


Abstract
While it has been long recognized that genes are not randomly positioned along the genome, the degree to which its 3D structure influences the arrangement of genes has remained elusive. In particular, several lines of evidence suggest that actively transcribed genes are spatially co-localized, forming transcription factories; however, a generalized systematic test has hitherto not been described. Here we reveal transcription factories using a rigorous definition of genomic structure based on Saccharomyces cerevisiae chromosome conformation capture data, coupled with an experimental design controlling for the primary gene order. We develop a data-driven method for the interpolation and the embedding of such datasets and introduce statistics that enable the comparison of the spatial and genomic densities of genes. Combining these, we report evidence that co-regulated genes are clustered in space, beyond their observed clustering in the context of gene order along the genome and show this phenomenon is significant for 64 out of 117 transcription factors. Furthermore, we show that those transcription factors with high spatially co-localized targets are expressed higher than those whose targets are not spatially clustered. Collectively, our results support the notion that, at a given time, the physical density of genes is intimately related to regulatory activity.
INTRODUCTION
The cell’s regulatory state is, to a large extent, reflected by the particular conformation that the genome assumes at any given particular instance (1–5). This has been observed at the level of pairs of genes whose proximity in the nucleus is dependent on the developmental stage (6,7). Particular loci have also been shown to be associated with many distantly located genomic loci (8,9), demonstrating the plasticity of the genome. Recently developed experimental methods (10) enable the systematic study of these phenomena. In particular, chromosome conformation capture (3C) followed by high-throughput sequencing greatly improves our ability to globally model genomic structure. Using this approach and its derivatives, the genomic structures of Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and human have been determined for particular conditions. The initial analyses of these datasets have already led to insights into the structure of the genome, including the fractal nature of the human genome (11), the centromere co-localization and Rabl conformation in brewer’s yeast (12), the proximity of functionally related genes in fission yeast (13) and the physical demarcation of chromosomal domains in Drosophila (14). The ability to measure genomic architecture in three dimensions provides an opportunity to address long-standing questions involving how genomic structure encodes the phenotype, and addressing these will require new computational tools with an appropriate framework for analysis.
Of particular interest is the notion of nuclear transcription factories, and their role in establishing the regulatory states that underlie physiological stages. Most gene targets of S. cerevisiae transcription factors (TFs) have been determined with high confidence, revealing an average of 70 gene targets per TF (15,16). Coupling this data with genome structure enables the study of the co-localization of TF targets. For example, are the targets of the same TF co-localized to the same spatial arrangement as the transcription factory model suggests? Under which conditions does such co-localization occur? Previous analyses have addressed this question leading to contradictory results. Dai and Dai compared the number of interactions in different gene sets and observed statistical enrichment under the hypergeometric null model for interactions among TF targets (17). However, Witten and Noble argued that edges in the 3C interaction graph are not statistically independent, as was assumed by Dai and Dai, and as such co-localization events would be over-counted (18). To correct for this, Witten and Noble applied a re-sampling methodology under which no signal for TF target co-localization was detected.
Importantly, while the previous studies treated genomic proximity differently than spatial proximity, this was done by examining only inter-chromosomal distances. In additional, the spatial organization of the genome was not directly compared with the primary gene order in terms of their respective functional enrichment. This latter point is important because genomic analyses have revealed that neighboring genes tend to have similar expression profiles (19). Furthermore, genes with housekeeping functions in particular tend to be co-positioned along chromosomes (20). In particular, gene targets of the same TF are enriched for proximity in their genomic order (21). Thus, controlling for the genomic clustering is crucial for unbiased evidence regarding the degree to which the spatial clustering contributes to regulating functionally related genes.
Here we introduce a statistical framework for modeling chromatin structure relying on a minimum set of assumptions and assaying the spatial proximity of functionally related genes while controlling for effects from linear co-localization along the genome. Our analysis is more subtle and flexible in refining gene sets for detecting the optimally clustered subset and defines enrichment environments more loosely based on this subset. Additionally, we apply a direct approach for controlling against results that may have emerged primarily from genomic proximity, thereby focusing our results on the phenomenon of spatial co-localization. Notably, our approach uses the hypergeometric test for assessing spatial co-localization at a particular locus, thereby disentangling dependencies that arose in previous analyses (17). We applied this approach on a parsimoniously interpolated 3C contact matrix. Our results indicate that for most TFs, the targets are significantly more co-localized in space than they are co-localized in genomic loci. We further found that TFs with spatially co-localized targets are also expressed higher under the same measurement condition, suggesting that regulatory activity is correlated with the presence of transcription factories. As more genomic structures are produced, our method promises to be of importance to the study of transcription factories.
MATERIALS AND METHODS
Natural neighbor interpolation of 3C data
The raw frequency measurements provided by the yeast 3C experiment of Duan et al. (12)—using the HindIII libraries filtered at P < 10−3 False discovery rate (FDR)-corrected—was represented as a scattered sparse block matrix, where each block corresponds to a pair of chromosomes. Each read of a mapped paired-end insert was assigned to the mid-base of a restriction enzyme fragment in its corresponding unique location along the genome. Each block of the raw data matrix was subjected to interpolation using a continuously differentiable C1 interpolant. The natural neighbor interpolation method (22) was implemented at 1-kb resolution using the TriScatteredInterp function in Matlab with the following modifications. First, the frequency of each position with itself was set to the highest observed frequency in the dataset. These measurements are not captured by the 3C method for technical reasons (12), but are required for the multi-dimensional scaling (MDS) to preserve positive definiteness. The results are robust to a wide range of different set diagonal frequencies (Supplementary Figure S3). For each diagonal block matrix, ‘ghost points’ (23) were added at a distance equivalent to 10% the distance of the chromosome size and set to a frequency of zero. This enabled extrapolation near telomeres where there are little to no data. Finally, due to rounding errors in the interpolation the resulting matrix was non-symmetric, which is resolved by averaging it with its transpose. The Voronoi tessellation, on which natural neighbor interpolation relies, is shown in Figure 1A, where the colored domains are Voronoi cells. Each cell is generated by the intersect of all half-spaces imposed by the orthogonal separating planes between the point inside the cell and every other point separately. The EcoRI library used in the original experiment (12) was used for comparison and validation of the resulting interpolation.
Figure 1.

Studying genome structure using 3C at 1-kb interpolated resolution. (A) 3C data for the S. cerevisiae chromosome I superimposed on the estimated chromosomal relationships (tessellation cells) they represent. Black dots represent pairs of restriction fragment mid-points with evidence of cross-linking. Cell color indicates the observed frequency (effectively identical to a nearest neighbor interpolant). The diagonal areas are artificially inserted to overcome inherent lack of self-contacts in the method (see also Supplementary Figure S3). (B) Natural neighbor interpolation of the 3C data at 1-kb resolution. The colors indicate the likelihood of proximity of the genomic loci. (C) A 3D model of chromosome I generated using non-linear dimensionally reduction on the interpolated dataset shown in B. Color indicates proximity to the mid-point of the chromosome—marked with a red arrow. Note that the distance is not equivalent to the distance on the primary sequence (indicated by the left color bar) as the shape projects inwards. (D) A model of the yeast genome by non-linear dimensionally reduction as in C but extended to all chromosomes by sampling (see ‘Materials and Methods’ section). Note that the chromosomes lie at the periphery in a spherical fashion with the ends extended and centromeres joined.
Modeling genome structure
The interpolated contact frequency matrix was used as input for modeling the structure. The matrix was embedded to coordinates in an arbitrary 3D Euclidean space using non-linear metric MDS (also referred to as principal coordinate analysis) (24). The three principal dimensions from the linear embedding were used as a starting reference for the genomic coordinates. Next, the isotonic least-squares optimization was used to minimize the deviation of distances between coordinates to that of the input matrix while preserving the order of pairwise distances. The target function was the Kruskal stress-1 criterion (24), which measures relative deviations from the input matrix:
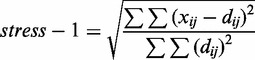 |
where 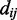 is the distance between coordinates
is the distance between coordinates 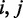 in the original input data, and
in the original input data, and 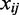 is the distance between coordinates
is the distance between coordinates 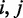 in the resulting model. For the whole-genome embedding, we re-sampled the genome using 5-kb resolution per coordinate. This lower resolution allowed the embedding process to converge at the whole-genome scale. To visualize this model at 1-kb resolution, we use piecewise cubic Hermite interpolation, a C1 interpolant for univariate data (25).
in the resulting model. For the whole-genome embedding, we re-sampled the genome using 5-kb resolution per coordinate. This lower resolution allowed the embedding process to converge at the whole-genome scale. To visualize this model at 1-kb resolution, we use piecewise cubic Hermite interpolation, a C1 interpolant for univariate data (25).
Functional enrichment of 3D and 1D loci
For each gene g, we compute the functional enrichment in 3D and 1D environments according to the following method. All other genes are ordered separately according to the following:
Their interpolated contact frequency with respect to g (3D proximity to g),
Their genomic distance (1D) from g.
For any given TF, we compute the minimum hypergeometric statistic (mHG) (26,27) for the enrichment of its target in both the 1D and 3D neighborhoods of g. Annotation data for TF targets were taken from a previous analysis [orfs_by_factor_p0.005_cons1 from (15)]. Briefly, for a given ranked list of genes (for an example see Figure 2A), mHG finds a prefix of the list that maximizes the statistical enrichment of genes pertaining to an annotation set. The mHG P-value represents the likelihood of observing such an enrichment, at some prefix, under a null model [see (26,27)]. We obtain a bound on the mHG P-value, per annotation term, and per centered gene g by multiplying the calculated mHG statistic by the number of genes in the annotation term. To correct for multiple testing, these are later Bonferroni-corrected across the different annotation terms. Because the process is applied on both the genomic and spatial orderings of genes, we limit the threshold search to the size of g’s chromosome, which results in comparable P-values for the most enriched spatial and genomic environments centered on g. Hence, this implementation of mHG is partition limited as previously described (26,27). Peaks of enrichment (Figure 3A) were detected using Matlab’s findpeaks function. We limited the peak calling to a minimum distance of 10 loci from one another and a height of −log10(0.05). As a supplement to the present work, we are providing the software package INSP3CT (Interpolation and Statistical Proximity of 3C Tables) as an implementation for similar datasets to identify and compare spatial and genomic co-localization of genomic annotated markers.
Figure 2.

Comparing functional enrichment between the genomic and spatial regions of the genome. (A) Two genomic distances. The schematic shows the gene neighborhood surrounding a particular gene (red). The neighboring genes may be ranked by their genomic proximity (left) or their spatial proximity (right). (B) Detecting areas of enrichment for TF-cohorts. In ranked gene lists, generated by either genomic or spatial proximity, the genes annotated as targets of a particular TF are indicated as black lines. The P-value of the enrichment of the targets for each threshold is indicated on the right. The threshold with the best P-value is indicated by the dashed line (see ‘Materials and Methods’ section). This analysis is shown for two genomic loci surrounding both YHL050C and YHL050W-A (top) and YCL012C (bottom) genes respectively, and querying for targets of GLN3. (C) Local structures of the two loci examined in B. Colors indicate distinct yeast chromosomes. The red circles indicate the center gene around which co-localization was tested. The content shown in each sphere is the environment that corresponds to the mHG threshold, dictated by the most enriched spatial environment for GLN3 targets. Bars on the right mark the loci along the linear genome, which participate in the most enriched environment by both the genomic and spatial rankings. Black dots, both in the bars and the visualized structure, indicate gene targets of GLN3. Scale bars were calculated according to an average size estimate for 1 kb of chromatin  0.33 µm. Chromosomes are colored as indicated in the legend.
0.33 µm. Chromosomes are colored as indicated in the legend.
Figure 3.

Gene targets of the same TF generally spatially cluster in the yeast genome. (A) For each position in genome (x-axis, chromosomes are separated by vertical dashed lines), the P-value of the enrichment for GLN3 gene targets is shown (y-axis, −log10 of the mHG corrected P-value, see ‘Materials and Methods’ section). The enrichment values are shown for both the 3D (red) and 1D (blue) distances. Dotted boxes correspond to the environments shown in Figure 2B. Points in the grayed out region are below the significance threshold (P > 0.05, mHG, corrected). Peaks over the significance threshold are indicated by arrows. Top left inset shows the effect of running the same analysis on one random permutation of the target genes of GLN3 (B and C) Analysis on the gene targets of 107 TFs. GLN3 is marked in red. (B) A comparison between the maximal −log10 P-value for 3D and 1D enrichments for each examined TF. (C) A comparison of the number of significant spatial (3D) and genomic (1D) regions (peaks; marked with arrows in A, see ‘Materials and Methods’ section) for each examined TF. The line indicates a unity relationship.
To compare the observed enrichment results, for a fixed given TF, to a background model, the genes were first sorted according to the log odds ratio of the 3D and 1D enrichments. Next, the same quantities were computed for each of 100 shuffled genomes (with gene identities randomly permuted), thus yielding Z-scores for each rank in the list of genes sorted by the actually observed log-odds. This comparison is exemplified in Figure 4A.
Figure 4.
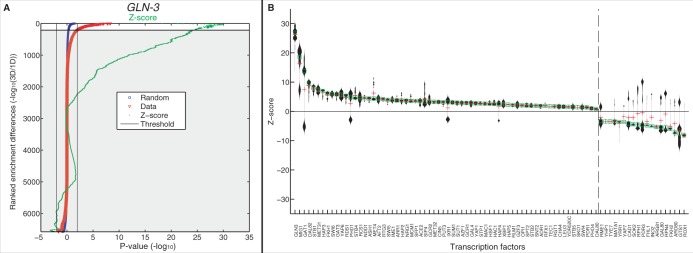
Comparing enrichment significance against a random model. (A) Ranking of the relative 3D to 1D enrichment (-log odds) for GLN3 targets in the regions surrounding all genes is shown in red. The same is also shown in blue for mean value of 100 gene order permutations. The Z-score is shown in green. Ranked indices with log odds that cross a significance threshold are used in downstream analysis (see ‘Methods and Materials’ section). (B) Showing the distribution of selected Z-scores for each TF. The dashed line separates the TFs that have positive median Z-score values from those with negative ones. TFs left of the line have more 3D enrichment than expected at random, whereas TFs right of the line are ones with more 1D enrichment than expected by random.
RESULTS
An unconstrained 1-kb resolution model of the yeast genome using natural neighbor interpolation and embedding
The systematic analysis of genome structure and of 3D features of genome organization requires a coherent and comprehensive representation of the contacts between genomic loci. However, actual data resulting from 3C measurement assays are scattered across irregular genomic intervals. Thus, our first goal was to use the previously determined dataset (12) to study the characteristics of the yeast genomic structure as it relates to function. To accomplish this, we first set out to regularize and provide a uniformly spaced contact matrix. For this purpose, we used a natural neighbor interpolation to arrive at a 1-kb resolution frequency matrix.
Because the median size of the intervals in the primary data is 1800 bp (median restriction fragment length) (12), we chose to interpolate at a 1-kb interval. This choice stemmed from the notion that the interpolated resolution must not greatly exceed that inherent in the primary data. We thus effectively binned the linear yeast genome to 12 071 regularly spaced 1-kb coordinates. Figure 1A shows a representation of the raw data from the 3C measurement assay (12) such that each measured data point (pair of observed restriction fragments, represented by a black dot in Figure 1A) is mapped to the respective genomic loci in chromosome I. We note the sparseness of the data at some loci, as reflected by the large and irregular domains for many of the data points (see ‘Methods’ section), indicating the limited resolution of the data for the interaction between the respective loci. Related to this sparse sampling are the sharp discontinuities present in the data (Figure 1A). Figure 1B shows our implementation of a natural neighbor interpolation (see ‘Materials and Methods’ section) on the same data for chromosome I, which addresses this sparseness and sharpness by setting the local contact behavior to what would be expected of a continuously differentiable (smooth) curve. From the perspective of its differential geometry, a chromosome is expected to behave continuously owing to its polymer structure and be differentiable owing to the mechanical angular limitations imposed by its chemistry. The resulting interpolated contact frequency map was compared with that corresponding to a library generated using a second restriction enzyme (EcoRI) in the original dataset (12). The high correlation (R = 0.98, P < 10−300, Supplementary Figure S9) provided validation of the quality of the interpolation.
To model the structure of the genome using the interpolated frequency matrix, we invoked a non-linear MDS (24). This method is grounded in the well-established algebraic method of non-classical dimensionality reduction and yields a deterministic 3D view of the yeast genome using an unconstrained and unsupervised methodology (see ‘Materials and Methods’ section). The linear embedding reduced the dimensionality of the dataset to orders-of-magnitude-more dimensions than is expected of a shape measured in 3D space, reflecting the biological and measurement noise inherent in the 3C method (Supplementary Figure S2). Applying this method on the intra-chromosomal interaction data of chromosome I resulted in a crescent-like curve, crumpled near the centromere (Figure 1C). Figure 1D shows the application of the method to the entire genome, resulting in a ‘water-lily’ conformation of the chromosomes, consistent with other models proposed in the literature (12), with centromeres somewhat interwoven in one end, and chromosome arms extending outward. The quality of this embedding was quantified using the Kruskal stress-1 criterion (28). The resulting stress value of our model is 0.28, which we propose as a measure of the noisiness of the 3C data. This model is stable under small perturbations, as we show in Supplementary Figure S3. In summary, our natural neighbor interpolation coupled with non-linear MDS provides a natural 3D model of the genome at 1-kb resolution.
Statistical assessment of spatial functional enrichment controlled by genomic order
Using the structural model of the genome, we asked whether genes regulated by the same TF cluster together spatially along the genome. For this we developed a method for assessing the functional enrichment in a 3D environment. We designed the method based on three principles: (i) Direct comparison of any spatial enrichment with that observed for the linear genomic ordering; (ii) Detection of enrichment of a subset rather than of correlation for the entire set (26,27); and (iii) Detecting enrichment for variable-size environments, as the exact size of enriched regions was not known. The first was done to correct for the known functional co-localization of genes along the chromosomes (21). In the comparison, enrichment was favored over correlation, as it is more sensitive at detecting signals at individual genomic locations, whereas genome-wide correlation methods will be dominated by noise and by effects outside of the scope of a possible transcription factory. As a statistical method, we invoked the robust, sensitive and threshold-free mHG method that has been successfully applied in other contexts (26,27,29–31). For each gene in the yeast genome, our method proceeds by ranking all other genes by either their genomic (linear) or their spatial (3D) distance to the gene (Figure 2A). Given a specific TF of interest, the mHG test is then applied to both of these two rankings to test whether the targets of that TF are enriched in the genomic and spatial neighborhoods of that gene (see ‘Methods’ section). Of particular interest are the most enriched environments, both in the genomic and in the spatial perspective, centered around a gene, as they can be compared on an equal setting. For any given locus, we quantify whether the spatial enrichment of targets is more significant than the genomic enrichment; for example, by examining the log odds ratio of the 3D and 1D enrichment P-values.
We demonstrate the method in Figure 2B with two specific loci in the yeast genome. In the first example (Figure 2B, top), we compare the enrichment of the targets of the TF GLN3 in the linear genomic and spatial neighborhoods centered at YHL050C and YHL050W-A, whose transcription start sites map to the same 1-kb region. For the first 140 genes added according to either genomic or spatial distance, the enrichment is similar. However, as the spatial distance is allowed to increase, the enrichment sharply increases in contrast to the respective genomic enrichment (Figure 2B, bottom). The analysis is terminated at 200 genes, as the end of the chromosome is reached (chromosome III) and so the comparison with the linear genomic ordering is no longer possible for large neighborhoods.
A similar pattern is observed in the other example of GLN3 targets when considering neighborhoods centered around YCL012C on chromosome VIII. The spatial enrichment, measured by the hypergeometric P-value, of the targets of GLN3 increases (Figure 2B, blue line) as the radius of the ball examined (centered at YCL012C) is expanded (i.e. more genes at greater distances are included). In the close neighborhood of YCL012C, the enrichment is the same for both spatial and genomic proximity, suggesting that the genes most spatially proximate to YCL012C are identical to those proximate to it in the linear genomic order. Interestingly, as the number of genes included exceeds the first 100, the spatial enrichment becomes even more significant, surpassing the linear genomic enrichment. This enrichment then peaks for an environment containing ∼125 genes (hypergeometric P < 10−12), after which the addition of more distant genes diminishes the statistical significance. In comparison, the most significant enrichment based on the genomic order alone is P < 10−5 obtained at a neighborhood that includes the nearest 80 genes. Thus, we conclude that for the environment centered on YCL012C, GLN3 targets are significantly more highly enriched in space than along the linear genome. We note that when randomly shuffling the genomic positions of the genes we did not find any significant enrichment of co-localization, spatial or genomic, such as those shown in Figure 2B.
Examining the structural environments of the two genomic loci described above (Figure 2B) provided insight into the detected enrichments. Figure 2C shows the environments along with the corresponding genomic regions that are mapped to them. In both cases, regions from different chromosomes contribute to the significant spatial enrichment. The thin part of the chromosome on which the center gene (marked in red) is located indicates the interval with the most significant linear genomic enrichment around the center gene.
Widespread spatial regions enriched for TF targets
Our method allowed us to systematically test the spatial and genomic enrichments of TF targets surrounding each gene in the genome, as shown for GLN3 targets in YCL012C (Figure 2B). The genomic landscape depicted in Figure 3A highlights the most significant spatial enrichment results surrounding each locus (marked in red) as well as the most significant linear genomic enrichment (marked in blue). The two specific regions shown in Figure 2C are noted with dashed boxes. Strikingly, in many loci we observe significant spatial enrichment that is higher than that obtained for genomic order enrichment. To evaluate this result, we used two controls. First, we tested whether a shuffled genomic ordering—maintaining the locations of the genes but randomizing their identities—would still lead to enrichment results, and found that, as expected, it does not (Figure 3A, inset). We also tested cyclic permutations of gene locations along each chromosome by cyclically shifting gene locations by selecting the shift size to be 10–90% of the chromosome size. Such shifted data maintain all 1D gene density properties of the genome. We observed that the linear genomic enrichment is conserved (as clearly expected), while the spatial enrichment is eliminated (Supplementary Figure S1). Finally, we compared the hypergeometric P-values with those resulting from a shuffled null model and found significant differences (Supplementary Figure S11) indicating that our use of the hypergeometric test does not produce spurious results.
To further quantify the observed higher spatial enrichment, compared with that obtained in linear genomic order, we first examined for each TF, the region with maximum enrichment at the 3D level and compared it with the 1D region that is most enriched. For GLN3, the most significant 3D region has an associated P-value of 10−9, while the most significant 1D region has a P-value of 10−8 (Figure 3A). Examining all 116 TFs, we found that 32 TFs have a more significant 3D region, while six have a more significant 1D region (Figure 3B). This indicates that when examining neighborhoods of genes, the 3D region captures more significant enrichment than an examination of solely the 1D order.
Next, we deployed a peak-detection algorithm on the genomic landscape to identify distinct regions of locally maximal enrichment. We assigned each peak to either the 3D or 1D enrichment depending on which is more significant, delineated to both in the case of a tie. Using GLN3 again as an example, we detected 70 and 5 for the 3D- and 1D-enriched peaks, respectively (Figure 3A, black arrows). A paired t-test on the 3D and 1D enrichment peaks indicated the significance of spatial enrichment (P < 10−6). Thus, for this TF, more enrichment is detected at the spatial level than in the genomic level, providing evidence for the tendency of the genome to co-localize its targets in transcription factories. Expanding these analyses to the rest of the TFs, we found an overall preponderance of 3D clusters relative to 1D clusters (P < 10−30 Kolmogorov–Smirnov test between the distributions of the number of peaks in 3D versus those in 1D). For some TFs, this effect is particularly strong (Figure 3C), while for three TFs—ROX1, YRR1 and ARG81—the signal is reversed, a more significant 1D clustering than 3D. SIP4 shows the most extreme spatial co-localization relative to genomic order (84–5, respectively, Supplementary Figure S4). Of 117 TFs, 64 show a significant (P < 0.05, FDR-corrected, one-tailed two-sample t-test) enrichment of spatial (and 10 of 117, a significant enrichment of genomic) co-localization of their targets. We found that this result is also observed in a second replicate of the dataset (Supplementary Figure S7) as well on the dataset following correction for potential biases using a recently proposed method (32) (Supplementary Figure S8).
The peak analysis may be biased because we filter out genomically consecutive signals (1D) but not potentially overlapping 3D signals. To address this, we compared our observed enrichments to a suite of 100 genomes whose gene order has been shuffled using a ranking-based approach (see ‘Materials and Methods’ section). Comparing with the randomly annotated genomes has the additional feature of direct P-value estimations without recourse to multiple testing corrections and parametric distribution assumptions. For GLN3, filtering for genes with two orders of magnitude more significant 3D to 1D and vice versa (non-grey region), the Z-scores indicate strong significance relative to the shuffled genomes (Figure 4A). Repeating this analysis for all of the available TFs, we found that for most TFs the Z-scores are positive, indicating that 3D enrichment is significantly greater than 1D enrichment when comparing with the random background model. Interestingly, some TFs show a wide bimodal distribution, indicating that the TF has both significant 1D and 3D regions of significant enrichment. We conclude that for most TFs we detect significant spatial co-localization of the targets.
TFs whose gene targets are spatially enriched are more highly expressed
If the targets of a particular TF show significant co-localization in the genome, one would expect that TF to be functional under the conditions sampled for the genomic structure. A proxy for the function of a TF is its expression level, and thus we asked whether those TFs showing the strongest signals of co-localized targets are also more highly expressed (33).
We sorted TFs according to the ratio of spatial to genomic co-localization of their targets, an indication of their 3D co-localization. The expression of the top 50 TFs was then compared with that of the bottom 50. We detected a significant difference in expression levels (P < 10−2, Kolmogorov-Smirnov test, Figure 5A). Overall, the correlation between the degree of co-localization (target co-localization P-value) and the average gene expression level was r = 0.25 (P < 10−2 Supplementary Figure S6). We further validate that this result is not confounded by the number of targets of the particular TFs and the choice of threshold (Supplementary Figure S10). While not highly significant, this correlation between expression levels and large-scale target co-localization supports the possible role of genomic configuration in accommodating different transcription factories.
Figure 5.

Gene expression is higher for genes in regions of functional co-localization. (A) Violin plots of expression levels of TFs with and without spatially co-localized gene targets. The expression values are compared for the 50 TFs with the highest and lowest spatial localization score (-log of the ratio of t-test P-value comparing genomic and spatial enrichment co-localization). TFs with spatial co-localized targets have a significantly higher expression (P < 0.01, Kolmogorov–Smirnov test). (B) Spatial locations of transcriptional factories. Superimposed on the genome structure, for each gene the color indicates the number of instances that the 3D structure is more significantly enriched in gene targets with respect to the linear order.
Finally, we queried for the spatial location of the apparent transcriptional factories. For each gene, we computed the number of instances in which a spatial region including that gene is enriched for TF gene targets more than for the genomic order, across the set of 107 TFs. Figure 5B shows these locations superimposed on the genomic structure. We found that regions that are enriched for such ‘transcriptional factories’ indeed form distinct clusters. In particular, we observe a high degree of association of genes with transcription factories in the periphery, mainly located on chromosome II, and also on chromosome XV and chromosome XVI (Figure 5B). Comparing the expression of the set of genes highly associated with factories (>25 TF sets) relative to the genes only weakly associated with factories (<25 TF sets), we find that the former genes are more highly expressed (P < 0.05, Kolmogorov–Smirnov, Supplementary Figure S5). This provides further evidence that transcriptional factories generally correspond to transcriptionally active regions.
DISCUSSION
Any advancement of biological methods to identify the precise structure of the genetic material throughout the life of an organism must be matched in rigor by the computational and statistical platforms that are used to interpret their measurement results. 3C has emerged as the most generalized method for establishing the structure of the genome in a systematic fashion (10). However, the statistical methods to make the most of the resulting data are only starting to be developed (11,12,32,34). Here, we report a novel approach to several aspects of the analysis of spatial conformation data. We model the structure of the S. cerevisiae genome without the previously imposed assumptions (see below), thus capturing an unbiased representation of the data in 3D. Our method is based on standard approaches in computational geometry, statistics and linear algebra (24), invoked here for the first time to the problem of genomic structure. We use the resulting contact matrix to ask whether functionally related genes are co-localized in the 3D structure. Using a rigorous and controlled statistical approach, we provide evidence for this notion. In this section, we consider the advantages and limitations of all aspects of our methodology including the choice of interpolation and embedding procedures, internal reference to the 1D gene order as a control. Finally we discuss the notion of widespread transcriptional control by spatially defined factories.
Existing literature that addresses directly the problem of contact map completion in the context of 3C data relies either on a convolution with a fixed environment size (12,13,35) or a statistical background model to estimate either enrichment or depletion of observed contacts (14,34). Convolution-based approaches lead to locally smoothed regions, while disproportionately distorting structures in data-sparse or outlier-rich regions. Both of these previously used approaches are dependent on a subjective choice of parameters such as the environment size and latent variables for statistical model. Because our method is fully reliant on a complete contact map, we established a robust approach to generate a full contact map by interpolating missing data. We propose that the most appropriate interpolation method for completing 3C data is a modification of natural neighbor interpolation (C1 family of interpolants). Natural neighbor interpolation is immune to the disadvantages inherent in nearest neighbor interpolation, where different genomic loci may optimally occupy the same position in space and tie-breaking scenarios are typically addressed in an arbitrary fashion. Further, natural neighbor is not as simplistic as bilinear interpolation, where only the two flanking data points in each dimension contribute to the interpolated value. Additionally, natural neighbor interpolation has been previously applied successfully for problems of smooth surface reconstruction (36), which relate to our problem in nature. Based on a tessellated view of the data (see ‘Methods’ section), natural neighbor interpolation computes the weighted average of all the neighboring data points that can contribute to the information of the contact between the locations under interpolation. We note that our interpolation approach—and likewise all interpolations—does not necessarily yield inter-point contacts that mathematically qualify as a metric, and as such, the resulting contact map does not necessarily describe a structure residing in a Euclidean space precisely. To visualize the resulting interpolated contact map, we attempted to generate a structural model that best captures the data. Our analysis was performed at a 1-kb binning of the resulting interpolation; however, as the resolution improves in future studies, we expect our method to have greater statistical potential, as less genes will be co-binned.
Previous studies attempting to generate a structural model for chromatin used supervised rule sets, a random starting conformation, and optimization algorithms to fix each coordinate pair in its expected distance (if available) from one another (12,13,35). We propose that because such methods rely on an underdetermined process, they cannot be rigorously applied to explore the most likely conformation. Our approach uses metric dimensionality reduction as a starting point, which sets as a starting conformation the principle 3D outline of the shape. This outline is expected to capture the essence of the underlying geometry of the data. The optimization process preserves the order among contacts, maintaining the coherence of contacts in the resulting structure. MDS is a classical algebraic and statistical approach that is well established in the literature (24). MDS relies on a practical assumption and attempts to minimize the square error of inter-point distances while maintaining their order when comparing the input data with the resulting model. Our approach thus minimally intervenes with the underlying measurements applying a parsimonious genome modeling preferences.
We provide a solid statistical framework to determine enrichment in the spatial co-localization of genomic elements and apply it to detect a significant co-localization of TF targets. We also show a correlation between co-localization and higher expression of the targeting TF. Our results are thus consistent with previous studies, attempting to link gene organization with control and regulation of transcription (6,7,9,37–41), and further extends previous systematic approaches to provide the imperative comparison to the genomic proximity of co-regulated genes. Collectively, these results indicate that genome remains poised for the expression of co-regulated genes by adjusting their conformation to enrich for their co-localization. This conformation may likely have benefits in terms of the operations of an activated TF, which if shuttled to a region with enriched targets, it will have a reduced number of possible gene targets to interact with by diffusion. This scenario would suggest that the mechanism for co-localization (whether active, or passively selected for), along with higher expression for the active TF, work in concert to regulate gene circuits, and the interplay between them is crucial to understanding expression regulation.
Future directions will no doubt include a comprehensive analysis of co-localization of genomic elements to detect functional partitioning and to better characterize transcription factories. Additionally, it will be interesting to examine the extent to which these findings will be conserved across organisms and tissues. Single-cell–based 3C methods—currently unavailable but sorely needed—will be able to produce a more accurate picture of genome structure, rather than a population-mean approach. Using sophisticated statistics for the detection of co-enrichment of ordinal measurements, similar methodology will surely be applied directly to non-binary or thresholded experimental results, such as the ones from chromatin immunoprecipitation (ChIP) experiments to provide more unbiased views on annotated features.
AVAILABILITY
A Matlab software package called INSP3CT is provided to analyse contact frequency datasets and genomic annotations by performing spatial and genomic enrichment on selected loci. INSP3CT takes as input files describing restriction sites, inter- and intra-chromosomal contact frequencies, the genomic sequence, loci of interest along the genome (for example genes) in bin coordinates and vectors of annotation with the number of co-binned loci of interest per bin. INSP3CT outputs a figure for each vector of annotation comparing 3D with 1D enrichment across loci. INSP3CT also provides access to the interpolated contact frequency matrix, the corrected enrichment scores per loci and the size of enrichment environment. INSP3CT is available at http://shayben.github.com/INSP3CT.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–11.
FUNDING
The Technion Lokey Interdisciplinary Center for Life Science and Engineering and Israel Science Foundation. Funding for open access charge: Technion - Israel Institute of Technology.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Eitan Yaffe, Israel Steinfeld, Florian Wagner, David Silver, Yael Mandel-Gutfreund, Nir Ailon and Amit Singer for advice and critical readings of the manuscript.
REFERENCES
- 1.Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008;4:e1000039. doi: 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meister P, Towbin BD, Pike BL, Ponti A, Gasser SM. The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev. 2010;24:766–782. doi: 10.1101/gad.559610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, Rinn J, Schier AF. Chromatin signature of embryonic pluripotency is established during genome activation. Nature. 2010;464:922–926. doi: 10.1038/nature08866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chambeyron S, Da Silva NR, Lawson KA, Bickmore WA. Nuclear re-organisation of the Hoxb complex during mouse embryonic development. Development. 2005;132:2215–2223. doi: 10.1242/dev.01813. [DOI] [PubMed] [Google Scholar]
- 7.Junier I, Dale RK, Hou C, Kepes F, Dean A. CTCF-mediated transcriptional regulation through cell type-specific chromosome organization in the beta-globin locus. Nucleic Acids Res. 2012;40:7718–7727. doi: 10.1093/nar/gks536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmer C, Fabre E. Principles of chromosomal organization: lessons from yeast. J. Cell. Biol. 2011;192:723–733. doi: 10.1083/jcb.201010058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sajan SA, Hawkins RD. Methods for Identifying Higher-Order Chromatin Structure. Annu. Rev. Genomics Hum. Genet. 2012;13:59–82. doi: 10.1146/annurev-genom-090711-163818. [DOI] [PubMed] [Google Scholar]
- 11.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. A three-dimensional model of the yeast genome. Nature. 2010;465:363–367. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanizawa H, Iwasaki O, Tanaka A, Capizzi JR, Wickramasinghe P, Lee M, Fu Z, Noma K. Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res. 2010;38:8164–8177. doi: 10.1093/nar/gkq955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 15.MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai Z, Dai X. Nuclear colocalization of transcription factor target genes strengthens coregulation in yeast. Nucleic Acids Res. 2012;40:27–36. doi: 10.1093/nar/gkr689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witten DM, Noble WS. On the assessment of statistical significance of three-dimensional colocalization of sets of genomic elements. Nucleic Acids Res. 2012;40:3849–3855. doi: 10.1093/nar/gks012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen BA, Mitra RD, Hughes JD, Church GM. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- 20.Lercher MJ, Urrutia AO, Hurst LD. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet. 2002;31:180–183. doi: 10.1038/ng887. [DOI] [PubMed] [Google Scholar]
- 21.Janga SC, Collado-Vides J, Babu MM. Transcriptional regulation constrains the organization of genes on eukaryotic chromosomes. Proc. Natl Acad. Sci. USA. 2008;105:15761–15766. doi: 10.1073/pnas.0806317105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bobach TA. 2008. Natural Neighbor Interpolation - Critical Assessment and New Results. PhD. Thesis, TU Kaiserslautern. [Google Scholar]
- 23.Bobach T, Farin G, Hansford D, Umlauf G. Natural neighbor extrapolation using ghost points. Comput. Aided Des. 2009;41:350–365. [Google Scholar]
- 24.Seber GAF. Multivariate Observations. Wiley, New York; 2004. [Google Scholar]
- 25.Carlson RE, Fritsch FN. Monotone piecewise bicubic interpolation. SIAM J. Numer. Anal. 1985;22:386–400. [Google Scholar]
- 26.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 2007;3:e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruskal JB. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika. 1964;29:1–27. [Google Scholar]
- 29.Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, Cedar H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009;16:564–571. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 30.Leibovich L, Mandel-Gutfreund Y, Yakhini Z. A structural-based statistical approach suggests a cooperative activity of PUM1 and miR-410 in human 3′-untranslated regions. Silence. 2010;1:17. doi: 10.1186/1758-907X-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avraham R, Sas-Chen A, Manor O, Steinfeld I, Shalgi R, Tarcic G, Bossel N, Zeisel A, Amit I, Zwang Y, et al. EGF decreases the abundance of microRNAs that restrain oncogenic transcription factors. Sci. Signal. 2010;3:ra43. doi: 10.1126/scisignal.2000876. [DOI] [PubMed] [Google Scholar]
- 32.Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 2011;43:1059–1065. doi: 10.1038/ng.947. [DOI] [PubMed] [Google Scholar]
- 33.James N, Landrieux E, Collart MA. A SAGA-independent function of SPT3 mediates transcriptional deregulation in a mutant of the Ccr4-not complex in Saccharomyces cerevisiae. Genetics. 2007;177:123–135. doi: 10.1534/genetics.107.076299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseau M, Fraser J, Ferraiuolo MA, Dostie J, Blanchette M. Three-dimensional modeling of chromatin structure from interaction frequency data using Markov chain Monte Carlo sampling. BMC Bioinformatics. 2011;12:414. doi: 10.1186/1471-2105-12-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tjong H, Gong K, Chen L, Alber F. Physical tethering and volume exclusion determine higher-order genome organization in budding yeast. Genome Res. 2012;22:1295–1305. doi: 10.1101/gr.129437.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boissonnat J, Cazals F. Smooth surface reconstruction via natural neighbour interpolation of distance functions. Proceedings of the Sixteenth Annual Symposium on Computational Geometry. 2000:223–232. [Google Scholar]
- 37.de Laat W, Grosveld F. Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003;11:447–459. doi: 10.1023/a:1024922626726. [DOI] [PubMed] [Google Scholar]
- 38.Ferrai C, de Castro IJ, Lavitas L, Chotalia M, Pombo A. Gene positioning. Cold Spring Harb. Perspect. Biol. 2010;2:a000588. doi: 10.1101/cshperspect.a000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palstra RJ. Close encounters of the 3C kind: long-range chromatin interactions and transcriptional regulation. Brief Funct. Genomic Proteomic. 2009;8:297–309. doi: 10.1093/bfgp/elp016. [DOI] [PubMed] [Google Scholar]
- 40.Steinfeld I, Shamir R, Kupiec M. A genome-wide analysis in Saccharomyces cerevisiae demonstrates the influence of chromatin modifiers on transcription. Nat. Genet. 2007;39:303–309. doi: 10.1038/ng1965. [DOI] [PubMed] [Google Scholar]
- 41.Taddei A, Schober H, Gasser SM. The budding yeast nucleus. Cold Spring Harb. Perspect. Biol. 2:a000612. doi: 10.1101/cshperspect.a000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.