting to regulate its own activity. These include PTP1B (Ptpn1), a protein tyrosine phosphatase reported to dephosphorylate JAK2 and TYK2 and also phospho-STAT3 (39). Additionally, SOCS3 is part of the ubiquitin pathway that negatively regulates JAK-STAT signaling by degrading both JAKs and STATs (40). A third mechanism of negative feedback is the suppressive function of the RNA-binding protein Zfp36 (Tristetraprolin) which negatively regulates IL-10 signaling in macrophages (41). Zfp36 could thus be playing an identical role in all four cell types described here. The remaining genes include over a dozen TFs, such as Stat3, Stat1, Bcl3, Bcl6, Tcf4, Cic, Sbno1 and the AP-1 family members Fos and Junb (GO:0003676: ‘nucleic acid binding’, GREAT FDR q = 3.6 × 10−2), which deserve further characterization. In addition to these, our STAT3 target set comprises several genes encoding proteins involved in DNA replication and repair, protein translation, cytoskeletal reorganization and protein trafficking and notably six genes that encode proteins involved in metabolism (summarized in Supplementary Table S5). Our findings on the STAT3 transcriptional program common to all four cellular types indicate that STAT3 establishes its own regulatory network by the following: (i) perpetuating its own transcription, (ii) being a master regulator of other TFs working downstream of it, (iii) stimulating the transcription of cytoplasmic enzymes that control STAT3’s activity and (iv) ensuring an efficient and robust cellular division program and the maintenance of a stable cell type. The STAT3 transcriptional program involves many levels of cellular control from basic DNA replication and chromatin remodeling, to the cell metabolic pathways producing key metabolites needed for increasing the transcriptional and translational processes essential in cell division and maintenance.
A distinct TRM defines cell type-independent STAT3 binding across all four cellular types
Since STAT3 binds to 35 identical sites across four distinct cellular types, it is reasonable to assume that it does so by assembling around a TRM that is common to all cellular types. To reconstruct the putative TRM that directs the expression of the genes regulated by the 35 STAT3-binding events, we integrated over-represented TF-binding sites co-occurring with STAT3-binding sites, protein–protein interaction data and expression data (as detailed in the ‘Materials and Methods’ section). The resulting TRM (Figure 4A and Supplementary Figure S6) is unique to the 35 STAT3-binding events shared by all cellular types and contains a number of co-factors and other proteins that are known to bind to STAT3 experimentally and whose corresponding genes are expressed in all four cellular types. The co-TFs that appear to work together with STAT3 in this TRM include many ‘general’ TFs known to operate in a variety of biological contexts. The cell type-independent TRM contains the TFs MYC, E2F1 and KLF4, all of which have been profiled by ChIP-seq in ESCs (4). These ChIP-seq libraries were re-analyzed as before (full details included in Supplementary Table S1) and we determined the co-occupancy of the 35 STAT3-binding events by MYC, E2F1 and KLF4 in ESCs. We found that 34 of 35 (97%) of the STAT3 peaks are co-occupied (within 800 bp of each other) by n-MYC, E2F1 or KLF4 and often by several of them (Figure 4B and Supplementary Figure S7), but only 19 of 35 (54%) are co-occupied by one of the ESC-specific factors ESRRB, SOX2, OCT4 or NANOG (Figure 4B). It must be noted that co-occupancy is not a factor of the number of peaks of the ChIP-seq libraries as ESRRB has the largest number of peaks (56 136) as opposed to E2F1 (11 448 peaks) (Supplementary Table S1). Finally, for a limited number of STAT3-binding sites in the shared overlap, we also probed their occupancy in IL-10 treated macrophages by ChIP-qPCR (Figure 4C). As expected, STAT3 is specifically recruited to all of the 14 sites we probed by ChIP-qPCR and remarkably E2F1 is not only bound at these same sites but is actually pre-bound at these sites close to STAT3 binding (with the exception of Diap1, Figure 4C). STAT3 is therefore specifically recruited to genomic loci that already have E2F1 bound. This may explain why several of these genes show very rapid induction of expression upon stimulation with IL-10 (14,42) since they are poised for expression by the presence of E2F1. In summary, STAT3 appears to use a TRM, binding close to E2F1 and possibly MYC, to regulate a core set of genes that tune the JAK-STAT pathway and to promote cell proliferation while counteracting differentiation.
Figure 4.

The shared overlap of STAT3 binding in all four cell types forms a cell type-independent regulatory network with MYC and E2F1. (A) HOMER was used to generate de novo motifs from the list of 35 STAT3-binding sites common to all four cell types. Motifs resembling STAT3 or a STAT3 half-site were removed and over-represented motifs were collected and annotated to genes. Interaction networks were constructed by interrogating the PPI network for proteins interacting with those representing the enriched motifs. TFs were clustered together by motif similarity and coloured by the cluster they belong to: white-colored nodes do not have a representative motif in the databases or do not bind to DNA directly; proteins with a bold circle have a motif enriched in that cell type, while proteins with no bolded circle have no discovered motif but were linked to STAT3 through the PPI network. Proteins in the network were filtered by gene expression and here we present the union of the network in all four cell types (the separate networks are presented in Supplementary Figure S6) (B) ChIP-seq data from ESCs (GSE11431) were re-analyzed and binding sites overlapping within 400 bp were collected. The heatmap shows the 35 STAT3-binding sites together with the other TFs bound in the vicinity of STAT3. (C) We designed primers for 14 STAT3-binding sites shared between all four cell types and performed ChIP-qPCR. Macrophages were treated for 4 h with IL-10 (black bars) or left untreated (white bars) and chromatin was harvested. ChIP was performed using antibodies against STAT3 (left panel), E2F1 (right panel) or GFP (as a control in both panels). Each group of bars represents a STAT3-binding site and is labeled with the name of the nearest gene.
Cell type-specific binding events determine the various functions of STAT3 in ESCs, CD4+ T cells, macrophages and AtT-20 cells
Since the 35 STAT3-binding events shared across all four cell types encode a self-regulatory program for STAT3 that is cell type-independent, the cell type-specific STAT3-binding events should be related to the specific functions of STAT3 in each of the four distinct cell types. GO analysis of the cell type-specific STAT3-binding events using the ‘Biological Process’ category shows that the most over-represented terms in ESCs include distinctive ESC functions such as ‘stem-cell maintenance’ and ‘stem-cell differentiation’ (Figure 5A). Likewise, in CD4+ T cells and macrophages terms pertaining to relevant processes were recovered. For AtT-20 cells, as a pituitary epithelial cell line, there are several terms related to epithelial cell function, including ‘cell–cell junction organization’, ‘cellular response to radiation’, ‘plasma membrane organization’ and particularly ‘regulation of insulin receptor signaling pathway’, indicating the role that the pituitary plays in responding to insulin. The over-represented terms from the Mouse Genome Database genotypes::phenotypes (43) are also indicative of specific STAT3 functions in the four cellular types, including ‘abnormal cytokine secretion’ (macrophages), ‘abnormal adaptive immunity’ (CD4+ T cells), ‘complete embryonic lethality’ (ESCs) and ‘increased body temperature’ (AtT-20 cells, since one of the functions of the pituitary is to regulate body temperature) (Figure 5B). Collectively, the GO analyses of the cell type-specific STAT3-binding events suggest that the functional specificity of STAT3 in each cell type is contained within the cell type-specific lists of STAT3-binding events and not within the shared overlap of peaks common to all four cellular types, which as we have previously shown has diverse cell type-independent functions.
Figure 5.

STAT3 biological specificity is found within the cell type-specific lists of genomic binding sites. Gene Ontology term enrichment analysis was done using GREAT with default parameters. Over-represented terms displayed here are from the ‘Biological Process’ category (A) and the Mouse genome informatics phenotype::genotype category (B). A significant q-value of 0.05 is represented by a solid black line and a q-value of 0.01 by a dotted gray line.
Non-canonical DNA binding is prevalent across distinct cellular types but is not sufficient to explain STAT3’s divergent functions
The DNA binding preferences for paralogous classes of TF tend to be rather uniform, making it difficult to attribute biological specificity to DNA base changes in TF-binding motifs. A clear example is provided by homeodomain TFs, all of which have strikingly similar DNA binding preferences despite encompassing all of mammalian development (2). The case of STAT3 is particular because although STAT3 has a well-defined canonical motif (TTCnnnGAA), it has nevertheless been reported to use a unique variant motif (TTAnnnGAA) to regulate the Prdm1 gene in CD4+ T cells (12).
The general motif recovered de novo in each set of cell type-specific STAT3-binding events is the prototypical STAT3 motif (Figure 6A), which is shared by other STAT factors too (44). Since alternative motifs appear to be important for STAT3 function, we investigated the presence of all possible STAT3 motif variants in the set of STAT3 peaks in each cellular type. This was done by counting STAT3 motifs where single base pairs were individually mutated and z-scores were derived by comparing against randomly selected sets of background sites drawn from the respective ChIP-seq control libraries. As expected, the STAT3 canonical binding site was found in all four cellular types and the shared overlap (Figure 6B). The variant TTAnnnGAA previously characterized in CD4+ T cells (12) was over-represented not only in CD4+ T cells but also in ESCs and AtT-20 cells (but not in macrophages). The variant STAT3 motif TGCnnnGAA is over-represented in all four cellular types, suggesting that this is a common alternative mode of DNA binding by STAT3. Finally, the motif CTCnnnGAA appears to be a variant used by STAT3 exclusively in AtT-20 cells (Figure 6B).
Figure 6.

STAT3 uses alternative (non-canonical) modes of binding to DNA. (A) de novo generated motifs from HOMER are very similar in the six lists of STAT3 binding. Motifs were generated from the entire lists of STAT3-binding sites for each category, except for CD4+ T cells where the top 1000 sites were used (as for the entire list we could only identify a STAT3 half-site). (B) z-score heatmap to show over representation of variant STAT3 motifs in the STAT3-binding sites in the various cell types. Variant motifs are presented here as all single base pair mutations of one-half of the STAT3 heterodimeric motif. (C) Pie charts showing the frequency of the DNA words TTCnnnGAA (canonical STAT3) or a non-canonical STAT3 binding: TTAnnnGGA and TGCnnnGGA for ESCs, CD4+ T cells, any two cell types and any three cell types; TGCnnnGAA for macrophages and the shared overlap; and TTAnnnGGA, TGCnnnGGA and CTCnnnGAA for AtT-20 cells. (D) Cartoon representation of the Asn466 amino acid of STAT3 making contact with the DNA base pairs [PDB entry 1bg1 (46)].
Moreover, we quantified the usage of the various canonical and variant motifs for each set of STAT3 peaks (Figure 6C). All STAT3 peaks contain putative half-sites (TTCC), often several of them per peak. At most 28% of STAT3 peaks harbor a perfect canonical site (in ESCs) while non-canonical sites are especially prevalent in AtT-20 cells (48% of sites). The set of 35 STAT3-binding events shared by all four cellular types turned out to be especially conservative in the use of canonical motifs, with 43% containing a canonical STAT3 motif. The binding of STAT3 to variant motifs could be linked to specific functions: for instance, the binding of STAT3 to canonical TTCnnnGAA might facilitate the recruitment of generic co-activators, such as p300, whereas the binding to the variant motif TTAnnnGAA (as is the case in CD4+ T cells) might lead to the recruitment of specific regulatory complexes. The differential recruitment of co-activators could be allosterically induced by different DNA ligands, as demonstrated for the glucocorticoid receptor (45), a model that provides a conceptual framework for understanding the functional plasticity of pleiotropic TFs such as STAT3. Here, the binding of STAT3 to variant DNA motifs induces conformational changes on the binding proteins, which in turn affect the recruitment of co-activators. Indeed, in the case of STAT3, specific base pairs that vary between the canonical and the variant motifs TTAnnnGAA and TGCnnnGAA (i.e. T[TC] versus T[TA] or T[GC]) are directly contacted by Asn466 of the connector domain of STAT3, in which case Asn466 must structurally rearrange to accommodate the altered chemical environment introduced by these variant motifs (Figure 6D) (46). These structural rearrangements could translate into global structural changes to the connector domain as well as the neighboring and C-terminal SH2 domain that mediates interactions with other proteins by recognizing phosphotyrosine residues. A further effect of specific variant motifs and specific co-factor interactions might be the stabilization of the TRM and the tethering of STAT3 to DNA. Indeed, Husby et al. (47) recently proposed that the two halves of the STAT3 homodimer do not make identical base pair contacts on a TTAGnGGAA variant motif. We found no clear link between the presence of variant motifs and specific biological functions, although there were several prominent examples including the aforementioned Prdm1, Il17a and Il17ra (both of which have nearby STAT3-binding sites with the variant motif TTAnnnGAA) and Cd28, which is associated with the STAT3 variant motif TGCtggGAA. The presence of alternative STAT3-binding motifs highlights the usage of variant motifs in regulating specific genes, although no clear pattern that explains how variant motifs encode the cell type-specific functions of STAT3 could be discerned.
Distinct TRMs determine STAT3’s cell type-specific functions
In the absence of a clear contribution by variant DNA motifs to explain STAT3’s cell type-specific functions, an attractive model providing biological specificity is one where various factors cluster together in a cooperative manner to provide biological specificity (48). This ‘piling-up’ of TFs and co-factors has been demonstrated by ChIP-seq experiments in ESCs, B cells and blood stem-cell precursors (4,5,29). One limitation of this approach, however, is that the identities of biologically relevant TFs need to be known in advance before performing any ChIP-seq experiment that will eventually allow one to reconstruct a transcriptional regulatory network (49).
By employing the same data integration and analysis techniques that we used to reconstruct the cell type-independent TRM regulating the shared overlap of 35 STAT3-binding events (Figure 4A), we generated models for the cell type-specific binding events of STAT3 in ESCs, CD4+ T cells, macrophages and AtT-20 cells (Figure 7A–D). We managed to recover the well-characterized ESC transcriptional regulatory network (4) comprising OCT4 (represented here by the motif POU2F2), SOX2, ESRRB, KLF4 and SMAD1 and a number of homeodomain proteins that may correspond to NANOG (Figure 7A). In addition, we identified TEAD1, a protein recently shown to be essential to maintain ESC pluripotency (50) and also identified in human ESCs as a potential key TF (51), and REST, whose role is to repress specific genes in ESCs and so block differentiation (52). This network is reminiscent of the ESC networks determined by mass spectrometry (53,54), although it is only a partial match possibly because STAT3 was not used as a bait in the generation of those networks.
Figure 7.

Discrete transcriptional regulatory modules (TRM) determine STAT3 cell type-specific functions. TRMs for each cell type were generated by collecting de novo motifs generated by HOMER and excluding motifs resembling STAT3 or a STAT3 half-site. Motifs were then scanned again and compared to a random background generated from the control background libraries to generate a z-score. Motifs with a z-score < 5.0 were removed and finally motifs were annotated to genes. These genes were filtered by expression data derived from the appropriate cell type and interaction networks were constructed by interrogating the PPI space. TFs were clustered together by motif similarity and coloured by the cluster they belong to. Proteins with a bold circle have an motif enriched in that particular cell type, while proteins without a bold circle have no enriched motif but were linked to STAT3 through the PPI network. White-coloured nodes do not have a representative motif in the motif databases or do not bind to DNA directly. The networks are divided into the cell type-specific lists for ESCs (A), CD4+ T cells (B), macrophages (C) and AtT-20 cells (D). (E) Pairwise correlation heatmap of the ChIP-seq peaks from ESCs, CD4+ T cells and the STAT3-binding sites described here. The lists of peaks were assessed for overlap in an all-against-all comparison and Pearson correlation scores were used to measured the frequencies of overlapping peaks when compared to all other overlaps.
The transcriptional regulatory network of CD4+ T cells is much less well characterized, but we managed to identify the only known STAT3-binding partner in the CD4+ T cells, IRF4 (12) (Figure 6B). Our model of the TRM of CD4+ T cells is completed by members of the TF families AP-1, several ETS-related TFs that are known to play important roles in T-cell biology (55) and a diversity of other TFs. ATF1 is recruited to the Ifng promoter in CD4+ T cells and with CREB represses Ifng expression in naive CD4+ T cells (56). Tantalizingly, STAT3 is also bound at the Ifng promoter and may co-operate with ATF1 to regulate Ifng expression. Finally, we also identify multiple GATA factors associated with STAT3 (Figure 7B), of which GATA3 is known to have multiple roles in T-cell development (10).
In macrophages, several classes of TF were identified (Figure 7C), the most important of which are members of the AP-1 family (FOS, NFE2L2 and JUN), where STAT3 might in turn be regulating the expression of FOS and NFE2L2 as it binds very close to their TSS and CEBPA, which is known to reprogram pre-B cells into macrophages (57).
The STAT3 transcriptional regulatory network of AtT-20 cells is less well characterized than those of ESCs and CD4+ T cells, and our TRM model suggests many potential partners for STAT3 in AtT-20 cells (Figure 7D). We did not recover any motif matching the glucocorticoid receptor-binding site, which although it is linked to the function of STAT3 in AtT-20 cells, it co-localizes with STAT3 in only 21% of STAT3-binding sites, of which ∼50% require the presence of glucocorticoid to co-recruit GR and STAT3 (15).
Finally, a pairwise correlation among the TF-specific binding events derived from ChIP-seq libraries done in ESCs, and all of the STAT3-binding events derived likewise (across all four cellular types) show the distinct TRMs characteristic of ESCs and T cells (Figure 7E). These TRMs contain both the previously reported TRM specific to ESCs (OCT4, SOX2, ESRRB and NANOG) and the ‘MYC-regulated’ block (MYC, E2F1). Additionally, a new TRM consisting of STAT3, IRF4 and a more loosely associated GATA3 emerged in various T-cell subsets. STAT3 binding in macrophages and AtT-20 cells have no other partner TFs from the published ChIP-seq libraries and hence form isolated clusters. These results demonstrate that STAT3 forms distinct cell type-specific TRMs to execute a diversity of gene expression programs.
DISCUSSION
In this study, we have investigated the factors that determine the functions of STAT3 in various cell types. STAT3 binding is predominantly cell type-specific, with just a small but significant number (35) of binding sites shared among ESCs, CD4+ T cells, macrophages and AtT-20 cells. This shared overlap appears to be a mode of auto-regulation for STAT3 function and targets many genes that are all directly up-regulated by STAT3 binding. Additionally, STAT3 also co-binds with E2F1, which is present at the same STAT3-binding sites in at least ESCs and macrophages, and we demonstrate that at least in macrophages E2F1 is even pre-bound to the future STAT3-binding sites. This may explain why so many of these STAT3-regulated genes are rapidly induced in <1 h upon stimulation with a cytokine. The recruitment of STAT3 to DNA via variant motifs does not appear to be a major factor regulating STAT3’s functions across diverse cell types. Nevertheless, a more plausible explanation is provided by the assembly of distinct TRMs around STAT3 in the four cell types. We produced models of TRMs by integrating TF-binding site data, protein–protein interaction and expression data that provide an explanation for STAT3’s specific biological functions across distinct cell types. Using just the STAT3-binding events in ESCs, we succeeded in recovering the ESCs-regulatory network and also propose STAT3-based TRMs for CD4+ T cells, macrophages and AtT-20 cells.
The cell type-specific TRM models of STAT3 predict an important level of epigenetic regulation, as these contain many histone-modifying enzymes, several histone deacetylases (HDAC2, HDAC3) histone acetyltransferases (EP300) and remodeling enzymes (SMARCE1, SMARCA4, SMARCC1) (Figure 7A–D). Additionally, in human ESCs, SIN3A, EP300 and HDAC2 are known to be associated with the ESC regulatory network (51), of which we identified SIN3A and EP300 (Figure 7A). SIN3A has been proposed as a master regulator of STAT activity, particularly in guiding STAT3 to the Socs3 gene (58). Histone modifications likely play a major role in determining cell type specificity either by blocking other STAT3-binding sites from becoming available or by pre-marking sites that STAT3 can be recruited to. Vallania et al. (59) computationally predicted ∼1.3 million STAT3-binding sites in the mouse genome. We nevertheless know from ChIP-seq experiments that STAT3 is binding to a fraction of these sites in distinct cell types. Although other members of the STAT family that share a very similar DNA-binding motif with STAT3 (44) might actually be occupying these other sites, in our opinion this possibility does not entirely explain why all those potential binding sites are unavailable to STAT3. Therefore, epigenetic regulation is probably a major factor controlling STAT3 transcriptional programs and has previously been shown for members of the STAT family of TFs (17). It remains to be determined whether chromatin is initially available for STAT3 recruitment, whether STAT3 binds to inaccessible sites but recruits chromatin modifiers to locally open the chromatin or a combination of both.
A number of TFs have been profiled by ChIP-seq in multiple cell types and a spectrum is emerging that goes from the exclusively cell type-invariant TFs to other TFs that are exclusively cell type-specific and a number of TFs that lie in between these two extremes. Among the remarkably cell type-invariant TFs is CTCF (60), whose primary role appears to be in maintaining the architecture of the genome. However, CTCF can also act as a typical cell type-specific TF. REST is another role appears to be in maintaining the architecture of the genome. REST is another cell type-independent TF with important roles in ESCs and neural precursors (52). Among the cell type-specific TFs are SMAD3, which only shares three genomic binding sites across mouse ESCs, myotubes and pro-B cells and only 1% of sites appear in two or more cell types (35). Additionally, SMAD1 and TCF7L2 show only a small overlap in binding sites in two different human cell lines in the erythroid lineage (61) and TCF7L2 similarly to STAT3 shows only a small overlap in common between six cells lines (62). MYC lies somewhere in the center of the specificity spectrum (60), thus reflecting its dual role as a cell type-specific TF and a ‘global’ regulator of transcription potentially regulating ∼15% of human genes (63). Finally, GATA3 binding was explored in multiple CD4+ T-cell subtypes and shown to be predominantly cell type-specific even within closely related cells of the T-cell lineage (10). Therefore, we could say that both GATA3 and STAT3 are predominantly cell type-specific although they share a limited number of binding sites that are common to all cell types where they have been profiled. These observations place GATA3 and STAT3 between MYC and SMAD1/SMAD3/TCF7L2.
CONCLUSIONS
Our analyses lead us to propose a dual model for STAT3 transcriptional regulation: (i) a limited, cell type-independent and evolutionarily conserved mode whereby STAT3 binds to DNA close to (and may co-operate with) MYC and E2F1 (and possibly other factors too) to regulate the expression of a set of genes in multiple cell types that regulate STAT3’s own signaling activity and (ii) a broader set of cell type-specific binding modes controlled by distinct cell type-specific TRMs that form around STAT3 to enact cell type-specific functions (Figure 8). This dual model of transcriptional regulation by means of assembling distinct TRMs around a master TF could be a general mechanism to regulate the transcriptional programs of other pleiotropic TFs.
Figure 8.
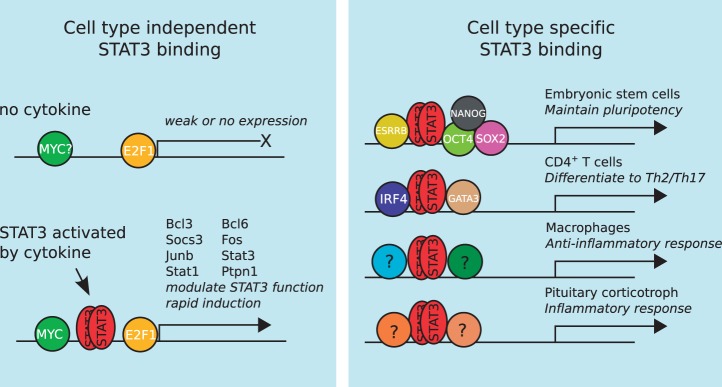
A putative model explaining how STAT3 can perform both cell type-independent and cell type-specific functions by assembling around distinct TRMs. STAT3 binding to the genome occurs in two distinct ways: (i) a cell type-independent mode that is primarily concerned with the regulation of STAT3’s own activity and (ii) a number of cell type-specific modes that execute distinct transcriptional programmes in various cell types.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–5, Supplementary Figures 1–7 and Supplementary References [64–78].
FUNDING
Japan Society for the Promotion of Science (JSPS) through the WPI-IFReC Research Program and a Kakenhi grant; Kishimoto Foundation; ETHZ-JST Japanese-Swiss Cooperative Program [to D.M.S.]; Jeanne and Jean-Louis Lévesque Chair in Cancer Research and Canadian Cancer Society Research Institute [#700922 to M.L.T.]. Funding for open access charge: JSPS.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Mr Shaq Liu for excellent systems support and Ms Mineko Tanimoto for secretarial support.
REFERENCES
- 1.Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Pena-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvey A, Agius P, Noble WS, Leslie C. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Res. 2012;22:1723–1734. doi: 10.1101/gr.127712.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 5.Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat. Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kidder BL, Palmer S. Examination of transcriptional networks reveals an important role for TCFAP2C, SMARCA4, and EOMES in trophoblast stem cell maintenance. Genome Res. 2010;20:458–472. doi: 10.1101/gr.101469.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 9.Nakae J, Kitamura T, Kitamura Y, Biggs WH, 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell. 2003;4:119–129. doi: 10.1016/s1534-5807(02)00401-x. [DOI] [PubMed] [Google Scholar]
- 10.Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, Narlikar L, Northrup DL, Tang Q, Paul WE, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011;35:299–311. doi: 10.1016/j.immuni.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150:1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon H, Thierry-Mieg D, Thierry-Mieg J, Kim HP, Oh J, Tunyaplin C, Carotta S, Donovan CE, Goldman ML, Tailor P, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. 2009;31:941–952. doi: 10.1016/j.immuni.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119:e110–e119. doi: 10.1182/blood-2011-09-381483. [DOI] [PubMed] [Google Scholar]
- 15.Langlais D, Couture C, Balsalobre A, Drouin J. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell. 2012;47:38–49. doi: 10.1016/j.molcel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 16.Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat. Rev. Immunol. 2011;11:239–250. doi: 10.1038/nri2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, Travers J, Nguyen ET, Levy DE, Kaplan MH. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011;34:39–49. doi: 10.1016/j.immuni.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 21.Langlais D, Couture C, Balsalobre A, Drouin J. Regulatory network analyses reveal genome-wide potentiation of LIF signaling by glucocorticoids and define an innate cell defense response. PLoS Genet. 2008;4:e1000224. doi: 10.1371/journal.pgen.1000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho L, Miller EL, Ronan JL, Ho WQ, Jothi R, Crabtree GR. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 2011;13:903–913. doi: 10.1038/ncb2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramadoss P, Chiappini F, Bilban M, Hollenberg AN. Regulation of hepatic six transmembrane epithelial antigen of prostate 4 (STEAP4) expression by STAT3 and CCAAT/enhancer-binding protein alpha. J. Biol. Chem. 2010;285:16453–16466. doi: 10.1074/jbc.M109.066936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009;37:D77–D82. doi: 10.1093/nar/gkn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–D110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullen AC, Orlando DA, Newman JJ, Loven J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell. 2011;147:565–576. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouyang Z, Zhou Q, Wong WH. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc. Natl Acad. Sci. USA. 2009;106:21521–21526. doi: 10.1073/pnas.0904863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, Handunnetthi L, Handel AE, Disanto G, Orton SM, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010;20:1352–1360. doi: 10.1101/gr.107920.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu F, Dube N, Kim JW, Cheng A, Ibarra-Sanchez Mde J, Tremblay ML, Boisclair YR. Protein tyrosine phosphatase 1B attenuates growth hormone-mediated JAK2-STAT signaling. Mol. Cell. Biol. 2003;23:3753–3762. doi: 10.1128/MCB.23.11.3753-3762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 41.Gaba A, Grivennikov SI, Do MV, Stumpo DJ, Blackshear PJ, Karin M. Cutting edge: IL-10-mediated tristetraprolin induction is part of a feedback loop that controls macrophage STAT3 activation and cytokine production. J. Immunol. 2012;189:2089–2093. doi: 10.4049/jimmunol.1201126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El Kasmi KC, Smith AM, Williams L, Neale G, Panopoulos AD, Watowich SS, Hacker H, Foxwell BM, Murray PJ. Cutting edge: a transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J. Immunol. 2007;179:7215–7219. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- 43.Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE. The Mouse Genome Database genotypes::phenotypes. Nucleic Acids Res. 2009;37:D712–D719. doi: 10.1093/nar/gkn886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, Takahashi H, Liang J, Gutierrez-Cruz G, Zang C, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32:840–851. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 47.Husby J, Todd AK, Haider SM, Zinzalla G, Thurston DE, Neidle S. Molecular dynamics studies of the STAT3 homodimer:DNA Complex: relationships between STAT3 mutations and protein-DNA recognition. J. Chem. Inform. Model. 2012;52:1179–1192. doi: 10.1021/ci200625q. [DOI] [PubMed] [Google Scholar]
- 48.Ogata K, Sato K, Tahirov TH. Eukaryotic transcriptional regulatory complexes: cooperativity from near and afar. Curr. Opin. Struct. Biol. 2003;13:40–48. doi: 10.1016/s0959-440x(03)00012-5. [DOI] [PubMed] [Google Scholar]
- 49.Miranda-Saavedra D, Gottgens B. Transcriptional regulatory networks in haematopoiesis. Curr. Opin. Genet. Dev. 2008;18:530–535. doi: 10.1016/j.gde.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 50.Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24:1106–1118. doi: 10.1101/gad.1903310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson R, Teh CH, Kunarso G, Wong KY, Srinivasan G, Cooper ML, Volta M, Chan SS, Lipovich L, Pollard SM, et al. REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol. 2008;6:e256. doi: 10.1371/journal.pbio.0060256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pardo M, Lang B, Yu L, Prosser H, Bradley A, Babu MM, Choudhary J. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell. 2010;6:382–395. doi: 10.1016/j.stem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, Orkin SH. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 55.Russell L, Garrett-Sinha LA. Transcription factor Ets-1 in cytokine and chemokine gene regulation. Cytokine. 2010;51:217–226. doi: 10.1016/j.cyto.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 56.Zhang F, Wang DZ, Boothby M, Penix L, Flavell RA, Aune TM. Regulation of the activity of IFN-gamma promoter elements during Th cell differentiation. J. Immunol. 1998;161:6105–6112. [PubMed] [Google Scholar]
- 57.Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- 58.Icardi L, Mori R, Gesellchen V, Eyckerman S, De Cauwer L, Verhelst J, Vercauteren K, Saelens X, Meuleman P, Leroux-Roels G, et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl Acad. Sci. USA. 2012;109:12058–12063. doi: 10.1073/pnas.1206458109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vallania F, Schiavone D, Dewilde S, Pupo E, Garbay S, Calogero R, Pontoglio M, Provero P, Poli V. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: the case of Stat3. Proc. Natl Acad. Sci. USA. 2009;106:5117–5122. doi: 10.1073/pnas.0900473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee BK, Bhinge AA, Battenhouse A, McDaniell RM, Liu Z, Song L, Ni Y, Birney E, Lieb JD, Furey TS, et al. Cell-type specific and combinatorial usage of diverse transcription factors revealed by genome-wide binding studies in multiple human cells. Genome Res. 2012;22:9–24. doi: 10.1101/gr.127597.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trompouki E, Bowman TV, Lawton LN, Fan ZP, Wu DC, DiBiase A, Martin CS, Cech JN, Sessa AK, Leblanc JL, et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell. 2011;147:577–589. doi: 10.1016/j.cell.2011.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frietze S, Wang R, Yao L, Tak YG, Ye Z, Gaddis M, Witt H, Farnham PJ, Jin VX. Cell type-specific binding patterns reveal that TCF7L2 can be tethered to the genome by association with GATA3. Genome Biol. 2012;13:R52. doi: 10.1186/gb-2012-13-9-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin. Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 64.Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunolog. Rev. 2012;247:172–183. doi: 10.1111/j.1600-065X.2012.01112.x. [DOI] [PubMed] [Google Scholar]
- 65.Boulon S, Bertrand E, Pradet-Balade B. HSP90 and the R2TP co-chaperone complex: building multi-protein machineries essential for cell growth and gene expression. RNA Biol. 2012;9:148–154. doi: 10.4161/rna.18494. [DOI] [PubMed] [Google Scholar]
- 66.Diani-Moore S, Ram P, Li X, Mondal P, Youn DY, Sauve AA, Rifkind AB. Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem. 2010;285:38801–38810. doi: 10.1074/jbc.M110.131573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jimenez G, Shvartsman SY, Paroush Z. The Capicua repressor—a general sensor of RTK signaling in development and disease. J. Cell Sci. 2012;125:1383–1391. doi: 10.1242/jcs.092965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kannan Y, Yu J, Raices RM, Seshadri S, Wei M, Caligiuri MA, Wewers MD. IkappaBzeta augments IL-12- and IL-18-mediated IFN-gamma production in human NK cells. Blood. 2011;117:2855–2863. doi: 10.1182/blood-2010-07-294702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lessard L, Stuible M, Tremblay ML. The two faces of PTP1B in cancer. Biochim. Biophys. Acta. 2010;1804:613–619. doi: 10.1016/j.bbapap.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 70.Lores P, Visvikis O, Luna R, Lemichez E, Gacon G. The SWI/SNF protein BAF60b is ubiquitinated through a signalling process involving Rac GTPase and the RING finger protein Unkempt. FEBS J. 2010;277:1453–1464. doi: 10.1111/j.1742-4658.2010.07575.x. [DOI] [PubMed] [Google Scholar]
- 71.Maldonado V, Melendez-Zajgla J. Role of Bcl-3 in solid tumors. Mol. Cancer. 2011;10:152. doi: 10.1186/1476-4598-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Passon DM, Lee M, Rackham O, Stanley WA, Sadowska A, Filipovska A, Fox AH, Bond CS. Structure of the heterodimer of human NONO and paraspeckle protein component 1 and analysis of its role in subnuclear body formation. Proc. Natl Acad. Sci. USA. 2012;109:4846–4850. doi: 10.1073/pnas.1120792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perez-Leal O, Merali S. Regulation of polyamine metabolism by translational control. Amino Acids. 2012;42:611–617. doi: 10.1007/s00726-011-1036-6. [DOI] [PubMed] [Google Scholar]
- 74.Takano A, Zochi R, Hibi M, Terashima T, Katsuyama Y. Expression of strawberry notch family genes during zebrafish embryogenesis. Dev. Dyn. 2010;239:1789–1796. doi: 10.1002/dvdy.22287. [DOI] [PubMed] [Google Scholar]
- 75.Walker W, Zhou ZQ, Ota S, Wynshaw-Boris A, Hurlin PJ. Mnt-Max to Myc-Max complex switching regulates cell cycle entry. J. Cell Biol. 2005;169:405–413. doi: 10.1083/jcb.200411013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, Tewari M. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 2011;21:1450–1461. doi: 10.1101/gr.118059.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou L, Zhang Z, Zheng Y, Zhu Y, Wei Z, Xu H, Tang Q, Kong X, Hu L. SKAP2, a novel target of HSF4b, associates with NCK2/F-actin at membrane ruffles and regulates actin reorganization in lens cell. J. Cell. Mol. Med. 2011;15:783–795. doi: 10.1111/j.1582-4934.2010.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.