


Abstract
DNA methylation has been implicated in a number of diseases and other phenotypes. It is, therefore, of interest to identify and understand the genetic determinants of methylation and epigenomic variation. We investigated the extent to which genetic variation in cis-DNA sequence explains variation in CpG dinucleotide methylation in publicly available data for four brain regions from unrelated individuals, finding that 3–4% of CpG loci assayed were heritable, with a mean estimated narrow-sense heritability of 30% over the heritable loci. Over all loci, the mean estimated heritability was 3%, as compared with a recent twin-based study reporting 18%. Heritable loci were enriched for open chromatin regions and binding sites of CTCF, an influential regulator of transcription and chromatin architecture. Additionally, heritable loci were proximal to genes enriched in several known pathways, suggesting a possible functional role for these loci. Our estimates of heritability are conservative, and we suspect that the number of identified heritable loci will increase as the methylome is assayed across a broader range of cell types and the density of the tested loci is increased. Finally, we show that the number of heritable loci depends on the window size parameter commonly used to identify candidate cis-acting single-nucleotide polymorphism variants.
INTRODUCTION
The identification of genetic markers that impact the phenotype of an individual is an important step towards identifying the genetic basis of disease. Replicated findings of such associations have become increasingly common (1). However, a formidable remaining challenge is finding the mechanisms through which these identified markers act to ultimately drive phenotypic variation. The epigenome is now recognized as playing a critical role in developmental processes and is also likely to be involved in ultimately determining phenotypic traits (2). For example, DNA methylation of CpG dinucleotides can exert regulatory influence on gene expression levels, which in turn can influence phenotype (3). Methylation levels vary between cell types (4), between individuals (5), and they are known to be influenced by both environmental and genetic factors (6). Importantly, methylation has been implicated in a wide range of diseases, including cancers, autism-spectrum disorders (2), as well as several auto-immune diseases (7). It, therefore, stands to reason that finding and characterizing the genetic determinants of methylation could yield insight into mechanisms of disease and the functional consequences of genetic variation.
Genetic sequence has been implicated as a determinant of DNA methylation in a number of contexts. Individuals who are heterozygous at a gene locus can exhibit allele-specific methylation that is dependent on DNA sequence and leads to differential gene expression patterns between the alleles (i.e. allele-specific gene expression) (8–11). Hellman and Chess (12) found that individuals who shared more parental chromosomes (i.e. are more related) tend to exhibit more similar methylation patterns. Single-nucleotide polymorphisms (SNPs) may also disrupt CpG dinucleotides (i.e. causing them to no longer be CpG), thereby preventing methylation there or at neighbouring loci (8). Several methylome-wide studies have identified individual SNPs that are correlated with specific methylation loci (13–15).
Despite these findings, the extent to which differences in stretches of cis-DNA sequence (i.e. multivariate SNP signal) explain differences in methylation of a given CpG dinucleotide between individuals, and, correspondingly, to what extent methylation is deemed heritable when estimated from SNP data from unrelated individuals, remains unclear. Recently, Bell et al. (14) used the differences in correlation between monozygotic and dizygotic twins to estimate the heritability of methylation in blood samples, finding a genome-wide mean heritability of 18%. Twin-based analyses are important in shedding light on an upper bound of heritability, but yield no information as to the mechanism of action underlying heritability, a critical piece of the story. Before this, several more focused studies have been conducted on examining the heritability of methylation in particular contexts, such as between cell divisions of cancer cells (16), for a particular gene (17) or for the major histocompatibility complex region in a twin study focused only on immune cells (18).
Herein, we identify ‘heritable methylation loci’—those loci for which cis-SNPs explain more of the phenotypic variance than expected by chance—within the human methylome, in four distinct brain regions across 150 unrelated individuals from publicly available data. Our goals were to investigate what role stretches of cis-DNA sequence plays in influencing methylation, what is an optimum definition of cis (i.e. locality) in this context, whether the additive effects of measured SNPs could explain the twin-based estimates of heritability previously reported and whether CpG dinucleotides with heritable methylation were more likely to be within or neighbouring particular classes of genes or genomic features.
MATERIALS AND METHODS
Individual SNP data and chromosomal coordinates were downloaded from dbGAP Study Accession phs000249.v1.p1. Normalized methylation levels across four brain regions [cerebellum (CRBLM), frontal cortex (FCTX), caudal pons (PONS) and temporal cortex (TCTX)] from 150 individuals were obtained from the Gene Expression Omnibus (GEO) database (accession GSE15745). This data profiled methylation levels of 27 578 CpG loci assayed using an Illumina HumanMethylation27 BeadChip. Methylation locus chromosome coordinates were obtained from GEO (GPL8490). SNP data for the same individuals were generated from tissue collected in the cerebellum brain region. All SNPs missing in >1% of the individuals, or those whose minor allele frequency was <0.01 were discarded. All individuals missing >5% of their SNP data were removed. Several methylation loci and individual samples were removed because of data quality concerns [see Supplementary Information of Gibbs et al. (13)].
Initially, we found that our estimates of heritability were significantly correlated with the number of SNPs within the methylation probe region. Thus, to avoid erroneously identifying methylation loci as heritable from such artefacts, we filtered out any methylation loci whose respective probe overlapped a SNP with minor allele frequency ≥0.05 (using the highest reported minor allele frequency from dbSNP, and the list of probe SNPs as provided by Illumina). This filter further removed 5816 methylation loci, leaving 21 000 methylation loci for our analysis. Individual covariate data were obtained from Supplementary Table S1 from Gibbs et al. (13) and converted to a 1-of-(M-1) encoding for discrete variables. Table 1 reports the final number of individuals and SNPs for each of the four brain regions.
Table 1.
Number of individuals and SNPs used in analyses for each of the four brain regions
| Region | Number of individuals | Number of SNPs |
|---|---|---|
| CRBLM | 106 | 495 788 |
| FCTX | 132 | 495 873 |
| PONS | 124 | 495 870 |
| TCTX | 125 | 495 866 |
Identification of heritable methylation loci
We used linear mixed models (LMMs) to assess the narrow-sense heritability of each methylation locus (19). Let the vector yi,t of length N represent the methylation levels of locus i at brain region t across N individuals. Using LMMs, we can decompose the variance associated with yi,t as the sum of a linear additive genetic (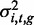 ) and residual (
) and residual (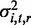 ) component,
) component,
| (1) |
where X is the N×Q matrix of Q individual covariates (gender, age, post-mortem interval, region source and methylation assay batch) and offset term, β is the Q×1 vector of covariate effects, I is the N×N identity matrix and K is the realized relationship matrix (RRM) (20) of size N×N. Note that K factors as 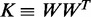 , where W of dimension N×s contains the s SNPs in our window local to the gene and that when s<N, parameter estimation and computation of the log likelihood becomes extremely efficient (21). We used the method of Lippert et al. (21) to compute restricted maximum likelihood estimates of
, where W of dimension N×s contains the s SNPs in our window local to the gene and that when s<N, parameter estimation and computation of the log likelihood becomes extremely efficient (21). We used the method of Lippert et al. (21) to compute restricted maximum likelihood estimates of 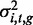 and
and 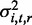 . Narrow-sense heritability for a particular methylation locus i in brain region t was then estimated as (19)
. Narrow-sense heritability for a particular methylation locus i in brain region t was then estimated as (19)
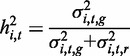 |
(2) |
To compute a P-value for whether a methylation locus, yi,t, was heritable—that is, to compute the significance of the genetic variance component in the model—we set 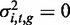 to obtain the null model, and then used a modified likelihood ratio test, which accounted for the fact that the parameter being tested was on the boundary of the allowed space in the null model (22,23). That is,
to obtain the null model, and then used a modified likelihood ratio test, which accounted for the fact that the parameter being tested was on the boundary of the allowed space in the null model (22,23). That is, 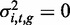 in the null model, and
in the null model, and 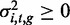 in the alternative model because it is a variance parameter. However, on checking the calibration of P-values by way of permutation tests, we discovered the P-values to be conservative, owing partly to the small sample size, but also to the approximation of the null distribution in this case [e.g. (24)], and thus used the permutation-based P-values instead (using 420 000 permutations of the individuals in the methylation data, and using the same permutations for each methylation locus). We defined a ‘heritable locus’ as one in which the P-value of association was smaller than a significance level of 0.05 after Bonferroni correction. Note that this test can be viewed as a test for association between the SNPs in the set and the phenotype in question, and it has been used in a similar manner in (25,26).
in the alternative model because it is a variance parameter. However, on checking the calibration of P-values by way of permutation tests, we discovered the P-values to be conservative, owing partly to the small sample size, but also to the approximation of the null distribution in this case [e.g. (24)], and thus used the permutation-based P-values instead (using 420 000 permutations of the individuals in the methylation data, and using the same permutations for each methylation locus). We defined a ‘heritable locus’ as one in which the P-value of association was smaller than a significance level of 0.05 after Bonferroni correction. Note that this test can be viewed as a test for association between the SNPs in the set and the phenotype in question, and it has been used in a similar manner in (25,26).
Determining an optimum cis window size
To find an optimal window size across all methylation loci for inclusion of cis-acting SNPs, we systematically varied the window size through 10 kb, 50 kb, 100 kb, 500 kb and 1 Mb, use of the entire chromosome that the locus fell on, and all SNPs assayed in the genome. We then deemed the optimum window to be the one yielding the largest number of heritable methylation loci, where we identified heritable loci by the permutation strategy previously described, but limiting the number of permutations to 10 000 for each window size for computational efficiency. Note the final set of heritable loci we report is based on the 420 000 permutations. We used a window that was symmetric around the methylation locus of interest. That is, we defined a window of size X kb centred at a methylation locus at position i as the DNA sequence within the region 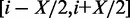 kb, inclusive. Closely related individuals can be problematic when estimating heritability (27) because of confounding owing to shared environmental factors. For example, Visscher and colleagues require removal of all individuals with RRM similarity >0.05 (27). In our data set, no two individuals were related this closely; thus, we did not filter any individuals by this criterion. Furthermore, a univariate scan of various methylation loci, randomly chosen, did not show significant deviation of the genomic control factor (28),
kb, inclusive. Closely related individuals can be problematic when estimating heritability (27) because of confounding owing to shared environmental factors. For example, Visscher and colleagues require removal of all individuals with RRM similarity >0.05 (27). In our data set, no two individuals were related this closely; thus, we did not filter any individuals by this criterion. Furthermore, a univariate scan of various methylation loci, randomly chosen, did not show significant deviation of the genomic control factor (28),  , from 1.0, suggesting that hidden confounders were not present in this data set.
, from 1.0, suggesting that hidden confounders were not present in this data set.
When scanning cis window sizes, we restricted our comparison with the 15 179 methylation loci for which we could find at least one SNP within each of the window sizes considered.
Assigning methylation loci to gene sets
We first assigned methylation loci to genes, based on proximity, and then assigned genes to gene sets. Methylation loci were assigned to their closest neighbouring genes as reported by the Illumina HumanMethylation27 BeadChip annotation files. Next, we associated genes to gene sets, considering all genes that were associated with at least one methylation locus under study. Gene sets were obtained from the Gene Ontology (GO) (29), which yields gene sets organized by biological process, from the Molecular Signatures Database (MSigDB) (30) that defines canonical biological pathways and from the Pharmacogenomics Knowledgebase (PharmGKB) (31) that defines known pathway targets of drugs. GO annotations for humans were obtained from GO on 14 May 2012. We tested the 2464 GO sets for which there were between 20 and 500 member genes, inclusive. Canonical pathway definitions from MSigDB version 3.0 were used, totalling 880 gene sets. All pathways for which at least one drug was known to target it were downloaded from PharmGKB on 21 July 2012, totalling 263 gene sets. In total, there were 3607 sets tested.
Computing correlation of heritable loci with open chromatin regions and known regulatory elements
We explored whether heritable loci were enriched for loci lying in open chromatin regions or in known regulatory elements. To do so, we used Fisher’s exact test (FET) (32), using our results of which loci were deemed heritable, in conjunction with external data sources which could be used to annotate the loci. In particular, we obtained open chromatin regions from data published by the encyclopedia of DNA elements (ENCODE) Project Consortium (33). Briefly, the University of North Carolina at Chapel Hill has collected formaldehyde-assisted isolation of regulatory elements (FAIRE) evidence of open chromatin and has made this data available through the University of California, Santa Cruz Genome Browser (34), from which we obtained it on 19 November 2012. In particular, we obtained all 273 110 open chromatin annotations for normal human astrocyte cells (cell type NH-A), the only cell type relevant to brain tissue that was available at this time, and used the LiftOver tool to map the coordinates to build hg18. For computing the overlap of heritable loci with known regulatory elements, we used publicly available data obtained from the ORegAnno database containing 23 206 known regulatory elements (35) downloaded on 19 November 2012. The majority (17 744 of 23 206, or 76%) of regulatory elements stored in ORegAnno are binding sites of CTCF; therefore, we restricted the regulatory elements to CTCF sites only. Determination of overlap between methylation loci and genomic annotations was computed using the BEDTools software (36).
Gene set enrichment testing
We performed gene set enrichment testing using FET, which tests whether the proportion of heritable methylation loci belonging to a gene set is larger than that expected by chance. We hypothesized that the FET P-values may be inaccurate because FET treats loci as independent and, therefore, does not account for correlated loci (2). Thus, we computed permutation-based P-values (using permutations of individuals) for the FET and found that the closed-form FET P-values were inflated. Consequently, we used the permutation-based P-values, from 200 000 permutations of the individuals, calling those with Bonferroni-corrected P-values <0.05 as significant.
Identification of genes preferentially expressed in brain regions using the same individuals
To identify genes that were preferentially expressed in each brain region (those expressed more highly in that region as compared with other regions), we used the matching gene expression data from our publicly available data set (GEO accession GSE15745). For this analysis, only individuals for whom all four brain regions were profiled (and were done so within the same batch) were kept, leaving 122 individuals. For each probe and each individual, the ranks of the probe intensities across the four brain regions were computed. Then, for each brain region and each probe, the ranks across all individuals were summed, resulting in a matrix of Rx4 summations, one for each of the four brain regions and each of the R probes. By the central limit theorem, each summation of ranks is normally distributed with mean 305.0 and variance 203.333, as there are 122 terms in each sum, and each term is sampled from a distribution with mean 2.5 and variance 1.667, assuming all ranks (1, 2, 3, 4 because we have four tissues) are equally likely. Probes were then mapped to genes using the Illumina probeset information file, and only those genes assayed by exactly one probe were retained. Finally, using FET, we measured correlation between whether a methylation locus was heritable and whether the gene associated with the locus was preferentially expressed in the relevant brain region. Only the set of genes both profiled in the expression data, and linked to a methylation locus, were considered.
Identification of genes preferentially expressed in brain regions using independent data
To identify genes that were highly expressed in brain tissue in general, we downloaded the raw gene expression profiles collected by Su et al. (37) for multiple cell types from GEO accession GSE1133. We used the robust multi-array average algorithm in Bioconductor (38) with R version 2.15.1 to both pre-process the array data and map probes to gene Entrez ID (39) using an updated annotation file hgu133ahsrefseqcdf_15.1.0. We kept only samples of normal tissues and cell types, leaving 73 samples profiled in duplicate. We then performed a one-sided Wilcoxon rank sum test to identify preferential expression in brain cell types relative to all other profiled cell types. Similarly to the previous section, FET was used to look for associations between a methylation locus being heritable, and whether the gene associated with that locus was preferentially expressed.
RESULTS
The number of heritable loci depended on the window size for defining cis-acting SNPs
To find an optimal window size across all methylation loci for inclusion of cis-acting SNPs, we centred a window symmetrically around each methylation locus, extending the size of this window through 10 kb, 50 kb, 100 kb, 500 kb and 1 Mb, and we also tried the entire local chromosome, as well as the entire genome. We then deemed the optimum window to be the one yielding the largest number of heritable methylation loci among those loci which had at least one SNP for every window size. As shown in Figure 1a, a window size of 50 kb led to the highest number of heritable methylation loci. After more permutations to obtain more accurate P-values (see ‘Materials and Methods’ section), we found 654, 812, 600 and 636 heritable methylation loci for FCTX, TCTX, PONS and CRBLM, respectively. Although the number of heritable loci is similar for both the 50- and 100 kb windows, it is clear that using too large of a window (e.g. the entire genome), or too small of a window (e.g. ≤10 kb), dramatically reduced the number of heritable loci.
Figure 1.

Number of heritable methylation loci in the four brain regions: TCTX, FCTX, CRBLM and PONS. (a) Number of methylation loci passing a Bonferroni-corrected P-value threshold of 0.05, as a function of DNA sequence window size, when using only methylation loci analysed for all window sizes (so as to make them comparable). (b) Histogram of the number of SNPs found within the 50 kb window of each of the 21 000 methylation loci.
We believe that our loss of power to detect heritable loci when the window size was extended beyond 50 kb is related to the loss of power we observed when using LMMs to correct for confounding variables in genome-wide association studies (40,41), although we now have a better understanding of this effect (http://research.microsoft.com/apps/pubs/default.aspx?id=178646). In particular, in the present context, most SNPs influencing a methylation locus are expected to be physically near to the locus (i.e. are cis-acting); therefore they can be captured by a relatively small window such as the 50 kb window we identified in Figure 1a. Below this window size, many influential SNPs are likely to be missed, causing a downwards bias in the estimate of  and, therefore, of heritability. With increasing window sizes, more and more extraneous SNPs are included in the RRM, causing an increase in the variance of the estimate of heritability. This bias-variance trade-off is perhaps best understood in light of the fact that an LMM with no fixed effects, using genetic similarities constructed from a set of SNPs, is equivalent to a form of linear regression of those SNPs on the phenotype. Thus, using extraneous SNPs in the estimation of the RRM is equivalent to using them as additional covariates in this form of linear regression, which increases the variance of the estimate of
and, therefore, of heritability. With increasing window sizes, more and more extraneous SNPs are included in the RRM, causing an increase in the variance of the estimate of heritability. This bias-variance trade-off is perhaps best understood in light of the fact that an LMM with no fixed effects, using genetic similarities constructed from a set of SNPs, is equivalent to a form of linear regression of those SNPs on the phenotype. Thus, using extraneous SNPs in the estimation of the RRM is equivalent to using them as additional covariates in this form of linear regression, which increases the variance of the estimate of  , diminishing our power to detect heritable loci (
, diminishing our power to detect heritable loci ( ). Therefore, in our analysis, as we included more and more SNPs up to and including a window which contained most influential SNPs (i.e. the 50 kb window), the downwards bias on heritability decreased (and the estimate of heritability increased). As we went beyond this optimal window size, an increasing proportion of extraneous SNPs were included in the RRM, up until the point where the variance of the estimate of heritability almost completely diminished our power to detect significantly heritable loci. This bias-variance effect would be mitigated by a larger sample size.
). Therefore, in our analysis, as we included more and more SNPs up to and including a window which contained most influential SNPs (i.e. the 50 kb window), the downwards bias on heritability decreased (and the estimate of heritability increased). As we went beyond this optimal window size, an increasing proportion of extraneous SNPs were included in the RRM, up until the point where the variance of the estimate of heritability almost completely diminished our power to detect significantly heritable loci. This bias-variance effect would be mitigated by a larger sample size.
Figure 1b illustrates the number of SNPs included in the local sequence window, for all methylation loci, at the selected optimal 50 kb window size. Our locality result is similar to that found by Price et al. (42), where it was found that heritability of gene expression was primarily because of SNPs at cis loci. In the univariate SNP-methylation association analysis of Bell et al. (14), they examined SNPs within 100 kb, but found that most associations were actually within a few kilobases, whereas Gibbs et al. (13) reported finding a peak at 45 kb. However, as noted in (19), use of a stringent, multiple-testing correction to select significantly associated univariate SNPs, as done in these two studies, is likely to miss much of the weaker signal that the LMM can capture. Thus, it is not surprising that our analysis finds an optimal local window which is slightly larger than what one might have speculated from stringent univariate analyses.
We also found that heritable methylation loci tended to have larger number of SNPs within their windows than non-heritable loci: for CRBLM, the median number of SNPs in the 50 kb window was nine versus seven, whereas for all other tissues, the median number was eight versus seven (all P < 10−8, Wilcoxon rank-sum test). This result suggests that with more SNPs, there is more power to uncover heritable methylation loci. The number of SNPs in each window are provided in Supplementary Table S1.
Figure 2 illustrates the distribution of estimated narrow-sense heritability over all 21 000 methylation loci for all four regions (region-specific distributions were similar to one another). The mean estimated heritability of all methylation loci deemed heritable (aggregated across all four brain regions) was 29.9%, indicating the extent to which local sequence alone can account for variation in methylation at those loci. Across all loci (including those not deemed heritable), the mean estimated heritability was 2.8%.
Figure 2.

Narrow-sense heritability estimates over all methylation loci in all four brain regions. Loci were divided based on whether they were located in (a) CpG islands (15 469 loci) or (b) not in CpG islands (5531 loci). Within each plot, loci are then further grouped based on whether they were identified as heritable. The smoothed histograms were constructed using density estimation with a Gaussian kernel with default parameters in R, and the y-axis is scaled to a maximum of 1. The number of individuals used in this analysis is reported in Table 1.
Concordance of heritable loci across brain regions and with eQTL and methQTL
We next assessed the extent to which heritable methylation loci were shared across regions when using the 50 kb window size. We found that 181 loci were heritable across all four regions with mean estimated heritability of 41.4%, whereas 207 loci were heritable across at least three regions (Figure 3a). The estimated narrow-sense heritability shows generally good agreement among FCTX, TCTX and PONS (Figure 3b). Supplementary Table S2 reports the list of all methylation loci, their estimated heritability and the significance of association with their 50 kb cis-sequence window.
Figure 3.

Concordance of heritable loci across the four brain regions and with eQTL and mQTL from Gibbs et al. (a) Number of heritable loci found to be overlapping in each of the four regions, using the 50 kb window size. (b) Correlation of the estimated narrow-sense heritability for each methylation locus, between tissue regions, using only the 1451 loci that were significant in at least one region. (c) Breakdown of heritable methylation loci according to whether a locus was also found to have at least one cis-mQTL in the Gibbs study—‘common’ refers to a locus we identified as heritable and for which Gibbs found at least one mQTL; ‘Quon-specific’ means the locus was found to be heritable but did not have an mQTL in the Gibbs study; and ‘Gibbs-specific’ means Gibbs et al. found at least one mQTL for a locus that we did not find to have heritable methylation. (d) Percentage of heritable loci for which at least one eQTL was reported by Gibbs et al. for the gene nearest to the heritable methylation locus (and where the eQTL was within the 50 kb window of the heritable locus), as compared with the number for all methylation loci. An asterisk indicates significant to a threshold of 0.05, as determined by a FET.
We compared the set of heritable loci to the set of methylation loci identified by Gibbs et al. as being associated with at least one cis-methylation quantitative trait locus (methQTL). We found that on average, 43% of each tissue’s set of heritable loci was identified as being associated with at least one cis-methQTL in the Gibbs study, indicating that we identified overlapping but distinct loci from that of Gibbs et al. (Figure 3c). We also identified on average 54% more methylation loci (with cis association) than did Gibbs et al. in their univariate scan. Note that their multiple-testing burden was larger because they also looked for trans-methQTLs. We next cross-referenced our list of heritable loci with the expression quantitative trait loci (eQTLs) reported by Gibbs et al. (first restricting the set of eQTLs to those within the 50 kb window of the 21 000 methylation loci and whose target gene is the same gene as the one we assigned to the respective methylation locus). We observed that in three of the four tissues (all but PONS), the heritable methylation loci were enriched for genomic regions containing cis-eQTLs [Figure 3d; P = 1.05 × 10−3 (FCTX), P = 3.16 × 10−3 (TCTX), P = 0.076 (PONS), P = 0.0202 (CRBLM); FET].
To explore the relationship between heritable methylation loci in each of the four brain regions and levels of gene expression in these brain regions, we again used the expression data corresponding to our samples, now to identify genes preferentially expressed in each region—genes expressed higher in that region than in others (see ‘Materials and Methods’ section). We found that the genes assigned to heritable loci identified in the frontal cortex and cerebellum brain regions were significantly depleted in genes preferentially expressed in that region [P = 0.024 (FCTX), P = 6.20 × 10−5 (CRBLM), P = 0.55 (TCTX), P = 0.90 (PONS), FET].
For a more general investigation of heritable loci and brain-specific expression, we obtained genome-wide expression profiles for 73 different cell types so as to identify those genes preferentially expressed in the brain compared with all other tissues. We found that heritable loci identified in three of the four brain regions (frontal cortex, temporal cortex and cerebellum) were significantly depleted near genes expressed more highly in the brain compared with other tissues [P = 4.64 × 10−4 (FCTX), P = 4.44 × 10−3 (TCTX), P = 2.32 × 10−4 (CRBLM), P = 0.19 (PONS), FET]. These results suggest that heritable loci are not regulating genes highly expressed in either brain-specific regions or whole-brain tissue, both of whom may be critical to brain function.
Heritable methylation loci were enriched for genomic locations containing regulatory elements
To assess the potential role of heritable methylation loci in gene regulation, we checked to see whether our heritable loci lay in regions previously annotated with genomic features that are indicative of gene regulatory elements. Using data from the ENCODE project (see ‘Materials and Methods’ section), we found that the heritable loci for all four brain regions were enriched in open chromatin regions [P = 9.43 × 10−3 (CRBLM), P = 0.02 (PONS), P = 0.0122 (FCTX), P = 0.018 (TCTX) FET]. Furthermore, when comparing our heritable loci with known CTCF binding sites [by way of ORegAnno (35), see ‘Materials and Methods’ section] we also found significant enrichment for overlap between the heritable loci and these regulatory elements [P = 0.031 (CRBLM), P = 0.035 (PONS), P = 0.035 (FCTX), P = 0.027 (TCTX), FET]. CTCF is implicated in both diverse genomic regulatory functions (activation, repression, insulation) and the global organization of chromatin architecture (43). Furthermore, DNA methylation of CTCF’s binding site is the best understood mechanism for modulating CTCF binding (43). As an example, methylation of CpG loci within the CTCF binding site eliminates binding of CTCF in vivo and has been demonstrated to disrupt its regulatory activity at the methylated binding site (44). These results suggest that heritable loci may play a regulatory role in the expression of neighbouring genes by modulating binding and activity of regulators, such as CTCF.
We also investigated whether those methylation loci found to be heritable favoured any particular position relative to the nearest transcription start site (TSS). We found the heritable methylation loci for each brain region were enriched for loci lying outside of CpG-islands (all P < 1.84 × 10−4, FET). Furthermore, as illustrated in Figure 4, we found that the heritable loci in the PONS tissue region were preferentially located downstream of the TSS relative to other methylation loci (median position relative to TSS was 72 versus −2 bp, P = 2.6 × 10−3, Wilcoxon rank-sum test); we did not find similar preferences for the other three tissue regions (all other P > 0.57). Heritable loci located much farther downstream from the TSS indicate possible genetic influence over alternative splicing events (45).
Figure 4.
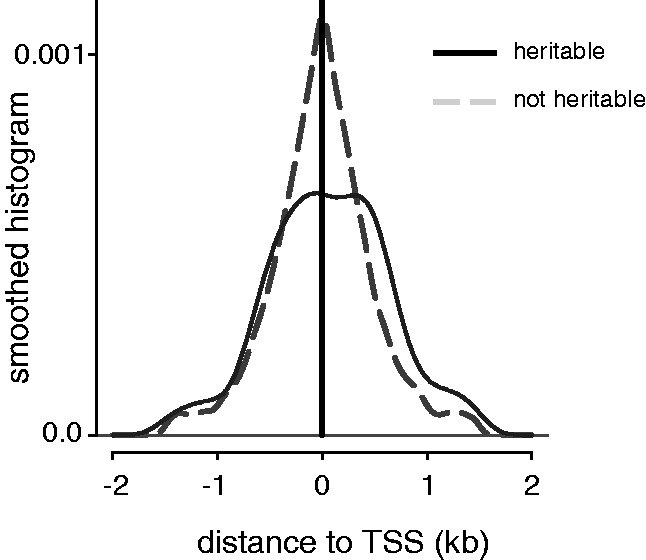
Relative position of heritable and non-heritable loci identified in the PONS tissue region with respect to the TSS of the gene to which they were closest. The x-axis has been thresholded at a distance of 2 kb.
Genes proximal to heritable methylation loci are involved in a variety of processes
One of the primary roles of DNA methylation is to control gene expression of particular genes. We next identified whether heritable loci seemed to be controlling any specific classes of genes. To do so, we first assigned methylation loci to genes, based on proximity (see ‘Materials and Methods’ section). We then performed a gene set enrichment analysis on all genes assigned to heritable methylation loci, using 3607 gene sets from the Gene Ontology (GO) Process hierarchy, canonical biological pathways from the Molecular Signatures Database (MSigDB) and drug-targeted pathways from the Pharmacogenomics Knowledgebase (PharmGKB). Supplementary Table S3 shows which methylation loci are assigned to which gene sets.
Among the gene sets found significant (Figure 5), two involved neurotransmitters (agmatine and dopamine), another involved neurotransmitter transporters (SLC transporters) and another involved nicotinamide salvaging (an anti-inflammatory pathway), suggesting candidate epigenetic mechanisms through which genotype may play an important role in drug efficacy. Other gene sets associated with heritable methylation loci involved regulation of energy production and the immune system. Supplementary Table S4 reports the results of the enrichment analysis on all categories tested. Enrichment tests were performed using only genes that were assigned to at least one of the 21 000 methylation loci assayed. This ‘background’ set of genes was not itself significantly enriched for any specific brain functions, although it was enriched for 55 GO categories (of the 2464 tested) across a variety of processes (Supplementary Table S5).
Figure 5.

Gene set enrichment of the heritable loci in each of the four brain regions. (a) A black rectangle indicates significant enrichment ( ) for the specified set and brain region combinations. (b) Network illustration of gene sets found to be enriched for heritable loci. Each node represents one gene set, whereas each edge represents an overlap of at least one methylation locus between the two gene sets. The size of each node is proportional to the number of methylation loci assigned to the respective gene set, and the width of each edge is proportional to the gene set coherence, defined as the number of loci in the overlap divided by the smaller size of the two gene sets. The legend depicts the minimum and maximum node sizes, as well as the edge width corresponding to the minimum gene set coherence (0.05) and maximum gene set coherence (1.0).
) for the specified set and brain region combinations. (b) Network illustration of gene sets found to be enriched for heritable loci. Each node represents one gene set, whereas each edge represents an overlap of at least one methylation locus between the two gene sets. The size of each node is proportional to the number of methylation loci assigned to the respective gene set, and the width of each edge is proportional to the gene set coherence, defined as the number of loci in the overlap divided by the smaller size of the two gene sets. The legend depicts the minimum and maximum node sizes, as well as the edge width corresponding to the minimum gene set coherence (0.05) and maximum gene set coherence (1.0).
DISCUSSION
Epigenetic mechanisms, such as DNA methylation, play a critical role in controlling the gene expression programme of cells, which in turn is thought to have significant impact on phenotype (3). Epigenetic markers, therefore, represent a potential mechanism through which genetic variation can affect phenotype. Herein, we examined how cis-DNA sequence influences methylation across the human genome in four phenotypically normal brain regions from unrelated individuals. We found that between 3 and 4% of the tested loci were heritable with respect to an empirically selected optimal cis DNA window of size 50 kb. Furthermore, the heritable loci were shown to be enriched in open chromatin regions, and also enriched in locations of known binding sites of CTCF, suggesting a functional role for at least some of these heritable loci in disrupting or modulating binding of transcription factors, such as CTCF. Also, genes associated with heritable loci in some of the brain regions were enriched in several pathways, including those involved in neurotransmitter processing, regulation of energy production and the immune system. None of the enriched gene sets are clearly brain region specific, suggesting the heritable loci we identified may be heritable in a wide range of tissues rather than brain-specific.
The number of heritable methylation loci depended on how large of a window of SNPs was considered local. We found that a window size of 50 kb was optimum in achieving a maximal number of heritable loci across all regions, and informs on a window in which the mechanistic action through which SNPs alter CpG methylation could be investigated. As the window size was extended beyond an optimal range, we hypothesize that the variance in the estimate of heritability became extremely high (especially with such a small cohort), and, therefore, that our ability to detect significance was diminished (http://research.microsoft.com/apps/pubs/?id=178646).
Our estimates of heritability were less than that reported in the twin-based study of Bell et al. (14), who found a mean genome-wide heritability of 18% from blood samples, as compared with our 3%. In Gervin et al. (18), heritability of the major histocompatibility complex region in cultured lymphocyte cells was investigated using a twin-based approach and was found to be low (2–16%). The discrepancy between our estimates and the twin-based estimates could be explained by unmeasured SNPs (46), the cohort or tissue in which measurements were performed, the upwards bias of twin-based studies (47–49) and limited sample size. Further studies should shed more light on this issue.
There are a number of reasons to suspect that the fraction of CpG dinucleotides whose methylation status is heritable is larger than what we have reported here. First, our study only included individuals with phenotypically healthy brains, and we expect that analysis of a wider range of tissues may uncover genetic dependencies that are tissue or condition specific. Second, our Bonferroni correction of P-values is likely ignoring weakly heritable loci. Third, use of more dense SNP and methylation assays will allow for a more refined exploration of the genetic basis of methylation. Finally, if heritable loci were tissue specific, we would lose power to detect them when analysing mixed tissues as we have here.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–5.
FUNDING
Microsoft Research. Funding for open access charge: Microsoft Research.
Conflict of interest statement. J.L., C.L. and D. H. own stock in Microsoft.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Jonathan Carlson and Carl Kadie for help with tools to manage and analyse the data, Jim Jernigan and the MSR HPC team for cluster support and anonymous reviewers for their suggestions. Division of Aging Biology and the Division of Geriatrics and Clinical Gerontology, NIA; Intramural Research Program, NIA.
REFERENCES
- 1.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 4.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 5.Rakyan VK, Hildmann T, Novik KL, Lewin J, Tost J, Cox AV, Andrews TD, Howe KL, Otto T, Olek A, et al. DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2004;2:e405. doi: 10.1371/journal.pbio.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 7.Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2011;7:263–271. doi: 10.1038/nrrheum.2011.16. [DOI] [PubMed] [Google Scholar]
- 8.Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20:883–889. doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008;40:904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Rohde C, Reinhardt R, Voelcker-Rehage C, Jeltsch A. Non-imprinted allele-specific DNA methylation on human autosomes. Genome Biol. 2009;10:R138. doi: 10.1186/gb-2009-10-12-r138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, Kucera KS, Willard HF, Myers RM. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7:e1002228. doi: 10.1371/journal.pgen.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellman A, Chess A. Extensive sequence-influenced DNA methylation polymorphism in the human genome. Epigenetics Chromatin. 2010;3:11. doi: 10.1186/1756-8935-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibbs JR, Van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629. doi: 10.1371/journal.pgen.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teng M, Balch C, Liu Y, Li M, Huang TH, Wang Y, Nephew KP, Li L. The influence of cis-regulatory elements on DNA methylation fidelity. PLoS One. 2012;7:e32928. doi: 10.1371/journal.pone.0032928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breton CV, Salam MT, Gilliland FD. Heritability and role for the environment in DNA methylation in AXL receptor tyrosine kinase. Epigenetics. 2011;6:895–898. doi: 10.4161/epi.6.7.15768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gervin K, Hammerø M, Akselsen HE, Moe R, Nygård H, Brandt I, Gjessing HK, Harris JR, Undlien DE, Lyle R. Extensive variation and low heritability of DNA methylation identified in a twin study. Genome Res. 2011;21:1813–1821. doi: 10.1101/gr.119685.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayes BJ, Visscher PM, Goddard ME. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 2009;91:47–60. doi: 10.1017/S0016672308009981. [DOI] [PubMed] [Google Scholar]
- 21.Lippert C, Listgarten J, Liu Y, Kadie CM, Davidson RI, Heckerman D. FaST linear mixed models for genome-wide association studies. Nat. Methods. 2011;8:833–835. doi: 10.1038/nmeth.1681. [DOI] [PubMed] [Google Scholar]
- 22.Self SG, Liang KY. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J. Am. Stat. Assoc. 1987;82:605–610. [Google Scholar]
- 23.Dominicus A, Skrondal A, Gjessing HK, Pedersen NL, Palmgren J. Likelihood ratio tests in behavioral genetics: problems and solutions. Behav. Genet. 2006;36:331–40. doi: 10.1007/s10519-005-9034-7. [DOI] [PubMed] [Google Scholar]
- 24.Pinheiro J, Bates D. Mixed-Effects Models in S and S-PLUS. New York: Springer-Verlag; 2000. [Google Scholar]
- 25.Listgarten J, Lippert C, Heckerman D. An efficient group test for genetic markers that handles confounding. 2012 doi: 10.1093/bioinformatics/btt177. ArXiv:1205.0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 29.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE. From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark. Med. 2011;5:795–806. doi: 10.2217/bmm.11.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeeberg B, Feng W, Wang G, Wang M, Fojo A, Sunshine M, Narasimhan S, Kane D, Reinhold W, Lababidi S, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The ENCODE Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffith OL, Montgomery SB, Bernier B, Chu B, Kasaian K, Aerts S, Mahony S, Sleumer MC, Bilenky M, Haeussler M, et al. ORegAnno: an open-access community-driven resource for regulatory annotation. Nucleic Acids Res. 2008;36:D107–D113. doi: 10.1093/nar/gkm967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Listgarten J, Lippert C, Kadie CM, Davidson RI, Eskin E, Heckerman D. Improved linear mixed models for genome-wide association studies. Nat. Methods. 2012;9:525–526. doi: 10.1038/nmeth.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Listgarten J, Lippert C, Heckerman D. Fast-LMM-Select for confounding from spatial structure and rare variants. Nat. Genet. 2012 doi: 10.1038/ng.2620. in press. [DOI] [PubMed] [Google Scholar]
- 42.Price AL, Helgason A, Thorleifsson G, McCarroll SA, Kong A, Stefansson K. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genet. 2011;7:e1001317. doi: 10.1371/journal.pgen.1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 45.Kwan T, Benovoy D, Dias C, Gurd S, Serre D, Zuzan H, Clark TA, Schweitzer A, Staples MK, Wang H, et al. Heritability of alternative splicing in the human genome. Genome Res. 2007;17:1210–1218. doi: 10.1101/gr.6281007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era—concepts and misconceptions. Nat. Rev. Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- 47.Benjamin DJ, Cesarini D, Van der Loos MJ, Dawes CT, Koellinger PD, Magnusson PK, Chabris CF, Conley D, Laibson D, Johannesson M, et al. The genetic architecture of economic and political preferences. Proc. Natl Acad. Sci. USA. 2012;109:8026–8031. doi: 10.1073/pnas.1120666109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charney E. Genes and ideologies. Perspect. Polit. 2008;6:299–319. [Google Scholar]
- 49.Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: genetic interactions create phantom heritability. Proc. Natl Acad. Sci. USA. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.